

Abstract
Background and Purpose
We sought to understand why (−)‐cannabidiol (CBD) and (−)‐cannabidiol‐dimethylheptyl (CBD‐DMH) exhibit distinct pharmacology, despite near identical structures.
Experimental Approach
HEK293A cells expressing either human type 1 cannabinoid (CB1) receptors or CB2 receptors were treated with CBD or CBD‐DMH with or without the CB1 and CB2 receptor agonist CP55,940, CB1 receptor allosteric modulator Org27569 or CB2 receptor inverse agonist SR144528. Ligand binding, cAMP levels and βarrestin1 recruitment were measured. CBD and CBD‐DMH binding was simulated with models of human CB1 or CB2 receptors, based on the recently published crystal structures of agonist‐bound (5XRA) or antagonist‐bound (5TGZ) human CB1 receptors.
Key Results
At CB1 receptors, CBD was a negative allosteric modulator (NAM), and CBD‐DMH was a mixed agonist/positive allosteric modulator. CBD and Org27569 shared multiple interacting residues in the antagonist‐bound model of CB1 receptors (5TGZ) but shared a binding site with CP55,940 in the agonist‐bound model of CB1 receptors (5XRA). The binding site for CBD‐DMH in the CB1 receptor models overlapped with CP55,940 and Org27569. At CB2 receptors, CBD was a partial agonist, and CBD‐DMH was a positive allosteric modulator of cAMP modulation but a NAM of βarrestin1 recruitment. CBD, CP55,940 and SR144528 shared a binding site in the CB2 receptor models that was separate from CBD‐DMH.
Conclusion and Implications
The pharmacological activity of CBD and CBD‐DMH in HEK293A cells and their modelled binding sites at CB1 and CB2 receptors may explain their in vivo effects and illuminates the difficulties associated with the development of allosteric modulators for CB1 and CB2 receptors.
Linked Articles
This article is part of a themed section on 8th European Workshop on Cannabinoid Research. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.10/issuetoc
Abbreviations
- 2‐AG
2‐arachidonoylglycerol
- CB1 receptor
type 1 cannabinoid receptor
- CB2 receptor
type 2 cannabinoid receptor
- CBD
(−)‐cannabidiol
- CBD‐DMH
(−)‐cannabidiol‐dimethylheptyl
- CRE
cAMP response element
- FSK
forskolin
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- Rluc
Renilla luciferase
- THC
∆9‐tetrahydrocannabinol
Introduction
Compounds that target the type 1 and type 2 cannabinoid receptors (CB1 and CB2 receptors) have been touted as possible treatments for a wide range of conditions including addiction, anxiety, depression, epilepsy, neurodegenerative diseases, chronic pain, inflammation, obesity and diabetes (Ross, 2007; Pertwee, 2008; Piscitelli et al., 2012). The orthosteric site of GPCRs is defined as the receptor site where the endogenous ligand binds. The majority of drugs that target GPCRs act at the orthosteric site (Christopoulos and Kenakin, 2002; Ross, 2007; Pertwee, 2008; Piscitelli et al., 2012). Allosteric sites are defined as distinct receptor domains that modulate orthosteric ligand binding and receptor activity via changes in receptor conformation (Wootten et al., 2013; Congreve et al., 2017). Conformational changes are integral to the function of GPCRs as GPCRs engage in allosteric interactions with other GPCRs, G proteins, βarrestins and small allosteric ligands (Christopoulos and Kenakin, 2002; Wootten et al., 2013). Some allosteric ligands act as positive allosteric modulators (PAMs) that enhance the binding, potency and efficacy of orthosteric ligand‐directed signalling, while others act as negative allosteric modulators (NAMs) that diminish the binding, potency and efficacy of orthosteric ligand‐directed signalling (Christopoulos and Kenakin, 2002; Wootten et al., 2013). The risk of adverse effects for allosteric modulators is reduced because allosteric compounds lack intrinsic agonism and can only modify the effects of endogenous or co‐administered orthosteric ligands (Christopoulos and Kenakin, 2002; Wootten et al., 2013). In the case of cannabinoid receptors, allosteric modulators may be especially useful because the psychomimetic and depressant effects of orthosteric ligands limit their potential therapeutic utility (Ignatowska‐Jankowska et al., 2015; Cairns et al., 2017; Slivicki et al., 2017).
(−)‐Cannabidiol (CBD) is the second most abundant phytocannabinoid present in Cannabis (Mechoulam et al., 2007). CBD acts as a partial agonist at CB2 receptors (Mechoulam et al., 2007), as well as the PPARγ (Campos et al., 2012), 5‐HT1A receptors (Russo et al., 2005) and the transient receptor potential cation channel subfamily V member 1 (TRPV1; Campos et al., 2012). CBD is a competitive antagonist of GPR55, which is described as a cannabinoid receptor and a lysophosphatidyl inositol receptor (Ryberg et al., 2007). CBD can also act as a PAM of the opioid μ‐ and δ‐receptors (Kathmann et al., 2006). Importantly, CBD can mediate NAM and antagonistic effects at CB1 receptors at concentrations well below the reported affinity (K i) values of CBD at the orthosteric site of CB1 receptors, suggesting that a high affinity CBD binding site, distinct from the orthosteric site on CB1 receptors, exists (Pertwee et al., 2002; Thomas et al., 2007; Hayakawa et al., 2008; Laprairie et al., 2015; McPartland et al., 2015; Sabatucci et al., 2017; Straiker et al., 2018). Given the diverse range of activities of CBD, modified synthetic CBD derivatives were synthesized as tools to elucidate the structure–activity relationship of CBD at CB1 and CB2 receptors. (−)‐Cannabidiol‐dimethylheptyl (CBD‐DMH) is one such synthetic derivative of CBD (Figure 1) that displays partial agonist activity in vivo at CB1 receptors and competes with CP55,940 for binding to CB1 and CB2 receptors. Yet the mechanism of action for this compound at CB1 and CB2 receptors remains unknown (Fride et al., 2005; Ben‐Shabat et al., 2006).
Figure 1.

CBD derived compounds used in this study: (A) CBD; (B) CBD‐DMH.
The aim of this study was to determine how CBD and CBD‐DMH – two structurally similar compounds – can differ in their function at CB1 and CB2 receptors. Cell culture assays and in silico ligand docking to the newly described CB1 receptor antagonist‐bound crystal structure (CB1 receptor‐5TGZ) (Hua et al., 2016) and AM11542 agonist‐bound crystal structure (CB1 receptor‐5XRA) (Hua et al., 2017) of CBD and CBD‐DMH were used to study how these ligands interact with, and affect signalling through, CB1 and CB2 receptors.
Methods
Cell culture
HEK293A cells were provided by Dr Denis Dupré (Dalhousie University, NS, Canada) and originally obtained from the American Type Culture Collection (Manassas, VI, USA). HEK293A cells were transfected with 400 ng of CB1 receptor‐GFP2 or CB2 receptor‐GFP2 expressing plasmid as described below, using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Burlington, ON, Canada). HEK293A cells used in this study were maintained between passage 3–12 at 37°C, 5% CO2 in DMEM supplemented with 10% FBS and 100 U·mL−1 Pen/Strep.
HEK293A Cignal Lenti cAMP response element (HEK‐CRE) reporter cells were provided by Dr Christopher J Sinal (Dalhousie University, NS, Canada). The HEK‐CRE cells stably express the firefly luciferase gene driven by tandem repeat elements of the cAMP transcriptional response element (CRE) (Qiagen, Toronto, ON, Canada). Thus, luciferase activity is directly proportional to the level of cAMP/PKA pathway activation or inhibition. HEK‐CRE cells used in this study were maintained at 37°C, 5% CO2 in DMEM supplemented with 10% FBS, 100 U·mL−1 Pen/Strep and 200 μg·mL−1 puromycin.
Plasmids
Human CB1 and CB2 receptors, and βarrestin1 were cloned and expressed as either GFP2 or Renilla luciferase (Rluc) fusion proteins at the C‐terminus. CB1 receptor‐GFP2 and CB2 receptor‐GFP2 were generated using the pGFP2‐N3 plasmid (PerkinElmer, Waltham, MA, USA) as described previously (Hudson et al., 2010). βarrestin1‐RlucII was generated using the pcDNA3.1 plasmid (Laprairie et al., 2014). The GFP2‐RlucII fusion plasmid was used as a positive control to calculate BRETMax, and the RlucII plasmid used as a negative control to calculate BRETMin has also been described previously (Laprairie et al., 2014).
Radioligand binding
Cell membranes were harvested by scraping and centrifugation (Hua et al., 2016). Pellets were stored at −80 °C until required. For radioligand binding assays, cells were defrosted and diluted in homogenization buffer (50 mM Tris, 100 mM NaCl, 1 mM EDTA, pH 7.4) and homogenized with a teflon‐on‐glass homogenizer. The assays were carried out with [3H]‐CP55,940 and Tris binding buffer (75 mM Tris, 12.5 mM MgCl2, 1 mM EDTA, 0.1% BSA, pH 7.4), total assay volume 200 μL. Binding was initiated by the addition of transfected human CB1 receptor or CB2 receptor HEK293A cell membranes (25 μg of protein per well). All assays were performed at room temperature for 2 h before termination by adding ice‐cold Tris binding buffer followed by vacuum filtration using a Millipore Sigma 12‐well sampling manifold and Brandel GF/B filters. Each reaction well was washed three times with 2 mL aliquots of Tris binding buffer. The filters were dried then placed in 5 mL of scintillation fluid overnight (Ultima Gold F, PerkinElmer, Billerica, MA, USA). Radioactivity was quantified by liquid scintillation spectrometry. Binding was defined as the difference between the binding that occurred in the presence and absence of non‐transfected HEK293A cell membranes (i.e. non‐specific). The concentration of [3H]‐CP55,940 used in displacement assays was 1 nM. The compounds under investigation were stored as stock solutions of 10 mM in DMSO, the vehicle concentration in all assay wells being 0.1% DMSO. Data are presented as % CP55,940 bound relative to ∆9‐tetrahydrocannabinol (THC) E max in cells expressing CB1 receptor‐GFP2 or CB2 receptor‐GFP2.
cAMP luciferase reporter assay
Forty‐eight hours after transfection of HEK‐CRE cells (grown in 6‐well plates) with hCB1 receptor‐GFP2 or hCB2 receptor‐GFP2‐expressing plasmid, cells were washed twice with cold 1× PBS [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 (pH 7.4)] and suspended in BRET buffer [1× PBS supplemented with glucose (1 mg·mL−1), benzamidine (10 mg·mL−1), leupeptin (5 mg·mL−1) and a trypsin inhibitor (5 mg·mL−1)]. Cells were dispensed into 96‐well plates (10 000 cells per well) and treated as indicated for 4 h. Cells were then lysed with passive lysis buffer (Promega, Oakville, ON, Canada) for 20 min at room temperature. Twenty microlitres of cell lysate were mixed with luciferase assay reagent (50 μM; Promega), and light emissions were measured at 405 nm using a Luminoskan Ascent plate reader (Thermo Scientific, Waltham, MA, USA), with an integration time of 10 s and a photomultiplier tube voltage of 1200 V. For measurements of cAMP inhibition, all cells were treated with 10 μM forskolin ± compounds and data are presented as % cAMP inhibition relative to CP55,940 E max in cells expressing CB1 receptor‐GFP2 or CB2 receptor‐GFP2. For cAMP accumulation, cells were treated with compounds used and data are presented as % cAMP accumulation relative to SR144528 E max in cells expressing CB1 receptor‐GFP2 or CB2 receptor‐GFP2.
BRET2
Direct interactions between CB1, CB2 receptors and βarrestin1 were quantified via BRET2. Cells were grown in a 6‐well plate and transfected with the indicated GFP2 (400 ng) and RlucII (200 ng) constructs using Lipofectamine 2000, according to the manufacturer's instructions (Invitrogen) and as previously described (Laprairie et al., 2014). Forty‐eight hours post‐transfection cells were washed twice with cold 1× PBS and suspended in BRET buffer. Cells were dispensed into white 96‐well plates (10 000 cells per well) and treated as indicated (PerkinElmer). Coelenterazine 400a substrate (50 μM; Biotium, Hayward, CA, USA) was added, and light emissions were measured at 460 nm (Rluc) and 510 nm (GFP2) using a Luminoskan Ascent plate reader (Thermo Scientific), with an integration time of 10 s and a photomultiplier tube voltage of 1200 V. BRET efficiency (BRETEff) was calculated as shown in Equation (1) and as described previously (Laprairie et al., 2014).
| (1) |
Data analysis and curve fitting
Data are presented as the mean ± the SEM from at least six independent experiments. No data collected were excluded from analysis. Microplate layout was by randomized block design to control for plate effects for all biochemical assays. Data recording was not blinded because of technical requirements, but data analyses were blinded for biochemical analyses. All data analysis and curve fitting was carried out using GraphPad Prism (v. 6.0). Radioligand data were fit with one site total binding (saturation data) or one site – fit K i (competition data) models. Radioligand competition data were fit with the concentration of [3H]‐CP55,940 at 1 nM and K D = 2.6 nM. Concentration‐response curves (CRC) were fit with the non‐linear regression with variable slope (four parameters), Gaddum/Schild EC50 shift model or allosteric operational model (Equation (2)) (Keov et al., 2011). All curves are shown according to the best‐fit model as determined by the R 2 value (GraphPad Prism v. 6.0). Pharmacological parameters were obtained from non‐linear regression models as indicated in figures and tables. To fit data to the Gaddum/Schild EC50 shift model, all variables were shared, and the Schild slope was constrained to 1.0. To fit data to the allosteric operational model (Equation (2)), all variables were shared except for E max. Logα (potency co‐operativity factor) and logβ (efficacy co‐operativity factor) were constrained between 0 and 1 (Keov et al., 2011; Hudson et al., 2014). Data in tables were determined by first fitting curves from individual experiments and then averaged (mean ± SEM) for those individual fits. Figures represent mean data, and non‐linear regressions in those figures are derived from mean data. Data collection and statistical analysis were conducted in adherence to the guidelines of the British Journal of Pharmacology (Curtis et al., 2018).
| (2) |
Receptor modelling and ligand docking
The 2.8 Å agonist‐bound (PDB ID: 5XRA) (Hua et al., 2017) and antagonist‐bound (PDB ID: 5TGZ) (Hua et al., 2016) human CB1 receptor crystal structures were selected as the best models for both CB1 and CB2 receptors. Whereas previous studies have chosen active (R*) and inactive (R) structures of bovine rhodopsin (1F88) (Hurst et al., 2006; Marcu et al., 2013) to model CB1 receptors, we were able to access the crystal structure of the active and inactive conformers of CB1 receptor (Hua et al., 2016, 2017). The amino acid sequence of the human CB2 receptor [Accession: NP_001832] was downloaded from GenBank (Benson et al., 2013). Previous studies have conducted homology modelling of CB2 receptors using different β‐adrenoceptor structure templates (2RH1, 3SN6) (El Bakali et al., 2010; Kusakabe et al., 2013; Renault et al., 2013); in this study, 5XRA and 5TGZ were chosen because the sequence similarity and predicted secondary structure were most similar between CB2 receptors and 5XRA and 5TGZ (human CB1 receptor) (Hua et al., 2016, 2017). For Table 3, amino acid position is indicated according to the Ballesteros and Weinstein (1995) method of residue numbering [i.e. single letter amino acid abbreviation, transmembrane helix number, the residue position relative to the most conserved position and the amino acid position as a superscript (e.g. F2.6291)]. Extracellular and intracellular loops and amino and carboxy termini of the modelled receptors were not constrained relative to the template receptors and as such were not constrained to the structures of their template molecules.
Ligand structure files
Ligand ‘.mol2’ structure and formula files for CP55,940 (zinc025648244), THC (zinc01530625), CBD‐DMH (zinc01530831), CBD (zinc04097406) and Org27569 (zinc35636065) were downloaded from ZINC (Irwin et al., 2012). The ligand ‘.mol2’ file for SR144528 was downloaded from PubChem (CID 3081335).
Homology modelling and model validation
Three‐dimensional models of human CB1 and CB2 receptors were generated in Swiss‐MODEL from the template structures (5XRA, 5TGZ) (Arnold et al., 2006; Kiefer et al., 2009). All settings were kept at default.
Ligand docking
Ligands were docked to model receptors using AutoDock 4.2.6 (Morris et al., 2009) by Lamarkian genetic algorithm (Hurst et al., 2006). AutoDock uses a Monte Carlo simulated annealing algorithm to explore a defined grid within the virtual space of a protein model with a selected ligand. The ligand is used to probe the defined grid space via molecular affinity potentials in various conformations of ligand and receptor. The binding site of the models was defined using the AutoGrid program within AutoDock, and the grid box was set to dimensions of 35 × 35 × 35 Å in order to include the entire extracellular surface and transmembrane regions of the model receptors. The rigidity parameters were set for the receptor, and the ligands were kept flexible. All other parameters were set to default. The AutoDock algorithm AutoDock Vina 1.1.2 (Morris et al., 2009; Trott and Olson, 2010) was used to fit the ligand to the template. The best conformation for each ligand–receptor is based on lowest binding energy among eight bioactive conformations generated by eight repeated program iterations.
Materials
CP55,940, Org27569, SR144528 and (−)‐CBD were purchased from Tocris Bioscience (Bristol, UK). THC was purchased from Sigma‐Aldrich (Mississauga, ON, Canada). CP55,940 is a commonly used reference compound to study cannabinoid receptor signalling (Pertwee, 2008). CP55,940 is a full agonist for inducing cAMP inhibition and βarrestin recruitment by stimulating both CB1 and CB2 receptors (Pertwee, 2008). (−)‐CBD‐DMH was synthesized and generously gifted from Dr Raphael Mechoulam (Hebrew University, Jerusalem). All compounds were dissolved in DMSO (final concentration of 0.1% in assay media for all assays) and added directly to the media at the concentrations and times indicated. All experimental protocols and design were conducted in adherence to the guidelines of the British Journal of Pharmacology (Curtis et al., 2018).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
The effect of CBD and CBD‐DMH at CB1 receptors
The effect of CBD and CBD‐DMH on CB1 receptor‐dependent CRE inhibition was measured in HEK‐CRE cells expressing CB1 receptor‐GFP2 and treated with 10 μM forskolin and 1 nM–10 μM CP55,940 ± 1 nM–10 μM CBD or CBD‐DMH (Figure 2A–C). CBD reduced orthosteric ligand potency and efficacy (hereafter referred to as α and β, respectively) with an estimated K B of 545 nM in the presence of CP55,940 (Figure 2A, Table 1). CBD displayed minimal agonist activity in the cAMP assay at 10 μM (Figure 2C). CBD‐DMH enhanced α and β of CP55,940 with an estimated K B of 121 nM (Figure 2B, Table 1). CBD‐DMH was a partial agonist in the cAMP assay (Figure 2C). Therefore, CBD displayed activity consistent with the effects of a NAM of CB1 receptor‐dependent CRE inhibition. In contrast, CBD‐DMH displayed activity consistent with both an agonist and PAM (i.e. ago‐PAM) for CB1 receptor‐dependent CRE inhibition.
Figure 2.

Activity of CBD and CBD‐DMH at CB1 receptors in the presence of CP55,940. cAMP inhibition (A–C) was quantified relative to CP55,940 E max (100%) in HEK‐CRE cells expressing CB1 receptor‐GFP2 treated with 1 nM–10 μM CP55,940 ± 1 nM–10 μM CBD (A) or CBD‐DMH (B), or CBD or CBD‐DMH alone (C), for 4 h. βarrestin1 recruitment (D–F) was measured via BRET2 in HEK293A cells expressing CB1 receptor‐GFP2 and βarrestin1‐Rluc treated with 1 nM–10 μM CP55940 ± 1 nM–10 μM CBD (D) or CBD‐DMH (E), or CBD or CBD‐DMH alone (F), for 30 min. Data were fit with the allosteric operational model [A (R 2 = 0.88), B (R 2 = 0.97), D (R 2 = 0.94), E (R 2 = 0.82)] or dose–response with variable slope (C, F). Data are mean ± SEM; n = 6 independent experiments.
Table 1.
Operational model analysis of CBD and CBD‐DMH in the presence of CP55,940
| Modulator | cAMP | βarrestin1 | ||
|---|---|---|---|---|
| CBD | CBD‐DMH | CBD | CBD‐DMH | |
| CB1 receptor | ||||
| logα | −0.36 ± 0.04 | 0.41 ± 0.15 | −0.15 ± 0.14 | 0.18 ± 0.02 |
| logβ | −0.37 ± 0.07 | 0.44 ± 0.15 | −0.19 ± 0.13 | 0.25 ± 0.08 |
| K B b (nM) | 545 ± 4.2 | 121 ± 88 | 547 ± 27 | 237 ± 64 |
| αβ | 0.19 | 20 | 0.50 | 3.0 |
| CB2 receptor | ||||
| logα | – | 0.56 ± 0.07 | – | −0.24 ± 0.12 |
| logβ | – | 0.62 ± 0.06 | – | 0.02 ± 0.10 |
| K B (nM) | 641 ± 4.0a | 38 ± 0.27 | 420 ± 13a | 156 ± 79 |
| αβ | – | 16 | – | 0.54 |
Data are mean ± SEM of six independent experiments.
Values estimated using the allosteric operational model in GraphPad Prism v. 6.0. ‘–’ means CRCs could not be fit to the operational model of allosterism.
Estimated using Gaddum/Schild EC50 shift equation.
βarrestin1 recruitment to CB1 receptors was measured using BRET2 in HEK293A cells expressing βarrestin1‐Rluc and CB1 receptor‐GFP2 and treated with 1 nM–10 μM CP55940 ± 1 nM–10 μM CBD or CBD‐DMH (Figure 2D–F). Data were analysed using the operational model of allosterism (eq. (2), Figure 2D, E). In the presence of CP55,940, CBD reduced ligand α and β with an estimated K B of 547 nM, (Figure 2D, Table 1). CBD displayed no agonist activity in the BRET2 assay at 10 μM in HEK293A cells (Figure 2F). CBD‐DMH enhanced the α and β of CP55,940 and an estimated K B of 237 nM (Figure 2E, Table 1). CBD‐DMH was a partial agonist in the BRET2 assay (Figure 2F). Therefore, the activity of CBD was consistent with a NAM of CB1 receptor‐dependent βarrestin1 recruitment, whereas CBD‐DMH displayed activity consistent with an ago‐PAM for CB1 receptor‐dependent βarrestin1 recruitment.
Org27569 is a CB1 receptor allosteric modulator that also functions as an inverse agonist of CB1 receptors in the cAMP assay (Price et al., 2005). We hypothesized that if CBD and CBD‐DMH were allosteric modulators that interacted with the same CB1 receptor site as Org27569, then CBD and CBD‐DMH should compete with Org27569 for the allosteric binding site, producing a rightward shift in a CRE CRC and a Schild regression with slope = −1. This hypothesis assumes that Org27569, CBD and CBD‐DMH share the same allosteric binding site, which has not yet been demonstrated. Org27569‐dependent CRE activation was measured in HEK‐CRE cells expressing CB1 receptor‐GFP2 and treated with 1 pM–10 μM Org27569 ± 1 nM–10 μM CBD or CBD‐DMH (Figure 3A, B). The % accumulation was calculated as described in the Methods, and data were analysed using the Gaddum/Schild EC50 shift model (Figure 3A, B). CBD shifted the Org27569 CRC rightward without affecting E max (Figure 3A, Table 2). CBD‐DMH also produced a rightward shift in the CRC for Org27569‐dependent cAMP accumulation (Figure 3B; Table 2). The observed Schild slope for CBD in the presence of Org27569 was not different from −1, whereas the Schild slope for CBD‐DMH in the presence of Org27569 was ≠−1 (Figure 3C). CBD‐dependent inhibition of Org27569 was consistent with competitive antagonism. CBD‐DMH did not appear to be competitive with Org27569 at CB1 receptors.
Figure 3.
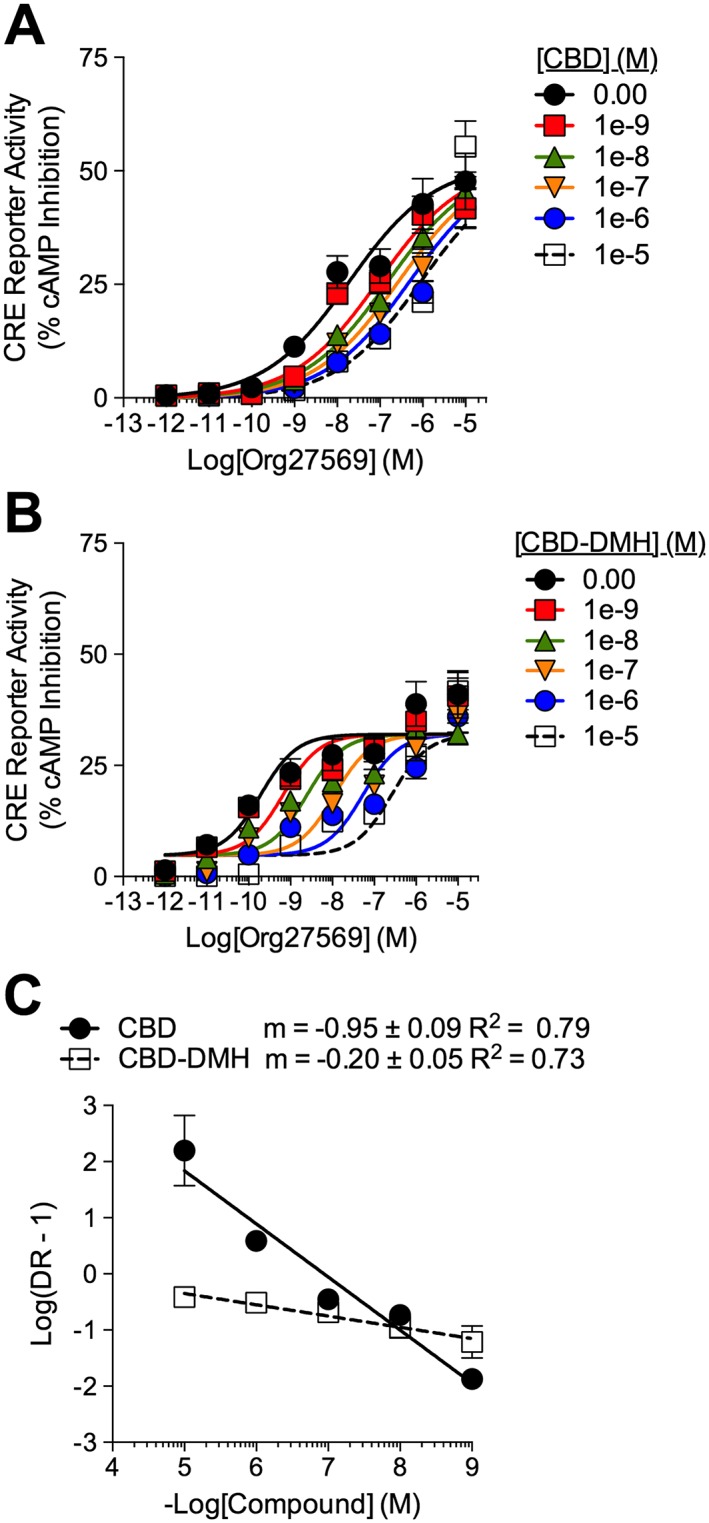
Activity of CBD and CBD‐DMH at CB1 receptors in the presence of Org27569. cAMP accumulation was quantified relative to 10 μM forskolin (100%) in HEK‐CRE cells expressing CB1 receptor‐GFP2 treated with 1 nM–10 μM CBD (A) or CBD‐DMH (B) ±1 pM–10 μM for 4 h. (C) Schild regressions for CBD and CBD‐DMH in the presence of Org27569. Data (mean ± SEM) from n = 6 independent experiments were fit with the Gaddum/Schild EC50 shift model [A (R 2 = 0.94), B (R 2 = 0.83)].
Table 2.
Operational model analysis of CBD and CBD‐DMH in the presence of ORG27569 at CB1 receptors or SR144528 at CB2 receptors
| CB1 receptor: Org27569 | CB2 receptor: SR144528 | |||
|---|---|---|---|---|
| Modulator | CBD | CBD‐DMH | CBD | CBD‐DMH |
| logα | – | – | – | 0.16 ± 0.04 |
| logβ | – | – | – | 0.15 ± 0.03 |
| K B (nM) | 674 ± 79a | 38 ± 11a | 9.9 ± 2.6a | 3.3 ± 0.12 |
| αβ | – | – | – | 2.1 |
Data are mean ± SEM of six independent experiments.
Values estimated using the allosteric operational model model in GraphPad Prism v. 6.0. “–” means CRCs could not be fit to the operational model of allosterism.
Estimated using Gaddum/Schild EC50 shift equation.
Radioligand binding was conducted to determine the abundance of CB1 receptors in transfected HEK293A cells and determine whether CBD and CBD‐DMH competed with [3H]‐CP55,940 for CB1 receptor binding. [3H]‐CP55,940 displayed an affinity of 2.6 ± 0.8 nM (Figure 4A). THC competed with [3H]‐CP55,940 for CB1 receptor binding (K i 16 ± 4.8 nM) (Figure 4B). Org27569 enhanced [3H]‐CP55,940 binding to CB1 receptors (K i 81 ± 23 nM), consistent with previous findings (Ahn et al., 2012, 2013) (Figure 4B). CBD reduced [3H]‐CP55,940 binding to CB1 receptors (K i 3300 nM ± 250) (Figure 4B). CBD‐DMH enhanced [3H]‐CP55,940 binding to CB1 receptors at concentrations <1 μM (K i 120 ± 7.3 nM) but reduced ligand binding above 1 μM (Figure 4B).
Figure 4.

Cannabinoid ligand binding and docking to CB1 receptor 5XRA (agonist‐bound) and 5TGZ (antagonist‐bound) models. (A) Saturation binding of [3H]‐CP55,940 in HEK293A cells expressing hCB1 receptor‐GFP2. (B) Competition binding with 1 nM [3H]‐CP55,940. Data were fit with the one site model. Data are mean ± SEM; n = 6–14 independent experiments. (C–H) The perspective is from the lipid bilayer. Helices are blue (I), light blue (II), turquoise (III), seafoam (IV), green (V), gold (VI) and orange (VII). (C) CP55,940 (black), (D) 2‐AG (purple), (E) THC (light green), (F) CBD (pink), (G) CBD‐DMH (grey) and (H) Org27569 (yellow). Interacting residues described in Table S1.
In silico ligand docking to CB1 receptor‐5XRA and CB1 receptor‐5TGZ was modelled in AutoDock 4.2.6. to predict and compare the possible binding sites of 2‐arachidonoylglycerol (2‐AG), THC, CP55,940, Org27569, CBD and CBD‐DMH (Figure 4C). 2‐AG, THC and CP55,940 interacted with a similar subset of amino acid residues in the agonist‐bound (5XRA) CB1 receptor model (Figure 4C, Table S1). 2‐AG, THC and CP55,940 also interacted with a similar subset of amino acid residues in the antagonist‐bound (5TGZ) CB1 receptor model, which differed from the agonist‐bound model (Figure 4C, Table S1). CBD interacted with a unique subset of amino acids compared to 2‐AG, THC and CP55,940 in the 5TGZ model but a similar subset of amino acids in the 5XRA model (Figure 4C, Table S1). CBD‐DMH interacted with amino acid residues from the 2‐AG/THC/CP55,940 binding sites in the 5XRA and 5TGZ models (Figure 4C, Table S1).
Ligand affinity was estimated for the 5XRA‐ and 5TGZ‐CB1 receptor models in AutoDock 4.2.6. for 2‐AG, THC, CP55,940, CBD, CBD‐DMH and Org27569 (Table 3). The estimated K A values for 2‐AG, THC, CP55,940 and Org27569 were not different from previously published data using [3H]‐CP55,940 in dissociated cell membranes expressing CB1 receptors (reviewed in Pertwee, 2008) and THC or Org27569 used in this study (Figure 4B), which supports the predictive power of these CB1 receptor models. The K A of CBD to the antagonist‐bound CB1 receptor (5TGZ, 146 nM), but not agonist‐bound (5XRA), was similar to functional data (K B, Table 1). The estimated K i value for CBD in radioligand binding (Figure 4B) was similar to the K A of agonist‐bound CB1 receptor (5XRA, 9930 nM) (Table 3). The K A values for CBD‐DMH to the CB1 receptor models (5XRA, 5TGZ) were similar to functional data (K B; Table 1) and radioligand binding data (K i; Figure 4B).
Table 3.
Estimated binding affinities for ligands docked to CB1 receptor and CB2 receptor models 5XRA and 5TGZ
| Ligand | CB1 receptor‐5XRA | CB1 receptor‐5TGZ | Ligand | CB2 receptor‐5XRA | CB2 receptor‐5TGZ | ||||
|---|---|---|---|---|---|---|---|---|---|
| Estimated Affinity (kcal·mol−1) | Estimated K A (nM) | Estimated Affinity (kcal·mol−1) | Estimated K A (nM) | Estimated Affinity (kcal·mol−1) | Estimated K A (nM) | Estimated Affinity (kcal·mol−1) | Estimated K A (nM) | ||
| CP55,940 | −10.9 | 20.8 | −11.2 | 12.8 | CP55,940 | −10.6 | 33.9 | −9.8 | 124 |
| 2‐AG | −8.4 | 1200 | −9.6 | 171 | 2‐AG | −8.7 | 740 | −8.7 | 740 |
| THC | −8.7 | 741 | −10.4 | 46.9 | THC | −10.5 | 39.9 | −8.8 | 629 |
| CBD | −7.1 | 9930 | −9.7 | 146 | CBD | −9.4 | 238 | −8.9 | 535 |
| CBD‐DMH | −8.9 | 535 | −9.4 | 238 | CBD‐DMH | −9.4 | 238 | −9.7 | 146 |
| Org27569 | −10.2 | 64.9 | −9 | 454 | SR144528 | −10.4 | 46.9 | −11.5 | 7.87 |
The effect of CBD and CBD‐DMH at CB2 receptors
CB2 receptor‐dependent CRE inhibition was measured in HEK‐CRE cells expressing CB2 receptor‐GFP2 and treated with 10 μM forskolin and 1 nM–10 μM CP55940 ± 1 nM–10 μM CBD or CBD‐DMH (Figure 5A–C). CBD treatment produced a concentration‐dependent rightward shift in CP55,940‐ and CB2 receptor‐dependent CRE inhibition with no change in E max and an estimated affinity (K B) of 641 nM (Figure 5A, Table 1). CBD was a partial CB2 receptor agonist (Figure 5C). CBD‐DMH enhanced α and β of CP55,940 (Figure 5B). CBD‐DMH displayed an estimated affinity (K B) of 38 nM (Figure 5B, Table 1). Therefore, CBD activity was consistent with orthosteric partial agonism at CB2 receptors, whereas CBD‐DMH was a PAM of CB2 receptor‐dependent CRE inhibition (Figure 5A–C).
Figure 5.
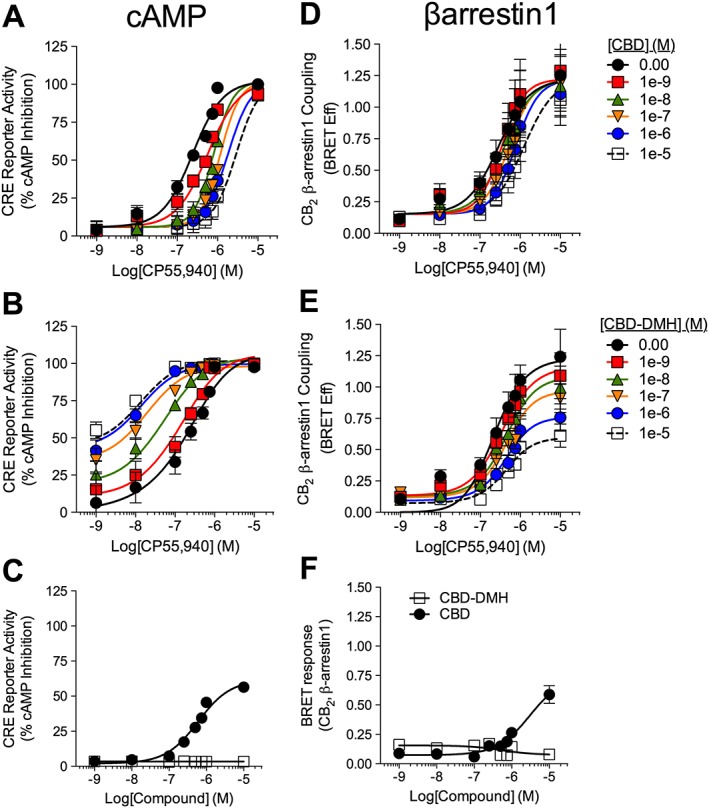
Activity of CBD and CBD‐DMH at CB2 receptors in the presence of CP55,940. cAMP inhibition (A–C) was quantified relative to CP55,940 E max (100%) in HEK‐CRE cells expressing CB2 receptor‐GFP2 treated with 1 nM–10 μM CP55,940 ± 1 nM–10 μM CBD (A) or CBD‐DMH (B), or CBD or CBD‐DMH alone (C), for 4 h. βarrestin1 recruitment (D–F) was measured via BRET2 in HEK293A cells expressing CB2 receptor‐GFP2 and βarrestin1‐Rluc treated with 1 nM–10 μM CP55,940 ± 1 nM–10 μM CBD (D) or CBD‐DMH (E), or CBD or CBD‐DMH alone (F), for 30 min. Data were fit with the Gaddum/Schild EC50 shift model [A (R 2 = 0.97), D (R 2 = 0.87)], allosteric operational model model [B (R 2 = 0.96), E (R 2 = 0.91)] or dose–response with variable slope (C, F). Data are mean ± SEM;. n = 6 independent experiments.
βarrestin1 recruitment to CB2 receptors was measured using BRET2 in HEK293A cells expressing βarrestin1‐Rluc and CB2 receptorR‐GFP2 and treated with 1 nM–10 μM CP55940 ± 1 nM–10 μM CBD or CBD‐DMH (Figure 5D–F). In the presence of CP55,940, CBD treatment produced a concentration‐dependent rightward shift in CP55,940‐ and CB2 receptor‐dependent βarrestin1 recruitment to CB2 receptors with an estimated K B of 420 nM (Figure 5D, Table 1). CBD was a partial agonist in the BRET2 assay at concentrations >10 μM (Figure 5F). CBD‐DMH reduced the α and β of CP55,940 (Figure 5E, F). CBD‐DMH displayed an estimated K B of 156 nM (Figure 5E, F, Table 1). Based on these results, we concluded that CBD was an orthosteric partial agonist at CB2 receptors, whereas CBD‐DMH was a NAM of CB2 receptor‐dependent βarrestin1 recruitment.
The orthosteric ligand, SR144528, is known to act as an inverse agonist of CB2 receptors in the cAMP assay (Portier et al., 1999). We hypothesized that if CBD‐DMH interacted with a unique receptor site compared to SR144528, then CBD‐DMH should not compete with SR144528 for the allosteric binding site. This hypothesis was tested by measuring inverse agonist activity of SR144528 at CB2 receptors in the presence of increasing concentrations of CBD‐DMH. The data support the hypothesis that CBD‐DMH was an allosteric compound, whereas SR144528 was an orthosteric compound because of the fit of these data to competitive versus allosteric non‐linear regression models. In contrast, CBD, as a partial agonist of CB2 receptors, should compete with SR144528 for receptor binding, producing a rightward shift in the CRC and a linear Schild regression. CB2 receptor inverse agonist‐dependent CRE activation was measured in HEK‐CRE cells expressing CB2 receptor‐GFP2 and treated with 1 pM – 10 μM SR144528 ± 1 nM – 10 μM CBD or CBD‐DMH (Figure 6). CBD treatment resulted in a concentration‐dependent rightward shift in SR144528‐mediated CRE activation, with an estimated K B of 9.9 nM (Figure 6A, Table 2). CBD‐DMH increased the E max and enhanced ligand α and β of SR144528‐dependent CRE activation with an estimated K B of 3.3 nM (Figure 6B, Table 2). The Schild slope for CBD in the presence of SR144528 was linear but ≠1 (Figure 6C). The Schild slope for CBD‐DMH in the presence of SR144528 was approximately 0 (Figure 6C). Based on these data, CBD did compete with SR144528 at CB2 receptors but do not indicate simple one‐site competition (i.e. Schild slope = 1), while CBD‐DMH was a PAM of SR144528‐mediated CRE activation at CB2 receptors.
Figure 6.
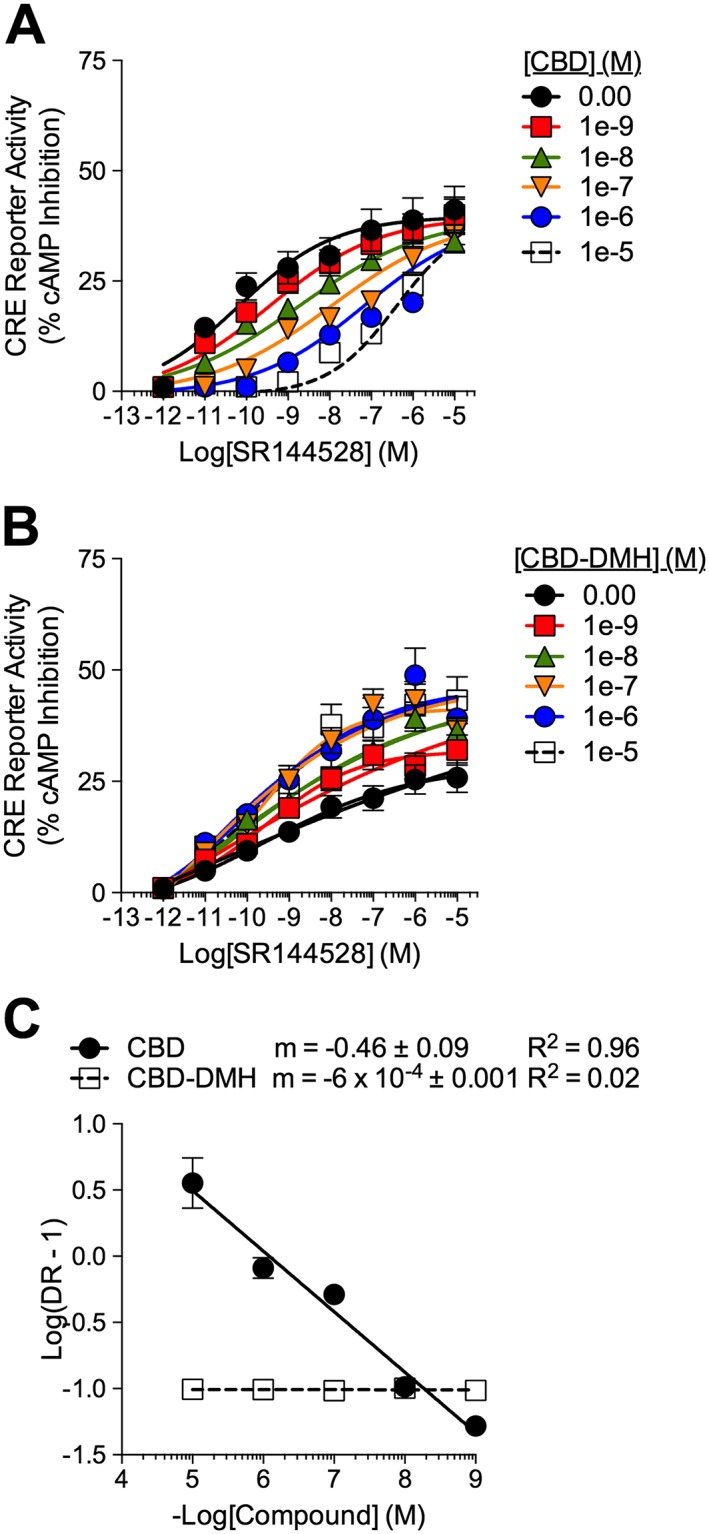
Activity of CBD and CBD‐DMH at CB2 receptors in the presence of SR144528. cAMP accumulation was quantified relative to 10 μM forskolin (100%) in HEK‐CRE cells expressing CB2 receptor‐GFP2 treated with 1 nM–10 μM CBD (A) or CBD‐DMH (B) ± 1 pM–10 μM SR144528 for 4 h. (C) Schild regression for CBD and CBD‐DMH in the presence of SR144528. Data were fit with the Gaddum/Schild EC50 shift model [A (R 2 = 0.94)] or Allosteric EC50 shift model [B (R 2 = 0.93)]. Data are mean ± SEM; n= 6 independent experiments.
Radioligand binding was conducted to determine the abundance of CB2 receptors in transfected HEK293A cells and determine whether CBD and CBD‐DMH competed with [3H]‐CP55,940 for CB2 receptor binding. [3H]‐CP55,940 displayed an affinity of 1.2 ± 0.4 nM (Figure 7A). THC competed with [3H]‐CP55,940 for CB2 receptor binding (K i 15 ± 5.2 nM) (Figure 7B). CBD reduced [3H]‐CP55,940 binding to CB2 receptors (K i 34 ± 12) (Figure 7B). CBD‐DMH enhanced [3H]‐CP55,940 binding between 0.1–10 nM and reduced [3H]‐CP55,940 binding above 10 nM (K i 130 ± 70 nM).
Figure 7.

Cannabinoid ligand binding and docking to CB2 receptor 5XRA (agonist‐bound) and 5TGZ (antagonist‐bound) models. (A) Saturation binding of [3H]‐CP55,940 in HEK293A cells expressing hCB2 receptor‐GFP2. (B) Competition binding with 1 nM [3H]CP55,940. Data were fit with the one site model. Data are mean ± SEM; n = 6–14 independent experiments. (C–H) The perspective is from the lipid bilayer. Helices are blue (I), light blue (II), turquoise (III), seafoam (IV), green (V), gold (VI) and orange (VII). (C) CP55,940 (black), (D) 2‐AG (purple), (E) THC (light green), (F) CBD (pink), (G) CBD‐DMH (grey) and (H) SR144528 (dark green). Interacting residues described in Table S2.
In silico ligand docking to the 5XRA‐ and 5TGZ‐based models of CB2 receptors was modelled in AutoDock 4.2.6. to predict and compare the possible binding sites of 2‐AG, THC, CP55,940, SR144528, CBD and CBD‐DMH (Figure 7C). 2‐AG, THC and CP55,940 all interacted with a similar subset of amino acids within the 5XRA and 5TGZ CB2 receptor models (Figure 7C, Table S2). With the exception of F2.6191 and A7.36282 in the 5XRA CB2 receptor model, CBD‐DMH interacted with a unique subset of amino acid residues compared to 2‐AG, THC, CP55,940 and CBD in the 5XRA and 5TGZ CB2 receptor models (Figure 7C, Table S2). Based on this model, CBD may interact with the orthosteric agonist site with a lower affinity that was not predicted in this model. For CBD‐DMH, these are the predicted results based on the observation that CBD‐DMH was an allosteric modulator of CP55,940‐dependent signalling (Figure 7C, Table S2).
Ligand affinity was estimated with the 5XRA and 5TGZ models of CB2 receptor in AutoDock 4.2.6. for CP55,940, 2‐AG, THC, CBD, CBD‐DMH and SR144528 (Table 3). The estimated K A values for CP55,940, 2‐AG, THC, SR144528 and CBD were in agreement with previously published results (summarized in Pertwee, 2008), and THC used in this study compared to the 5XRA agonist‐bound model (Figure 7B). The CBD K i measured by [3H]‐CP55,940 competition (Figure 7B) was greater than the observed K B (Table 1) or estimated K A (Table 3). The K i for CBD‐DMH measured by [3H]‐CP55,940 competition (Figure 7B) was similar to the observed K B in the βarrestin1 assay (Table 1) and estimated K A in the modelled receptor (Table 3). These data support the hypothesis that CBD‐DMH occupied a high affinity site on CB2 receptors separate from 2‐AG, THC, CP55,940 and CBD.
Discussion
The data presented here indicate that CBD was a NAM of CB1 receptor signalling. CBD and the known CB1 receptor NAM Org27569 both occupied a ligand binding site in the antagonist‐bound CB1 receptor model (5TGZ) that was separate from the orthosteric agonists tested. The results of in silico CB1 receptor modelling are in agreement with functional data demonstrating that residues such as F2.57170, F2.61174, F2.64177, L3.29193, F7.34379 and S7.39383 are critical for ligand binding (Hua et al., 2016, 2017). In the agonist‐bound CB1 receptor model (5XRA), CBD interacted with a similar subset of residues to the agonists 2‐AG, THC and CP55,940, albeit with a low affinity similar to that observed in [3H]‐CP55,940 binding assays. CBD appears to have fluid affinity for both allosteric (inactive state, R) and orthosteric (active state, R*) sites at CB1 receptors, depending on the receptor's conformation. Based on these data, we propose that CBD has high affinity for an allosteric site in the outer vestibule of the CB1 receptor in the inactive conformation. This hypothesis is supported by our earlier mutagenesis work with CB1 receptors (Laprairie et al., 2015), recent modelling of CB1 receptor 5TGZ with CBD (Sabatucci et al., 2017), electrophysiology data from cultural autaptic hippocampal neurons (Straiker et al., 2018) and accumulating data showing the class A GPCR outer vestibule to be a common binding region for allosteric modulators (Congreve et al., 2017).
Unlike the actions of CBD at CB1 receptors, CBD‐DMH enhanced the potency, efficacy and binding of CP55,940‐dependent signalling and reduced the potency of Org27569‐dependent inverse agonist activity. The reason for these differences is not clear but may have occurred because CP55,940 stabilized the active conformation of the CB1 receptor (R*), whereas Org27569 stabilized intermediate (R**) or inactive (R) receptor conformations (Hurst et al., 2006; Marcu et al., 2013; Shore et al., 2014). CBD‐DMH shared common interaction residues with agonists and the allosteric modulator Org27569 in the agonist‐ and antagonist‐bound CB1 receptor models, suggesting these amino acids may contribute to receptor activation. Therefore, the binding site for CBD‐DMH in the CB1 receptor models bridged the allosteric and orthosteric sites.
At the CB2 receptor, CBD was an orthosteric ligand, whereas CBD‐DMH was an allosteric ligand. Our data suggest that CBD binding competed, or blocked the binding of, CP55,940 and SR144528 to CB2 receptors. The binding site and regions of CB2 receptors that interacted with 2‐AG, THC and CP55,940 in our CB2 receptor models align with previous reports of orthosteric ligand binding at CB2 receptors (El Bakali et al., 2010; Renault et al., 2013). CBD‐DMH was an allosteric modulator of signalling that reduced [3H]‐CP55,940 binding and interacted with a unique subset of modelled residues compared to other ligands tested. We observed CBD‐DMH had a lower K i (130 nM) with [3H]‐CP55,940 binding than has been previously reported with [3H]‐HU243 (Fride et al., 2005). Reduced [3H]‐CP55,940 binding may have been allosteric as was observed in cAMP and βarrestin1 signalling. It is important to note that the lipophilicity and lateral movement of cannabinoids such as 2‐AG and presence of catabolic α/β hydrolases may produce in vitro affinities that are lower than those observed in in silico simulations (Fay and Farrens, 2013). The allosteric activity of CBD‐DMH at CB2 receptors was pathway‐specific. CBD‐DMH was a PAM of CP55,940‐dependent cAMP inhibition and a NAM of βarrestin1 recruitment. Although the exact mechanism for this was not determined, CBD‐DMH may promote a conformational change in CB2 receptors that promotes G protein‐dependent signalling at the expense of βarrestin1‐dependent signalling. Similarly, the CB1 receptor allosteric modulator Org27569 has been described as a NAM of CB1 receptor agonist‐induced cAMP inhibition and a PAM of CB1 receptor agonist‐induced ERK phosphorylation (Price et al., 2005; Ahn et al., 2013; Baillie et al., 2013). Therefore, a precedent exists for pathway‐specific allostery at the cannabinoid receptors.
The allosteric operational model was used to estimate ligand K B and co‐operativity where data could not be fit to conventional models of receptor signalling (Keov et al., 2011; Hudson et al., 2014). Ligand binding was also determined using conventional radioligand competition experiments (Christopoulos and Kenakin, 2002; Smith et al., 2011). CBD reduced [3H]‐CP55,940 binding with an apparent K i much lower than the affinity estimated in signalling assays, indicating reduced orthosteric ligand binding above 3 μM CBD and affirming higher affinity NAM signalling activity (Smith et al., 2011; Hudson et al., 2014; Congreve et al., 2017). We observed here, and previously (Laprairie et al., 2015), that CBD functioned as a NAM in several cell culture assays at concentrations lower than the reported concentrations at which CBD acts an orthosteric ligand of the CB1 receptor (Thomas et al., 2007; Hayakawa et al., 2008; McPartland et al., 2015; Sabatucci et al., 2017; Straiker et al., 2018). In contrast, CBD‐DMH may enhance the binding of the orthosteric probe to a maximum and, at higher concentrations, reduce orthosteric probe binding, producing a bell‐shaped curve. In the case of CB2 receptors, CBD competitively reduced [3H]‐CP55,940 binding. CBD‐DMH reduced orthosteric probe binding and βarrestin1 recruitment to CB2 receptors consistent with NAM activity but enhanced CP55,940‐ and SR144528‐dependent cAMP signalling consistent with PAM activity.
The orthosteric and allosteric binding sites of both CB1 and CB2 receptors appear to be highly fluid and flexible. In addition, CB1 and CB2 receptors bind highly lipophilic and flexible ligands. The allosteric binding sites and mixed effects observed here for CBD and CBD‐DMH are not without precedent: the endogenous peptide Pepcan‐12 is a CB1 receptor NAM and CB2 receptor PAM that may bind to similar structural motifs at the cannabinoid receptors to CBD and CBD‐DMH (Bauer et al., 2012; Straiker et al., 2015; Petrucci et al., 2017). For CBD and CBD‐DMH, these ligands may be capable of acting either as orthosteric agonists, allosteric modulators or a bridge between the two, depending on concentration and relative binding site affinity. Previously, fenofibrate has been described as a CB1 receptor agonist at low concentrations and NAM at higher concentrations – demonstrating the potential of cannabinoid ligands to act through multiple mechanisms of action rather than in ‘pure’ agonist or allosteric roles (Priestley et al., 2015). The N‐terminal Cys98 and Cys107 contribute to the NAM activity of CBD and Org27569 (Fay and Farrens, 2013; Laprairie et al., 2015). Sabatucci et al. (2017) recently confirmed the importance of the CB1 receptor N‐terminus for allosteric ligands – including CBD – in silico, and our findings here support their modelling. Further, the NAM activity of CBD was expanded upon in an autaptic hippocampal neuron model, suggesting it may play a role in depolarization‐induced suppression of excitation (Straiker et al., 2018). For the CB2 receptor, it remains unknown whether the N‐terminus affects ligand activity or binding (El Bakali et al., 2010).
Conclusions
Given the highly similar structures of CBD and CBD‐DMH, these data demonstrate that the orthosteric and allosteric site pharmacophores are potentially overlapping, yet the downstream effects on signalling differ markedly. These data also support the hypothesis that ligand interaction with the allosteric and orthosteric sites of cannabinoid receptors is highly fluid. Therefore, the design of pure CB1 /CB2 receptor allosteric modulators that lack agonist or inverse agonist activity at these receptors may be challenging (Iliff et al., 2011; Janero et al., 2015; Hua et al., 2016, 2017). Additional research is required to determine the specific residues that mediate negative versus positive allosteric modulation of orthosteric ligand binding and G protein‐ and βarrestin‐dependent signalling at CB1 and CB2 receptors.
Author contributions
M.T. conducted the experiments, analysed the data and edited the manuscript; O.Y. and M.A. wrote and edited the manuscript; M.E.M.K. provided essential materials and edited the manuscript; E.M.D.‐W. analysed the data and wrote and edited the manuscript; R.B.L. designed and conducted the experiments, analysed the data and wrote and edited the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Summary of CB1R agonist‐bound (5XRA) and antagonist‐bound (5TGZ) amino acid residues interacting with cannabinoid ligands in AutoDock.
Table S2 Summary of modelled CB2R agonist‐bound (5XRA) and antagonist‐bound (5TGZ) amino acid residues interacting with cannabinoid ligands in AutoDock.
Acknowledgements
We thank Dr Raphael Mechoulam for providing (−)‐CBD‐DMH used in this study and for critique of this manuscript. We thank Dr Brian D Hudson for critical evaluation of the data and figure presentation. This work was supported by a partnership grant from GlaxoSmithKline and the Canadian Institutes of Health Research (CIHR) (RN323670‐386247) to R.B.L., a CIHR operating grant (MOP‐97768) to M.E.M.K. and a Bridge funding grant from Dalhousie University to E.M.D.‐W. O.Y. is supported by an undergraduate student research award from the National Sciences and Engineering Research Council (NSERC).
Tham, M. , Yilmaz, O. , Alaverdashvili, M. , Kelly, M. E. M. , Denovan‐Wright, E. M. , and Laprairie, R. B. (2019) Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol‐dimethylheptyl at the type 1 and type 2 cannabinoid receptors. British Journal of Pharmacology, 176: 1455–1469. 10.1111/bph.14440.
References
- Ahn KH, Mahmoud MM, Kendall DA (2012). Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein‐independent ERK1/2 kinase activation. J Biol Chem 287: 12070–12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Shim JY, Kendall DA (2013). Distinct roles of β‐arrestin 1 and β‐arrestin 2 in Org27569‐induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J Biol Chem 288: 9790–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The concise guide to pharmacology 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006). The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 22: 195–201. [DOI] [PubMed] [Google Scholar]
- Baillie GL, Horswill JG, Anavi‐Goffer S, Reggio PH, Bolognini D, Abood ME et al (2013). CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol 83: 322–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H (1995). Integrated methods for the construction of three‐dimensional models and computational probing of structure‐function relations in G protein‐coupled receptors. Meth Neurosci 25: 366–428. [Google Scholar]
- Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B et al (2012). Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J Biol Chem 287: 36944–36967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Shabat S, Hanus LO, Katzavian G, Gallily R (2006). New cannabidiol derivatives: synthesis, binding to cannabinoid receptor, and evaluation of their antiinflammatory activity. J Med Chem 49: 1113–1117. [DOI] [PubMed] [Google Scholar]
- Benson DA, Cavanaugh M, Clark K, Karsch‐Mizrachi I, Lipman DJ, Ostell J et al (2013). GenBank. Nucl Acids Res 41: D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns EA, Szczesniak AM, Straiker AJ, Kulkarni PM, Pertwee RG, Thakur GA et al (2017). The in vivo effects of CB1‐positive allosteric modulator GAT229 on intraocular pressure in ocular normotensive and hypertensive mice. J Ocul Pharmacol Ther 33: 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS (2012). Multiple mechanisms involved in the large‐spectrum therapeutic potential of cannabidiol in psychiatric disorders. Philos Trans R Soc Lond B Biol Sci 367: 3364–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T (2002). G protein‐coupled receptor allosterism and complexing. Pharmacol Rev 54: 323–374. [DOI] [PubMed] [Google Scholar]
- Congreve M, Oswald C, Marshall FH (2017). Applying structure‐based drug design approaches to allosteric modulators of GPCRs. Trends Pharmacol Sci 38: 837–847. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bakali J, Muccioli GG, Renault N, Pradal D, Body‐Malapel M, Djouina M et al (2010). 4‐Oxo‐1,4‐dihydropyridines as selective CB2 cannabinoid receptor ligands: structural insights into the design of a novel inverse agonist series. J Med Chem 53: 7918–7931. [DOI] [PubMed] [Google Scholar]
- Fay JF, Farrens DL (2013). The membrane proximal region of the cannabinoid receptor CB1 N‐terminus can allosterically modulate ligand affinity. Biochemistry 52: 8286–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fride E, Ponde D, Breuer A, Hanus L (2005). Peripheral, but not central effects of cannabidiol derivatives: mediation by CB(1) and unidentified receptors. Neuropharmacology 48: 1117–1129. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, Hazekawa M, Sano K, Irie K, Orito K et al (2008). Cannabidiol potentiates pharmacological effects of Delta(9)‐tetrahydrocannabinol via CB(1) receptor‐dependent mechanism. Brain Res 1188: 157–164. [DOI] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y et al (2016). Crystal structure of the human cannabinoid receptor CB1. Cell 167: 750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L et al (2017). Crystal structures of agonist‐bound human cannabinoid receptor CB1. Nature 547: 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hudson BD, Hébert TE, Kelly ME (2010). Physical and functional interaction between CB1 cannabinoid receptors and beta2‐adrenoceptors. Br J Pharmcol 160: 627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Christiansen E, Murdoch H, Jenkins L, Hansen AH, Madsen O et al (2014). Complex pharmacology of novel allosteric free fatty acid 3 receptor ligands. Mol Pharmacol 86: 200–210. [DOI] [PubMed] [Google Scholar]
- Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M et al (2006). Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C‐3 carboxamide oxygen/lysine3.28(192) interaction. J Med Chem 49: 5969–5987. [DOI] [PubMed] [Google Scholar]
- Iliff HA, Lynch DL, Kotsikorou E, Reggio PH (2011). Parameterization of Org27569: an allosteric modulator of the cannabinoid CB1 G protein‐coupled receptor. J Comput Chem 32: 2119–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska‐Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA et al (2015). A cannabinoid CB1 receptor‐positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology 40: 2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger M, Bolstad ES, Coleman RG (2012). ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52: 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janero DR, Yaddanapudi S, Zvonok N, Subramanian KV, Shukla VG, Stahl E et al (2015). Molecular‐interaction and signaling profiles of AM3677, a novel covalent agonist selective for the cannabinoid 1 receptor. ACS Chem Nerosci 6: 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathmann M, Flau K, Redmer A, Tränkle C, Schlicker E (2006). Cannabidiol is an allosteric modulator at mu‐ and delta‐opioid receptors. Naunyn Schmiedebergs Arch Pharmacol 372: 354–361. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T (2009). The SWISS‐MODEL repository and associated resources. Nucleic Acids Res 37: D387–D392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P, Sexton PM, Christopoulos A (2011). Allosteric modulation of G protein‐coupled receptors: a pharmacological perspective. Neuropharmacology 60: 24–35. [DOI] [PubMed] [Google Scholar]
- Kusakabe K, Tada Y, Iso Y, Sakagami M, Morioka Y, Chomei N et al (2013). Design, synthesis, and binding mode prediction of 2‐pyridone‐based selective CB2 receptor agonists. Bioorg Med Chem 21: 2045–2055. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Dupré DJ, Kelly MEM, Denovan‐Wright EM (2014). Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J Biol Chem 289: 24845–24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly MEM, Denovan‐Wright EM (2015). Cannabidiol is a negative allosteric modulator of the type 1 cannabinoid receptor. Br J Pharmacol 172: 4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu J, Shore DM, Kapur A, Trzandel M, Markiyannis A, Reggio PH et al (2013). Novel insights into CB1 cannabinoid receptor signalling: a key interaction identified between the extracellular‐3 loop and transmembrane helix 2. J Pharmacol Exp Ther 345: 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland JM, Duncan M, Di Marzo V, Pertwee R (2015). Are cannabidiol and Δ9 ‐tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol 172: 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Peters M, Murillo‐Rodriguez E, Hanus LO (2007). Cannabidiol—recent advances. Chem Biodivers 4: 1678–1692. [DOI] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS et al (2009). Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 30: 2785–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucci V, Chicca A, Glasmacher S, Paloczi J, Cao Z, Pacher P et al (2017). Pepcan‐12 (RVD‐hemopressin) is a CB2 receptor positive allosteric modulator constituively secreted by adrenals and in liver upon tissue damage. Sci Rep 7: 9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA, Craib SJ, Thomas A (2002). (−)‐Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur J Pharmacol 456: 99–106. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2008). Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addict Biol 13: 147–159. [DOI] [PubMed] [Google Scholar]
- Piscitelli F, Ligresti A, La Regina G, Coluccia A, Morera L, Allarà M et al (2012). Indole‐2‐carboxamides as allosteric modulators of the cannabinoid CB1 receptor. J Med Chem 55: 5627–5631. [DOI] [PubMed] [Google Scholar]
- Portier M, Rinaldi‐Carmona M, Pecceu F, Combes T, Poinot‐Chazel C, Calandra B et al (1999). SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther 284: 644–650. [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R et al (2005). Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol 68: 1484–1495. [DOI] [PubMed] [Google Scholar]
- Priestley RS, Nickolls SA, Alexander SP, Kendall DA (2015). A potential role for cannabinoid receptors in the therapeutic action of fenofibrate. FASEB J 29: 1446–1455. [DOI] [PubMed] [Google Scholar]
- Renault N, Laurent X, Farce A, El Bakali J, Mansouri R, Gervois P et al (2013). Virtual screening of CB(2) receptor agonists from bayesian network and high‐throughput docking: structural insights into agonist‐modulated GPCR features. Chem Biol Drug Des 81: 442–454. [DOI] [PubMed] [Google Scholar]
- Ross RA (2007). Allosterism and cannabinoid CB(1) receptors: the shape of things to come. Trends Pharmacol Sci 28: 567–572. [DOI] [PubMed] [Google Scholar]
- Russo EB, Burnett A, Hall B, Parker KK (2005). Agonistic properties of cannabidiol at 5‐HT1a receptors. Neurochem Res 30: 1037–1043. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J et al (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152: 1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatucci A, Tortolani D, Dainese E, Maccarrone M (2017). In silico mapping of allosteric ligand binding sites in the type‐1 cannabinoid receptor. Biotechnol Appl Biochem 65: 21–28. [DOI] [PubMed] [Google Scholar]
- Shore DM, Baillie GL, Hurst DH, Navas F 3rd, Seltzman HH, Marcu JP et al (2014). Allosteric modulation of a cannabinoid G protein‐coupled receptor: binding site elucidation and relationship to G protein signaling. J Biol Chem 289: 5828–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki RA, Xu Z, Kulkarni PM, Pertwee RG, Mackie K, Thakur GA et al (2017). Positive allosteric modulation of cannabinoid receptor type 1 suppresses pathological pain without producing tolerance or dependence. Biol Psychiatry 8: 31761–31764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Ward RJ, Stoddart LA, Hudson BD, Kostenis E, Ulven T et al (2011). Extracellular loop 2 of the free fatty acid receptor 2 mediates allosterism of a phenylacetamide ago‐allosteric modulator. Mol Pharmacol 80: 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mitjavila J, Yin D, Gibson A, Mackie K (2015). Aiming for allosterism: Evaluation of allosteric modulators of CB1 in a neuronal model. Pharmacol Res 99: 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Dvorakova M, Zimmowitch A, Mackie K (2018). Cannabidiol inhibits endocannabinoids signaling in autaptic hippocampal neurons. Mol Pharmacol 94: 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG (2007). Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro . Br J Pharmacol 150: 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM (2013). Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 12: 630–644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary of CB1R agonist‐bound (5XRA) and antagonist‐bound (5TGZ) amino acid residues interacting with cannabinoid ligands in AutoDock.
Table S2 Summary of modelled CB2R agonist‐bound (5XRA) and antagonist‐bound (5TGZ) amino acid residues interacting with cannabinoid ligands in AutoDock.