

Abstract
CRISPR-Cas9 is the state-of-the-art technology for editing and manipulating nucleic acids. However, the occurrence of off-target mutations can limit its applicability. Here, all-atom enhanced molecular dynamics (MD) simulations—using Gaussian accelerated MD (GaMD)—are used to decipher the mechanism of off-target binding at the molecular level. GaMD reveals that base pair mismatches in the target DNA at distal sites with respect to the protospacer adjacent motif (PAM) can induce an extended opening of the RNA:DNA heteroduplex, which leads to newly formed interactions between the unwound DNA and the L2 loop of the catalytic HNH domain. These conserved interactions constitute a “lock” effectively decreasing the conformational freedom of the HNH domain and hampering its activation for cleavage. Remarkably, depending on their positions at PAM distal sites, DNA mismatches responsible for off-target cleavages are unable to “lock” the HNH domain, thereby leading to the unselective cleavage of DNA sequences. In consistency with the available experimental data, the ability to “lock” the catalytic HNH domain in an inactive “conformational checkpoint” is shown to be a key determinant in the onset of off-target effects. This mechanistic rationale contributes in clarifying a long lasting open issue in the CRISPR-Cas9 function and poses the foundation for designing novel and more specific Cas9 variants, which could be obtained by magnifying the “locking” interactions between HNH and the target DNA in the presence of any incorrect off-target sequence, thus preventing undesired cleavages.
Short abstract
Accelerated molecular dynamics simulations reveal the molecular basis of off-target effects in CRISPR-Cas9, disclosing a locking mechanism that can be a target of rational engineering.
Introduction
CRISPR-Cas9 (CRISPR, clustered regularly interspaced short palindromic repeats) is an adaptive immune system found in bacteria and archaea conferring protection from foreign DNA.1 By enabling deletion, insertion, or correction of DNA at specific targeted sites within an organism’s genome, CRISPR-Cas9 is used as a genome editing technology holding enormous promises for medical, pharmaceutical, and (bio)technological applications, while also being of invaluable impact for fundamental research.1,2 The CRISPR-Cas9 technology is based on a single protein—the endonuclease Cas9—programmed with single guide RNAs (sgRNAs) to site-specifically target any desired DNA sequence. The presence of a short sequence (i.e., a protospacer adjacent motif, PAM) in close proximity to the cleavage site enables recognition of the desired DNA sequence across the genome and allows programmable applications. Upon PAM recognition, the DNA binds Cas9 by base pairing the RNA guide with one strand (the target strand, TS) and forming an RNA:DNA hybrid, while the nontarget DNA strand (NTS) is unwound and also accommodated within the protein complex. Structures of the CRISPR-Cas9 complex indicate a recognition lobe, which mediates the binding of the nucleic acids through three REC1–3 regions, flanked by a PAM interacting (PI) domain and two nuclease domains, HNH and RuvC, which cleave the TS and NTS, respectively (Figure 1A).
Figure 1.

(A) Crystal structure of the S. pyogenes CRISPR-Cas9 system, including the endonuclease Cas9, a guide RNA (orange), the target DNA (TS, cyan), and nontarget DNA (NTS, violet) strands.26 Cas9 is shown in molecular surface, with protein domains in different colors. The X-ray structure captures the inactive state of the HNH domain, which is a “conformational checkpoint” between DNA binding and cleavage. The right panel highlights the PAM distal sites on the RNA:DNA hybrid and the conformational change of the HNH domain required for catalysis, which is shown with an arrow, indicating the movement of catalytic H840 toward the cleavage site on the TS. (B) Diagram of the DNA and RNA filaments in CRISPR-Cas9, showing the location of base pair mismatches (mm) associated with off-target effects, at PAM distal sites. The model systems considered for Gaussian accelerated MD (GaMD) simulations include the on-target DNA sequence (on-target) and DNA sequences containing base pair mismatches at PAM distal sites (i.e., mm@20, mm@19–20, mm@18–20, and mm@17–20).
In spite of the remarkable advantages of the CRISPR-based genome editing technology with respect to traditional therapies, safety and efficacy issues have to be fully addressed prior to clinical applications. The most severe issues limiting the applicability of CRISPR-Cas9 in vivo are the off-target DNA cleavages, which produce mutations at sites in the genome other than the desired target site, causing unwanted phenotypes.3
In this respect, a promising strategy to fight off-target cleavages is the molecular engineering of highly specific Cas9 proteins,4−6 which would enable safer and easy-to-use applications of the CRISPR technology on a genome-wide scale.7 However, rational design of CRISPR-Cas9 requires detailed knowledge of the molecular bases underlying off-target effects, which are not yet understood.8,9 At the molecular level, off-target effects are the unselective cleavages of DNA sequences that do not fully match the guide RNA, bearing base pair mismatches within the DNA:RNA hybrid (Figure 1). Extensive biophysical experiments have been performed to understand the molecular basis of off-target binding.5,10−13 Kinetic and single molecule (sm) Förster resonance energy transfer (FRET) experiments have shown that the occurrence of off-target cleavages is directly related to the conformational state adopted by the catalytic HNH domain.5,10 Upon DNA binding, HNH undergoes a conformational change from an inactive state, in which the catalytic H840 is far away from the cleavage site on the TS, to an activated state that is prone for catalysis (Figure 1A). The inactive state of the HNH domain has been identified as a “conformational checkpoint” between DNA binding and cleavage, in which the RNA:DNA complementarity is recognized before the HNH domain assumes an activated conformation.10 sm FRET experiments have shown that the presence of DNA mismatches at the PAM distal ends of the DNA:RNA hybrid can trap the HNH domain in the “conformational checkpoint” state, whose population increases by augmenting the number of mismatches at PAM distal sites. Early experimental characterizations also revealed that the presence of base pair mismatches at PAM distal ends leads to the formation of a stable CRISPR-Cas9 complex.13 In this scenario, however, it is unknown how the presence of DNA mismatches at these sites can favor the inactivation of HNH. Detailed molecular knowledge of this mechanism is of major importance for developing more specific Cas9 variants, in which a single base pair mismatch is sufficient for trapping HNH in the inactive state, thus preventing the cleavage of any incorrect DNA sequence.
Here, we make use of extensive molecular dynamics (MD) simulations to characterize at the atomic level the molecular determinants of off-target binding, providing critical insights on the mechanism of off-target effects in CRISPR-Cas9. MD simulations have been shown to be a valuable tool for understanding the molecular basis of the CRISPR-Cas9 function.14−18 Among other studies, our group has successfully applied MD simulations to disclose a mechanism for the conformational activation of Cas9, clarifying the activation process by which the HNH domain is repositioned for cleavage, in good agreement with structural and sm FRET experiments.19 Remarkably, MD simulations as well as experimental studies have indicated that the conformational changes underlying the CRISPR-Cas9 function occur over and beyond micro-to-millisecond time scales.15,20 Such long time scales require the use of computational methods that enable enhanced sampling of the configurational space, such as accelerated MD (aMD) simulations.21 The aMD method adds a boost potential to certain regions of the potential energy surface, effectively decreasing the energy barriers and thus accelerating transitions between low-energy states. In this way, aMD simulations can capture biological processes occurring over milliseconds (and in some cases beyond), allowing the study of complex conformational transitions in folded or unstructured systems.22 Recent advances have led to the development of a robust aMD methodology—i.e., Gaussian accelerated MD (GaMD)23—that extends the use of aMD to larger and more complex biological systems. Besides enabling the description of the activation mechanism of the Cas9 protein,19 GaMD simulations aided to the disclosure of a catalytically active CRISPR-Cas9 complex.14 As well, GaMD has been used to determine ligand binding in G-protein-coupled receptors24 and, remarkably, the mechanism of a G-protein mimetic nanobody binding of a medically important GPCR with intracellular signaling proteins.25
By using GaMD we investigate the mechanistic basis of binding of off-target sequences at PAM distal sites. GaMD simulations reveal that the presence of base pair mismatches at specific PAM distal sites of the RNA:DNA hybrid can reduce the conformational mobility of the HNH domain. Indeed, depending on the positions of base pair mismatches at PAM distal sites, newly formed interactions are shown to “lock” the catalytic HNH domain in an inactive “conformational checkpoint”, preventing its activation toward DNA cleavages. In consistency with the available experimental data, the ability to “lock” the catalytic HNH domain in the “conformational checkpoint” is shown to be a key determinant in the onset of off-target effects. Overall, this study characterizes at the atomic level the molecular features of off-target binding in CRISPR-Cas9. This information poses the foundations for future biophysical investigations and structure-based design of the system toward improved genome editing.
Results
To determine how off-target sequences affect the conformational activation of the HNH domain, we performed all-atom GaMD simulations of the Cas9 protein in the “conformational checkpoint” state of the HNH domain (i.e., PDB 4UN3).26 GaMD simulations have already been used to successfully describe the activation process of the HNH domain19 and are therefore ideal to characterize the effects caused by base pair mismatches in the conformational dynamics of CRISPR-Cas9 and its HNH domain. For this purpose, the CRISPR-Cas9 complex has been simulated in complex with the fully matched RNA:DNA hybrid (considered the reference on-target system) and in the presence of base pair mismatches at the PAM distal ends of the RNA:DNA hybrid (Figure 1B). Specifically, we introduced 1–4 mismatches (mm) at PAM distal sites (i.e., at positions 20–16 of the RNA:DNA hybrid), resulting in the following models: mm@20, mm@19–20, mm@18–20, mm@17–20. These systems are consistent with experimental models for which the DNA cleavage rates have been measured.5 Indeed, experimental characterization has shown that the mm@20, mm@19–20, and mm@18–20 systems cleave their DNA substrates with rates similar to the on-target system and thus can be considered “productive” for DNA cleavage. Contrariwise, the mm@17–20 system cleaves the DNA substrates at a significant slower rate and is “unproductive” for DNA cleavage. Each GaMD run has been carried out for >1 μs, with simulation conditions well-suited for protein/nucleic acid complexes27 and acceleration parameters that allow for sufficiently broad exploration of the conformational space and consistent observation of the molecular consequences of off-target DNA binding.
DNA:RNA Conformational Dynamics in the Presence of Mismatches
During GaMD simulations, we observe an opening of the RNA:DNA hybrid at PAM distal sites with disruption of the Watson–Crick base pairing in the systems including off-target DNA sequences (Figure 2). Contrariwise, in the system containing the on-target DNA, the DNA:RNA hybrid stably maintains its Watson–Crick base pairing, in good agreement with previous conventional and aMD simulations of CRISPR-Cas9.14−19 Visual inspection of the trajectories reveals that all systems containing base pair mismatches display ill-behaved base pairs at the very end of the DNA-RNA hybrid (positions 20–18), whereas the on-target system remains well-behaved throughout the entire DNA:RNA hybrid (Figure 2). The difference between the “productive” systems (i.e., mm@20, mm@19–20, and mm@18–20, which include one to three base pair mismatches at the RNA:DNA ends) and the “unproductive” system (i.e., mm@17–20, with four mismatches) lies at position 17 of the RNA:DNA hybrid, where a remarkable loss of the Watson–Crick base pairing is observed in the “unproductive” system (Figure 2, right panel). With the aim of exploring more in-depth the effect of base pair mismatches around position 17, as well as to confirm the outcomes of the mm@17–20 system, a further replica of the system has been simulated including base pair mismatches at positions 16–17. As a result, this simulation confirms the remarkable loss of base pairing at position 17 (Figure S1). At position 15, all systems keep their Watson–Crick base pairing intact, regardless of mismatches (Figure 2 and Figure S1). It is important to note that transient openings at the end of a DNA duplex are not unusual in long time scale MD simulations, as well as that base flipping can occur in well-matched DNAs, as shown by independent research groups including ours.28−32 However, in the simulations of the on-target CRISPR-Cas9 system, the RNA:DNA hybrid maintains the Watson–Crick base pairing, most likely stabilized by the protein framework, as previously observed in conventional and aMD simulations of this system.14−19 Clearly, the opening of the RNA:DNA hybrid at PAM distal sites only occurs in the presence of base pair mismatches.
Figure 2.
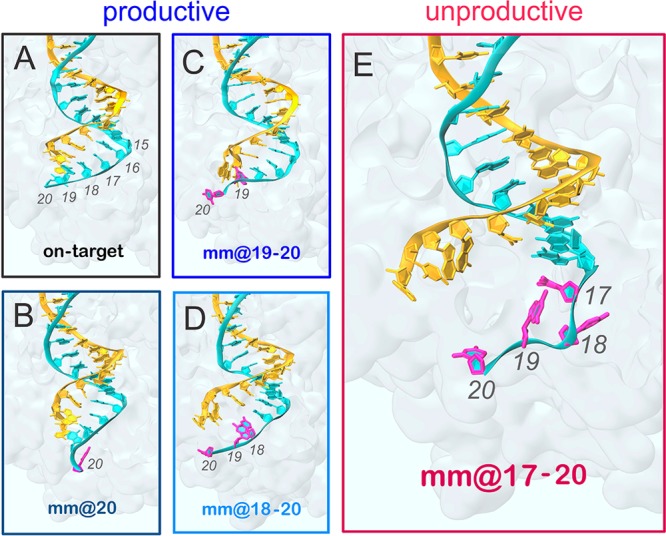
Conformations adopted by the RNA:DNA hybrid along GaMD simulations. Representative snapshots extracted from GaMD simulations of the CRISPR-Cas9 system, including the on-target DNA (A) and base pair mismatches (mm) at different positions of the hybrid: mm@20 (B), mm@19–20 (C), mm@18–20 (D), and mm@17–20 (E). “Productive” systems, which efficiently cleave their DNA substrate at rates similar to the on-target Cas9, are highlighted using cool colors (black for the “on-target” CRISPR-Cas9 and blue for the “off-target” systems), whereas the “unproductive” mm@17–20 system, which slowly cleaves the DNA substrate, is highlighted in red (warm color).5 The RNA (orange) and the target DNA (TS, cyan) are shown as ribbons. Mismatched bases on the TS are highlighted in magenta. The protein environment is shown as a molecular surface. These configurations are representative of the conformational changes detailed in Figures S3–S5 and in Figure 3.
To better estimate the extent and the precise location of the nucleic acid distortions promoted by mismatches, we performed an in-depth analysis of the minor groove width at the PAM distal region of the DNA:RNA hybrid. In detail, the minor groove width has been computed at six different levels (i–vi, from positions 20 to 16 of the TS, Figure 3A), perpendicularly to the global helical axis (full details are in the Materials and Methods section). As an effect of the instability promoted by the loss of base pairing, we detect an increase of the minor groove width at PAM distal ends of the RNA:DNA hybrid in the systems containing off-target DNA sequences (Figure S2). Interestingly, while “productive” systems promote the minor groove widening only at the very end of the hybrid (i.e., levels i and ii, Figure S2), the “unproductive” system mm@17–20 shows a remarkable increase of the minor groove at more upstream regions of the hybrid, as shown by a shift of the probability distribution of the minor groove width toward larger values at the levels iii–v (Figure 3A). To complement the conformational analysis of the hybrid, the geometrical descriptors defining the base pair complementarity (shear, propeller, stagger, buckle, stretch, opening) have been computed. At position 17 (Figure 3B), the broadly distributed scatter plots produced by the “unproductive” system mm@17–20 are indicative of significant loss of base pairing. Contrariwise, systems that are “productive” for DNA cleavage (on-target, mm@20, mm@19–20, and mm@18–20) display confined distributions of scattered dots at position 17, as consistent with well-matched base pairs. Figures S3–S5 report data for all simulated systems computed at positions 20–15, showing that the “productive” systems lose the complementarity only at the very end of the hybrid (i.e., positions 20–18), while the “unproductive” mm@17–20, as well as the simulation replica including base pair mismatches at positions 16–17, also shows remarkable loss of the complementarity at upstream positions. Indeed, in these systems, the opening and distortion of the RNA:DNA hybrid includes position 17 and reaches position 16 (Figures S3–S5). At position 15 all systems keep their Watson–Crick base pairing intact, consistently with the visual inspection of the trajectory (Figure 2 and Figure S1).
Figure 3.

RNA:DNA geometrical properties along GaMD simulations. (A) The RNA:DNA minor groove width has been computed in the PAM distal region at six different levels (i–vi, from positions 20 to 16 of the TS), which are schematically shown on the 3D structure of the RNA:DNA hybrid. The probability distributions of the RNA:DNA minor groove width at the iii–v levels are shown for the on-target CRISPR-Cas9 and for the systems including base pair mismatches at different positions of the hybrid (i.e., mm@20, mm@19–20, mm@18–20, and mm@17–20). A vertical bar indicates the experimental minor groove width (i.e., ∼11 Å from X-ray crystallography, enlarged by ∼1 Å if NMR data are considered).29 Full data are reported in Figure S2. (B) Scatter plots of the geometrical base pair descriptors, computed at position 17 of the RNA:DNA hybrid for all studied systems (full data are in Figures S3–S5). Translational (i.e., shear, stretch, and stagger) and angular (i.e., buckle, propeller, and opening) descriptors are expressed in Å and degrees, respectively. The RNA:DNA hybrid is shown on the right for the on-target CRISPR-Cas9 (gray) superposed to the mm@17–20 (red) system.
Taken together, the base pair geometrical descriptors well agree with the analysis of the minor groove widths, indicating for the “unproductive” mm@17–20 system a significant minor groove widening at levels iii–v (Figure 3A) and the loss of base pairing at position 17 (Figure 3B), which is not observed in the “productive” systems. This pinpoints that the conformational dynamics of the RNA:DNA hybrid is affected by the presence of base pair mismatches at PAM distal ends, and that the extent and location of the distortions in the hybrid are related to the DNA cleavage activity. Indeed, by taking together the outcomes from the here presented GaMD simulations and the available experimental DNA cleavage assays,5 mismatches that perturb the hybrid downstream of position 18 are “productive” for DNA cleavage, while mismatches whose distortions occur (or are propagated) up to position 17 are “unproductive” for DNA cleavage, as is the case of mismatches mm@17–20 (Figure 2).5
Effect of off-Target Binding on the Catalytic Domains
The observed opening of the RNA:DNA hybrid causes novel interactions with the protein framework. Here, we detail the interactions established by the RNA:DNA hybrid with the catalytic domains HNH and RuvC. Specifically, we measure the tight contacts (i.e., within 4 Å radius) established along the dynamics by the hybrid and the neighboring residues.33 As a result, we detect a remarkable increase of interactions between the hybrid and the HNH domain in the “unproductive” system (mm@17–20), which is particularly relevant with the polar and charged residues (Figure 4 and Figure S6). Remarkably, the relevance of these newly formed contacts is confirmed by the statistical error nonoverlapping with the “productive” systems. An increase of interactions for the “unproductive” system is also observed with the RuvC domain. Importantly, the HNH domain connects RuvC through two flexible loops: L1 (residues 765–780) and L2 (residues 902–918), which are part of HNH and are important in the function of CRISPR-Cas9 (Figure 4A, right panel). Indeed, L1/L2 intervene in the repositioning of HNH from the inactivated (i.e., “conformational checkpoint”) to the activated state, by changing configuration.19,34 As well, L1/L2 exert an allosteric control on the activity of HNH and RuvC, enabling the information transfer for concerted cleavages of the two DNA strands.17,35 To understand the role of these two loops in the interaction with the hybrid, we specifically measured the interactions established by L1/L2 and the DNA:RNA hybrid. As a result, the interactions of L1 do not show relevant differences among the investigated systems (Figure S6B). Indeed, L1 is a disordered loop that is highly flexible, resulting in overlapping error bars. Contrariwise, the interactions established by the RNA:DNA hybrid and L2 are remarkably increased in the “unproductive” mm@17–20 system (Figure 4 and Figure S6B). As noted above, the increase in interactions involving L2 in these systems has nonoverlapping error bars with the “productive” systems and is particularly relevant for polar and charged residues.
Figure 4.
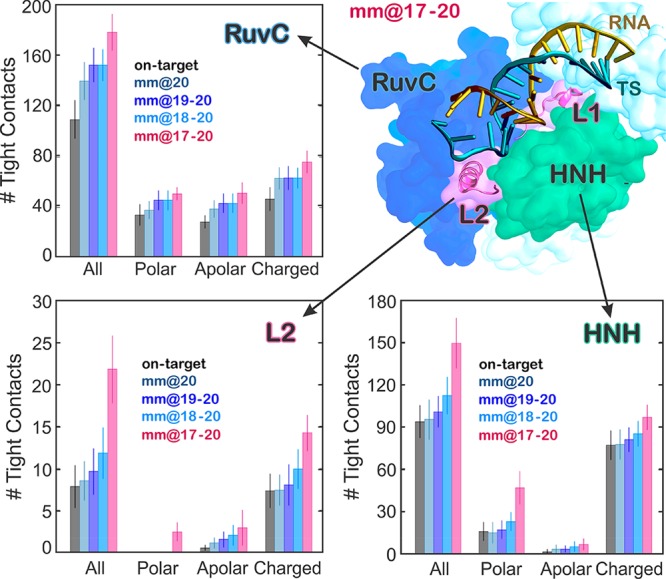
Quantitative evaluation of the interactions between the RNA:DNA hybrid and the Cas9 protein. The number of tight contacts (i.e., within 4 Å radius) established along the dynamics by the RNA:DNA hybrid with the neighboring residues of the HNH and RuvC domains, as well as with the L2 loop connecting HNH to RuvC at PAM distal ends, has been computed for the simulated systems (i.e., on-target, mm@20, mm@19–20, mm@18–20, and mm@17–20). The polar, apolar, and charged groups of residues have been considered. A cartoon of the mm@17–20 system, highlighting the RNA:DNA hybrid and its interactions with HNH and RuvC, as well as with the L1/L2 loops, is shown (right panel).
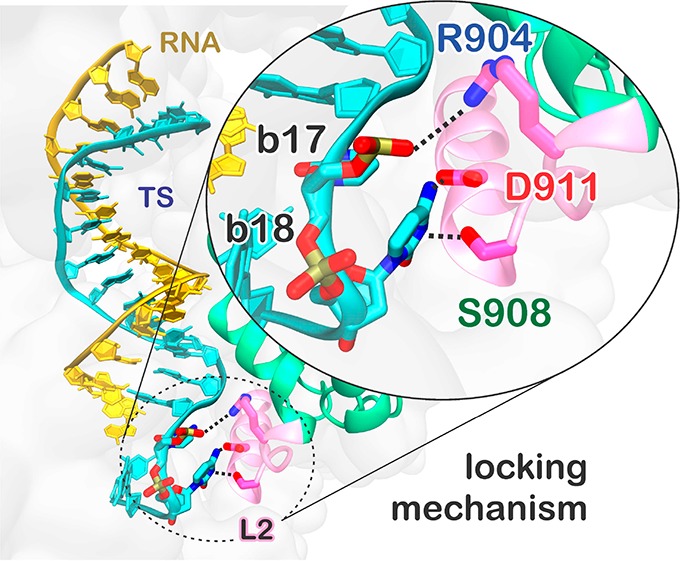
Analysis of the interactions between the hybrid and the HNH domain excluding the residues belonging to L2 corroborates that the increase of interactions between HNH and the hybrid occurs at the level of L2—practically vanishing when L2 is excluded from the analysis—and mainly involves polar and charged residues (Figure S6B). A more detailed inspection of the trajectories reveals specific interactions that are formed in the “unproductive” mm@17–20 system and could be key for explaining the effect of base pair mismatches on the capability of Cas9 to cleave off-target sequences.5 We detect the formation of a salt bridge between R904 of L2 and the TS backbone at position 17 (Figure 5; Movie S1). This interaction with the TS backbone is also formed along the simulation of the system including base pair mismatches at positions 16–17, whereas it does not form in the on-target Cas9 or in the “productive” off-target systems (Figure S7). In the “unproductive” mm@17–20 system, additional interactions are also established upon ∼0.45 μs between the D911 and S908 residues of L2 and the base 18 of the TS, which is flipped out of the hybrid (Figure 5). Remarkably, the interaction with the TS backbone is conserved along the dynamics of the “unproductive” system and also observed in the presence of base pair mismatches at positions 16–17 (Figure S7), as it is favored by the local unwinding of the DNA:RNA hybrid at position 17, which is in turn promoted by the presence of base pair mismatches at this level (Figures 2 and 3). Contrariwise, when the hybrid is well-formed at this level (i.e., position 17), as in the case of the “productive” mismatches and for the on-target DNA, the TS backbone is not free to approach and bind the L2 loop.
Figure 5.

Locking interactions between the DNA target strand (TS) and the L2 loop of the HNH domain, which decrease the HNH conformational flexibility in the presence of 4 base pair mismatches at PAM distal sites. (A) Representative snapshot of the mm@17–20 system, showing the interactions established by the RNA:DNA hybrid with the residues of the L2 loop. (B) The time evolution of the interactions established by R904, D911, and S908 and the TS at positions 17 (R904) and 18 (D911 and S908) is reported. Data for the on-target Cas9 (black) are compared to the mm@17–20 system (red).
When formed, these interactions constitute a “lock” for the L2 loop, decreasing its conformational flexibility. This is confirmed by a lower root-mean-square deviation (RMSD) of the L2 loop in the “unproductive” mm@17–20 system as compared to the “productive” systems (Figure S8). Considering that the activation of the HNH domain requires a substantial conformational rearrangement of L2,19,34,35 which moves away from the PAM distal region of the hybrid, the newly formed interactions observed in the “unproductive” system can prevent HNH from undergoing its conformational activation. This mechanism is consistent with several experimental studies showing that the presence of base pair mismatches up to positions 17 trap HNH in a “conformational checkpoint” state, preventing it from reaching the active conformation.5,10,36 On the contrary, the presence of up to 3 mismatches at positions 20–18 still allows HNH to correctly reposition for cleavage, although with slightly slower rates. Remarkably, this experimental evidence is consistent with the outcomes of GaMD, showing that 3 mismatches at positions 20–18 are unable to “lock” the HNH domain (Figure S7). In light of this experimental evidence, our simulations reveal the interactions—formed between a locally unwound TS and the L2 loop—by which specific base pair mismatches prevent HNH activation for cleavage.
Effect of Off-Target Binding on the Hybrid Recognition
The formation of the RNA:DNA hybrid has been shown to be a prerequisite for the on-target selectivity.5 Single molecule and bulk experiments have shown that the REC lobe of CRISPR-Cas9 exerts a key role in the recognition. Specifically, the REC3 region, which directly contacts the very end of the hybrid, would “sense” the formation of the DNA:RNA hybrid and allow for HNH nuclease activation.5,15
To understand the role of REC3 in the hybrid recognition and in the on-target selectivity, we monitored the interactions established by the RNA:DNA hybrid with REC3 in the simulated systems. In the “unproductive” mm@17–20 system, we observe a significant increase of interactions between the hybrid and REC3 (involving polar, apolar, and charged residues; Figure 6A and Figure S9). In this system, due to the extended opening of the RNA:DNA hybrid, the 692–700 α-helix inserts within the heteroduplex, causing novel electrostatic interactions and steric contacts mediated by apolar residues (Figure 6B). On the contrary, in the on-target Cas9, as well as in the “productive” systems including off-target sequences, the 692–700 α-helix does not insert within the hybrid (Figure 6C). The 692–700 α-helix includes a set of key residues (N692, M694, Q695, and H698), whose mutation to alanine confers increased selectivity.5 Indeed, the hyper accurate Cas9 (HypaCas9) variant, including the N692A, M694A, Q695A, and H698A mutations, cleaves the on-target DNA with rates similar to the wt-Cas9, but the cleavage is reduced in the presence of mismatches. The specific interactions of these residues have been monitored during GaMD. As a result, in the on-target Cas9, N692 and Q695 bind the TS backbone throughout the dynamics, while M694 and H698 establish additional interactions (Figure S10), contributing also to the stabilization of the fully matched RNA:DNA hybrid (Figure 2). Remarkably, the interactions established by N692 and Q695 with the TS are also observed in the “productive” off-target systems, but are lost in the “unproductive” mm@17–20 system, as also shown by including base pair mismatches at positions 16–17 (Figure S11). Indeed, in the presence of “unproductive” mismatches, the 692–700 α-helix inserts within the heteroduplex, contributing in the opening of the RNA:DNA hybrid. These data indicate that the 692–700 α-helix is a key element for the recognition of the complementarity of the RNA:DNA hybrid and for the selection of the “productive” systems. Indeed, in the “unproductive” mm@17–20 system, the insertion of the α-helix 692–700 within the heteroduplex contributes to the observed opening of the RNA:DNA hybrid and, in turn, to the establishment of interactions between the TS and the L2 loop (Figure 5). This establishes a mechanism of selectivity where, in the “productive” systems, REC3 “senses” the formation of a formed hybrid through the 692–700 α-helix, in agreement with the experimental hypothesis that the REC3 region would “sense” the formation of the DNA:RNA hybrid and allow for HNH nuclease activation.5,15 Indeed, in the on-target Cas9 and in the presence of 1–3 mismatches at PAM distal ends, the N692 and Q695 stably bind the TS backbone, contributing also to the stabilization of the hybrid itself. Contrariwise, in the presence of an extended opening of the RNA:DNA hybrid, as including base pair mismatches at positions 17–16, the 692–700 α-helix moves apart losing its interaction with the TS backbone (Figure S11) and results in the insertion within the hybrid.
Figure 6.

(A) Number of tight contacts (i.e., within 4 Å radius) established along the dynamics by the RNA:DNA hybrid and the neighboring residues of the REC3 region computed for the simulated systems (i.e., on-target, mm@20, mm@19–20, mm@18–20, and mm@17–20). The polar, apolar, and charged groups of residues have been considered. (B) Representative snapshot from GaMD simulations of the mm@17–20 system, showing the extended opening of the RNA:DNA hybrid and the insertion of the 692–700 α-helix (gray) within the heteroduplex. (C) Snapshot from GaMD of the on-target CRISPR-Cas9, showing a well-behaved RNA:DNA hybrid and the conserved interactions established by Q965, N692, and the DNA TS. The RNA (orange) and the DNA TS (cyan) are shown as ribbons. The 692–700 α-helix (gray) is shown as a cartoon; interacting protein residues are shown as sticks.
These computational outcomes also provide a rationale for the “energy excess” hypothesis37 that has been used as a foundation for the recent engineering of Cas9 systems with improved on-target specificity, such as HypaCas9.4,5,7 Accordingly to this hypothesis, the disruption of the contacts between the RNA:DNA hybrid and the neighboring residues of the REC3 region might alter the energetics of the complex, such that it might somehow retain a diminished ability to cleave mismatched off-target sequences. GaMD simulations show that the residues belonging to REC3, which are responsible for the “energy excess” in HypaCas9 (N692, M694, Q695, and H698), have a key role in the discrimination between “productive” and “unproductive” sequences. Indeed, while these residues tightly bind the TS backbone in the “productive” systems conferring stabilization to the RNA:DNA hybrid, in the presence of “unproductive” mismatches the residues contribute to the widening and destabilization of the RNA:DNA hybrid. This indicates a tight electrostatic and steric control at the level of the RNA:DNA hybrid, which is exerted by the residues of REC3. As such, the substitution of these residues by alanine, as in HypaCas9,5 would reduce the electrostatic and steric interactions with the hybrid, altering the mechanism depicted above and the energetics of the complex.
Overall, these observations provide a rationale for the role of REC3 in the on-target selectivity, revealing how, by “sensing” the RNA:DNA hybrid, REC3 discriminates “productive” from “unproductive” sequences for cleavage. As well, by taking together these observations and the “energy excess” hypothesis,37 the mutation of the N692, M694, Q695, and H698 residues to alanine, as in HypaCas9,5 would reduce the electrostatic and steric interactions with the hybrid, altering the capability of REC3 to bind and “sense” the formation of the RNA:DNA hybrid. In light of the above, and to fully understand the role of the REC3 residues responsible for increased specificity, future GaMD studies of HypaCas9 will be required to further understand and ultimately capture the role of alanine substitution in the mechanism of altered specificity.
Discussion
Off-target effects represent a severe issue hindering a full exploitation of the CRISPR-Cas9 technology.3−7 At the molecular level, off-target effects are the unselected cleavage of DNA sequences that do not fully match the guide RNA, bearing base pair mismatches within the RNA:DNA hybrid. Here, we investigated the molecular basis for the binding of base pair mismatches at PAM distal sites, by using accelerated MD simulations. A Gaussian accelerated MD methodology23—enabling the capture of long time scale conformational changes that are not accessible via conventional MD simulations—has been applied to characterize the binding of off-target DNA sequences, compared to a fully matching on-target DNA. The simulations reveal that while the on-target DNA remains fully matched to its complementary RNA, the presence of base pair mismatches induces an opening of the RNA:DNA hybrid (Figure 2). We observe that 1–3 mismatches up to position 18 perturb the hybrid at its very end, whereas base pair mismatches up to positions 17–16 produce an extended opening of the RNA:DNA heteroduplex, which includes position 17 and reaches position 16 (Figures S3–S5). The mechanical distortion of the DNA in the presence of mismatches has been very recently also observed through optical tweezers and fluorescence experiments, further supporting the conformational changes observed through molecular simulations.47 Remarkably, the presence of 1–3 base pair mismatches up to position 18 at PAM distal sites has been experimentally shown to still enable productive cleavages of the DNA substrate at similar rates of the on-target Cas9.5 Contrariwise, by increasing the number of base pair mismatches up to positions 17, Cas9 is rendered unproductive, exhibiting slower DNA cleavage rates.5 In light of this experimental evidence, mismatches that perturb the hybrid downstream of position 18 are “productive” and lead to unselective DNA cleavages, while mismatches whose distortions occur (or are propagated) up to positions 17 are “unproductive” for DNA cleavage.
As an effect of the extended opening observed in the presence of base pair mismatches at positions including 17, the DNA TS engages in conserved interactions with the L2 loop of the HNH domain (Figure 5 and Figure S7). In particular, R904 of the L2 loop stably binds the TS backbone at position 17 in the “unproductive” mm@17–20 system (Movie S1). This stable interaction is confirmed in the mm@16–17 system, which also displays an extended opening of the RNA:DNA hybrid (Figures S3–S5). Additional interactions with the TS bases also involve D911 and S908. These interactions constitute a “lock” for the L2 loop, which decreases its conformational freedom in the presence of “unproductive” mismatches (Figure S8). Considering that the activation of the HNH domain requires the conformational rearrangement of L2,19,34,35 which enables HNH to properly relocate for the TS cleavage, the newly formed interactions prevent HNH from undergoing its conformational activation. This suggests that base pair mismatches up to (or including) position 17 of the RNA:DNA hybrid can “lock” the conformational activation of the catalytic HNH domain by binding at the level of the L2 loop. This is consistent with the experimental evidence that 4 base pair mismatches at PAM distal ends (up to position 17) trap HNH in an inactive “conformational checkpoint” state, preventing it from reaching the active conformation.5,10,36 Contrariwise, GaMD simulations show that the presence of up to 3 base pair mismatches at positions 20–18 does not result in stable interactions “locking” L2 (Figure S7). This computational finding is also in agreement with the experimental fact that 3 mismatches (at positions 20–18) still allow the repositioning of HNH and are “productive” for DNA cleavage.5,10,36 In light of this experimental evidence, GaMD simulations suggest a mechanism for the binding of off-target sequences at PAM distal sites, capturing the interactions—formed between a locally unwound TS backbone and the L2 loop—by which base pair mismatches including position 17 prevent HNH activation for cleavage. GaMD also shows that the REC3 region of the Cas9 recognition lobe undergoes a significant conformational rearrangement in the presence of base pair mismatches including position 17 of the RNA:DNA hybrid (Figure 6). Indeed, the 692–700 α-helix of REC3 inserts within the heteroduplex, further contributing to the observed opening of the RNA:DNA hybrid and, in turn, to the establishment of interactions between the TS and the L2 loop. Contrariwise, in the presence of an on-target DNA, as well as including up to 3 base pair mismatches at PAM distal sites, the 692–700 α-helix does not insert within the hybrid, while the N692 and Q695 residues stably bind the TS backbone. This clarifies the mechanism by which REC3 “senses” the formation of a formed hybrid through the 692–700 α-helix,5,15 contributing also to the stabilization of the hybrid itself.
Conclusions
Off-target effects are a severe issue limiting the applicability of the CRISPR-Cas9 technology.3−7 In this paper, we investigated the molecular basis for the binding of DNA off-target sequences at the molecular level, by using all-atom Gaussian accelerated MD (GaMD) simulations. The simulations reveal that, by introducing up to 4 base pair mismatches in the target DNA at PAM distal sites, an extended opening of the RNA:DNA hybrid is observed, which leads to newly formed interactions between the unwound DNA and the L2 loop of the catalytic HNH domain. These conserved interactions constitute a “lock” effectively decreasing the conformational freedom of the HNH domain and its activation for cleavage. Contrariwise, up to 3 base pair mismatches at PAM distal ends are unable to stably “lock” HNH in an inactivated state, still allowing its repositioning for cleavage.5,10,36 This mechanism agrees with the experimental evidence that base pair mismatches up to position 17 of the hybrid trap HNH in an inactive “conformational checkpoint” state, while up to 3 base pair mismatches (up to position 18) allow HNH to reach its active conformation for cleavage.5,10,36 Therefore, the ability to “lock” the catalytic HNH domain in an inactive “conformational checkpoint” is shown to be a key determinant in the onset of off-target effects. Overall, the outcomes of the here presented simulations provide a mechanistic rationale that contributes in clarifying a long lasting open issue in the CRISPR-Cas9 function. Building on this study, novel computational investigations are ongoing in our laboratories to fully characterize the dynamic and energetic features of a large database of off-target sequences, exploring the effect of base pair mismatches along the DNA:RNA hybrid11,13 and implementing key point mutations.4,5,7 This will provide a more comprehensive understanding of the CRISPR-Cas9 specificity. Finally, the findings reported here also pose the basis for novel rational engineering of the system toward improved genome editing. Indeed, the structural modifications of the L2 loop—by using for instance non-natural amino acids—could be implemented with the goal of magnifying the “locking” interactions with the TS in the presence of off-target sequences. This could help the development of more specific Cas9 variants, in which a single base pair mismatch is sufficient for trapping HNH in its “conformational checkpoint”10 state, thus preventing the cleavage of any incorrect DNA sequence.
Materials and Methods
Structural Models
MD simulations have been based on the X-ray structure of the Streptococcus pyogenes Cas9 in complex with RNA and DNA (PDB 4UN3), solved at 2.58 Å resolution,26 which identifies the inactivated state of the HNH domain (i.e., the so-called “conformational checkpoint”).10 Based on this X-ray structure, five model systems have been built, including the on-target DNA sequence (on-target) and base pair mismatches at different positions of the RNA:DNA (namely, mm@20, mm@19–20, mm@18–20, mm@17–20, as in Figure 1B). Base pair mismatches within the RNA:DNA hybrid have been introduced by substituting the nucleobases in the DNA TS crystallized in the PDB 4UN3 (i.e., 3′–TATT–5′, Figure 1B), with nucleobases that do not match the RNA guide, resulting in the following base pair mismatches within the RNA:DNA hybrid: A:C (at position 20), U:G (at position 19), A:C (at position 18), and A:C (at position 17). These systems are consistent with the experimental models for which the DNA cleavage rates have been measured.5 Moreover, to further explore the effect of base pair mismatches around position 17, as well as to confirm the outcomes of the mm@17–20 system, a further replica of the system has been simulated including base pair mismatches at positions 16–17 (namely, mm@16–17). At this position, the C:A base pair mismatch has been introduced in the RNA:DNA hybrid. All model systems have been embedded in explicit waters, while Na+ ions were added to neutralize the total charge, leading to an orthorhombic periodic simulation cell of ∼145·110·145 Å,3 for a total of ∼220 000 atoms.
Conventional Molecular Dynamics (MD) Simulations
MD simulations have been performed to equilibrate the systems and to provide starting points for GaMD simulations. A simulation protocol tailored for RNA/DNA endonucleases has been adopted,27 embracing the use of the Amber ff12SB force field, which includes the ff99bsc0 corrections for DNA31 and the ff99bsc0+χOL3 corrections for RNA.38,39 The Ȧqvist40 force field has been employed for Mg ions, as in previous studies on similar Mg-aided RNA/DNA nucleases.27,41 An integration time step of 2 fs has been employed. All bond lengths involving hydrogen atoms were constrained using the SHAKE algorithm. Temperature control (300 K) has been performed via Langevin dynamics,42 with a collision frequency γ = 1. Pressure control was accomplished by coupling the system to a Berendsen barostat,43 at a reference pressure of 1 atm and with a relaxation time of 2 ps. The system has been subjected to energy minimization to relax water molecules and counterions, keeping the protein, the RNA, DNA, and Mg ions fixed with harmonic position restraints of 300 kcal/(mol Å2). Then, the system has been heated up from 0 to 100 K in the canonical ensemble (NVT), by running two simulations of 5 ps each, imposing position restraints of 100 kcal/(mol Å2) on the above-mentioned elements of the system. The temperature was further increased up to 200 K in ∼100 ps of MD in the isothermal–isobaric ensemble (NPT), reducing the restraint to 25 kcal/(mol Å2). Subsequently, all restraints were released, and the temperature of the system was raised up to 300 K in a single NPT simulation of 500 ps. After ∼1.1 ns of equilibration, ∼10 ns of NPT runs were carried out allowing the density of the system to stabilize around 1.01 g/cm3. Finally, the production run was carried out in the NVT ensemble, collecting ∼100 ns. Simulations have been performed using the GPU version of AMBER 16.44 The well-equilibrated systems have been used as a starting point for GaMD simulations.
Gaussian Accelerated MD (GaMD) Simulations
Accelerated MD (aMD) is an enhanced sampling method that works by adding a non-negative boost potential to smoothen the system potential energy surface (PES), thus effectively decreasing the energy barriers and accelerating transitions between the low-energy states.21 Here, aMD simulations have been performed using the novel and more robust Gaussian aMD (or GaMD)23 implementation, in which the boost potential follows Gaussian distribution. This allows reconstructing the original shape of the potential energy surface, therefore obtaining the canonical ensemble, through accurate reweighting using cumulant expansion to the second order. As such, even with biasing potential, the same low-energy physical states are sampled in the GaMD simulations. Hence, reweighting can allow for quantitative recovery of conformational distributions, while un-reweighted results, as here, capture the low-energy physical states and also provide a useful semiquantitative ranking of their probabilities. The capability of the method in accurately describing the canonical distribution has been shown for a large biomolecular system, such as CRISPR-Cas914,19 and G-protein-coupled receptors,24,25 in agreement with the available experimental data. GaMD therefore extends the use of aMD to large biological systems, for which it has been difficult to attain the canonical ensemble, given the large statistical noise.45
Considering a system with N atoms at positions 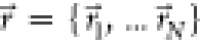 , when the system
potential
, when the system
potential 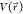 is lower than a threshold energy E, the energy
surface is modified by adding a boost potential
as
is lower than a threshold energy E, the energy
surface is modified by adding a boost potential
as
| 1 |
| 2 |
where k is the harmonic force
constant. The two adjustable parameters E and k are automatically determined by applying the following
three criteria. First, for any two arbitrary potential values 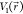 and
and 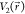 found on the original energy surface,
if
found on the original energy surface,
if 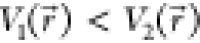 , ΔV should be a
monotonic function that does not change the relative order of the
biased potential values, i.e.,
, ΔV should be a
monotonic function that does not change the relative order of the
biased potential values, i.e., 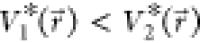 . Second, if
. Second, if 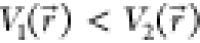 , the potential difference observed on the
smoothened energy surface should be smaller than that of the original,
i.e.,
, the potential difference observed on the
smoothened energy surface should be smaller than that of the original,
i.e., 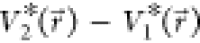 <
<  . By combining the first two criteria and
plugging in the formula of
. By combining the first two criteria and
plugging in the formula of 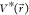 and ΔV,
we obtain
and ΔV,
we obtain
| 3 |
where Vmin and Vmax are the system minimum and maximum potential energies. To ensure that eq 4 is valid, k has to satisfy k ≤ 1/Vmax – Vmin. By defining k ≡ k0(1/Vmax) – Vmin, then 0 < k ≤ 1. Third, the standard deviation of ΔV needs to be small enough (i.e., narrow distribution) to ensure accurate reweighting using cumulant expansion to the second order: σΔV = k(E – Vavg)σV ≤ σ0, where Vavg and σV are the average and standard deviation of the system potential energies, σΔV is the standard deviation of ΔV, and σ0 is a user-specified upper limit (e.g., 10 kBT) for accurate reweighting. When E is set to the lower bound, E = Vmax, according to eq 4, k0 can be calculated as
| 4 |
Alternatively, when the threshold energy E is set to its upper bound E = Vmin + 1/k, k0 is
| 5 |
if k0″ is calculated between 0 and 1. Otherwise, k0 is calculated using eq 4 instead of being set to 1 directly as described in the original paper.23
Based on extensive testing, performed in our previous study on the CRISPR-Cas9 conformational dynamics,19 the system threshold energy has be set to E = Vmax for all GaMD simulations. The boost potential has been applied in a dual-boost scheme, in which two acceleration potentials are applied simultaneously to the system: (i) the torsional terms only and (ii) across the entire potential. A time step of 2 fs has been used. Given an average system size of ∼220 K atoms, the maximum, minimum, average, and standard deviation values of the system potential (Vmax, Vmin, Vavg and σV) has been obtained from an initial ∼100 ns NVT simulation with no boost potential (see details above). Each GaMD simulation proceeded with a ∼50 ns run, in which the boost potential has been updated every 1.6 ns, thus reaching equilibrium values. Finally, ∼1 μs of GaMD simulations have been carried out in the NVT ensemble. For each system, ∼1 μs (GaMD production) + 50 ns (GaMD equilibration) + 100 ns (pre-equilibration conventional MD) have been carried out. Since GaMD has been applied on 6 model systems, including on- and off-target DNAs, a total of ∼7 μs (i.e., ∼1 μs of production + 150 ns of equilibration × 6 systems) of simulations has been produced. These simulations have been performed with the GPU version of AMBER 16.44 Importantly, GaMD simulations have been preceded by the equilibration of the structures by means of conventional MD simulations, as described above. These runs have been performed for ∼100/120 ns, for a total of ∼0.7 μs (i.e., ∼120 ns × 6 systems).
Analysis of the Results
Analysis of the RNA:DNA conformational dynamics has been done over the GaMD production runs using the CURVES+46 code. Specifically, we computed the geometrical parameters defining the base pair complementarity (i.e., shear, stretch, stagger, buckle, propeller, and opening) and the minor groove width at the PAM distal sites of the RNA:DNA hybrid. As standard in Curves+, the minor groove has been measured between cubic spline curves running through the phosphorus atoms of the nucleic backbone and then reduced by 5.8 Å (2 × 2.9 Å) to discount the average radius of two adjacent phosphodiester backbones. Hence, the computed widths do not correspond to the phosphorus–phosphorus distance between the base pair on the RNA:DNA hybrid, but are a measure of the groove width with respect to (i.e., perpendicularly to) the global helical axes. This measurement enables a reliable description of the groove widths, which is superior to the simple measurement of the distance between phosphates on different strands. The minor groove widths have been measured at six different levels (i–vi, from positions 20 to 16 of the TS), as illustrated in Figure 3A and Figure S2.
The interactions established by the RNA:DNA hybrid and the surrounding protein domains (HNH, RuvC, REC3, and the L1/L2 loops interconnecting HNH and RuvC) have been characterized by calculating the statistical distribution of the direct contacts.33 In detail, the number of tight contacts (i.e., within 4 Å radius) established by the hybrid and the neighboring residues has been measured and averaged over the last ∼400 ns of GaMD simulations. This allowed us to consider fully established interactions. Indeed, as shown in Figure 5B, D911 and S908 engage in interactions with the TS after ∼0.45 μs. Data are reported in Figures 4 and 6A and Figure S6 with the associated statistical errors.
Acknowledgments
G.P. thanks Alexis Komor and Samuel H. Sternberg for useful discussions. G.P. thanks XSEDE for computer time awarded through Grant TG-MCB160059. J.S.C. thanks NSF for Graduate Research Fellowship. J.A.M. thanks NIH, NBCR, and SDSC for support. J.A.D. is supported by NIH, NSF and HHMI.
Supporting Information Available
Movie S1: The movie discloses the The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00020.
Conformations, RNA:DNA minor groove width, geometric parameters, interactions between the RNA:DNA hybrid and the Cas9 protein, and stability of the L2 loop (PDF)
Movie S1: “locking mechanism” that prevents the activation of the HNH nuclease for cleavage in the presence of 4 base pair mismatches at PAM distal ends of the DNA target strand (TS, cyan), wherein during the dynamics, the DNA establishes newly formed interactions with the L2 loop (pink) of the catalytic HNH domain (green), therefore decreasing the conformational flexibility of HNH and hampering its activation for DNA cleavage (MPG)
The authors declare the following competing financial interest(s): J.A.D. is a co-founder of Caribou Biosciences, Editas Medicine, Intellia Therapeutics, Scribe Therapeutics and Mammoth Biosciences, and a Director of Johnson & Johnson. J.A.D. is a scientific advisor to Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Synthego, Metagenomi, Mammoth Biosciences and Inari. J.A.D. has research projects sponsored by Biogen and Pfizer.
Supplementary Material
References
- Jinek M.; Chylinski K.; Fonfara I.; Hauer M.; Doudna J. A.; Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J. A.; Charpentier E. Genome editing. The new Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Foden J. A.; Khayter C.; Maeder M. L.; Reyon D.; Joung J. K.; Sander J. D. High-Frequency Off-target Mutagenesis Induced by CRISPR-Cas Nucleases in Human Cells. Nat. Biotechnol. 2013, 31, 822–826. 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I. M.; Gao L.; Zetsche B.; Scott D. A.; Yan W. X.; Zhang F. Rationally Engineered Cas9 Nucleases with Improved Specificity. Science 2016, 351, 84–88. 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. S.; Dagdas Y. S.; Kleinstiver B. P.; Welch M. M.; Harrington L. B.; Sternberg S. H.; Joung J. K.; Yildiz A.; Doudna J. A. Enhanced Proofreading Governs CRISPR-Cas9 Targeting Accuracy. Nature 2017, 550, 407–410. 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casini A.; Olivieri M.; Petris G.; Montagna C.; Reginato G.; Maule G.; Lorenzin F.; Prandi D.; Romanel A.; Demichelis F.; Inga A.; Cereseto A. A Highly Specific spCas9 Variant is Identified by in vivo Screening in Yeast. Nat. Biotechnol. 2018, 36, 265–271. 10.1038/nbt.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver B. P.; Pattanayak V.; Prew M. S.; Tsai S. Q.; Nguyen N. T.; Zheng Z. L.; Joung J. K. High-fidelity CRISPR-Cas9 Nucleases with no Detectable Genome-Wide Off-target Effects. Nature 2016, 529, 490–495. 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. S.; Doudna J. A. The Chemistry of Cas9 and its CRISPR Colleagues. Nat. Rev. Chem. 2017, 1, 78. 10.1038/s41570-017-0078. [DOI] [Google Scholar]
- Raper A. T.; Stephenson A. A.; Suo Z. Sharpening the Scissors: Mechanistic Details of CRISPR/Cas9 Improve Functional Understanding and Inspire Future Research. J. Am. Chem. Soc. 2018, 140, 11142–11152. 10.1021/jacs.8b05469. [DOI] [PubMed] [Google Scholar]
- Dagdas Y. S.; Chen J. S.; Sternberg S. H.; Doudna J. A.; Yildiz A. A Conformational Checkpoint between DNA Binding and Cleavage by CRISPR-Cas9. Sci. Adv. 2017, 3, eaao0027 10.1126/sciadv.aao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D.; Wang Y.; Mallon J.; Yang O.; Fei J.; Poddar A.; Ceylan D.; Bailey S.; Ha T. Mechanisms of Improved Specificity of Engineered Cas9s Revealed by Single-Molecule FRET Analysis. Nat. Struct. Mol. Biol. 2018, 25, 347–354. 10.1038/s41594-018-0051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczelkun M. D.; Tikhomirova M. S.; Sinkunas T.; Gasiunas G.; Karvelis T.; Pschera P.; Siksnys V.; Seidel R. Direct Observation of R-loop Formation by Single RNA-Guided Cas9 and Cascade Effector Complexes. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9798–9803. 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D.; Sternberg S. H.; Fei J.; Doudna J. A.; Ha T. Real-Time Observation of DNA Recognition and Rejection by the RNA-Guided Endonuclease Cas9. Nat. Commun. 2016, 7, 12778. 10.1038/ncomms12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G., Structure and Dynamics of the CRISPR-Cas9 Catalytic Complex. J. Chem. Inf. Model. 2019, in press. 10.1021/acs.jcim.8b00988 [DOI] [PubMed] [Google Scholar]
- Palermo G.; Chen J. S.; Ricci C. G.; Rivalta I.; Jinek M.; Batista V. S.; Doudna J. A.; McCammon J. A. Key Role of the REC Lobe During CRISPR-Cas9 Activation by “Sensing”, “Regulating” and “Locking” the Catalytic HNH Domain. Q. Rev. Biophys. 2018, 51, e9 10.1017/S0033583518000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Miao Y.; Walker R. C.; Jinek M.; McCammon J. A. Striking Plasticity of CRISPR-Cas9 and Key Role of Non-target DNA, as Revealed by Molecular Simulations. ACS Cent. Sci. 2016, 2, 756–763. 10.1021/acscentsci.6b00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Ricci C. G.; Fernando A.; Rajshekhar B.; Jinek M.; Rivalta I.; Batista V. S.; McCammon J. A. PAM-Induced Allostery Activates CRISPR-Cas9. J. Am. Chem. Soc. 2017, 139, 16028–16031. 10.1021/jacs.7b05313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z.; Liu J. Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. Sci. Rep. 2016, 6, 37584. 10.1038/srep37584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Miao Y.; Walker R. C.; Jinek M.; McCammon J. A. CRISPR-Cas9 Conformational Activation as Elucidated from Enhanced Molecular Simulations. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7260–7265. 10.1073/pnas.1707645114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M.; Nishimasu H.; Kodera N.; Hirano S.; Ando T.; Uchihashi T.; Nureki O. Real-Space and Real-Time Dynamics of CRISPR-Cas9 Visualized by High-Speed Atomic Force Microscopy. Nat. Commun. 2017, 8, 1430. 10.1038/s41467-017-01466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D.; Mongan J.; McCammon J. A. Accelerated Molecular Dynamics: a Promising and Efficient Simulation Method for Biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- Pierce L. C. T.; Salomon-Ferrer R.; de Oliveira C. A. F.; McCammon J. A.; Walker R. C. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Feher V. A.; McCammon J. A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. Graded Activation and Free Energy Landscapes of a Muscarinic G Protein-Coupled Eeceptor. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12162–12167. 10.1073/pnas.1614538113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. Mechanism of the G-Protein Mimetic Nanobody Binding to a Muscarinic G-Protein-Coupled Receptor. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3036–3041. 10.1073/pnas.1800756115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C.; Niewoehner O.; Duerst A.; Jinek M. Structural Basis of PAM-dependent Target DNA Recognition by the Cas9 Endonuclease. Nature 2014, 513, 569–573. 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Cavalli A.; Klein M. L.; Alfonso-Prieto M.; Dal Peraro M.; De Vivo M. Catalytic Metal Ions and Enzymatic Processing of DNA and RNA. Acc. Chem. Res. 2015, 48, 220–228. 10.1021/ar500314j. [DOI] [PubMed] [Google Scholar]
- Mura C.; McCammon J. A. Molecular Dynamics of a kappaB DNA Element: Base Flipping via Cross-Strand Intercalative Stacking in a Microsecond-Scale Simulation. Nucleic Acids Res. 2008, 36, 4941–4955. 10.1093/nar/gkn473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A.; Lankas F.; Luque F. J.; Orozco M. Towards a Molecular Dynamics Consensus View of B-DNA Flexibility. Nucleic Acids Res. 2008, 36, 2379–2394. 10.1093/nar/gkn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A.; Luque F. J.; Orozco M. Dynamics of B-DNA on the Microsecond Time Scale. J. Am. Chem. Soc. 2007, 129, 14739–14745. 10.1021/ja0753546. [DOI] [PubMed] [Google Scholar]
- Perez A.; Marchan I.; Svozil D.; Sponer J.; Cheatham T. E. 3rd; Laughton C. A.; Orozco M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of alpha/gamma Conformers. Biophys. J. 2007, 92, 3817–3829. 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci C. G.; de Andrade A. S.; Mottin M.; Netz P. A. Molecular Dynamics of DNA: Comparison of Force Fields and Terminal Nucleotide Definitions. J. Phys. Chem. B 2010, 114, 9882–9893. 10.1021/jp1035663. [DOI] [PubMed] [Google Scholar]
- Palermo G.; Favia A. D.; Convertino M.; De Vivo M. The Molecular Basis for Dual Fatty Acid Amide Hydrolase (FAAH)/Cyclooxygenase (COX) Inhibition. ChemMedChem 2016, 11, 1252–1258. 10.1002/cmdc.201500507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F. G.; Taylor D. W.; Chen J. S.; Kornfeld J. E.; Zhou K. H.; Thompson A. J.; Nogales E.; Doudna J. A. Structures of a CRISPR-Cas9 R-loop Complex Primed for DNA Cleavage. Science 2016, 351, 867–871. 10.1126/science.aad8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg S. H.; LaFrance B.; Kaplan M.; Doudna J. A. Conformational Control of DNA Target Cleavage by CRISPR-Cas9. Nature 2015, 527, 110–113. 10.1038/nature15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Peng S.; Sun R.; Lin J.; Wang N.; Chen C. The Conformational Dynamics of Cas9 Governing DNA Cleavage Are Revealed by Single-Molecule FRET. Cell Rep. 2018, 22, 372–382. 10.1016/j.celrep.2017.12.048. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Sander J. D.; Reyon D.; Cascio V. M.; Joung J. K. Improving CRISPR-Cas Nuclease Specificity using Truncated Guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton M. D.; Taylor B. J.; Driessen R. P. C.; Roos L.; Cvetesic N.; Allyjaun S.; Lenhard B.; Cuomo M. E.; Rueda D. S. DNA stretching induces Cas9 off-target activity. Nat. Struct. Mol. Biol. 2019, 26, 185. 10.1038/s41594-019-0188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banas P.; Hollas D.; Zgarbova M.; Jurecka P.; Orozco M.; Cheatham T. E.; Sponer J.; Otyepka M. Performance of Molecular Mechanics Force Fields for RNA Simulations: Stability of UUCG and GNRA Hairpins. J. Chem. Theory Comput. 2010, 6, 3836–3849. 10.1021/ct100481h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgarbova M.; Otyepka M.; Sponer J.; Mladek A.; Banas P.; Cheatham T. E.; Jurecka P. Refinement of the Cornell et al. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. 10.1021/ct200162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqvist J. Ion-Water interaction Potentials Derived from Free Energy Perturbation Simulations. J. Phys. Chem. 1990, 94, 8021–8024. 10.1021/j100384a009. [DOI] [Google Scholar]
- Casalino L.; Palermo G.; Abdurakhmonova N.; Rothlisberger U.; Magistrato A. Development of Site-Specific Mg2+-RNA Force Field Parameters: A Dream or Reality? Guidelines from Combined Molecular Dynamics and Quantum Mechanics Simulations. J. Chem. Theory Comput. 2017, 13, 340–352. 10.1021/acs.jctc.6b00905. [DOI] [PubMed] [Google Scholar]
- Turq P.; Lantelme F.; Friedman H. L. Brownian Dynamics - its Application to Ionic-Solutions. J. Chem. Phys. 1977, 66, 3039–3044. 10.1063/1.434317. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Case D. A.; Betz R. M.; Botello-Smith W.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; Izadi S.; Janowski P.; Kaus J.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Luchko T.; Luo R.; Madej B.; Mermelstein D.; Merz K. M.; Monard G.; Nguyen H.; Nguyen H. T.; Omelyan I.; Onufriev A.; Roe D. R.; Roitberg A.; Sagui C.; Simmerling C. L.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 2016; University of California: San Francisco, 2016.
- Miao Y.; McCammon J. A. Unconstrained Enhanced Sampling for Free Energy Calculations of Biomolecules: A Review. Mol. Simul. 2016, 42, 1046–1055. 10.1080/08927022.2015.1121541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery R.; Moakher M.; Maddocks J. H.; Petkeviciute D.; Zakrzewska K. Conformational Analysis of Nucleic Acids Revisited: Curves. Nucleic Acids Res. 2009, 37, 5917–5929. 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.