

Abstract
Background and Purpose
Numerous claims are made for cannabis' therapeutic utility upon human seizures, but concerns persist about risks. A potential confounder is the presence of both Δ9‐tetrahydrocannabinol (THC), variously reported to be pro‐ and anticonvulsant, and cannabidiol (CBD), widely confirmed as anticonvulsant. Therefore, we investigated effects of prolonged exposure to different THC/CBD cannabis extracts on seizure activity and associated measures of endocannabinoid (eCB) system signalling.
Experimental Approach
Cannabis extract effects on in vivo neurological and behavioural responses, and on bioanalyte levels, were measured in rats and dogs. Extract effects on seizure activity were measured using electroencephalography telemetry in rats. eCB signalling was also investigated using radioligand binding in cannabis extract‐treated rats and treatment‐naïve rat, mouse, chicken, dog and human tissue.
Key Results
Prolonged exposure to cannabis extracts caused spontaneous, generalized seizures, subserved by epileptiform discharges in rats, but not dogs, and produced higher THC, but lower 11‐hydroxy‐THC (11‐OH‐THC) and CBD, plasma concentrations in rats versus dogs. In the same rats, prolonged exposure to cannabis also impaired cannabinoid type 1 receptor (CB1 receptorhttp://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=56)‐mediated signalling. Profiling CB1 receptor expression, basal activity, extent of activation and sensitivity to THC suggested interspecies differences in eCB signalling, being more pronounced in a species that exhibited cannabis extract‐induced seizures (rat) than one that did not (dog).
Conclusions and Implications
Sustained cannabis extract treatment caused differential seizure, behavioural and bioanalyte levels between rats and dogs. Supporting radioligand binding data suggest species differences in eCB signalling. Interspecies variations may have important implications for predicting cannabis‐induced convulsions from animal models.
Linked Articles
This article is part of a themed section on 8th European Workshop on Cannabinoid Research. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.10/issuetoc
Abbreviations
- CBD
cannabidiol
- EEG
electrocorticography
- eCB
endocannabinoid
- PCA
principal component analysis
- PSD
power spectrum density
- THC
Δ9‐tetrahydrocannabinol
Introduction
Recent legal and regulatory change in the USA and elsewhere has increased awareness, and use of, cannabis (marijuana) for recreational and potential medicinal purposes, including treatment‐resistant paediatric epilepsies (Devinsky et al., 2014; 2015). Such cannabis preparations typically contain significant amounts of Δ9‐tetrahydrocannabinol (THC), a high potency, low intrinsic efficacy, CB1 receptor partial agonist; however, there is little evidence of THC efficacy or safety in epilepsy (Press et al., 2015). Moreover, reports are tempered by psychiatric complications of cannabis in adolescents (Volkow et al., 2014) and medical and psychiatric emergencies, including seizures and mortality, among recreational users of novel synthetic CB1 receptor high intrinsic efficacy agonists (Castaneto et al., 2014; Gurney et al., 2014). Short‐term exposure to CB1 receptor partial or full agonists typically exerts anticonvulsant effects in animal models of seizure and epilepsy (Rosenberg et al., 2015). By contrast, sustained THC administration is reported to cause convulsions in rats and mice (Chan et al., 1996; NTP, 1996) and, anecdotally, chickens. The other most common cannabinoid, cannabidiol (CBD), is not psychoactive, is widely confirmed as anticonvulsant in animal models of seizure and epilepsy and lacks reported proconvulsant effects (Rosenberg et al., 2015). CBD reduces convulsive seizures in children and young adults with Dravet syndrome and with Lennox–Gastaut syndrome (Devinsky et al., 2016; 2017). A meta‐analysis found that CBD behavioural pharmacology is unrelated to direct effects at CB1 receptors (McPartland et al., 2015), although indirect CBD effects on the endocannabinoid (eCB) system, as well as negative allosteric modulation of CB1 receptors in vitro (Laprairie et al., 2015) have been reported; rather, CBD has several potential non‐CB1 receptor‐mediated actions (Hill et al., 2012).
Despite this knowledge, it remains unknown whether sustained cannabis‐induced convulsions are spontaneous and/or epileptiform in nature, with reports of both depressed and enhanced epileptogenesis in animal models (Rosenberg et al., 2017). Moreover, no relationship between convulsion incidence and other aberrant behaviours has been established. The extent to which changes in eCB signalling that may be involved in cannabis‐induced convulsions in rodents are recapitulated in other species also remains to be elucidated. With these points in mind, we examined the effects of standardized extracts containing different doses of THC and CBD on in vivo behaviour and seizure activity in rats and dogs, species reportedly prone and resistant to cannabis‐induced seizure respectively. We demonstrate for the first time that prolonged exposure to cannabis extracts produces dose‐related motor convulsions subserved by epileptiform activity and associated seizure‐related behaviours in rats. By contrast, cannabis extracts never caused seizures in dogs, which exhibited reduced THC but higher 11‐OH‐THC and CBD plasma concentrations than rats. Across several species, the eCB system signalling profile was highest in the rat but lowest in the dog. These data clarify several apparent inconsistencies in the field and suggest that choice of model species has important implications in the study of cannabis‐induced convulsions.
Methods
Animals
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Rodent behavioural studies were conducted under contract by Covance Laboratories Ltd (Leeds, UK) according to the authors' experimental design and in accordance with the UK Animals (Scientific Procedures) Act 1986. Sixty‐nine adult (240–280 g at study start) female Wistar–Han rats were used. Female rats were used here since they are reported to have an increased frequency of THC‐induced convulsions compared to male rats (NTP, 1996). Rats, habituated for 16 days prior to the start of any experimental procedures, were singly housed in standard laboratory cages with environmental enrichment and provided access to food (RM1.(E).SQC.; SDS Ltd., Witham, UK) and water ad libitum throughout in an environment of 20–24°C, 45–65% humidity and a 12:12 h light : dark period.
Canine behavioural studies were conducted in accordance with good laboratory practice standards (US FDA Good Laboratory Practice Regulations 21 CFR Part 58) under contract by CIT Safety and Health Laboratories (Évreux, France) in accordance with EU Directive 86/609/EEC and to the authors' experimental design. A total of 40 [20 male (mean weight: 9.5 kg) and 20 female (mean weight: 7.8 kg)] adult (8 months) beagle dogs (Marshall Farms, NY, USA) were habituated for 2 weeks and maintained at 20°C, 30–70% humidity and a 12 h:12 h light : dark period in individual kennels containing wood shavings (SICSA, Leon, France) and provided free access to water plus ~300 g·day−1 pelleted diet (125 C3; SAFE, Augy, France). At the end of each behavioural study, animals were humanely killed 24 h after the final treatment, using an appropriate method, as described below. Adult, male C57BL/6 mice (n = 12), chickens (n = 12) and Wistar–Han rats (n = 6) were obtained from Charles River Ltd (Harlow, UK) and humanely killed in accordance with the UK Animals (Scientific Procedures) Act 1986 and associated guidelines for the humane use of experimental animals and approved by the University of Reading Animal Welfare and Ethical Review Body, to provide brain tissue for use in radioligand binding assays. Male beagle and male human cerebellae were supplied by Charles River (UK) and Asterand Bioscience (Herts, UK), respectively, and stored at −80°C until use. No distinct ethical approval was required for the use of beagle or human tissue since each was obtained from a licensed supplier.
Drugs and formulation
Standardized cannabis extracts (1.08:1 ratio of THC and CBD) were supplied by GW Research Ltd (river, UK). The extract's composition complied with the US Food and Drug Administration guidelines for botanical drug products.
Experimental design
Rat behavioural experiment
Rats were randomly allocated into three groups. One group received low‐dose (1.08 mg·kg−1 THC + 1 mg·kg−1 CBD in sesame oil; n = 25), while another received high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD in sesame oil; n = 25) cannabis extract. The dose levels used in rats were designed to lead to reported effective plasma concentrations (Deiana et al., 2012). A third group received vehicle (sesame oil; n = 19). Drugs (or vehicle) were administered once daily via p.o. gavage for 13 weeks (constant dose volume = 10 mL·kg−1 based on weekly animal weights. Five animals per group were humanely killed by a schedule 1 method (e.g. overdose of anaesthetic followed by cervical dislocation) at the end of each of day 2 and weeks 4, 8 and 13 for bioanalyte assessment and assessment of CB1R function.
Canine behavioural experiment
Dogs were randomized to five groups each containing eight animals (four males and four females) that received, via p.o. gavage daily, vehicle (ethanol (sufficient quantity), propylene glycol 50% v.v‐1 and peppermint oil 0.05% v.v‐1), sham treatment (purified water), low‐dose (2.7 mg·kg−1 THC + 2.5 mg·kg−1 CBD), intermediate‐dose (13.5 mg·kg−1 THC + 12.5 mg·kg−1 CBD) or high‐dose (27 mg·kg−1 THC + 25 mg·kg−1 CBD) cannabis extract treatment for 56 weeks (up to 4 weeks habituation + 52 weeks steady‐state treatment). Habituation to treatment in the intermediate‐ and high‐dose groups was as follows: high dose (doses expressed as mg·kg−1·day−1 THC/CBD): 5.4/5.0 (days 1–44), 8.1/7.5 (days 5–9), 10.8/10.0 (days 10–14), 13.5/12.5 (days 15–19), 16.2/15.0 (days 20–24) and 21.6/20.0 (days 25–28); intermediate dose: 2.7/2.5 (days 10–14), 5.4/5.0 (days 15 to 19), 8.1/7.5 (days 20–24) and 10.8/10.0 (days 25–28). We believe that this is the first study to investigate the effects of prolonged cannabis extract exposure on seizures in dogs; dose levels were selected on the basis of the results of a previous internal study in dogs with a similar route of administration and similar dose levels, which resulted in good systemic exposure via p.o. gavage administration. While acute cannabis intoxication in dogs has not been reported to cause seizures, there is some suggestion that longer‐term, higher‐dose cannabis may do so (Fitzgerald et al., 2013); therefore, we tested the effects over a 52 week period in dogs to ensure maximum possibility of detecting cannabis extract‐induced seizure activity. On completion of the treatment or treatment‐free period, all surviving dogs were anaesthetized by an i.v. injection of thiopental sodium and killed by exsanguination.
Collection of behavioural and telemetry measures
In rats, a subgroup (vehicle: n = 4; low dose: n = 10; high dose: n = 10) was assessed by researchers trained to identify, code and discriminate between convulsive behaviours according to conventionally used rodent welfare criteria (Wolfensohn & Lloyd, 2013). Behaviours associated with generalized seizures in rodents included tonic or clonic convulsions, myoclonic jerk, forelimb paddling, forelimb clonus, forelimb flickering, popping (involuntary movement characterized by repeated and typically rhythmic jumping and/or twitching that can range from stationary hiccough‐like movements to vigorous jumping) (Mastropaolo et al., 2004), wet dog shakes, tremor, twitching and chewing (Luttjohann et al., 2009). Behaviours not typically associated with seizure: piloerection, ptosis, digit biting, increased grooming, increased scratching, mouth rubbing, behavioural arrest, fasciculations, writhing, licking, salivation, hind limb extension, head searching, hunched posture and exophthalmos. Since THC‐induced convulsions in rodents have been suggested to be associated with the act of drug administration and/or handling (Chan et al., 1996; NTP, 1996), rats were observed undisturbed in the home cage for at least 5 min and for a further 5 min after removal from the home cage before final observation for at least 10 min after treatment had been administered each day. Behaviours were grouped into two categories: ‘acute’ (during the 10 min observation period following daily dosing) and ‘persistent’ (during the 10 min in the home cage prior to daily treatment). The subgroup of rats was obtained with F40‐EET (DSI, New Brighton, MN, USA) telemetry transmitters and electrocorticography (EEG) electrodes already surgically implanted by Charles River (Cambridge, UK). Electrodes comprised two subcranial (dural) wires (frontal cortex AP +4.7 mm and ML −0.5 mm; parietal cortex AP −3.8 mm and ML −3.0 mm, c.f. bregma), and EEG data were collected for 22 h periods on each of 10 pre‐specified days during the study (day −1 plus 1 day from each of the following day pairs: 28/29, 35/36, 42/43, 56/57, 63/64, 70/71, 77/78, 84/85 and 90/91). No animals exhibited unusual EEG activity in recordings taken during the 22 h prior to first treatment. Full details of EEG recording and signal processing approaches are described in the Supporting Information.
Dogs were examined for mortality, signs of morbidity and conventional clinical signs, including seizure behaviour, twice daily throughout the study period.
Analysis of drug and metabolite levels
In rats, terminal venous blood (~0.5 mL) was obtained into a heparin‐containing polypropylene tube. Samples were mixed for ~2 min, placed on ice, centrifuged (~2300 g, 4°C, 10 min) within 30 min of collection and plasma stored in polypropylene tubes (−20°C) until analysis. Each rat brain was rapidly removed after death, cerebrum and cerebellum separated, flash frozen in liquid nitrogen and stored at −80°C. Venous canine blood was sampled immediately prior to the animals being killed in heparin‐containing polypropylene tubes, centrifuged (~2300 × g, 4°C, 10 min) and plasma stored in polypropylene tubes at −20°C until analysis. Prior to analysis, rodent (cerebellum only) and canine brain samples were homogenized (Lysing Matrix D, MP Biomedical, Santa Ana, California, USA) in methanol and water (20:80 v.v‐1) on ice using a FastPrep (MP Biomedical) for ~60 s. Plasma (rat and dog) and brain (rat) concentrations of THC, 11‐OH‐THC (Lemberger et al., 1973) and CBD were determined, while plasma and brain concentrations of 6‐hydroxy‐CBD (6‐OH‐CBD) and 7‐hydroxy‐CBD (7‐OH‐CBD) were also determined for rats, using UPLC‐MS/MS in all cases. Details of sample preparation and analysis are included in the Supporting Information.
Radioligand binding
Detailed methodology for membrane preparation, [3H]‐SR1416717A saturation binding and GTPγS assays are included in the Supporting Information. Membranes were prepared from all cerebellae tissue used in the rat cannabis extract treatment study; cerebellae tissue was used due to bioanalyte levels being measured from cerebellae tissue in these experiments and also due to high CB1 receptor expression in rat cerebellum (Tsou et al., 1998). Standardized cerebellar membrane preparations were also produced from male treatment‐naïve C57BL/6J mice, chickens, Wistar–Han rats, beagles and humans, also due to uniformly high CB1 recepor expression in cerebellar tissue across different mammalian species (Herkenham et al., 1990).
Saturation binding
The high affinity antagonist [3H]‐SR1416717A (pK D 8.9–10, Alexander et al., 2017) was used, assays were conducted in triplicate and three separate assays were performed in each case. Radioactivity bound to cortex membranes was quantified in disintegrations per minute (dpm) before conversion to pmol·mg−1. Analyses of saturation binding data were conducted by non‐linear regression and fitted to a one‐binding site model to determine the equilibrium K D (nM) and the maximal number of binding sites Bmax (pmol·mg−1) using GraphPad Prism software (GraphPad Software Inc., San Diego, CA, USA).
[35S]‐GTPγS binding assay
Assays were carried out in triplicate, and three separate assays were performed in each case. [35S]‐GTPγS assay data were analysed using GraphPad Prism. Concentration–response data were analysed using a sigmoidal concentration–response model or linear regression and compared using an F‐test to select the appropriate model. On this basis, best fits to sigmoidal curves were obtained with Hill slopes of unity, and no other constraints applied. For curves showing no concentration‐related increases, linear regression was performed to determine if slopes differed significantly from zero. Values for EC50 were derived from fitted curves to mean data and Emax expressed as percentage over basal or as percentage of the mean maximal response following stimulation with the highest concentration of the CB1 receptor full agonist, WIN55,212‐2 (10 μM). In experiments that examined the effects of agonist stimulation in membranes prepared from drug‐treated animals and where mathematically possible, data were fitted to an operational model of ligand binding (Black & Leff, 1983). Here, dpm were plotted, and the tissue‐agonist combination that yielded the largest maximal stimulation was identified (i.e. WIN55,212‐2 responses in vehicle‐treated animals). The magnitude of this highest maximal stimulation was used to scale (0–100%) other tissue–agonist combinations. Prior to scaling, basal stimulation was subtracted to constrain the bottom of all derived curves to zero. In experiments that examined differences between tissues from different species using [35S]‐GTPγS assays, prior to normalization, data expressed as dpm were plotted in order to assess any differences arising from expression and sensitivity levels.
We produce a descriptive representation of the overall profile of THC‐mediated CB1 receptor‐mediated signalling for each species (which we term the ‘eCB signalling footprint’) by normalizing (i) CB1 receptor expression (Bmax; ‘Expression’), (ii) basal G‐protein turnover (dpm at the lowest concentration of THC; ‘Basal’), (iii) sensitivity (EC50; ‘Sensitivity’) and (iv) activation (Emax; ‘Extent’) in response to agonist stimulation, to the species with the highest value for each measure.
Drugs
The following drugs were used: WIN55,212‐2 (Tocris, Bristol, UK), [3H]‐SR141716A and [35S]‐GTPγS (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK).
Randomization and blinding
Canine and rat behavioural studies and canine and rat bioanalyte studies were conducted in accordance with industry‐standard good laboratory practice and additional regulatory compliances as detailed above. Such compliance ensures randomization of animals to each specified group and appropriate blinding. For canine studies (CIT Safety and Health Laboratories), a computerized randomization procedure (using validated CIT software) was used. For rat studies (Covance, Leeds, UK), animals were identified by numbered tail marks and electronic ID; prior to the start of the study, animals were randomly allocated to treatment groups and individually tattooed by Charles River. In all cases, operators were blinded to treatment. For in vitro binding studies, membrane preparations were randomly selected by the operator; here, all parameters stated are measured numerical values, which were not influenced by any observer‐related bias, and therefore, blinding was not considered to be necessary.
Statistics
Data subjected to statistical comparisons did not violate assumptions of normality (D'Agostino–Pearson omnibus test) and are expressed as mean ± SEM. Group sizes for data subjected to statistical comparisons were designed on the basis of power calculations to identify differences between cannabis extract doses. Groups were compared by one‐ or two‐way ANOVA tests followed by Tukey's post hoc tests as appropriate using GraphPad Prism; post hoc tests were run only if F achieved P < 0.05 and there was no significant variance in homogeneity. A variance–covariance principal component analysis (PCA) of animal behavioural data was undertaken using XLSTAT (New York, NY, USA); this analysis used daily data normalized to the proportion of animals per group exhibiting any given behaviour before calculation of a group mean value for each behaviour for the 13 week treatment period. Interpretation of the variance described by the first and second principal components was undertaken by examination of the squared cosines and percentage contributions of each variable to the total variance (see Supporting Information). In accordance with the journal policy, P < 0.05 was reported as level of significance. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Effects of cannabis extract treatment on plasma and brain cannabinoid bioanalyte levels
Concentrations of THC and its active metabolite, 11‐OH‐THC, plus CBD and its metabolites, 6‐OH‐CBD and 7‐OH‐CBD, in plasma and cortical homogenate from rats that received 13 weeks' vehicle, low‐dose (1.08 mg·kg−1 THC and 1 mg·kg−1 CBD) or high‐dose (40.5 mg·kg−1 THC and 37.5 mg·kg−1 CBD) cannabis extract treatment via p.o. gavage were measured (Table 1). With the exception of 6‐OH‐CBD, which was not detected in any samples from any group, all other bioanalytes were identified in at least one group of rats that received low‐ or high‐dose cannabis extract treatment. One‐way ANOVA tests revealed a significant effect of group upon THC, 11‐OH‐THC, CBD and 7‐OH‐CBD concentrations in brain and plasma, which arose from a significant increase in level of each cannabinoid and metabolite present in the high‐dose, in comparison with low‐dose, cannabis extract‐treated groups (Table 1). THC, 11‐OH‐THC and CBD (but not 6‐OH‐CBD and 7‐OH‐CBD) were present at detectable levels in the low‐dose group.
Table 1.
Cannabis extract treatment phytocannabinoid and metabolite concentrations in plasma from rats and dogs and cortex homogenate from rats
| THC | 11‐OH‐THC | CBD | 6‐OH‐CBD | 7‐OH‐CBD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Plasma (ng·mL−1) | Brain (ng·g−1) | Plasma (ng·mL−1) | Brain (ng·g−1) | Plasma (ng·mL−1) | Brain (ng·g−1) | Plasma (ng·mL−1) | Brain (ng·g−1) | Plasma (ng·mL−1) | Brain (ng·g−1) | |
| Rat | ||||||||||
| Vehicle | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Low cannabis dose | 1.25 ± 0.30 | 2.55 ± 0.24 | 0.75 ± 0.19 | 3.12 ± 0.51 | 0.31 ± 0.06 | 0.09 ± 0.09 | 0 | 0 | 0 | 0 |
| High cannabis dose | 226.10 ± 17.24* | 614.30 ± 50.86* | 24.07 ± 5.04* | 63.77 ± 7.46* | 97.81 ± 9.85* | 161.7 ± 38.97* | 0 | 0 | 28.80 ± 2.81* | 7.42 ± 0.94* |
| Dog | ||||||||||
| Sham | 0 | – | 0 | – | 0 | – | – | – | – | – |
| Vehicle | 0 | – | 0 | – | 0 | – | – | – | – | – |
| Low cannabis dose | 22.02 ± 6.16 | – | 6.66 ± 4.48 | – | 50.76 ± 30.10 | – | – | – | – | – |
| Intermediate cannabis dose | 60.48 ± 9.01* | – | 31.85 ± 10.1* | – | 201.3 ± 39.03* | – | – | – | – | – |
| High cannabis dose | 70.40 ± 11.49* | – | 45.87 ± 11.14* | – | 318.3 ± 58.30* | – | – | – | – | – |
Phytocannabinoid and selected metabolite concentrations in plasma from rats and dogs and cortex homogenate from rats, following daily p.o. administration of vehicle/sham or cannabis extract treatment. Rats received vehicle (n = 4), low‐ (1.08 mg·kg−1 THC + 1 mg·kg−1 CBD; n = 10) or high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD; n = 10) cannabis extract for 13 weeks. Dogs (n = 8 per group) received vehicle, sham treatment, low‐dose (2.7 mg·kg−1 THC + 2.5 mg·kg−1 CBD), intermediate‐dose (13.5 mg·kg−1 THC + 12.5 mg·kg−1 CBD) or high‐dose (27 mg·kg−1 THC + 25 mg·kg−1 CBD) cannabis extract for 56 weeks. Values are mean ± SEM. Values shown as zero reflect results below the limit of quantification.
P < 0.05 for planned pairwise comparisons between low‐dose, intermediate‐dose (where used) and/or high‐dose groups using Tukey's post hoc tests following a one‐way ANOVA test.
Concentrations of THC, 11‐OH‐THC and CBD in canine plasma samples from sham, vehicle, low‐dose (2.7 mg·kg−1 THC and 2.5 mg·kg−1 CBD), intermediate‐dose (13.5 mg·kg−1 THC and 12.5 mg·kg−1 CBD) and high‐dose (27 mg·kg−1 THC and 25 mg·kg−1 CBD) cannabis extract‐treated groups were measured (Table 1). All bioanalytes were identified in at least one group that received low‐, intermediate‐ or high‐dose treatment; one‐way ANOVA tests revealed a significant effect of group upon THC, 11‐OH‐THC and CBD concentrations in plasma, which arose from significant increases in cannabinoid levels in the intermediate‐ and high‐dose, in comparison with low‐dose, cannabis extract‐treated groups. THC, 11‐OH‐THC and CBD were detectable in the low‐dose group in canines. No evidence for any cannabinoids was found in sham‐ or vehicle‐treated samples. Overall, relative to dose administered, the THC plasma concentration was higher in rats than dogs; although the active metabolite 11‐OH‐THC, and CBD, was higher in dogs. Together, these data demonstrate that p.o. administration of higher dose cannabis extracts is effective in producing increased physiologically relevant levels of major cannabinoids and metabolites and that bioanalyte profiles differed between rats and dogs. These data provided a validated basis to study dose‐dependency of seizure induction by cannabis extracts and permit qualitative comparisons between species.
Effects of cannabis extract treatment upon behaviours in rats and dogs
Low‐dose cannabis extract treatment produced acute (observed during the 10 min period following daily dosing) behavioural signs in rats from day 17, which continued throughout the 13 week treatment (Figure 1). The most frequently observed acute effects (descending order of magnitude of the median proportion exhibiting a behaviour during the treatment period) were mouth rubbing, forelimb paddling, increased scratching, wet dog shakes, forelimb flickering, increased grooming, ptosis and chewing; less frequent effects (median incidence of zero but with non‐zero interquartile range) were writhing and salivation (Table 2 and Supporting Information Table S1). Persistent (observed during the 10 min prior to daily treatment) behavioural effects of low‐dose cannabis extract in rats occurred on day 18 and continued throughout the 13 week treatment (Figure 2). Here, the most frequently observed effects were increased grooming, increased scratching and ptosis; less frequent effects were wet dog shakes, forelimb flickering, forelimb paddling and writhing (Table 2 and Supporting Information Table S1). In the high‐dose group, acute behavioural signs in rats were observed from day 17 and continued throughout the 13 week treatment (Figure 1). The most frequently observed acute effects were forelimb paddling, mouth rubbing, ptosis, increased scratching, piloerection, wet dog shakes, forelimb flickering, chewing, salivation and increased grooming; less frequently, behavioural arrest and twitch were observed (Table 2 and Supporting Information Table S1). The first persistent behavioural effects of high‐dose treatment in rats were seen from day 18 and also continued throughout the 13 week treatment (Figure 2). Here, the most frequently observed behaviours were increased scratching, increased grooming, piloerection and wet dog shakes; less frequently, ptosis and forelimb paddling were observed (Table 2 and Supporting Information Table S1). No vehicle‐treated animals exhibited any of the behaviours coded (results from this group are omitted for clarity). The mean values for the normalized incidence of each of the behaviours exhibited by cannabis extract‐treated rats, prior to and after treatment administration (Figures 1 and 2 and Supporting Information Table S2), were subjected to PCA. The first three components accounted for 100% of the variability of which 90.1 and 8.4% of variability was accounted for by the first and second principal components respectively. Mouth rubbing and forelimb paddling behaviours made a cumulative contribution of 68.3% to the first principal component with corresponding squared cosine values >0.9 (Supporting Information Table S2). A biplot of the first two principal components revealed a positive correlation between ‘acute’ behaviours and the first principal component, while the converse applied to ‘persistent’ behaviours (Figure 3A); the first principal component therefore represents behaviours associated with administration of the drug. The second principal component positively correlated with high‐dose cannabis extract treatment, indicating that this measure is dose related. A circle plot of these data (Figure 3B) revealed that several behaviours were strongly associated with acute exposure to cannabis extract, irrespective of dose. The circle plot revealed that several behaviour characteristics of generalized seizures such as popping, convulsion, myoclonic jerk and twitch were positively correlated with the high‐dose group, while the converse applied to increased grooming and writhing (Figure 3B). Notably, myoclonic jerk and convulsion were independent, while popping, increased grooming and writhing were negatively associated, with acute measurement of behaviour. The remaining behaviours showed no overt dose‐dependency.
Figure 1.
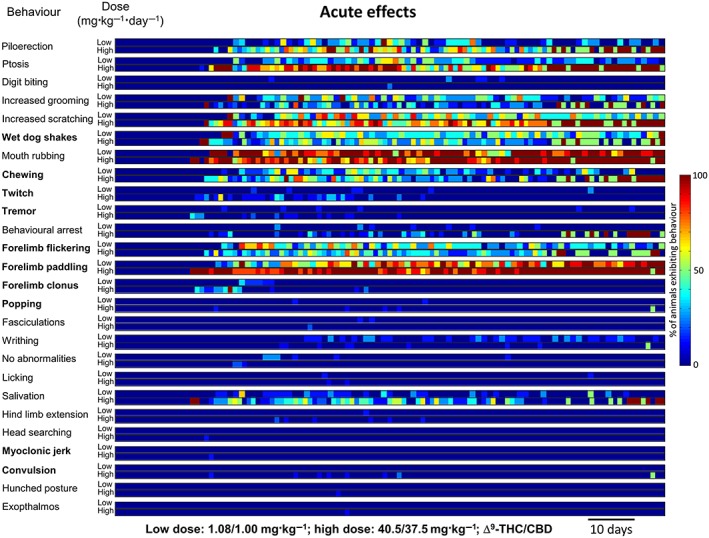
Temporal representation of acute behaviours in rats (n = 10 per group) 10 min after daily p.o. low‐dose [1.08 mg·kg−1 THC (Δ9‐THC) + 1 mg·kg−1 CBD] or high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD) cannabis extract treatment for 13 weeks. Behavioural events associated with generalized seizures in rodents are highlighted in bold.
Table 2.
Incidence of acute and persistent cannabis extract‐induced behaviours in rats
| Rank | Low dose (1.08/1.00 mg·kg−1 THC/CBD) | High dose (40.5/37.5 mg·kg−1 THC/CBD) | ||||||
|---|---|---|---|---|---|---|---|---|
| Acute | Median % (IQR) | Persistent | Median % (IQR) | Acute | Median % (IQR) | Persistent | Median % (IQR) | |
| 1 | Mouth rubbing | 87.5 (62.5–100) | Increased grooming | 50 (23–67) | Forelimb paddling | 90 (80–100) | Increased scratching | 40 (25–60) |
| 2 | Forelimb paddling | 62.5 (31–88) | Increased scratching | 37.5 (12.5–50) | Mouth rubbing | 87.5 (62.5–100) | Increased grooming | 25 (11–44) |
| 3 | Increased scratching | 37.5 (12.5–50) | Ptosis | 12.5 (0–12.5) | Ptosis | 62.5 (32.5–90) | Piloerection | 10 (0–22) |
| 4 | Wet dog shakes | 37.5 (12.5–50) | Wet dog shakes | 0 (0–12.5) | Increased scratching | 62.5 (37.5–80) | Wet dog shakes | 10 (0–12.5) |
| 5 | Forelimb flickering | 37.5 (9.5–50) | Forelimb flickering | 0 (0–12.5) | Piloerection | 37.5 (8–60) | Ptosis | 0 (0–20) |
| 6 | Increased grooming | 25 (0–40.5) | Forelimb paddling | 0 (0–12.5) | Wet dog shakes | 25 (10–40) | Forelimb paddling | 0 (0–11) |
| 7 | Ptosis | 12.5 (0–37.5) | Writhing | 0 (0–12.5) | Forelimb flickering | 25 (12.5–37.5) | – | – |
| 8 | Chewing | 12.5 (0–27) | – | – | Chewing | 22.2 (0–40) | – | – |
| 9 | Writhing | 0 (0–12.5) | – | – | Salivation | 20 (0–40) | – | – |
| 10 | Salivation | 0 (0–12.5) | – | – | Increased grooming | 12.5 (0–25) | – | – |
| 11 | – | – | – | – | Behavioural arrest | 0 (0–11) | – | – |
| 12 | – | – | – | – | Twitch | 0 (0–10) | – | – |
Incidence of behaviours in rats (n = 10 per group) observed immediately after 10 min (acute) or ~23 h after (persistent) daily p.o. administration of low‐dose (1.08 mg·kg−1 THC + 1 mg·kg−1 CBD) or high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD) cannabis extract for 13 weeks. Behaviours are ranked by descending magnitude of median incidence over 13 weeks where the calculated median was non‐zero. Thereafter, behaviours are ranked by descending order of magnitude of the 75th percentile where the IQR was non‐zero. Behaviours exhibiting a median and IQR of zero are shown in Supporting Information Table S1. Behavioural events conventionally associated with generalized seizures in rodents are highlighted in bold. IQR, interquartile range.
Figure 2.
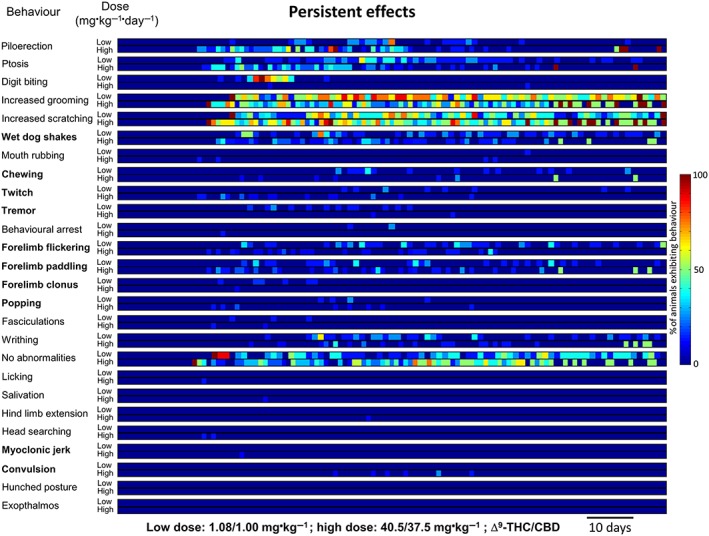
Temporal representation of persistent behaviours in rats (n = 10 per group) ~23 h after daily p.o. administration of low‐dose [1.08 mg·kg−1 THC (Δ9‐THC)+ 1 mg·kg−1 CBD] or high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD) cannabis extract treatment for 13 weeks. Behavioural events associated with generalized seizures in rodents are highlighted in bold.
Figure 3.
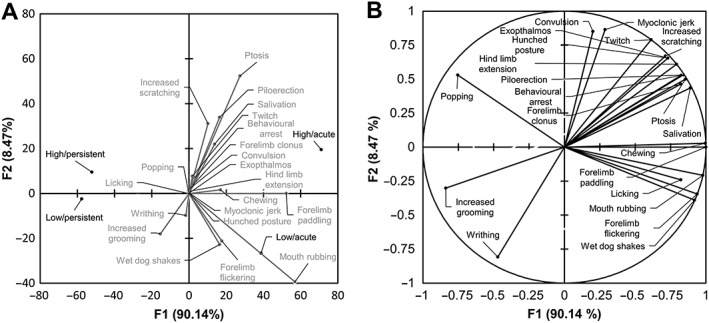
(A) Biplot of the first two principal components (F1 and F2) derived from daily behavioural data (see Methods). Positive values of the first principal component were positively correlated with observations made in the period shortly after dosing (acute), irrespective of dose, whereas the converse applied to observations made prior to daily treatment (persistent). Positive values of the second principal component were positively correlated with observations made in animals that received high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD; n = 10) cannabis extract, irrespective of dose timing, whereas the converse applied to observations made in animals that had received low‐dose (1.08 mg·kg−1 THC + 1 mg·kg−1 CBD; n = 10) cannabis extract. (B) Correlation plot showing association of behaviours with the first and second principal components.
In dogs, no behaviours associated with seizures were seen. There were seven unscheduled deaths across all groups except the sham control and low‐dose groups, which occurred as follows: vehicle group (1 female; day 361), intermediate group (3 male; days 31, 315 and 339) and high group (3 male; days 14, 30 and 221). The mortality pattern showed no measurable dose relationship, and post‐mortem examination attributed mortality to clinical complications following reflux and aspiration of stomach contents and/or formulated treatment into the lungs rather than a drug‐related effect. During the habituation period, ptyalism was observed in vehicle, intermediate‐ and high‐dose groups, which continued in the steady‐state period, during which time this sign was also noted in the low dose and, to the least extent, sham groups (Table 3). Incidence of ptyalism was not dose related and was attributed to vehicle excipients. Other clinical signs seen during habituation occurred predominantly in the high‐dose group and could be assigned to two categories: (i) hypoactivity, ataxia and tremor (from day 2) and (ii) abdominal breathing, tachypnoea, lateral recumbency, reflux at dosing, vomiting, soft or liquid faeces and dehydration (from day 18). During the steady‐state treatment period, dogs exhibited clinical signs (Table 3) divided into four categories: (i) dose‐related neurological signs (ataxia, tremor and hypoactivity) occurred primarily in cannabis extract‐treated groups but with decreased frequency compared with the habituation phase; (ii) thin appearance manifested without clear treatment, time, dose or sex relationship, and all dogs consumed ≥75% of food offered each day; (iii) gastrointestinal signs, occurred primarily in cannabis extract‐treated groups with dose‐related incidence; (iv) oro‐respiratory signs (ptyalism, dyspnoea and abdominal breathing). Ptyalism occurred in all groups at steady state but was highest in cannabis extract‐treated groups, suggesting attribution to the excipients. Overall, in dogs, cannabis extract, even at the highest dose tested, caused limited neurological, gastrointestinal and oro‐respiratory behavioural signs. Most importantly, convulsive episodes were never observed in dogs from any group, and repeated drug treatment was well tolerated.
Table 3.
Incidence of cannabis extract‐induced behaviours in dogs
| THC/CBD dose (mg·kg−1·day−1) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sham | Vehicle | 2.7/2.5 | 13.5/12.5 | 27/25 | ||||||||||||||||
| Sex | ||||||||||||||||||||
| M | F | M | F | M | F | M | F | M | F | |||||||||||
| Phase | ||||||||||||||||||||
| H | SS | H | SS | H | SS | H | SS | H | SS | H | SS | H | SS | H | SS | H | SS | H | SS | |
| Number of survivors | ||||||||||||||||||||
| 6 | 6 | 6 | 5 | 4 | 4 | 2 | 4 | 3 | 6 | |||||||||||
| Ptyalism | – | 2 | – | 2 | – | 5 | 2 | 4 | – | 4 | – | 4 | 5 | 2 | 2 | 4 | 5 | 3 | 4 | 6 |
| Reflux at dosing | – | – | – | – | – | – | – | – | – | – | – | – | 1 | – | – | – | 1 | – | – | – |
| Abdominal breathing | – | – | – | – | – | 1 | 1 | – | – | – | – | – | – | 1 | – | – | 1 | – | – | – |
| Dyspnoea | – | – | – | – | – | 1 | 1 | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Tachypnoea | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | – | – | – |
| Hypoactivity | – | – | – | – | – | 1 | 1 | – | 1 | – | – | 2 | 1 | 1 | 1 | 4 | 3 | 1 | – | 1 |
| Lateral recumbency | – | – | – | – | – | – | 1 | – | – | – | – | – | 1 | – | – | – | 2 | – | – | – |
| Ataxia | – | – | – | – | – | – | – | – | – | 2 | – | 1 | – | – | – | – | 5 | – | 2 | – |
| Tremor | – | – | – | – | – | – | – | – | – | – | – | – | 1 | – | 1 | – | – | – | 1 | – |
| Vomiting | – | – | – | – | – | 1 | 1 | – | – | 2 | – | 1 | 2 | 1 | – | 3 | 4 | 2 | 3 | 5 |
| Soft/liquid faeces | – | – | – | – | – | – | 1 | – | – | – | – | 1 | 2 | – | – | – | 2 | 3 | 1 | 3 |
| Dehydration | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | – | – | 1 |
| Thin appearance | – | 1 | – | – | – | 1 | – | – | – | – | – | 1 | – | – | – | 1 | – | 1 | – | 1 |
The incidence of clinical signs observed in dogs (n = 8 per group) treated (daily; p.o.) with sham (purified water), vehicle, low‐dose (2.7 mg·kg−1 THC + 2.5 mg·kg−1 CBD), intermediate‐dose (13.5 mg·kg−1 THC + 12.5 mg·kg−1 CBD) or high‐dose (27 mg·kg−1 THC + 25 mg·kg−1 CBD) cannabis extract during H (weeks 1–4) and SS (weeks 5–56) periods. – indicates no clinical sign noted for a given group. H, habituation; SS, steady‐state.
EEG and seizure analysis in rats
Visually identified motor convulsions (Racine stage: ≥3; Jones et al., 2010, 2012) occurred in 80% (8/10) of rats in the high‐dose cannabis extract group; by contrast, motor convulsions were never observed in the low‐dose or vehicle treatment groups. A total of 24 motor convulsions were observed in the high‐dose group where average time from start of treatment to first convulsion was 50.5 ± 7.5 days; convulsions continued until the end of the study (Supporting Information Table S3). Of the 24 convulsive events, 17 occurred during drug administration or the subsequent observation period, while the remaining 7 events occurred prior to animal handling or were detected after review of video data when EEG analysis revealed an epileptiform event. EEG recordings from the high‐dose group revealed 18 events exhibiting epileptiform activity (handling‐related artefacts rendered 2/18 recordings unsuitable for presentation and are omitted) (Figure 4 and Supporting Information Table S3). Video data or direct observation showed that 15/18 (~80%) of epileptiform events were accompanied by a motor convulsion (Racine stage: ≥3) from which each animal recovered without intervention. By contrast, only one animal (out of 10) from the low‐dose group exhibited an epileptiform event (Figure 5A and Supporting Information Table S3); although the electrophysiological profile of this event was consistent with events seen the high‐dose group (Figure 4 vs. Figure 5A, B), video data from this one animal did not reveal an accompanying motor convulsion. All epileptiform events exhibited rhythmic, large amplitude, sharp wave activity of increasing amplitude prior to spontaneous termination (c.f. pretreatment baseline activity; Figures 4 and 5A, B) that persisted for 55 ± 7.6 s (n = 17). Spectrograms showed that all epileptiform activities induced by high‐dose and the single low‐dose example dominated the 1–20 Hz range where accompanying measures of power spectrum density (PSD) revealed 2–7 Hz peaks [Figure 5B, panel b, C (inset)]. Mean PSD confirmed that epileptiform activity induced by high‐dose cannabis extract exhibited a signal profile (Figure 5C) with peaks present at 2, 3 and 4.5 Hz, consistent with primary generalized seizures (Luttjohann et al., 2009). Together, these data suggest that sustained high‐dose treatment reliably caused motor convulsions, subserved by spontaneous epileptiform activity in rat.
Figure 4.
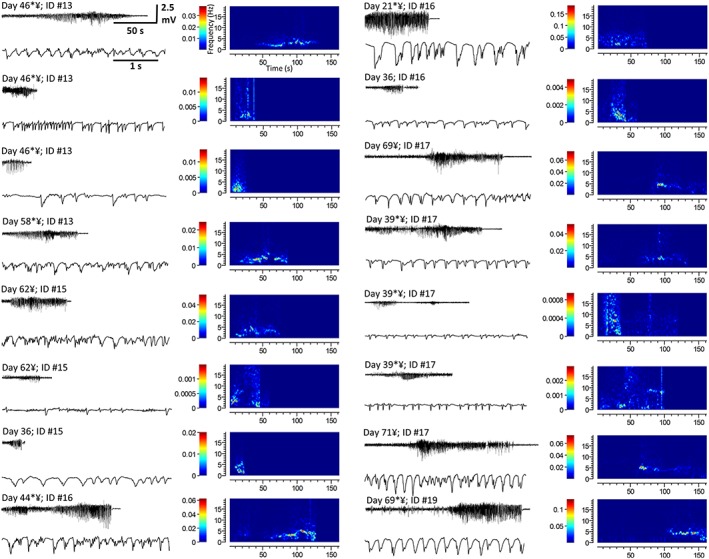
Epileptiform events recorded via EEG in rats treated with high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD) cannabis extract (Supporting Information Table S2). Each panel shows the EEG recording of the complete epileptiform event (top left), a shorter section of the event during the period of greatest amplitude activity represented on an extended timescale (bottom left) and a spectrographic representation of each event [right; x‐axis: time (s); y‐axis: frequency (Hz); and colour bar: power (dB·mV−2)]. * Indicates occurrence of a seizure during drug administration or within 10 min thereafter. ¥ Indicates an epileptiform event detected via EEG that was accompanied by a motor convulsion.
Figure 5.
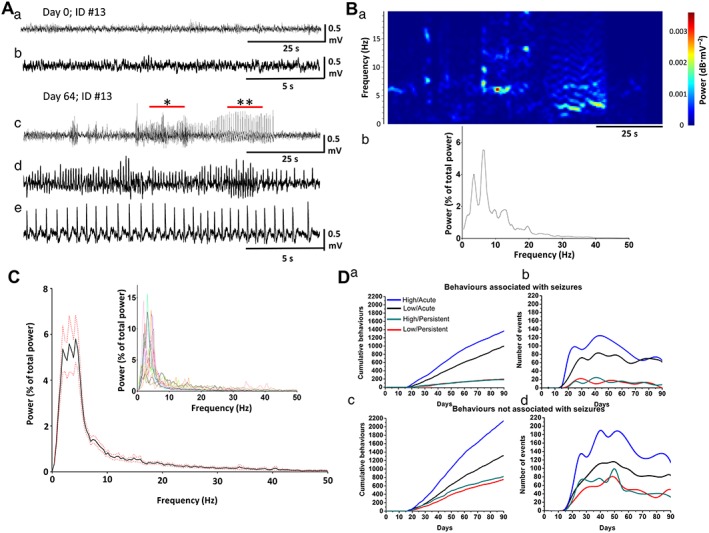
(A) EEG recordings from a rat treated with low‐dose (1.08 mg·kg−1 THC + 1 mg·kg−1 CBD) cannabis extract during (panels a, b) pretreatment baseline and (panels c, d) an epileptiform event. In panel c, * and ** show areas reproduced in panels d, e respectively. (B, panel a) Spectrographic representation of [A, panel c, right; x‐axis: time (s); y‐axis: frequency (Hz); and colour bar: power (dB·mV−2)] and (panel b) resulting PSD. (C) Mean (black) ± SEM (red dotted) PSD of epileptiform events recorded via EEG from animals treated with high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD; n = 10) cannabis extract. Inset shows overlay of individual PSD plots per event per animal. (D) (panel a) Cumulative incidence and (panel b) temporal distribution of coded behaviours associated with seizures in animals (see Methods) treated with low‐ or high‐dose cannabis extracts. (panel c) Cumulative incidence and (panel d) temporal distribution of coded behaviours not associated with seizures in animals (see Methods) treated with low‐ or high‐dose cannabis extracts.
Coded behaviours were re‐examined, and those consistent with primary generalized seizure in rats were pooled before calculation of cumulative incidence (Figure 5D, panel a) to reveal more frequent occurrence of acute than persistent seizure‐related behaviours in both low‐dose and high‐dose treatment groups. Irrespective of these acute effects, seizure‐related behaviours occurred more frequently in the high‐dose than the low‐dose group (Figure 3B). Further, when behaviours consistent with seizure (bold in Figures 1 and 2) were examined (Figure 5D, panel b), event incidence reached maximum levels at 40–50 days treatment before declining, irrespective of dose or time of observation (i.e. ‘acute’ or ‘persistent’). Some behaviours are not typically associated with seizure in rodents (not bold in Figures 1 and 2); nevertheless, their cumulative incidence (Figure 5D, panel c) and temporal distribution (Figure 5D, panel d) were similar to those of seizure‐associated behaviours (Figure 5D, panels a, b), suggesting a common underlying aetiology. These data suggest that behaviour signs in rat are increased during or immediately after handling/drug administration and that such variations should be considered when testing for drug effects in rodents.
Effects of cannabis extract treatment on CB1 receptor expression and G‐protein turnover in rat cerebellar membranes
CB1 receptor density was investigated in membranes from cerebellar brain tissue of all rats used in the above behavioural studies obtained at four time points (2 days and weeks 4, 8 and 13) during treatment with vehicle, low‐dose or high‐dose cannabis extract by saturation binding assay using the CB1 receptor antagonist, [3H]‐SR141716A, and expressed as Bmax (Figure 6A, panels a–d, B and Table 4). [35S]‐GTPγS binding was also examined in the same rat brain cerebellar membrane preparations from the 13 week treatment groups to assess CB1 receptor sensitivity to the partial agonist, THC, and the full agonist, WIN55,212‐2 (Figure 6C, panels a–d). Here, in membranes from vehicle‐treated animals, THC had a profile consistent with partial agonism (EC50: 69 nM; Emax: 27%), while WIN55,212‐2 exhibited a comparable EC50 but with a numerically greater Emax (EC50: 68 nM; Emax: 99%), consistent with its profile as a full agonist. In tissue from the low‐dose‐treated group, responses to THC (EC50: 40 nM; Emax: 31%) and WIN55,212‐2 (EC50: 53 nM; Emax: 93%) were similar to those seen in the vehicle‐treated group; this was in clear contrast to the high‐dose group, where the response to WIN55,212‐2 (EC50: 84 nM; Emax: 44%) was attenuated, and the THC response so markedly attenuated as to be too small for an EC50 value to be accurately derived, and Emax was depressed to ~7%. These results indicate that prolonged cannabis extract treatment clearly attenuates CB1 receptor‐mediated G‐protein signalling in rats with more profound effects in the high‐dose cannabis group.
Figure 6.
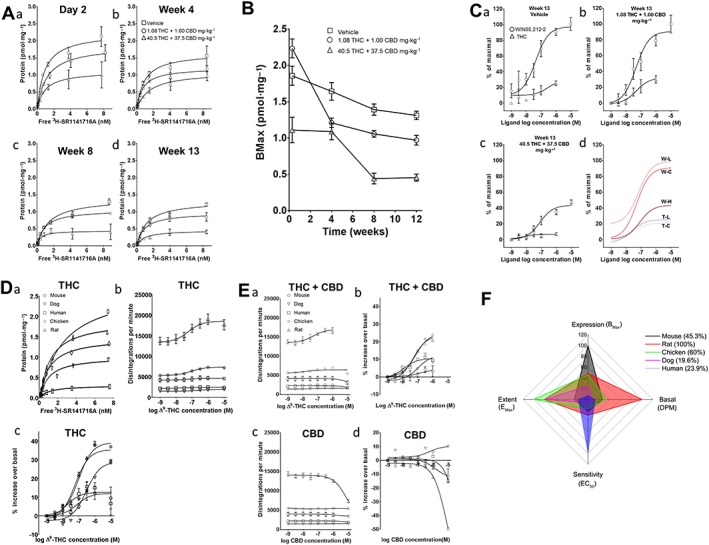
Saturation binding of [3H]‐SR141716A to cerebellar membranes from rats treated with vehicle, low‐dose (1.08 mg·kg−1 THC + 1 mg·kg−1 CBD) or high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD) cannabis extracts for (i) 2 days, (ii) 4, (iii) 8 and (iv) 13 weeks. (B) Temporal profile of Bmax (pmol·mg−1) derived from (A, panels a–d). (C) Log concentration–response best‐fit curves for stimulation of [35S]‐GTPγS binding by THC and WIN55 212‐2 in cerebellar membranes for week 13 data shown in (A). Data expressed as % maximal stimulation by 10 μM WIN55,212‐2 in vehicle group membranes fitted to an operational model of ligand binding (THC/high dose lack of response prevented valid curve derivation, and a subjectively assessed non‐linear fit was employed). (panel d) Overlay of best fit curves derived from panels a–c. W, WIN55,212‐2; T, THC; C, control (vehicle); L, low dose; H, high dose. No ‘T–H” curve presented. (D) (panel a) Saturation binding of [3H]‐SR141716A to human, chicken, dog, mouse and rat cerebellar membranes. (panel b) Log concentration–response curves for stimulation of [35S]‐GTPγS binding (dpm) by THC in cerebellar membranes from species indicated. (panel c) Log concentration–response curves for stimulation of [35S]‐GTPγS binding (normalized) by THC in cerebellar membranes from the species indicated. (E) (panel a) Equivalent (to D, panel b) curves for THC (Δ9‐THC)+ CBD (1.08:1.00). (panel b) Equivalent (to D, panel c) curves for THC + CBD (1.08:1.00). Data obtained from chicken, dog and human membrane samples were not amenable to sigmoidal curve fitting; here, subjective best fits are shown, but EC50 not calculated. Concentration expressed as THC. (panel c) Equivalent (to D, panel b) curves for CBD. (panel d) Equivalent (to D, panel c) curves for CBD. Data obtained were not amenable to sigmoidal curve fitting; here, subjective best fits are shown but EC50 not calculated. (F) Radar plot showing CB1 receptor (‘Expression’; D, panel a), basal G‐protein turnover (‘Basal’; D, panel b) and sensitivity to and extent of agonist stimulation (‘Sensitivity’ and ‘Extent’; D, panel c) by species based upon (A–E) scaled as percentage of the species exhibiting the highest value for a given measure. With the exception of (F), all values shown are mean ± SEM; n = 3 experiments of three technical replicates in all cases.
Table 4.
[3H]‐SR141716A saturation binding in cannabis extract‐treated rats
| Treatment group | Treatment period | |||||||
|---|---|---|---|---|---|---|---|---|
| 2 days | 4 weeks | 8 weeks | 13 weeks | |||||
| Bmax (pmol·mg−1) | K D | Bmax (pmol·mg−1) | K D | Bmax (pmol·mg−1) | K D | Bmax (pmol·mg−1) | K D | |
| Vehicle | 1.86 ± 0.13 | 1.19 ± 0.25 | 1.64 ± 0.12 | 0.93 ± 0.23 | 1.39 ± 0.08 | 1.43 ± 0.23 | 1.31 ± 0.06 | 1.18 ± 0.16 |
| Low dose | 2.23 ± 0.13 | 0.90 ± 0.17 | 1.21 ± 0.06 | 0.74 ± 0.13 | 1.06 ± 0.05 | 0.90 ± 0.13 | 0.97 ± 0.07 | 0.88 ± 0.20 |
| High dose | 1.11 ± 0.17 | 0.95 ± 0.47 | 1.10 ± 0.11 | 1.54 ± 0.45 | 0.44 ± 0.07 | 0.39 ± 0.25 | 0.45 ± 0.05 | 0.90 ± 0.31 |
Bmax (pmol·mg−1) and K D values for CB1 receptor expression using saturation binding of [3H]‐SR141716A to cerebellar membranes from animals that had been treated with vehicle, low‐dose (1.08 mg·kg−1 THC + 1 mg·kg−1 CBD) or high‐dose (40.5 mg·kg−1 THC + 37.5 mg·kg−1 CBD) cannabis extract for 2 days and 4, 8 and 13 weeks (Figure 6A, panels a–d, B). Values shown are mean ± SEM; experiments in triplicate in three separate preparations.
Inter‐species differences in CB1 receptor expression and effects of cannabinoids on G‐protein turnover in cerebellar membranes
CB1 receptor density was first investigated by saturation binding assay using [3H]‐SR141716A in membranes from treatment‐naïve mouse, rat, chicken, dog and human cerebellar tissue (Figure 6D, panel a, and Table 5). [35S]‐GTPγS binding assays were also conducted using the same membrane preparations to examine the effects of THC, THC + CBD (in the same 1:08:1.00 ratio used in in vivo rat study above) and CBD alone. For THC‐alone datasets, a range of basal activity (measured as actual dpm in the presence of the lowest concentration of agonist) between species was observed (Figure 6D, panel b). Following data normalization (Figure 6D, panel c), rats, mice and chickens were shown to have similar EC50 values, while humans showed the highest and dogs the lowest EC50; however, it is possible to have greater sensitivity but less consequence of activation, and THC Emax was numerically higher for chickens, rats and dogs than mice and humans (Table 5). For THC plus CBD, while a range of basal activity was again evident (Figure 6E, panel a), normalized treatment‐induced increases in stimulation were much more limited in comparison with THC alone (Figure 6E, panel b, vs. D, panel c) and, for the human, dog and mouse, fits could not be derived (Table 5). For CBD alone, a range of basal activity was again seen, and approximately negligible [35S]‐GTPγS binding was observed (Figure 6E, panels c, d), consistent with a lack of CB1 receptor agonist effect, as reported by us previously (Jones et al., 2010).
Table 5.
Species‐specific responses in radioligand binding assays
| Species | Saturation binding | [35S]‐GTPγS assays | ||||
|---|---|---|---|---|---|---|
| THC | THC + CBD (1.08:1.00) | |||||
| Bmax (pmol·mg−1) | K D (nM) | EC50 (nM) | Emax (% over basal) | EC50 (nM) | Emax (% over basal) | |
| Mouse | 3.94 ± 0.38 | 3.18 ± 0.59 | 76 | 9.4 ± 5.9 | N/A | N/A |
| Rat | 1.91 ± 0.04 | 1.14 ± 0.13 | 58 | 29.0 ± 1.2 | 52 | 20.8 ± 0.9 |
| Chicken | 1.80 ± 0.09 | 2.21 ± 0.98 | 97 | 37.0 ± 4.5 | 72 | 23.9 ± 2.1 |
| Dog | 1.01 ± 0.11 | 0.97 ± 0.36 | 281 | 28.8 ± 4.1 | N/A | N/A |
| Human | 0.30 ± 0.11 | 1.03 ± 0.18 | 17 | 6.6 ± 6.1 | N/A | N/A |
Bmax (pmol·mg−1) and K D (nM) derived from saturation binding of [3H]‐SR141716A (Figure 6D, panel a) and EC50 (nM) (derived from a single fit to group data) and Emax (% over basal) for THC (Figure 6D, panel c) and THC + CBD (1.08:1.00 ratio) (Figure 6E, panel b) derived from [35S]‐GTPγS assays conducted using membranes prepared from treatment‐naïve cerebellae. EC50 for THC + CBD (derived from a single fit to group data) is expressed against concentration of THC present in the assay. CBD alone was also examined in all species but revealed approximately negligible [35S]‐GTPγS binding (Figure 6E, panel d), as was also the case for THC + CBD in human, dog and mouse membranes. In these cases, EC50 and Emax could not be confidently estimated and are omitted and recorded as N/A. Values shown are mean ± SEM; experiments in triplicate in three separate preparations. N/A, not available.
To best assess our supporting in vitro data, we combined these observations to provide an overall profile of THC‐mediated, CB1 receptor‐mediated signalling for each species (Figure 6F) by normalizing CB1 receptor expression (Bmax; ‘Expression’) (Figure 6D, panel a), basal G‐protein turnover (dpm at the lowest concentration of THC; ‘Basal’; Figure 6D, panel b) and sensitivity (EC50; ‘Sensitivity’) and activation (Emax; ‘Extent’) in response to agonist stimulation (Figure 6D, panel c), to the species with the highest value for each measure. We term this measure the ‘eCB signalling footprint’; as discussed more fully below, together these data suggest species‐specific differences in this profile with the highest value in the rat, intermediate values in the mouse and chicken and comparably lower values for humans and dogs.
Discussion
We show that motor convulsions in rats, but not dog, can be induced by sustained treatment with cannabis extract in a dose‐related manner; effects of higher dose cannabis extract in rats are subserved by primary, generalized epileptiform discharges in vivo and are associated with impaired CB1 receptor‐mediated signalling. Furthermore, in vitro profiling experiments suggest that eCB signalling plays a more dominant role in rat, a species susceptible to cannabis extract‐induced seizures, than in dog, a species resistant to such seizures.
Prolonged cannabis extract treatment causes spontaneous convulsions due to primary, generalized epileptiform events and associated deficits in CB1 receptor signalling in rats
In behavioural analysis of cannabis extract‐treated rats, behaviours classically associated with primary generalized seizures, including myoclonic jerk and convulsions (Mastropaolo et al., 2004; Luttjohann et al., 2009), were positively correlated with high‐dose treatment; by contrast, rats exhibited limited peripheral symptoms following such treatment. Sustained cannabis extract exposure induced spontaneous convulsions in rats. Here, the frequency of convulsive episodes induced at the higher dose was lower than that reported in a previous study for comparable doses of THC (administered alone) in rats (Chan et al., 1996). Since the same route and administration frequency, plus similar formulation, were used, we suggest that CBD in our cannabis preparations may exert anticonvulsant effects (Rosenberg et al., 2015) that limited, but did not prevent, proconvulsant effects of THC. We also propose that measurements of acute cannabis extract effects in rats reflect handling and/or drug‐related stress effects; thus, when measured within 10 min of drug administration, several behaviours were exacerbated, and similar behavioural responses have been noted previously (Chan et al., 1996; NTP, 1996). As such, for measures of acute cannabis extract effects, it may be difficult to determine the relative influences of (i) interactions between handling and pharmacological effects of cannabinoids already present, (ii) acute responses to formulation palatability and/or gavage and (iii) rising plasma concentrations of cannabinoids after dosing (phytocannabinoid Tmax in rodents (p.o.): ~30–60 min (Deiana et al., 2012), upon positively correlated seizure‐associated behaviours in rats. Persistent behavioural symptoms only manifested after several days' sustained treatment in both rat and dog. Phytocannabinoid p.o. bioavailability is known to be poor, but lipophilicity is high, meaning that repeated administration for several days is needed to saturate the fat compartment and thereafter achieve higher plasma concentrations (Sharma et al., 2012). Moreover, adaptive responses by signalling systems (e.g. protein trafficking) targeted by phytocannabinoids may require several days to manifest (Silva et al., 2016).
We demonstrate for the first time that cannabis extract‐induced convulsions in rats are subserved by spontaneous epileptiform discharges. Such seizures in rodents are characterized by EEG abnormalities such as 6–10 Hz spike–wave discharges (behavioural arrest), 5–9 Hz spiking (facial clonus) and rising and falling frequency 2–3 to 6–7 Hz high‐amplitude events (clonic or tonic–clonic seizures) (Luttjohann et al., 2009). We routinely observed these associated behaviours in rat; moreover, power spectra revealed peaks in the equivalent frequency bands. Of further interest was that high‐dose cannabis extract reliably produced such seizures; however, only one rat with low‐dose treatment demonstrated epileptiform activity, and this was not accompanied with a motor convulsion.
Our radioligand binding results in cannabis extract‐treated rats demonstrate that THC effects on the endogenous cannabinoid system signalling were clearly impaired in a dose‐related manner. Thus, among the 13 week treatment groups, high‐dose cannabis extract treatment caused CB1 receptor‐mediated G‐protein signalling to be severely attenuated such that we were unable to fit curves to derive any EC50 value and Emax was clearly depressed. Overall, we propose that prolonged high‐dose cannabis extracts functionally impairs eCB system signalling and that this mechanism underlies the reported exacerbation of seizures in rat. It was of interest that the incidence of seizure‐associated behaviours in rats diminished from day ~50; these data are consistent with a long‐term suppression of seizures, as reported from day ~500 in rats treated with THC for 2 years (Chan et al., 1996). Temporal profiles of this sort are distinct from that associated with kindling, a commonly used model of human epilepsy (Bertram, 2007), and, we suggest, are manifestations of an adaptive response to a down‐regulated eCB system to restore physiological seizure threshold.
Inter‐species differences in susceptibility to cannabis extract‐induced convulsions
The predictive validity of non‐human models for cannabinoid effects is generally regarded as inconsistent; moreover, therapeutic cannabis benefits are predicted from animal models of anxiety, depression, schizophrenia and pain; conversely, evidence supports exacerbation of mental illness and contraindication of CB1 receptor ligands in people with a history of seizures (Hill et al., 2012). We therefore investigated the effects of different cannabis extracts on behaviours (including seizure activity) in an alternative (canine) species to rat. Of interest is that dogs (including beagles) are highly susceptible to epilepsy (Heske et al., 2014). Cannabinoid plasma concentrations detected were consistent with ranges associated with human recreational and medicinal use in both the rat and dog (Lee et al., 2015). Here, we report that, despite this general sensitivity, sustained cannabis extract exposure for up to 52 weeks in dogs is not a precipitating factor for seizures; although we cannot fully rule out the effects at higher cannabis extract doses than tested here, we demonstrate clear species differences in terms of lack of cannabis extract‐induced seizures and limited effects on CNS behavioural measures in dogs. Brain cannabinoid concentration levels were not measured in dogs; however, we reason that plasma cannabinoid concentration is proportional to brain concentration [as phytocannabinnoids readily penetrate the mammalian blood–brain barrier (Deiana et al., 2012) and there is no a priori reason to believe that the canine blood–brain barrier differentially affects this parameter] allowing us to extrapolate from plasma data. Dogs receiving intermediate‐ and high‐dose cannabis extract treatment also received a habituation phase to avoid potentially toxic effects in this higher species; however, no seizures were seen in this period. Moreover, by directly comparing cannabinoid plasma concentrations, it is clear that despite low‐dose cannabis extract‐treated rats having THC plasma levels ~70‐fold lower than high‐dose‐treated dogs, we saw more pronounced CNS behavioural effects in rats and were still able to report seizure‐related behaviours and (albeit rare) epileptiform events, which were never seen dog, even at high dose. It is also of note that, relative to dose administered, the plasma concentration of the active metabolite 11‐OH‐THC, which has reportedly higher in vivo potency than THC (Lemberger et al., 1973), was higher in dog than rat. Overall, these data are consistent with THC pharmacokinetic differences not being able to explain fully these differences in seizure behaviour. A pertinent difference was that plasma CBD levels were also consistently higher in the dog than rat; thus, our data are also consistent with increased CBD levels in dogs acting to ameliorate the proconvulsant effects of long‐term THC seen in rats, as reported recently for CBD prevention of chronic THC‐induced long‐term behavioural abnormalities in mice (Murphy et al., 2017).
Results from our supporting description of eCB signalling footprint between species suggest a profile whereby rat > chicken > mouse > human = dog. Of interest is that this profile was highest in rat, a species in which cannabis extracts induced reliable epileptiform convulsions and caused clear signalling down‐regulation, but was lowest in dog, a species in which epileptiform convulsions did not occur. These data also support previous meta‐analysis across different studies whereby THC was reported to show differential inhibitory constant (K i) values between human versus rat CB1 receptors (McPartland et al., 2007). Our data further confirms a lack of CB1 receptor signalling by CBD alone and, consistent with previous reports (McPartland et al., 2015), that the presence of higher concentrations of CBD reduced THC‐induced effects in our [35S]‐GTPγS assays. The latter may reflect an attenuation of THC's effects by CBD, and these data are further consistent with the lack of seizure‐related behaviour seen in dogs, as discussed above; alternatively, this may reflect CBD negative allosteric modulation (Laprairie et al., 2015) and/or non‐specific effects due to cannabinoid lipophilicity. Taken together, this description suggests that seizure activity in rats reflects THC proconvulsant effects and that the eCB system plays a greater role in the physiology of species susceptible to THC/cannabis‐induced seizures than species where seizures are not seen.
In terms of potential mechanism of action, CB1 receptor‐mediated signalling acts primarily to inhibit neurotransmitter release from excitatory and inhibitory presynapses in the CNS (Diana & Marty, 2004). We propose that eCB signalling plays a greater role in regulating neurotransmitter release in species susceptible to cannabis‐induced seizure; for example, a THC‐induced down‐regulation of eCB signalling may lead to a net loss of CB1 receptor‐mediated inhibition of excitatory neurotransmitter release in these species to allow seizures to manifest. It is possible that the lower eCB signalling footprint we identified in dogs is reflected by a resistance of CB1 receptors to down‐regulation or, for example, that CB1 receptors are more weakly coupled to inhibition of presynaptic neurotransmitter release.
Conclusions and future perspectives
Our data suggest that choice of model species to study cannabis‐induced convulsions may have important implications in extrapolation to the human condition. We reveal clear differences in seizure behaviour and in cannabinoid plasma concentrations in response to cannabis extract consumption in rats versus dogs and suggest differences in eCB signalling in rats compared with dogs and humans (and to a lesser extent to mice and chickens). In humans, the reported THC : CBD plasma concentration ratio following p.o. (Guy & Robson, 2003) or oromucosal (Karschner et al., 2011) administration of similar cannabis extracts better approximates values for rats, rather than dogs, reported here. When set beside our findings that prolonged cannabis extract treatment induced seizures in rats but not dogs, our study indicates a poor predictive validity for animal models when assessing cannabis‐mediated effects in humans. Thus, we propose a number of important caveats that must be considered in this context; specifically, irrespective of species, responses to acute exposure to eCB system modulators are unlikely to reflect responses to sustained exposure, the latter potentially due to a propensity for CB1 receptor down‐regulation. Further, the eCB system may play a more dominant role in the physiology of lower‐order species. As such, assertions of therapeutic benefits or risks from rodent data may be diminished in clinical conditions. It is therefore important that an investigation of comparative eCB system physiology between species is undertaken to determine model predictive validity. As such, greater use of acutely excised human tissue and characterization of the eCB system in human stem cell‐derived cultures may address these problems and better predict potential risks associated with an emerging new wave of cannabinoid therapeutics.
Author contributions
B.J.W., R.A.G., C.E.R., M.B. and G.J.S. wrote the manuscript, designed the experiments and analysed the data; H.L. conducted the radioligand binding; L.B. analysed the data; T.H. wrote the manuscript and designed the experiments; A.P. designed the experiments and analysed the data; and O.D. and C.M.W. wrote the manuscript and analysed the data.
Conflict of interest
T.H. is formerly an employee of GW Pharmaceuticals Ltd. B.J.W., A.P., R.A.G., C.E.R. and M.B. were employees of GW Pharmaceuticals Ltd at the time of article submission. O.D. has commercial interests in several companies involved in the production of medicinal cannabis products, including consultant work for GW Pharmaceuticals Ltd and equity interests in other companies involved in the production of cannabis‐related products to treat epilepsy, including Privateer Holdings, Tilray, Receptor Life Sciences and Egg Rock.
Correction note
This article was corrected on the 20th of February 2019 following original publication online Early View on the 25th of March 2018. The Conflict of interest section was updated to include interests mistakenly omitted from the original submission.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Incidence of behaviours in rats (n=10 per group) observed immediately after (10 min; ‘acute’) or ~23 hours after (‘persistent’) daily oral administration of low dose (1.08 mg kg‐1 Δ 9‐THC + 1 mg kg‐1 CBD) or high dose (40.5 mg kg‐1 Δ 9‐THC + 37.5 mg kg‐1 CBD) cannabis extract for 13 weeks that exhibited a median and IQR of zero. Behavioural events conventionally associated with generalised seizures in rodents are highlighted in bold.
Table S2 Table showing squared cosine and percentage contribution of each measured behaviour following variance‐covariance principal component analysis applied to all behaviours recorded in low dose (1.08 mg kg‐1 Δ 9‐THC + 1 mg kg‐1 CBD) and high dose (40.5 mg kg‐1 Δ 9‐THC + 37.5 mg kg‐1 CBD) cannabis extract treated animals. Behaviours highlighted in bold show those conventionally associated with primary generalised seizures in rodents (see also: Figures 1 & 2). Squared cosine values shown in bold highlight the principal component in which the value exhibited its highest value. Table also shows factor values for each observation for the first two principal components (F1 and F2).
Table S3 Incidence of all convulsive motor events and/or epileptiform events exhibited in rats treated with low dose (1.08 mg kg‐1 Δ 9‐THC plus 1 mg kg‐1 CBD) or high dose (40.5 mg kg‐1 Δ 9‐THC plus 37.5 mg kg‐1 CBD) cannabis extract for 13 weeks. Note that in some case, accompanying EEG recordings (see Figure 4) showed (*) multiple, discrete, epileptiform events during a single motor convulsion and (**) animal handling or severity of motor convulsion that prevented acquisition of valid EEG data.
Acknowledgements
The authors thank Dr T. Bushell and colleagues at The University of Strathclyde for insights into the interpretation of interactions between cannabis‐induced rodent behaviours using PCA. The work described was funded by GW Pharmaceuticals Ltd.
Whalley, B. J. , Lin, H. , Bell, L. , Hill, T. , Patel, A. , Gray, R. A. , Elizabeth Roberts, C. , Devinsky, O. , Bazelot, M. , Williams, C. M. , and Stephens, G. J. (2019) Species‐specific susceptibility to cannabis‐induced convulsions. British Journal of Pharmacology, 176: 1506–1523. 10.1111/bph.14165.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram E (2007). The relevance of kindling for human epilepsy. Epilepsia 48: 65–74. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P (1983). Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220: 141–162. [DOI] [PubMed] [Google Scholar]
- Castaneto MS, Gorelick DA, Desrosiers NA, Hartman RL, Pirard S, Huestis MA (2014). Synthetic cannabinoids: epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend 144: 12–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PC, Sills RC, Braun AG, Haseman JK, Bucher JR (1996). Toxicity and carcinogenicity of Δ9‐tetrahydrocannabinol in Fischer rats and B6C3F1 mice. Fundam Appl Toxicol 30: 109–117. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiana S, Watanabe A, Yamasaki Y, Amada N, Arthur M, Fleming S et al (2012). Plasma and brain pharmacokinetic profile of cannabidiol (CBD), cannabidivarine (CBDV), Δ9‐tetrahydrocannabivarin (THCV) and cannabigerol (CBG) in rats and mice following oral and intraperitoneal administration and CBD action on obsessive–compulsive behaviour. Psychopharmacology (Berl) 219: 859–873. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Cilio MR, Cross H, Fernandez‐Ruiz J, French J, Hill C et al (2014). Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 55: 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R et al (2017). Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engll J Med 376: 2011–2020. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J et al (2016). Cannabidiol in patients with treatment‐resistant epilepsy: an open‐label interventional trial. Lancet Neurol 15: 270–278. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Whalley BJ, Di Marzo V (2015). Cannabinoids in the treatment of neurological disorders. Neurotherapeutics 12: 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Marty A (2004). Endocannabinoid‐mediated short‐term synaptic plasticity: depolarization‐induced suppression of inhibition (DSI) and depolarization‐induced suppression of excitation (DSE). Br J Pharmacol 142: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KT, Bronstein AC, Newquist KL (2013). Marijuana poisoning. Top Companion Anim Med 28: 8–12. [DOI] [PubMed] [Google Scholar]
- Gurney SM, Scott KS, Kacinko SL, Presley BC, Logan BK (2014). Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 26: 53–78. [PubMed] [Google Scholar]
- Guy GW, Robson P (2003). A phase I, open label, four‐way crossover study to compare the pharmacokinetic profiles of a single dose of 20 mg of a cannabis based medicine extract (CBME) administered on 3 difference areas of the buccal mucosa and to investigate the pharmacokinetics of CBME per oral in healthy male and female volunteers. J Cannabis Ther 3: 79–120. [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR et al (1990). Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A 87: 1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heske L, Nødtvedt A, Jäderlund KH, Berendt M, Egenvall A (2014). A cohort study of epilepsy among 665,000 insured dogs: incidence, mortality and survival after diagnosis. Vet J 202: 471–476. [DOI] [PubMed] [Google Scholar]
- Hill AJ, Williams CM, Whalley BJ, Stephens GJ (2012). Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol Ther 133: 79–97. [DOI] [PubMed] [Google Scholar]
- Jones NA, Glyn SE, Akiyama S, Hill TDM, Hill AJ, Weston SE et al (2012). Cannabidiol exerts anti‐convulsant effects in animal models of temporal lobe and partial seizures. Seizure‐Eur J Epilepsy 21: 344–352. [DOI] [PubMed] [Google Scholar]
- Jones NA, Hill AJ, Smith I, Bevan SA, Williams CM, Whalley BJ et al (2010). Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo . J Pharmacol Exp Ther 332: 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschner EL, Darwin WD, Goodwin RS, Wright S, Huestis MA (2011). Plasma cannabinoid pharmacokinetics following controlled oral Δ9‐tetrahydrocannabinol and oromucosal cannabis extract administration. Clin Chem 57: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Denovan‐Wright EM (2015). Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol 172: 4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Bergamaschi MM, Milman G, Barnes AJ, Queiroz RH, Vandrey R et al (2015). Plasma cannabinoid pharmacokinetics after controlled smoking and ad libitum cannabis smoking in chronic frequent users. J Anal Toxicol 39: 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberger L, Martz R, Rodda B, Forney R, Rowe H (1973). Comparative pharmacology of Δ9‐tetrahydrocannabinol and its metabolite, 11‐OH‐Δ9‐tetrahydrocannabinol. J Clin Invest 52: 2411–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttjohann A, Fabene PF, van Luijtelaar G (2009). A revised Racine's scale for PTZ‐induced seizures in rats. Physiol Behav 98: 579–586. [DOI] [PubMed] [Google Scholar]
- Mastropaolo J, Rosse RB, Deutsch SI (2004). Anabasine, a selective nicotinic acetylcholine receptor agonist, antagonizes MK‐801‐elicited mouse popping behavior, an animal model of schizophrenia. Behav Brain Res 153: 419–422. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland JM, Duncan M, Di Marzo V, Pertwee RG (2015). Are cannabidiol and Δ9‐tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol 172: 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland JM, Glass M, Pertwee RG (2007). Meta‐analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol 152: 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Mills S, Winstone J Leishman E, Wager‐Miller J, Bradshaw H et al (2017). Chronic adolescent Δ9‐tetrahydrocannabinol treatment of male mice leads to long‐term cognitive and behavioral dysfunction, which are prevented by concurrent cannabidiol treatment. Cannabis Cannabinoid Res 2: 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTP (1996). National Toxicology Program – toxicology and carcinogenesis studies of 1‐trans‐Δ9‐tetrahydrocannabinol (CAS no. 1972‐08‐3) in F344 rats and B6C3F1 mice (gavage studies). Natl Toxicol Program Tech Rep Ser 446: 1–317. [PubMed] [Google Scholar]
- Press CA, Knupp KG, Chapman KE (2015). Parental reporting of response to oral cannabis extracts for treatment of refractory epilepsy. Epilepsy Behav 45: 49–52. [DOI] [PubMed] [Google Scholar]
- Rosenberg EC, Tsien RW, Whalley BJ, Devinsky O (2015). Cannabinoids and epilepsy. Neurotherapeutics 12: 747–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg EC, Patra PH, Whalley BJ (2017). Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy‐related neuroprotection. Epilepsy Behav 70: 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Murthy P, Bharath MM (2012). Chemistry, metabolism, and toxicology of cannabis: clinical implications. Iran J Psychiatry 7: 149–156. [PMC free article] [PubMed] [Google Scholar]
- Silva L, Black R, Michaelides M, Hurd YL, Dow‐Edwards D (2016). Sex and age specific effects of Δ9‐tetrahydrocannabinol during the periadolescent period in the rat: the unique susceptibility of the prepubescent animal. Neurotoxicol Teratol 58: 88–100. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo‐Pena MC, Mackie K, Walker JM (1998). Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83: 393–411. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Baler RD, Compton WM, Weiss SR (2014). Adverse health effects of marijuana use. N Engl J Med 370: 2219–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfensohn S, Lloyd M (2013). Handbook of Laboratory Animal Management and Welfare. Wiley & Sons: Oxford. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Incidence of behaviours in rats (n=10 per group) observed immediately after (10 min; ‘acute’) or ~23 hours after (‘persistent’) daily oral administration of low dose (1.08 mg kg‐1 Δ 9‐THC + 1 mg kg‐1 CBD) or high dose (40.5 mg kg‐1 Δ 9‐THC + 37.5 mg kg‐1 CBD) cannabis extract for 13 weeks that exhibited a median and IQR of zero. Behavioural events conventionally associated with generalised seizures in rodents are highlighted in bold.
Table S2 Table showing squared cosine and percentage contribution of each measured behaviour following variance‐covariance principal component analysis applied to all behaviours recorded in low dose (1.08 mg kg‐1 Δ 9‐THC + 1 mg kg‐1 CBD) and high dose (40.5 mg kg‐1 Δ 9‐THC + 37.5 mg kg‐1 CBD) cannabis extract treated animals. Behaviours highlighted in bold show those conventionally associated with primary generalised seizures in rodents (see also: Figures 1 & 2). Squared cosine values shown in bold highlight the principal component in which the value exhibited its highest value. Table also shows factor values for each observation for the first two principal components (F1 and F2).
Table S3 Incidence of all convulsive motor events and/or epileptiform events exhibited in rats treated with low dose (1.08 mg kg‐1 Δ 9‐THC plus 1 mg kg‐1 CBD) or high dose (40.5 mg kg‐1 Δ 9‐THC plus 37.5 mg kg‐1 CBD) cannabis extract for 13 weeks. Note that in some case, accompanying EEG recordings (see Figure 4) showed (*) multiple, discrete, epileptiform events during a single motor convulsion and (**) animal handling or severity of motor convulsion that prevented acquisition of valid EEG data.