

Abstract
Phosphatidylinositol 3-kinase signaling promotes cell growth and survival and is frequently activated in infiltrative gliomas. Activating mutations in PIK3CA gene are observed in 6–15% of glioblastomas, although their clinical significance is largely undescribed. The objective of this study was to examine whether PIK3CA mutations are associated with a specific clinical phenotype in glioblastoma. We retrospectively reviewed 157 consecutive newly diagnosed glioblastoma patients from December 2009 to June 2012 who underwent molecular profiling consisting of targeted hotspot genotyping, fluorescence in situ hybridization for gene amplification, and methylation-specific PCR for O6-methylguanine-DNA methyltransferase promoter methylation. Molecular alterations were correlated with clinical features, imaging and outcome. The Cancer Genome Atlas data was analyzed as a validation set. There were 91 males; median age was 58 years (range, 23–85). With a median follow-up of 20.9 months, median progression-free survival (PFS) and estimated overall survival (OS) were 11.9 and 24.0 months, respectively. Thirteen patients (8.3%) harbored PIK3CA mutation, which was associated with younger age (mean 49.4 vs. 58.1 years, p = 0.02). PIK3CA mutation correlated with shorter PFS (median 6.9 vs. 12.4 months, p = 0.01) and OS (median 21.2 vs. 24.2 months, p = 0.049) in multivariate analysis. A significant association between PIK3CA mutation and more disseminated disease at diagnosis, as defined by gliomatosis, multicentric lesions, or distant leptomeningeal lesions, was observed (46.2% vs. 11.1%, p = 0.004). In conclusion, despite the association with younger age, PIK3CA activating mutations are associated with earlier recurrence and shorter survival in adult glioblastoma. The aggressive course of these tumors may be related to their propensity for disseminated presentation.
Electronic supplementary material
The online version of this article (10.1186/s40478-019-0720-8) contains supplementary material, which is available to authorized users.
Keywords: Dissemination, Glioblastoma, Gliomatosis, Multicentric, PIK3CA
Introduction
Recent years have witnessed a remarkable transformation in our understanding of glioma development and classification. It is now recognized that gliomas consist of clinically distinct, molecularly-defined disease entities, with different entities often distinguished by somatic mutations in single genes [1–3]. These molecular subgroups are not only distinguishable by their biology and associations with outcome, but many have highly distinct clinical characteristics such as age at diagnosis, radiographic appearance, or location within the central nervous system [4–6]. However, there remains variability within glioma molecular subgroups, and identification of novel genotype-phenotype correlations may allow for further refinement of molecular classification and improve the development of novel therapies [7, 8].
Class IA phosphatidylinositol 3-kinase (PI3K) is activated in several cancer types by somatic activating hotspot mutations in the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene, which encodes the catalytic subunit p110α. In some cases, somatic truncations/in-frame deletions of the phosphatidylinositol-4,5-bisphosphate 3-kinase regulatory subunit 1 (PIK3R1) gene, which encodes the regulatory subunit p85α, are also observed [9, 10]. In vitro and in vivo, PIK3CA and PIK3R1 mutations constitutively increase PI3K pathway activity, and are oncogenic in several cancer models [11–14].
Recently, recurrent somatic mutations in PIK3CA and PIK3R1 were identified in 6–15 and 10% of glioblastomas, respectively [15, 16], which were accompanied by activated PI3K signaling [15]. However, the clinical impact of these mutations is largely undescribed in glioblastoma. Therefore, we sought to determine whether somatic mutations in PIK3CA are associated with a distinct phenotype in patients with newly diagnosed glioblastoma.
Patients and methods
Patients and tumor specimens
We retrospectively analyzed a consecutive cohort of adult patients with newly diagnosed glioblastoma that had been molecularly profiled in our center from December 2009 to June 2012 (n = 184). We included only patients with newly diagnosed glioblastoma with a Karnofsky performance status (KPS) score of at least 60 and who were treated with standard chemoradiation (n = 157) [17]. Patients with a known history of lower grade glioma were excluded.
Medical charts and images were retrospectively reviewed to gather data on patient and tumor characteristics as well as the type of surgery. The extent of resection was assessed based on T1-weighted images with gadolinium enhancement. We defined widespread disseminated disease as having either a diffuse gliomatosis-like growth pattern (tumor in three or more lobes, including the brain stem), multicentric lesions (lesions in different lobes or > 2 cm apart without intervening T2/fluid-attenuated inversion recovery changes), or distant leptomeningeal lesions.
Genotyping
Molecular profiling included SNaPshot genotyping, which interrogates 68 established hotspot loci from 15 oncogenes and tumor suppressors (AKT1, APC, BRAF, CTNNB1, epidermal growth factor receptor (EGFR), HER2, isocitrate dehydrogenase 1 (IDH1), KIT, KRAS, MEK1, NOTCH1, NRAS, PIK3CA, PTEN, TP53), fluorescence in situ hybridization (FISH) for EGFR, mesenchymal-epithelial transition (MET), and platelet-derived growth factor receptor alpha (PDGFRA) gene amplifications, and methylation-specific PCR for O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation [18]. PIK3R1, IDH2, and TERT promoter were not included in this genotyping platform.
Survival analysis
In this retrospective analysis, progressive disease was defined either by tissue diagnosis or when two of the following criteria were met: a) radiographic progression by central review that occurred after more than 3 months from the end of radiation, b) neurological decline related to the tumor (clinical progression) documented by the treating physician, and c) initiation of new anti-tumor therapy. Overall survival (OS) was calculated from the day of initial surgery. Patients were censored when they were lost to follow-up or died from causes unrelated to the disease.
Independent dataset validation
Glioblastoma mutation and copy number data from The Cancer Genome Atlas (TCGA) datasets were downloaded from www.cbioportal.org [19–21]. The mutation and progression-free survival (PFS) data of 291 sequenced glioblastomas in the TCGA project [19] were accessed on September 22, 2017 and used for survival analysis. Kaplan-Meier curves for PFS were calculated with stratification by PIK3CA mutation status.
Statistical analysis
Two-tailed Student’s t-test and Fisher’s exact test were used to compare continuous and categorical variables between two groups, respectively. The log-rank test was used in univariate analysis of factors associated with survival. The Cox hazards model was used in multivariate analysis. Age and KPS score were assessed as continuous variables. JMP 11 (SAS Institute, Cary, NC, USA) was used for statistical analysis. A probability (p) value of < 0.05 was considered significant.
Results
Patient characteristics, survival, and established prognostic factors in the cohort
A total of 157 consecutive adult patients with molecularly profiled glioblastoma who had a KPS score of at least 60 and were treated with standard chemoradiation were included in the analysis. The median age was 58 years (range, 23–85), there were 91 males (58.0%) and the median KPS score was 90 (Table 1). Most patients had undergone resection (136 patients, 86.6%) rather than only biopsy, and 69 (44.0%) patients underwent gross total resection (GTR) of enhancing disease. With a median follow-up of 20.9 months for the entire cohort, the median PFS and OS of the entire cohort were 11.9 and 24.0 months, respectively. As expected, established clinical prognostic factors such as age, KPS, and GTR were associated with longer PFS and/or OS in univariate analyses (age: PFS p = 0.0004, OS p = 0.0004; KPS: PFS p = 0.10, OS p = 0.047; GTR: PFS p = 0.02, OS p = 0.11) (Table 2). Of note, deep tumor location (i.e. basal ganglia or corpus callosum) compared to hemispheric location was not associated with either PFS (p = 0.50) or OS (p = 0.65), and therefore was not included in further survival analyses. Similarly, the known favorable molecular prognostic factors of IDH1 mutation and MGMT promoter methylation, observed in 7.6% (12/157 tumors) and 50.0% (60/120 tumors), respectively, were also associated with longer PFS and OS (IDH1: PFS p < 0.0001, OS p = < 0.0001; MGMT: PFS p < 0.0001, OS p < 0.0001) (Table 2, Additional file 1: Figure S1).
Table 1.
Patient and tumor characteristics
| Total | PIK3CA mutant | PIK3CA wildtype | P value | |
|---|---|---|---|---|
| Number of patients | 157 | 13 | 144 | |
| (male patient) | (91 male) | (9 male) | (82 male) | 0.56 |
| Age | median 58 (23–85) | mean 49.4 ± 15.9 | mean 58.1 ± 12.1 | 0.02* |
| KPS score | median 90 (60–100) | mean 92.3 ± 7.3 | mean 87.7 ± 10.4 | 0.048* |
| Gross total resection | 69 (44.0%) | 5 (38.5%) | 64 (44.4%) | 0.78 |
| IDH1 mutation | 12/157 (7.6%) | 0/13 (0.0%) | 12/144 (8.3%) | 0.60 |
| MGMT promoter methylation | 60/120 (50.0%) | 8/11 (72.7%) | 52/109 (47.7%) | 0.20 |
| EGFR amplification | 63/143 (44.1%) | 2/12 (16.7%) | 61/131 (46.6%) | 0.07 |
* Statistical significance: P ≤ 0.05
Abbreviations: KPS, Karnofsky performance status; IDH1, isocitrate dehydrogenase 1; MGMT O6-methylguanine-DNA methyltransferase
Table 2.
PIK3CA mutation was significantly associated with shorter survival after adjusting for age, KPS, gross total resection, IDH1 mutation, and MGMT promoter methylation
| Univariate analysis | Multivariate analysis | ||
|---|---|---|---|
| P value | P value | HR | |
| [PFS] | |||
| High age | 0.0004* | 0.005* | 1.03 (1.01–1.05)# |
| High KPS score | 0.10 | 0.28 | 0.99 (0.97–1.01)# |
| Gross total resection | 0.02* | 0.17 | 0.75 (0.49–1.13) |
| PIK3CA mutation | 0.03* | 0.01* | 2.89 (1.30–5.90) |
| IDH1 mutation | < 0.0001* | 0.002* | 0.10 (0.006–0.50) |
| MGMT promoter methylation | < 0.0001* | < 0.0001* | 0.33 (0.21–0.52) |
| [OS] | |||
| High age | 0.0004* | 0.003* | 1.03 (1.01–1.05)# |
| High KPS score | 0.047* | 0.32 | 0.99 (0.96–1.01)# |
| Gross total resection | 0.11 | 0.53 | 0.87 (0.55–1.35) |
| PIK3CA mutation | 0.19 | 0.049* | 2.32 (1.00–4.88) |
| IDH1 mutation | < 0.0001* | 0.02* | 0.15 (0.008–0.74) |
| MGMT promoter methylation | < 0.0001* | < 0.0001* | 0.27 (0.17–0.44) |
* Statistical significance: P ≤ 0.05
# HR per unit
Abbreviations: PFS, progression-free survival; OS, overall survival; KPS, Karnofsky performance status; IDH1, isocitrate dehydrogenase 1; MGMT, O6-methylguanine-DNA methyltransferase; HR, hazard ratio
Characteristics of PIK3CA mutant glioblastoma
We identified 13 patients (8.3%) with PIK3CA activating mutations by DNA sequence analysis (4 at position R88, 4 at hotspots 542–546, and 5 at position H1047) (Table 3). PIK3CA mutation was significantly associated with a younger age (p = 0.02) and better KPS (p = 0.048) (Table 1). Strikingly, it was also associated with decreased PFS (median 6.9 months for mutant vs. 12.4 months for wildtype, p = 0.03) (Fig. 1), an association that remained significant after adjusting for other known prognostic factors in a multivariate model, including age, KPS, GTR, IDH1 mutation, and MGMT promoter methylation (HR 2.89, p = 0.01). There was no difference in OS in univariate analysis (median 21.2 months vs. 24.2 months, p = 0.19) (Additional file 2: Figure S2); however, OS was significantly worse in PIK3CA mutant patients in multivariate analysis after adjusting for the above known prognostic factors (HR 2.32, p = 0.049) (Table 2). Other factors that were independently associated with survival in multivariate analysis were high age (PFS: HR 1.03, p = 0.005; OS: HR 1.03, p = 0.003), IDH1 mutation (PFS: HR 0.10, p = 0.002; OS: HR 0.15, p = 0.02), and MGMT promoter methylation (PFS: HR 0.33, p < 0.0001; HR 0.27, p < 0.0001) (Table 2).
Table 3.
Case description of PIK3CA-mutant glioblastomas
| Case | Age | Sex | Tumor at presentation | Type of surgery | KPS | PIK3CA mutation | MGMT | IDH1 | EGFR | Other alterations | PFS (mo.) |
OS (mo.) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 67 | M | multicentric | PR | 90 | H1047R | u | wt | n.t. |
TP53
R248W |
6.3 | 9.7 |
| 2 | 68 | M | gliomatosis | biopsy | 90 | R88Q | n.t. | wt | N | 3.2 | 5.1 | |
| 3 | 31 | M | leptomeningeal metastasis | biopsy | 90 | H1047R | n.t. | wt | N | 6.6 | 10.9 | |
| 4 | 42 | F | single lesion | PR | 100 | R88Q | m | wt | N |
TP53
R273C |
12.4 | 54.9 |
| 5 | 72 | F | single lesion | GTR | 90 | R88Q | m | wt | N | 5.3 | 10.6 | |
| 6 | 35 | F | multicentric | GTR | 90 | Q546R | u | wt | amp | 26.6 | 32.4 | |
| 7 | 25 | M | multicentric | biopsy | 80 | R88Q | m | wt | N | 6.9 | 19.1 | |
| 8 | 65 | F | single lesion | GTR | 100 | Q546K | m | wt | N | 7.6 | 36.9 | |
| 9 | 43 | M | single lesion | PR | 90 | E545K | u | wt | N | 5.7 | 11 | |
| 10 | 32 | M | single lesion | PR | 100 | H1047R | m | wt | N | 13.8 | 17.7* | |
| 11 | 54 | M | single lesion | GTR | 100 | E542K | m | wt | N | 34.8* | 34.8* | |
| 12 | 57 | M | gliomatosis | biopsy | 80 | H1047Y | m | wt | N | 6.6 | 21.2 | |
| 13 | 51 | M | single lesion | GTR | 100 | H1047R | m | wt | amp | 14.9 | 31.1 |
Abbreviations: M, male; F, female; PR, partial resection; GTR, gross total resection; KPS, Karnofsky performance status; n.t., not tested; IDH1, isocitrate dehydrogenase 1; MGMT, O6-methylguanine-DNA methyltransferase promoter methylation; m, methylated; u, unmethylated; wt, wild-type; amp, amplified; N, non-amplified; PFS, progression-free survival; OS, overall survival; mo., months; *censored at last follow-up
Fig. 1.
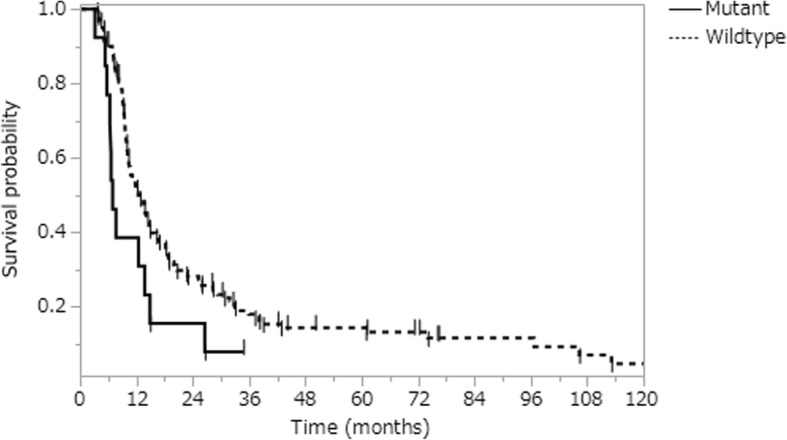
Kaplan-Meier curves of PFS stratified by PIK3CA mutation. PIK3CA mutant tumors (solid) and wildtype tumors (dashed) depicted
Given the known prognostic impact of IDH1 mutation on survival, we investigated the association of PIK3CA mutation with PFS and OS after excluding 12 IDH1 mutant glioblastomas. Within the remaining 145 IDH1 wildtype glioblastoma patients, the difference in PFS between PIK3CA mutant and PIK3CA wildtype tumors was statistically significant by Wilcoxon test (p = 0.02) and was a trend to significance by log-rank test (p = 0.11) (Additional file 3: Figure S3). In a multivariate analysis adjusting for other known prognostic factors such as age, KPS score, GTR, and MGMT promoter methylation, PIK3CA mutation was associated with decreased PFS (HR 2.85, p = 0.01) (Additional file 6: Table S1). With regards to OS, we observed no difference by PIK3CA mutation in univariate analysis (median 21.2 months vs. 22.2 months, p = 0.42); however in multivariate analysis there was a trend for PIK3CA mutant tumors to have worse OS (HR 2.25, p = 0.057) (Additional file 6: Table S1, Additional file 4: Figrue S4).
PIK3CA mutation in the TCGA dataset
To confirm the impact of PIK3CA mutation on patient survival observed in our discovery cohort, we analyzed the publicly available validation dataset derived from the TCGA project [20, 21]. PIK3CA mutations were observed in 26 of 291 (8.9%) glioblastomas in this cohort. PFS of the patients with PIK3CA mutant glioblastoma (6.1 months) was significantly shorter than that of patients with the wildtype counterpart (9 months) (p = 0.008) (Additional file 5: Figure S5), while OS was not significantly different (median 13.1 months vs. 13.3 months, p = 0.40). Of note, PIK3R1 mutations were noted in 25 of 256 (10.0%) glioblastomas; however, these mutations were not significantly associated with shorter PFS (median 6.2 months vs. 8.5 months, p = 0.14) or OS (median 13.7 months vs. 13.3 months, p = 0.73).
Imaging characteristics for PIK3CA mutant glioblastomas
To identify the putative causes of the apparent aggressive behavior of PIK3CA mutant tumors, we reviewed their clinical profiles. PIK3CA mutant glioblastoma often presented with widespread tumor dissemination relative to PIK3CA wildtype tumors. Representative contrast-enhanced MRI images of three PIK3CA mutant cases are shown in Fig. 2, with arrows indicating the tumor areas. We compared PIK3CA mutant and wildtype tumors for dissemination at diagnosis, defined as having either a diffuse gliomatosis-like growth pattern, multicentric lesions, or distant leptomeningeal lesions. We found a significant association between PIK3CA mutation and more disseminated disease at diagnosis, as 6 of 13 (46.2%) PIK3CA mutant glioblastomas presented with dissemination compared to 16 of 144 (11.1%) PIK3CA wildtype tumors (p = 0.004).
Fig. 2.

Representative cases of PIK3CA mutant glioblastomas demonstrating widespread disease at presentation. a The tumors were multicentric, affecting the left frontal and left parietal lobes without any intervening T2/fluid attenuated inversion recovery abnormality. b The tumor diffusely involved multiple lobes including the right frontal, parietal, and occipital lobes as well as the left parietal lobe. c The main tumor was located in the left posterior mesial temporal lobe with leptomeningeal involvement of the adjacent brain such as the midbrain and pons. Distant lesions were also observed on the ventral surface of the pons and the dorsal surface of the cervical spinal cord
We also examined the effects of other molecular alterations associated with glioblastoma migration. High-level EGFR amplification was previously detected in the invasive subpopulation within heterogeneously amplified glioblastomas [22, 23]. In our dataset, 63 of 144 (43.8%) tested tumors showed high-level EGFR amplification. We did not observe a significant association between EGFR-amplification and dissemination (20.6% of EGFR-amplified tumors vs. 16.0% of non-amplified tumors, p = 0.52). Half of the tumors tested had MGMT promoter methylation (59/118, 50%), and there was no association with dissemination (18.6% of methylated tumors vs. 13.6% of unmethylated tumors, p = 0.62). After standard chemoradiation, tumors with MGMT promoter methylation were reported to have higher rates of distant recurrence [24]. No molecular alterations were associated with dissemination in the newly diagnosed setting when tested among the PIK3CA wildtype tumors alone (data not shown).
Discussion
Here, we found that PIK3CA-activating mutations are associated with early recurrence and poor prognosis in glioblastoma. Our dataset included patients who received identical adjuvant treatment in a single center, and our early recurrence finding was validated in a large, multicenter independent dataset [19]. Additionally, we identified novel clinical associations with PIK3CA-activating mutations, including younger age at diagnosis and propensity to present with widespread disease. We demonstrated that PIK3CA-activating mutations were associated with clinically-apparent increased invasiveness and/or CNS dissemination in patients with newly diagnosed glioblastoma.
Increased PI3K signaling is well known to promote the survival, growth, and proliferation of cancer cells [9, 11], as well as their motility, invasion, and metastasis [9, 25, 26]. In vitro studies have similarly demonstrated that inappropriately activated PI3K signaling was associated with cell motility and invasion in glioma [27–30]. Recently, Lee et al. reported that multifocal glioblastomas were enriched for PIK3CA mutations [31], which is consistent with our findings. Therefore, we speculate that activated PIK3CA drives increased glioma cell migration, resulting in a disseminated malignant phenotype that may escape standard-of-care adjuvant involved-field radiation therapy. In the TCGA validation dataset, PIK3CA mutations were associated with shorter PFS, but the association with OS was not statistically significant. The greater likely patient variation in the larger TCGA dataset and wide variety of salvage therapies for the recurrent disease could partially account for the lack of significance seen in OS analysis.
There were some limitations to this study. The sample size of PIK3CA mutations was small; however, the mutation frequency in our large cohort was consistent with the findings of previous studies [15], and the demographic and molecular characteristics as well as treatment and outcomes in our entire cohort were largely comparable to those in other large published datasets on glioblastoma [17, 32], indicating the generalizability of our cohort. The progression of glioblastoma may be difficult to discern from treatment-related necrosis radiographically; however, all the progression events in this study were well-evidenced by standard response assessment criteria [33, 34] and confirmed by well-documented clinical and radiographic follow-up.
Another concern is the lack of assessment on other potentially important genes in the SNaPshot versions used in this study, including IDH2, PIK3R1 and TERT promoter [35]. IDH2 is present in only 2% of all diffuse gliomas and less than 1% in adult primary glioblastoma – the study population in this study [32], therefore including IDH2 would not likely impact the results. PIK3R1 mutations have been shown to induce a gain of PI3K enzymatic function and enhance PI3K signaling, which implies that PIK3R1 mutant glioblastomas may well have the similar impact as the PIK3CA mutant counterpart. However, the aforementioned study by Lee et al. demonstrated that PIK3CA mutations were enriched in multifocal glioblastomas, while PIK3R1 mutations were not [31]. PIK3CA mutations were associated with shorter PFS on the TCGA dataset, but PIK3R1 mutations were not. These observations may suggest a differential impact on clinical pictures of glioblastomas between the two PI3K pathway mutations. The prognostic implication of PIK3R1 mutation remains to be elucidated and definitely warrants further studies. A recent study by Izquierdo et al. reported on radiological characteristics of IDH wildtype lower grade astrocytoma [36]. TERT promoter mutations in this cohort were associated with glimatosis-like pattern at presentation, and apparently tended for poorer prognosis. Our recent study investigated on a more recent cohort from our institution demonstrated that TERT promoter wildtype glioblastoma, IDH wildtype harbored frequent PI3K pathway mutations as compared to the TERT promoter mutant counterpart [35]. This study lacked radiological assessment or survival follow-up. Therefore, the interactions between PIK3R1 mutation, TERT promoter mutation and PIK3CA mutation in adult glioblastoma clinical characteristics remain to be determined.
This study confirms our initial observation that PIK3CA mutant glioblastomas were associated with more widespread disease at presentation and shorter PFS [37] as well as a recent report by Lee et al. [31], who reported PIK3CA mutations were associated with multifocal/multicentric (versus solitary) enhancing tumors and decreased survival. In addition, we extend Lee et al.’s cohort to include distant leptomeningeal spread and non-enhancing gliomatosis. Based on these data, and the finding by Lee et al. that PIK3CA mutation is early and truncal in glioblastoma [31], knowledge of PIK3CA mutation status could aid in the decision of whether to utilize locally-directed therapies such as surgical resection and focal radiation therapy. Additionally, clinical trial designs may have to account for PIK3CA mutation status, as widespread or multifocal disease often precludes patients from participating in clinical trials. Inhibitors of PI3K and mammalian target of rapamycin tested in trials are theoretically the most effective for tumors addicted to PI3K signaling, such as PIK3CA mutant tumors, however patients with these tumors may be preferentially excluded from clinical trials because of their widespread disease. Therefore, our findings may have significant implications for interpreting the results of clinical trials of PI3K inhibitors [38] and for designing PI3K-specific clinical trials (NCT01339052).
Conclusions
This study indicates that PIK3CA-activating mutations identify a subset of glioblastomas associated with younger patient age, early progression, and propensity to present with widespread disease. Patients with PIK3CA mutant glioblastoma may require additional consideration in treatment planning and clinical trials.
Additional files
Figure S1. Kaplan-Meier curves of OS and PFS stratified by established molecular prognostic factors (IDH1 mutation and MGMT promoter methylation). PFS (A and C) and OS (B and D) are plotted stratified by IDH1 mutation (A and B) and MGMT promoter methylation (C and D). The curves shown in solid lines represent IDH1 mutant or MGMT promoter methylated and those in dashed lines represent IDH1 wildtype or MGMT promoter unmethylated. (TIF 158 kb)
Figure S2. Kaplan-Meier curves of OS stratified by PIK3CA mutation. PIK3CA mutant tumors (solid) and wildtype tumors (dashed) depicted. (TIF 493 kb)
Figure S3. Kaplan-Meier curves of PFS in IDH1 wildtype glioblastomas stratified by PIK3CA mutation. PIK3CA mutant tumors (solid) and wildtype tumors (dashed) depicted. (TIF 487 kb)
Figure S4. Kaplan-Meier curves of OS in IDH1 wildtype glioblastomas stratified by PIK3CA mutation. PIK3CA mutant tumors (solid) and wildtype tumors (dashed) depicted. (TIF 493 kb)
Figure S5. Kaplan-Meier curves of PFS stratified by PIK3CA mutation in the TCGA cohort. PIK3CA mutant (solid) and PIK3CA wildtype (dashed) glioblastoma patients depicted. (TIF 494 kb)
Table S1. Association of PIK3CA mutation with progression-free and overall survival in IDH1 wildtype glioblastoma. (DOC 55 kb)
Acknowledgements
Not applicable.
Funding
A.S.C. was supported by a Joan Ambriz American Brain Tumor Association Basic Research Fellowship and Early Career Research Award from the Ben and Catherine Ivy Foundation. D.P.C. is supported by Career Awards in Medical Scientists of Burroughs Wellcome Fund..
Availability of data and materials
The anonymized datasets used and/or analysed during the current study are available from the corresponding authors on reasonable request.
The TCGA datasets generated and/or analysed during the current study are available in the cBioPortal website (www.cbioportal.org). The data as of September 22, 2017 were obtained.
Abbreviations
- EGFR
Epithelial growth factor receptor
- FISH
Fluorescence in situ hybridization
- IDH1
Isocitrate dehydrogenase
- KPS
Karnofsky performance status
- MET
Mesenchymal-epithelial transition
- MGMT
O6-methylguanine-DNA methyltransferase
- OS
Overall survival
- PDGFR
Platelet-derived growth factor receptor alpha
- PFS
Progression-free survival
- PI3K
Phosphatidylinositol 3-kinase
- PIK3CA
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PIK3R1
Phosphatidylinositol-4,5-bisphosphate 3-kinase regulatory subunit 1
- TCGA
The Cancer Genome Atlas
Authors’ contributions
ST and ASC designed the study. ST, DY, DPC, and ASC collected the clinical and genetic data. AJI, DD, DRB, LWE, and DNL performed the clinical sequencing of the materials in the study. ST, DPC, and ASC analyzed and interpreted the data. ST, TTB, DPC, and ASC wrote the manuscript. TTB, DNL, DPC, and ASC supervised the study. All authors read and approved the final manuscript.
Ethics approval and consent to participate
An IRB-approved informed consent form was obtained from all individual patients included in this study.
Consent for publication
Not applicable.
Competing interests
A.S.C. is an employee of Neon Therapeutics and has served on the advisory board of Cota Healthcare. T.T.B. received consulting honoraria from Merck, GenomiCare, NXDC, UpToDate, Inc., and Champions Biotechnology. A.J.I. received equity from ArcherDx. The other authors declare no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Daniel P. Cahill, Email: cahill@mgh.harvard.edu
Andrew S. Chi, Phone: 617-337-4701, Email: achi@neontherapeutics.com
References
- 1.Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474. doi: 10.1038/nature26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. New England J Med. 2015;372:2499–2508. doi: 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reifenberger G, Wirsching HG, Knobbe-Thomsen CB, Weller M. Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol. 2017;14:434–452. doi: 10.1038/nrclinonc.2016.204. [DOI] [PubMed] [Google Scholar]
- 4.Ellingson BM, Cloughesy TF, Lai A, Nghiemphu PL, Pope WB. Cell invasion, motility, and proliferation level estimate (CIMPLE) maps derived from serial diffusion MR images in recurrent glioblastoma treated with bevacizumab. J Neuro-Oncol. 2011;105:91–101. doi: 10.1007/s11060-011-0567-8. [DOI] [PubMed] [Google Scholar]
- 5.Patel SH, Poisson LM, Brat DJ, Zhou Y, Cooper L, Snuderl M, Thomas C, Franceschi AM, Griffith B, Flanders AE, et al. T2-FLAIR mismatch, an imaging biomarker for IDH and 1p/19q status in lower-grade Gliomas: a TCGA/TCIA project. Clin Cancer Res. 2017;23:6078–6085. doi: 10.1158/1078-0432.CCR-17-0560. [DOI] [PubMed] [Google Scholar]
- 6.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, Koelsche C, Wefers A, Reinhardt A, Huang K, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136:153–166. doi: 10.1007/s00401-018-1849-4. [DOI] [PubMed] [Google Scholar]
- 9.Courtney Kevin D., Corcoran Ryan B., Engelman Jeffrey A. The PI3K Pathway As Drug Target in Human Cancer. Journal of Clinical Oncology. 2010;28(6):1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizoguchi M, Nutt CL, Mohapatra G, Louis DN. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol. 2004;14:372–377. doi: 10.1111/j.1750-3639.2004.tb00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 12.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104:5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16:463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research N Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallia GL, Rand V, Siu IM, Eberhart CG, James CD, Marie SK, Oba-Shinjo SM, Carlotti CG, Caballero OL, Simpson AJ, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. 2006;4:709–714. doi: 10.1158/1541-7786.MCR-06-0172. [DOI] [PubMed] [Google Scholar]
- 17.Stupp R, Mason W, van den Bent M, Weller M, Fisher B, Taphoorn M, Belanger K, Brandes A, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 18.Chi Andrew S., Batchelor Tracy T., Dias-Santagata Dora, Borger Darrell, Stiles Charles D., Wang Daphne L., Curry William T., Wen Patrick Y., Ligon Keith L., Ellisen Leif, Louis David N., Iafrate A. John. Prospective, high-throughput molecular profiling of human gliomas. Journal of Neuro-Oncology. 2012;110(1):89–98. doi: 10.1007/s11060-012-0938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada Y, Hurwitz EE, Esposito JM, Brower MA, Nutt CL, Louis DN. Selection pressures of TP53 mutation and microenvironmental location influence epidermal growth factor receptor gene amplification in human glioblastomas. Cancer Res. 2003;63:413–416. [PubMed] [Google Scholar]
- 23.Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20:810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Brandes AA, Tosoni A, Franceschi E, Sotti G, Frezza G, Amista P, Morandi L, Spagnolli F, Ermani M. Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: correlation with MGMT promoter methylation status. J Clin Oncol. 2009;27:1275–1279. doi: 10.1200/JCO.2008.19.4969. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt-Kittler O, Zhu J, Yang J, Liu G, Hendricks W, Lengauer C, Gabelli SB, Kinzler KW, Vogelstein B, Huso DL, et al. PI3Kalpha inhibitors that inhibit metastasis. Oncotarget. 2010;1:339–348. doi: 10.18632/oncotarget.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue G., Hemmings B. A. PKB/Akt-Dependent Regulation of Cell Motility. JNCI Journal of the National Cancer Institute. 2013;105(6):393–404. doi: 10.1093/jnci/djs648. [DOI] [PubMed] [Google Scholar]
- 27.Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, Thiemann M, Mueller W, Sykora J, Kuhn A, et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell. 2008;13:235–248. doi: 10.1016/j.ccr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Koul D, Shen R, Kim YW, Kondo Y, Lu Y, Bankson J, Ronen SM, Kirkpatrick DL, Powis G, Yung WK. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro-Oncology. 2010;12:559–569. doi: 10.1093/neuonc/nop058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pu P, Kang C, Li J, Jiang H. Antisense and dominant-negative AKT2 cDNA inhibits glioma cell invasion. Tumour Biol. 2004;25:172–178. doi: 10.1159/000081099. [DOI] [PubMed] [Google Scholar]
- 31.Lee JK, Wang J, Sa JK, Ladewig E, Lee HO, Lee IH, Kang HJ, Rosenbloom DS, Camara PG, Liu Z, et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet. 2017;49:594–599. doi: 10.1038/ng.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macdonald D, Cascino T, Schold SJ, Cairncross J. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 34.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 35.Williams EA, Miller JJ, Tummala SS, Penson T, Iafrate AJ, Juratli TA, Cahill DP. TERT promoter wild-type glioblastomas show distinct clinical features and frequent PI3K pathway mutations. Acta Neuropathol Commun. 2018;6:106. doi: 10.1186/s40478-018-0613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Izquierdo C, Barritault M, Poncet D, Cartalat S, Joubert B, Bruna J, Jouanneau E, Guyotat J, Vasiljevic A, Fenouil T et al (2018) Radiological characteristics and natural history of adult IDH-Wildtype Astrocytomas with TERT promoter mutations. Neurosurgery. 10.1093/neuros/nyy513 [DOI] [PubMed]
- 37.Tanaka S, Batchelor T, Iafrate AJ, Dias-Santagata D, Borger DR, Ellisen LW, Yang D, Louis DN, Cahill DP, Chi AS. Association of PIK3CA-activating mutations with more disseminated disease at presentation and earlier recurrence in glioblastoma. J Clin Oncol. 2013;31:2029–2029. doi: 10.1200/jco.2013.31.15_suppl.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pitz MW, Eisenhauer EA, MacNeil MV, Thiessen B, Easaw JC, Macdonald DR, Eisenstat DD, Kakumanu AS, Salim M, Chalchal H, et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro-Oncology. 2015;17:1270–1274. doi: 10.1093/neuonc/nou365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Kaplan-Meier curves of OS and PFS stratified by established molecular prognostic factors (IDH1 mutation and MGMT promoter methylation). PFS (A and C) and OS (B and D) are plotted stratified by IDH1 mutation (A and B) and MGMT promoter methylation (C and D). The curves shown in solid lines represent IDH1 mutant or MGMT promoter methylated and those in dashed lines represent IDH1 wildtype or MGMT promoter unmethylated. (TIF 158 kb)
Figure S2. Kaplan-Meier curves of OS stratified by PIK3CA mutation. PIK3CA mutant tumors (solid) and wildtype tumors (dashed) depicted. (TIF 493 kb)
Figure S3. Kaplan-Meier curves of PFS in IDH1 wildtype glioblastomas stratified by PIK3CA mutation. PIK3CA mutant tumors (solid) and wildtype tumors (dashed) depicted. (TIF 487 kb)
Figure S4. Kaplan-Meier curves of OS in IDH1 wildtype glioblastomas stratified by PIK3CA mutation. PIK3CA mutant tumors (solid) and wildtype tumors (dashed) depicted. (TIF 493 kb)
Figure S5. Kaplan-Meier curves of PFS stratified by PIK3CA mutation in the TCGA cohort. PIK3CA mutant (solid) and PIK3CA wildtype (dashed) glioblastoma patients depicted. (TIF 494 kb)
Table S1. Association of PIK3CA mutation with progression-free and overall survival in IDH1 wildtype glioblastoma. (DOC 55 kb)
Data Availability Statement
The anonymized datasets used and/or analysed during the current study are available from the corresponding authors on reasonable request.
The TCGA datasets generated and/or analysed during the current study are available in the cBioPortal website (www.cbioportal.org). The data as of September 22, 2017 were obtained.