

Abstract
At the beginning of the 21st century, the therapeutic management of neurodegenerative disorders remains a major biomedical challenge, particularly given the worldwide ageing of the population over the past 50 years that is expected to continue in the forthcoming years. This review will focus on the promise of cannabinoid‐based therapies to address this challenge. This promise is based on the broad neuroprotective profile of cannabinoids, which may cooperate to combat excitotoxicity, oxidative stress, glia‐driven inflammation and protein aggregation. Such effects may be produced by the activity of cannabinoids through their canonical targets (e.g. cannabinoid receptors and endocannabinoid enzymes) and also via non‐canonical elements and activities in distinct cell types critical for cell survival or neuronal replacement (e.g. neurons, glia and neural precursor cells). Ultimately, the therapeutic events driven by endocannabinoid signalling reflect the activity of an endogenous system that regulates the preservation, rescue, repair and replacement of neurons and glia.
Linked Articles
This article is part of a themed section on 8th European Workshop on Cannabinoid Research. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.10/issuetoc
Abbreviations
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- BBB
blood–brain barrier
- CBD
cannabidiol
- CBG
cannabigerol
- CB1
cannabinoid type 1 receptor
- CB2
cannabinoid type 2 receptor
- FAAH
fatty acid amide hydrolase
- HD
Huntington's disease
- iNOS
inducible NOS
- NPCs
neural progenitor cells
- OPCs
oligodendrocyte precursor cells
- PD
Parkinson's disease
- Δ9‐THC
Δ9‐tetrahydrocannabinol
The biomedical challenge of the pathological brain ageing
Brain ageing is a natural process that leads to numerous molecular, cellular and tissue changes and adaptations. These include local reductions in glucose metabolism, oxidative stress, impaired cellular calcium signalling, slower protein synthesis and degradation, which may in turn facilitate the accumulation of damaged proteins and neuronal atrophy driven by defective activity at synapses (e.g. receptors, transporters, ion channels and synthetic enzymes), as well as reduced neurogenesis and glial reactivity (Salat, 2011; Stranahan and Mattson, 2012; Grimm and Eckert, 2017). In normal conditions of ‘physiological brain ageing’, such ageing‐related responses are less predominant, clearly differing in magnitude to such events when associated with the so‐called pathological brain ageing characteristic of neurodegenerative disorders. However, the fact that ageing occurs over decades and that humans may now more frequently reach 80–90 years of age implies that the modest events typically found in brain ageing may aggravate and drive the physiological process towards a pathological condition. This fact is particularly evident when we consider the increase in the incidence of three representative and common neurodegenerative diseases with age; the incidence of Alzheimer's disease (AD), Parkinson's disease (PD) and Huntington's disease (HD), in the range of 60–64 to 85–89 years increase by approximately 15‐, 7‐ and 5‐fold respectively (see Figure 1 and Saba, 2015).
Figure 1.
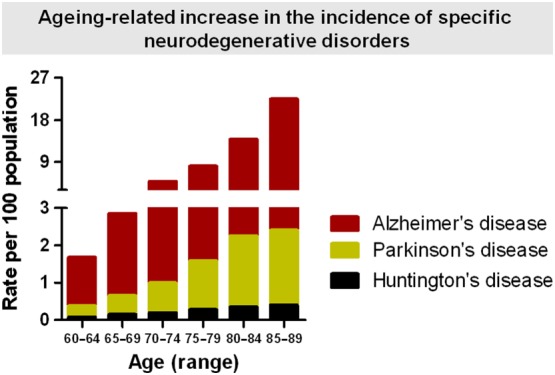
Progression of the incidence of AD, PD and HD in relation to age as a function of the rate of each disease per 100 subjects (source: Saba, 2015).
Similar findings can be derived from examining the life expectancy data and global population pyramids over the past 40–50 years, as well as from the predictions for the forthcoming decades (United Nations, 2015; World Health Organization, 2016; OECD, 2017). These data indicate that the population of individuals aged over 65 years has increased significantly in developed countries in the last 40–50 years (due to a greater life expectancy and a lower birth rate), both in absolute terms and also in proportion to the entire population. This increase has been paralleled with an exponential increase in the incidence of AD, PD and HD, as well as other neurodegenerative disorders (Mazucanti et al., 2015). For example, life expectancies in 1970 were 70–75 years in Japan, most European countries, New Zealand and Canada, yet by 2015, they had risen to above 80 years (see details for European countries in Table 1). Similar increases were also found in other countries like Russia (formerly the Soviet Union: from 65 years in 1970 to approximately 70 years in 2015), Australia (from 70 to more than 80) and the USA (from 70 to 79), whereas countries with a life expectancy below 60 years in 1970 reached values of approximately 75 years in 2015, such as China, Brazil and Mexico (OECD, 2017).
Table 1.
Life expectancy and % of people over 65 years in 2015 in European countries (source: OECD, 2017)
| Country | Life expectancy (years) | People over 65 years (%) |
|---|---|---|
| Spain | 83 | 19 |
| Switzerland | 83 | 18 |
| Italy | 82.6 | 22 |
| Iceland | 82.5 | 14 |
| France | 82.4 | 18 |
| Luxembourg | 82.4 | 14 |
| Norway | 82.4 | 16 |
| Sweden | 82.3 | 20 |
| Finland | 81.6 | 20 |
| The Netherlands | 81.6 | 18 |
| Ireland | 81.5 | 13 |
| Austria | 81.3 | 18 |
| Portugal | 81.2 | 20 |
| Belgium | 81.1 | 18 |
| Greece | 81.1 | 21 |
| UK | 81 | 18 |
| Slovenia | 80.9 | 18 |
| Denmark | 80.8 | 19 |
| Germany | 80.7 | 21 |
| Czech Republic | 78.7 | 18 |
| Turkey | 78 | 8 |
| Estonia | 77.7 | 19 |
| Poland | 77.6 | 15 |
| Slovak Republic | 76.7 | 14 |
| Hungary | 75.7 | 18 |
| Latvia | 74.6 | 20 |
| Lithuania | 74.5 | 19 |
| Russia | 71.3 | 14 |
The problem is that the expectations for the forthcoming years are no better, as the world population has increased, and a further twofold increase is expected by 2030 (United Nations, 2015; World Health Organization, 2016), with a particularly notable increase in elderly people. This elevation in the number of aged individuals will be concentrated in developed countries, although the association between living longer and a higher incidence of neurodegenerative disorders will also become evident in developing countries (see Figure 2 for a prediction of the life expectancy and the proportion of people over 65 years of age in 2050 compared with 2015). In developed countries with current life expectancies around 80 years of age or more, such as most of Europe (see details for specific countries in Table 1), Japan, New Zealand, Australia, the USA and Canada, lifespans close to (or higher than) 90 years of age may be reached by 2050, with a 28% rise in individuals over 65 years of age, which will double the incidence of neurodegenerative disorders (Dorsey et al., 2007; Kowal et al., 2013). However, countries with a shorter life expectancies (between 60 and 75 years) and a lower proportion of individuals over 65 years of age in 2015 (<21%) will also contribute to the higher incidence of neurodegenerative disorders, as the life expectancy in these countries is expected to rise between 1.5‐ and 3‐fold by 2050. This is the case of Russia (an exception among the European countries), China, Saudi Arabia, South Africa, Brazil and Argentina and particularly in Maghreb countries, Mexico, some Central American countries and India, where the individuals over 65 years will grow from the present 7 to nearly 21% (see Figure 2). Therefore, it is conceivable that neurodegenerative disorders will become an epidemic as the 21st century progresses, with their therapeutic control representing the major biomedical challenge that we will have to deal with in this century. Such challenges may be similar to the situation of other diseases that have had a significant impact on human health in the past century, like infectious diseases prior to the development of antibiotics, and cancer, which is currently evolving towards a chronic and better‐controlled disease with the improvement in antitumoural chemotherapies.
Figure 2.
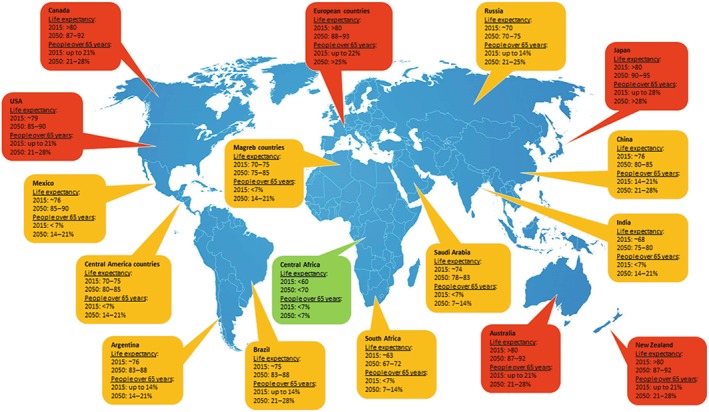
Comparison of life expectancy in 2015 and 2050 and the proportion of people over 65 years of age (predicted values), in different countries and geographical areas around the world (source: United Nations, 2015; World Health Organization, 2016; OECD, 2017). European countries, Japan, New Zealand, Australia, the USA and Canada will reach the highest life expectancies (close to or above 90 years), and the rate of people older than 65 will be at least 28% by 2050, with the highest incidence of neurodegenerative disorders (marked in red). Similarly, by 2050, the life expectancy and rates of people older than 65 will increase between 1.5‐fold and 3‐fold in Russia, China, Saudi Arabia, South Africa, Brazil, Argentina and particularly in Maghreb countries, Mexico, some countries in Central America and India, also in association with an important increase in the incidence of these disorders (marked in yellow).
The failure in developing neuroprotective therapies
The decade of the 1990s was declared by US Congress as the ‘Decade of the Brain’, with the expectation of generating neuroprotective and neurorepair strategies that would effectively control diseases like AD and other types of dementia, PD, amyotrophic lateral sclerosis (ALS) and HD by 2000 (Jones and Mendell, 1999). Unfortunately, the realization of this expectation was limited, and the situation persists almost 20 years later. Examples of this limited success were addressed in an interesting review, highlighting how despite the large number of therapeutic agents investigated to combat AD (agents that apparently reduce β‐amyloid production and aggregation, or tau protein aggregation and hyperphosphorylation, or that elevate the clearance of β‐amyloid), only four agents with modest efficacy have been approved to date. Indeed, the failure rate at the preclinical or clinical stages of evaluation has been particularly high (Mangialasche et al., 2010). The same situation can be found for other common neurodegenerative disorders like PD (Olanow et al., 2017), ALS (Petrov et al., 2017) and HD (Rodrigues and Wild, 2018: see Table 2 for an indication of some failed therapies for these four disorders).
Table 2.
Examples of families of compounds that have been investigated (or are being presently investigated) for their potential disease‐modifying properties in neurodegenerative disorders and that reached clinical testing
| Type of compounds classified according to their major mechanism of action | Disease | Result in clinical testing | |
|---|---|---|---|
| Antiexcitotoxic agents | Ceftriaxone | ALS | Failed |
| Riluzole | ALS | Approved for the first time in 1995 | |
| Riluzole | AD | Under investigation | |
| Riluzole | PD | Failed | |
| Talampanel | ALS | Failed | |
| Memantine | AD | Approved for the first time in 1989a | |
| Memantine | ALS | Failed | |
| Memantine | HD | Under investigation | |
| Antioxidant agents | Coenzyme Q10 | ALS, PD, HD | Failed |
| Creatine | ALS, PD | Failed | |
| Edavarone | ALS | Approved in 2015 in Japan | |
| Benfotiamine | AD | Under investigation | |
| Vitamin E | PD | Failed | |
| Anti‐inflammatory agents | Celecoxib | ALS | Failed |
| Glatiramer acetate | ALS | Failed | |
| Minocycline | ALS, PD | Failed | |
| Pioglitazone | ALS | Failed | |
| Pioglitazoneb | AD | Under investigation | |
| Rosiglitazoneb | AD | Failed | |
| Masitinib | ALS | Approved as orphan drug in 2015 | |
| Mitochondria‐targeted agents | Olexisome | ALS | Failed |
| SBT‐020 | HD | Under investigation | |
| Dexpramipexole | ALS | Failed | |
| Latrepirdine | AD | Failed | |
| Neurotrophic factors | BDNF | ALS | Failed |
| IGF‐1 | ALS | Failed | |
| CNTF | ALS | Failed | |
| NGF | AD | Failed | |
| GDNF | PD | Failed | |
| Neurturin | PD | Failed | |
| Autophagy enhancers | Trehalose | HD | Under investigation |
| Apoptosis inhibitors | THC 346 and CEPI 347 | PD | Failed |
| Cholinergic agents | Donepezil | AD | Approved for the first time in 1997a |
| Rivastigmine | AD | Approved for the first time in 2000a | |
| Galantamine | AD | Approved for the first time in 2001a | |
| Protein (β‐amyloid, tau, huntingtin, α‐synuclein, LRRK‐2)‐targeted agents | Immunotherapy (solanezumab) | AD | Failed |
| Immunotherapy (crenezumab) | AD | Under investigation | |
| β‐Secretase inhibitors (AZD3293) | AD | Under investigation | |
| γ‐Secretase inhibitors (semagacestat) | AD | Failed | |
| Inhibitors βA aggregation (tramiprosate) | AD | Failed | |
| Htt‐lowering ASO (ISIS‐443139) | HD | Under investigation | |
| tau inhibitor (methylene blue) | AD | Under investigation | |
| Inhibitors of GSK‐3β (tideglusib) | AD | Failed | |
| SNCA agents (NPT200‐11; nilotinib) | PD | Under investigation | |
| LRRK‐2 inhibitors (DNL 201) | PD | Under investigation | |
| Others (different mechanisms of action) | Lithium | ALS, AD | Failed |
| Valproate | ALS, AD | Failed | |
| Statins | AD | Failed | |
| Rasagiline | AD | Under investigation | |
| Rasagiline | PD | Failed | |
| Ambroxol | PD | Under investigation | |
| Laquinimod | HD | Under investigation | |
| Pridopidine | HD | Failed | |
| PF‐0254920 | HD | Failed | |
Symptom‐relieving agents with potential neuroprotective properties.
Can also promote β‐secretase inhibition.
βA, β‐amyloid; ASO, antisense oligonucleotides; BDNF, brain‐derived neurotrophic factor; CNTF, ciliary neurotrophic factor; GDNF, glial cell‐derived neurotrophic factor; GSK‐3β, glycogen synthase kinase‐3β; Htt, huntingtin; IGF‐1, insulin‐like growth factor 1; LRRK‐2, leucine‐rich repeat kinase‐2; NGF, nerve growth factor; SNCA, α‐synuclein.
What are the causes for these high failure rates when attempting to develop efficacious neuroprotective therapies? There is no clear answer to this question, yet over the years we have been able to identify some important problems that possibly condition the development of such therapies. Thus, as yet no clear pharmacological targets have been confirmed, either targets that require the use of combination therapies or those that would respond to selective compounds. Moreover, the relationships between the experimental models used and each of the specific diseases often remain unclear. Indeed, the predictive nature of current animal models is not always ideal, and there is still a clear need for new, more appropriate models. To date, translation from basic science to clinical practice is more than often inefficient. In addition, neurodegenerative diseases are particularly susceptible to delays in the initiation of clinical treatments, which is likely to have an important effect on the success of many therapies. Such delays are mainly due to the failure to diagnose the disease early on its development, and they reflect the urgent need for early biomarkers. Finally, the limited focus on neuroprotection rather than strategies that also encompass neurorepair has hindered progress towards successful treatment of these conditions.
Pharmacological targets
Regarding the problem of pharmacological targets, the potential benefits of numerous compounds have been studied over the past 10–20 years: antioxidant compounds, acting as mere ROS scavengers or modulating Nrf2 signalling (Khan et al., 2016); anti‐inflammatory agents capable of limiting glial reactivity and of inhibiting COX‐2, inducible NOS (iNOS) or the generation of cytokines (Baune, 2015); antagonists of glutamate receptors to limit excitotoxicity (Hoque et al., 2016); caspase inhibitors to reduce apoptosis (Kudelova et al., 2015); enhancers of autophagy to eliminate protein aggregates (Sarkar, 2013); neurotrophic factors to promote neuronal survival (Meldolesi, 2017); or other types of compounds that apparently favour neuronal preservation, rescue, repair and replacement and that may therefore possibly represent effective disease‐modifying therapies for neurodegenerative disorders (see Table 2 for a summary of already or ongoing investigated neuroprotective agents). However, the results obtained have not been as positive as expected, with some of these compounds failing in cellular or animal models and others failing to reproduce their positive effects in humans (Berk et al., 2014; Sampaio et al., 2014; Athauda and Foltynie, 2015). These poor outcomes may simply be related to the fact that these compounds target only one of the different mechanisms involved in degenerative cell death and that their efficacy was tested in cellular and in vivo models that specifically promote such mechanisms. However, the pathological context of the distinct human neurodegenerative disorders is quite different, with neurotoxic events combining to produce neuron and glia loss. In these conditions, efficacious therapies would need to be based on ‘polypharmacological strategies’, using broad‐spectrum agents capable of limiting different cytotoxic events at the same time or combining more selective agents for each process (Geldenhuys and Van der Schyf, 2013).
Experimental models
There are currently around 25 different transgenic mouse models of HD, although none of them fully reproduce all the pathological aspects of this disease. Such variability could be related to the extreme complexity of the human CNS, the regional vulnerability apparent in this disease and the variation in disease progression that is difficult to reproduce in classic laboratory species like rodents (Eaton and Wishart, 2017). This failure to establish adequate models may explain the poor outcome in the development of neuroprotective treatments, highlighting the need to develop more useful and predictive models that better reproduce neurodegenerative pathologies in cells and laboratory animals (see Figure 3 for a summary of currently available experimental models and tools). Such advances should aid drug development, as has been addressed in some key reviews that identified different problems and proposed some ideas that could be pursued (Menalled and Brunner, 2014; Mancuso and Navarro, 2015; Koprich et al., 2017; Sasaguri et al., 2017). For example, it has been proposed that experimental models need to adjust to the progressive course of the different diseases and better adapt to the time points at which these diseases become evident in humans. An interesting case is the R6/2 mice, a common model of HD frequently used to screen new neuroprotective agents (Morton and Howland, 2013). However, these mice represent a very aggressive model in which the different pathogenic events occur too early and develop too fast (huntingtin aggregation, transcriptional dysregulation, motor impairment or neuronal death), before the mice reach 12 weeks of age. By contrast, to fully reflect the human disease, these symptoms should appear at 16 months of age and progress over at least 6 months.
Figure 3.
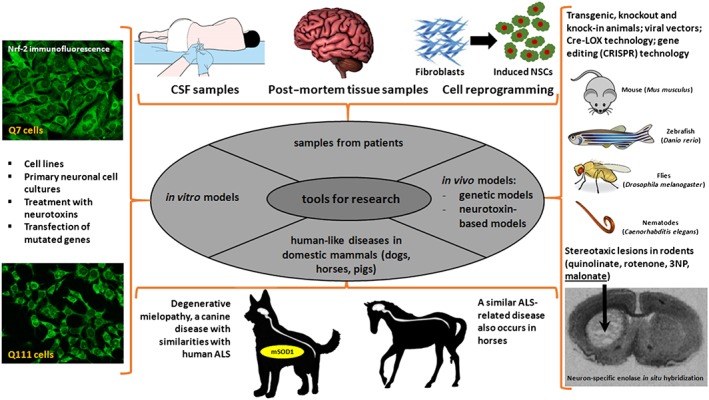
Diagram showing the different research tools used to study neurodegenerative disorders.
Genetic cases of neurodegenerative disorders are frequently reproduced in laboratory species by transgenesis or using viral vectors to drive overexpression of the mutant protein. However, when the disease mutation causes a loss of function, the response may be compensated for by the normal function of the wild‐type protein naturally produced by the host animal. This long‐standing problem may be overcome by the development of knock‐in models, in which the mutant gene inserted substitutes the wild‐type one, and more so with the more recent arrival of the novel gene editing tools (Whitelaw et al., 2016; Yang et al., 2016). These new tools (e.g. CRISPR/Cas9 technology) will soon facilitate site‐specific mutation within the genome, allowing animals carrying known human disease mutations to be generated (Whitelaw et al., 2016; Yang et al., 2016). In addition, the use of Cre/LoxP‐mediated recombination technology (McLellan et al., 2017) also makes cell‐ and region‐specific and/or time‐dependent expression of the mutant protein possible, such that alterations occur in specific cell substrates or CNS areas and/or they are restricted to specific ages or disease stages.
We also need to better classify the different experimental models in relation to their usefulness for the study of these diseases. While some models may be used to study specific pathogenic events, they may not detect the efficacy of potential neuroprotective agents, and vice versa. For example, models based on the exacerbation of one cytotoxic mechanisms (e.g. excitotoxicity) may be used to investigate the contribution of such events to the disease, although they will not be useful for drug screening other than for agents targeting that specific mechanism. By contrast, neurotoxin‐based models that are not useful to reproduce the progression of pathogenic events may nevertheless be useful to screen new drugs. In this sense, the initial screening of neuroprotective drugs frequently includes the use of simple cell models, or invertebrate (e.g. flies and nematodes) or vertebrate models not close to humans (e.g. zebrafish). These in vivo models have the advantage of being easier to use, and they are cheaper, although they generally have limited value for clinical translation, even though they may provide useful data. Hence, the compounds validated in the initial screens would need to be subjected to more detail preclinical studies in mammals. These should include small mammals like rodents and possibly species closer to humans in which new gene editing tools may already be used (e.g. sheep and pigs: Holm et al., 2016).
Another important characteristic of human neurodegenerative disorders is that they appear spontaneously, irrespective of the genetic or environmental factors that determine their aetiology. However, the reproduction of these disorders in laboratory animals, in general small species (e.g. rodents, fishes, flies and nematodes), implies inducing the pathology through different strategies (e.g. transgenesis, viral vectors, neurotoxins and CRISPR/Cas9 gene editing), and as already mentioned, such models are frequently associated with problems in terms of translation to a clinical scenario. Research into these disorders would benefit significantly from the use of species in which human‐like neurodegenerative disorders develop naturally (Youssef et al., 2016). This is the case of an ALS‐like disorder typical of dogs, so‐called degenerative myelopathy (Coates and Wininger, 2010), and its equivalent described in horses (Divers et al., 1997), these sharing many neuropathological features with human ALS including mutation of similar genes (e.g. SOD‐1: Coates and Wininger, 2010). Hence, using animal species that are phylogenetically closer to humans and in which the disease develops spontaneously represents a more promising approach to investigate such diseases.
Early disease detection
It is generally accepted that the poor outcome of neuroprotective treatments in clinical trials may be related to the fact that they are generally initiated at an advanced state of disease progression due to the late diagnosis of these conditions. The diagnosis of neurodegenerative disorders is mainly based on clinical criteria and the appearance of classic symptoms. It is also generally accepted that earlier diagnosis based on the identification of early disease biomarkers, and an early onset of treatment, would significantly improve therapeutic efficacy. This is critical in most neurodegenerative disorders, but a key example is PD, a disease that is predominantly of sporadic origin in which the first motor symptoms (e.g. akinesia, rigidity and tremor) appear when more than 50% of nigral neurons have been lost during the prodromal phase of the disease. In such conditions, neuroprotective therapies are doomed to fail, although they are necessary to preserve the surviving neurons. Hence, it is essential to identify early biomarkers based on imaging techniques, neurological recording or biochemical analysis of biological fluids, tissues or cells, to improve the efficacy of neuroprotective treatments (reviewed in Delenclos et al., 2016). Such biomarkers will help achieve an earlier diagnosis or identify subjects at risk of developing the disease, thereby enabling neuroprotective treatments to be initiated earlier and improve the chances that they may be efficacious.
Similar lines of investigation are being followed in the study of AD and ALS, aimed at detecting presymptomatic individuals at risk of progressing towards the symptomatic phase of the disease (Frisoni et al., 2017; Gordon and Meininger, 2011, respectively). In the case of AD (see details in Figure 4), different parameters in the so‐called preclinical stage of the disease have been proposed (Frisoni et al., 2017), these including earlier analysis of β‐amyloid aggregation with PET probes or in the CSF; the detection of synaptic dysfunction in specific CNS areas using metabolic recording with fluorodeoxyglucose‐PET or MRI; the analysis of tau levels in the CSF; and the recording of structural brain changes with volumetric MRI. Such approaches may be accompanied by evaluation with psychological scales (e.g. mini‐mental state examination), these possibly providing evidence of mild cognitive impairment, considered to be a pre‐Alzheimer state (Frisoni et al., 2017). With such early diagnosis, the possibilities of obtaining positive results in clinical testing should improve significantly.
Figure 4.
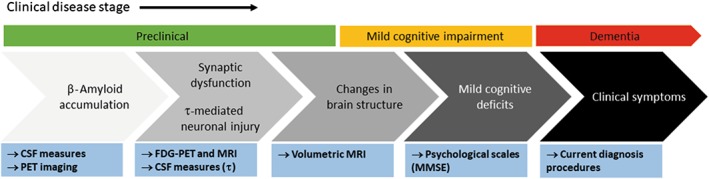
Diagram showing a series of potential biomarkers that have been proposed to detect the progression of AD before its clinical diagnosis and the biological samples/methodological tools for their analysis (source: Frisoni et al., 2017). MMSE, mini‐mental state examination.
The need for neurorepair
Since clinical diagnosis of many neurodegenerative disorders occurs when an important degree of neuronal injury and loss has already occurred, it is essential for disease‐modifying treatments to include, or be accompanied by, neurorepair strategies aimed at restoring the neurons lost (Ziemka‐Nałęcz and Zalewska, 2012; Demuth et al., 2017). This has been an important research challenge for these pathologies, particularly in the case of PD, the disease that has received most attention in terms of developing neuronal cell replacement procedures that use a different cell source that can potentially acquire dopaminergic characteristics. This was first attempted with chromaffin cells, followed by fetal dopaminergic neurons, and more recently with neural stem cells reprogrammed from human fibroblasts (Stoker et al., 2017). Similar strategies have been studied for other disorders like HD (Rosser and Svendsen, 2014), although numerous problems and side effects have been associated with these procedures. To date, these strategies have progressed to the use of natural cell replacement from neurogenic niches (Farzanehfar, 2016), although our understanding of these processes is still limited.
Cannabinoids as neuroprotectants
The first evidence that cannabinoids may offer neuroprotection was obtained in the 1990s working with HU‐211 (known as dexanabinol), which is the inactive enantiomer of the potent cannabinoid receptor agonist HU‐210. Unlike HU‐210, HU‐211 has negligible activity at classic endocannabinoid‐related targets but provided neuroprotection in experimental models of ischaemia and brain trauma. These preclinical data led to further clinical trials, but unfortunately, these were not successful (Pop, 2000; Maas et al., 2006). Later in that decade, a series of in vitro studies demonstrated neuroprotective effects of the phytocannabinoids Δ9‐tetrahydrocannabinol (Δ9‐THC) and cannabidiol (CBD), which proved to be cannabinoid receptor independent and generated by their activity as ROS scavengers (Hampson et al., 1998). This pioneering work was followed by a number of studies demonstrating how cannabinoids may preserve not only neurons but also some glial cell subpopulations (e.g. astrocytes, oligodendrocytes and their precursor cells) when exposed to different insults that jeopardize cell homeostasis and integrity (Fernández‐Ruiz et al., 2015a,b, for review). Moreover, these studies implicated targets within the endocannabinoid system (cannabinoid receptors or endocannabinoid enzymes) that may be activated or inhibited to produce neuroprotection, as well as other targets outside the endocannabinoid system (Fernández‐Ruiz et al., 2015a,b). These findings situated cannabinoids and the endocannabinoid system itself in a promising position to achieve neuroprotection and with notable advantages when compared with other neuroprotective agents studied. Hence, it became possible that cannabinoid‐based neuroprotective and neuroreparative strategies could be developed for neurodegenerative disorders with certain advantages over other therapies (see Figure 5 for a summary of these, which will be described in more detail below).
Figure 5.
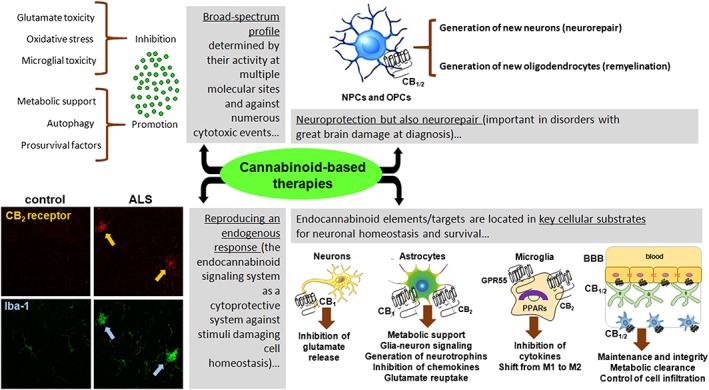
Diagram summarizing four important advantages of possible cannabinoid‐based therapies for the treatment of neurodegenerative disorders when compared with the other therapeutic strategies investigated to date. CB2 receptor‐immunolabelled cells (in red and marked with arrows in the left bottom corner) are much more frequent in ALS tissue than in control tissue, corresponding to reactive microglia (in green and marked with arrows).
Cannabinoids have a broad neuroprotective profile
An evident advantage of cannabinoids as neuroprotectants is their wide range of action (reviewed in Fernández‐Ruiz et al., 2015a,b). Hence, a single molecule, or a mixture of two or more cannabinoids can combine the ability to combat excitotoxicity, inflammation, oxidative stress and protein aggregation, as well as other processes that damage neuronal homeostasis and integrity. Given that these pathological events work in a cooperative manner to kill neurons, the protection of these cells must act on these targets and combine different therapeutic events: a ‘multi‐target strategy’ such as that employed by cannabinoids. The research carried out in HD is a good example of this concept, with antioxidant phytocannabinoids [e.g. CBD and cannabigerol (CBG)] acting on 3‐nitropropionate‐lesioned rodents, a model of HD priming oxidative damage (reviewed in Fernández‐Ruiz et al., 2015a,b, 2017). In addition, cannabinoids that selectively activate cannabinoid type 1 (CB1) receptors are neuroprotective in R6/2 mice, the classic genetic model of HD, and in quinolinate‐lesioned mice, a model driven by excitotoxic events. By contrast, cannabinoids that selectively activate the cannabinoid type 2 (CB2) receptors reproduced the same benefits in both models, and they were also active in rats lesioned with malonate, a model with strong glial reactivity and activation of the apoptotic machinery (Fernández‐Ruiz et al., 2015a,b, 2017, for review). Finally, cannabinoids with negligible activity at CB1/CB2 receptors but that act at the PPAR‐γ were also beneficial in R6/2 and 3‐nitropropionate‐lesioned mice (reviewed in Fernández‐Ruiz et al., 2015a,b, 2017). In general, these results support the idea that therapies for HD patients should be based on antioxidant cannabinoids acting through canonical cannabinoid receptors (CB1 and CB2 receptors) and non‐endocannabinoid‐related targets (e.g. PPAR‐γ), or on a combination of cannabinoids ensuring all these activities (reviewed in Fernández‐Ruiz et al., 2015a,b; 2017). Studies in experimental models of other disorders (e.g. AD, PD and ALS) indicate that similar approaches might also be beneficial, using cannabinoids with a broad activity profile (reviewed in Fernández‐Ruiz et al., 2015a,b, 2017; see Figure 6 for a graphical representation of this idea).
Figure 6.
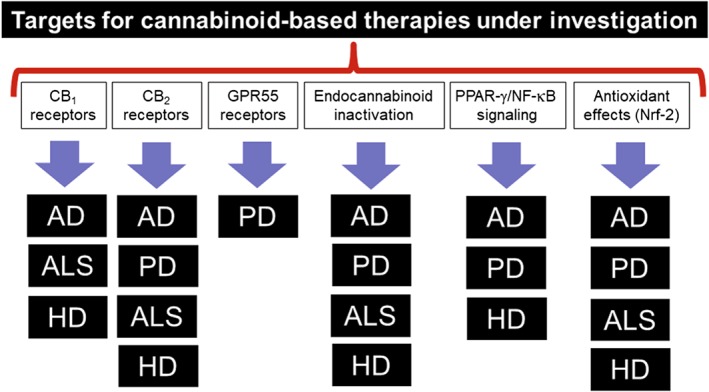
Summary of the different key targets and/or processes that are being investigated, and already showed some promise, for the development of cannabinoid‐based neuroprotective therapies in the four more representative chronic progressive neurodegenerative disorders (e.g. AD, PD, ALS and HD).
Such broad neuroprotective effects of cannabinoids are the natural consequence of their pleiotropic profile, which on the one hand is related to their direct actions on elements of the endocannabinoid system (e.g. CB1/CB2 receptors, inactivating enzymes and GPR55), and on the other hand, involves targets not directly related to endocannabinoid signalling (see Figures 5 and 6; reviewed in Fernández‐Ruiz et al., 2015a,b, 2017). Cannabinoids may limit excitotoxic damage by activating CB1 receptors, probably by inhibiting glutamate release and possibly by promoting glutamate reuptake, enhancing blood supply and activating autophagy to eliminate protein aggregates (see Figure 5), all events that will benefit neuronal homeostasis and integrity. In addition, by activating CB2 receptors, the orphan receptor GPR55 or the nuclear receptors of the PPAR family, cannabinoids may also attenuate glial toxicity in neurons (see Figure 5). This latter effect involves the regulation of NF‐κB and its downstream activators of pro‐inflammatory gene expression (e.g. iNOS, COX‐2, metalloproteases and cytokines).
Like their antioxidant effects, the other neuroprotective effects of cannabinoids are independent of cannabinoid receptors, as was the case of the phytocannabinoids Δ9‐THC, cannabinol, CBD, cannabidiolic acid, cannabidivarin and CBG, as well as the synthetic cannabinoids nabilone, levonantradol and dexanabinol (Hampson et al., 1998; Marsicano et al., 2002). However, further research into these antioxidant effects suggested the possible activation of nuclear receptors of the PPAR family or of other similar intracellular targets that might regulate gene responses associated with the activity of transcription factors like Nrf‐2 (Fernández‐Ruiz et al., 2017). In addition, CBD is active at numerous non‐endocannabinoid targets whose activation/inhibition has been linked to cytoprotective effects (Iuvone et al., 2009). For example, CBD can inhibit the mechanisms underlying endocannabinoid inactivation, thereby elevating the activity of endocannabinoids and enhancing their neuroprotective effects mediated by cannabinoid receptors (reviewed in Fernández‐Ruiz et al., 2017). This may be particularly relevant in certain pathological conditions (e.g. autosomal dominant inherited ataxias: Rodríguez‐Cueto et al., 2014, 2016) where dysregulation in these inactivating mechanisms has been seen to affect CNS structures [e.g. elevated fatty acid amide hydrolase (FAAH) activity], leading to excess degradation of endocannabinoids. These effects are associated with enhanced vulnerability to damaging stimuli and the development of degenerative processes. In such conditions, inhibiting FAAH may offer therapeutic benefits (Hwang et al., 2010).
Neurorepair strategies involving cannabinoids
As indicated above, the clinical diagnosis of most neurodegenerative disorders, in particular those of sporadic origin, is mainly based on the detection of clinical symptoms following standardized scales. However, when these symptoms become evident, the extent of neuronal injury is usually quite important. Hence, treatment needs not only to preserve the surviving neurons but also to promote the replacement of those neurons lost during the prodromal stage of the disease. Such replacement from neurogenic niches is something that the pharmacological manipulation of endocannabinoid signalling may also promote given the presence of key elements of this system (e.g. CB1 and CB2 receptors and FAAH) in neural progenitor cells (NPCs), cells that may differentiate in neurons, astrocyte or oligodendrocytes (reviewed in Galve‐Roperh et al., 2013). In these precursor cells, CB1 and/or CB2 receptor activation, and the inhibition of FAAH, has been associated with proliferation, as well as their maturation and/or differentiation to neurons or glia. Thus, such events could potentially be pharmacologically manipulated to repair lesioned areas (see Figure 5: reviewed in Galve‐Roperh et al., 2013). CB2 receptors have also been identified in oligodendrocyte precursor cells (OPCs), playing a key role in these glial cells that are in turn crucial for neurons (e.g. remyelination, see Figure 5: reviewed in Arévalo‐Martín et al., 2008).
Cannabinoid treatment mimics an endogenous protective response facilitated by the key cellular location of their targets
Years of research have provided sufficient experimental evidence to interpret the benefits that can be achieved with cannabinoids in terms of neuroprotection and neurorepair, principally as a natural consequence of two important properties of the endocannabinoid signalling system. On the one hand, cannabinoids have a broad neuroprotective profile, the endocannabinoid system providing endogenous protective responses against inflammatory, excitotoxic, infectious, traumatic or oxidant stimuli that potentially damage the CNS (Pacher and Mechoulam, 2011; Fernández‐Ruiz et al., 2015a,b). On the other hand, this protective/reparative effect is likely to be facilitated by the location of the elements of this system in cells/tissues that are crucial for the preservation, rescue, repair and/or replacement of neurons (see Figure 5 for details).
In terms of the former, it was first proposed that endocannabinoid signalling may function as an endogenous protective system in response to conditions that could damage the brain (e.g. inflammation, trauma, oxidative stress, excitotoxicity, protein and cell organelle aggregation) nearly a decade ago (Pacher and Mechoulam, 2011). This hypothesis was based on the responses of specific elements of this signalling system to lesions of structures associated with most neurodegenerative disorders, in particular the elevated generation of endocannabinoids and the up‐regulation of CB2 receptors detected (Pacher and Mechoulam, 2011). Accordingly, pharmacological enhancement of these responses proved to be potentially beneficial in these disorders (e.g. blocking endocannabinoid inactivation, administering endocannabinoids or selectively activating the CB2 receptor: Mechoulam and Shohami, 2007; reviewed in Fernández‐Ruiz et al., 2015a,b). This was particularly evident in the case of CB2 receptor up‐regulation in glial elements, mainly microglia (see Figure 5) and astrocytes, in parallel to their activation in response to damaging stimuli. By contrast, other responses experienced by the endocannabinoid signalling appear to be more closely related to a dysregulation of this system (e.g. reduced CB1 receptor signalling and elevated FAAH activity), which may be instrumental in the pathogenesis of these disorders (Fagan and Campbell, 2014). In this case, rather than being enhanced, these responses need to be pharmacologically corrected, as indicated above (e.g. FAAH inhibition: see Hwang et al., 2010).
A second determinant in the endogenous neuroprotective and neurorepair activity exerted by the endocannabinoid system is the location of its elements in cell substrates and structures that are strongly implicated in neuronal homeostasis, integrity and survival. This includes their location not only in specific neuronal subpopulations but also in astrocytes, resting and reactive microglia, perivascular microglial cells, oligodendrocytes, OPCs and NPCs, and even in key CNS structures like the blood–brain barrier (BBB: see details in Figure 5). As such, cannabinoids can selectively control the activity of these cells (or structures), particularly in processes related to survival, protection and/or repair (Galve‐Roperh et al., 2013; Fernández‐Ruiz et al., 2015a). For example, recent studies demonstrated that preservation of BBB integrity and function, which is essential in neuroprotection, is under the control of both CB1 and CB2 receptor‐mediated signals (Fujii et al., 2014). Both receptors are located in specific cells in the BBB, where they contribute to maintain the integrity of tight junctions, to inhibit leukocyte infiltration and to facilitate β‐amyloid clearance (Vendel and de Lange, 2014). Both receptors are also located in the brain microvasculature, where they help reduce endothelin‐1‐induced vasoconstriction and restore blood supply to the injured brain (Mechoulam and Shohami, 2007; Choi et al., 2013).
In terms of neuroprotection, the presence of CB1 receptors at the glutamatergic terminals of neurons can inhibit the influence of excess glutamate release, thereby avoiding excitotoxic damage (see Figure 5). In addition, post‐synaptic CB1 receptors in neurons containing glutamate receptors (e.g. NMDA receptors) may also facilitate the activity of cannabinoids by closing voltage‐dependent calcium channels, thereby controlling the intracellular levels of calcium and avoiding the overactivation of calcium‐dependent destructive pathways (reviewed in Fernández‐Ruiz et al., 2015a,b). Both these effects are completely dependent on the pre‐ and post‐synaptic location of CB1 receptors at glutamatergic synapses, and they aim to preserve correct synaptic homeostasis and avoid the consequences of excess excitatory transmission.
Reactive astrocytes are also key cell substrates for neuroprotection, which express not only CB2 but also CB1 receptors. Accordingly, both receptors may work in conjunction to regulate astrocyte behaviour and particularly its influence on neuronal homeostasis, integrity and survival, either positively or negatively (reviewed in Fernández‐Ruiz et al., 2015a,b; Stella, 2010). Their effects include enhancing the supply of metabolic substrates (e.g. lactate or ketone bodies: Duarte et al., 2012), glutamate reuptake and the generation of neurotrophins, anti‐inflammatory mediators (e.g. IL‐10 and IL‐1 receptor antagonist) or prosurvival factors (e.g. TGF‐β), which could potentially rescue damaged neurons (see Figure 5: Molina‐Holgado et al., 2003; Arévalo‐Martín et al., 2008; Fernández‐Ruiz et al., 2017).
Endocannabinoid signalling is also active in microglial cells, and CB2 receptors are up‐regulated upon activation of these cells in the CNS, as first seen in microglial cells surrounding senile plaques in post‐mortem AD brains (Benito et al., 2003). These microglial CB2 receptors play an important role in the proliferation of microglia and their migration to lesion sites, as well as in the shift from the M1 pro‐inflammatory microglial phenotype towards the anti‐inflammatory M2 phenotype. Activation of these receptors also limits the synthesis of different pro‐inflammatory factors by these cells, factors like TNF‐α and other pro‐inflammatory cytokines (Stella, 2010) that may damage neurons (Fernández‐Ruiz et al., 2015a,b). The CB2 receptor‐mediated inhibition of TNF‐α in microglial cells appears to involve the inhibition of NF‐κB signalling, and it can also be elicited by cannabinoids that activate GPR55 and/or PPAR‐γ, consistent with the broad profile of these compounds (reviewed in Fernández‐Ruiz et al., 2017).
Concluding remarks and futures perspectives
Compared with other neuroprotectants, cannabinoids display a broad spectrum of activity, a particularly important feature when considering neurodegenerative disorders in which the damage to neurons and glia is provoked by different cytotoxic events acting in conjunction. To combat such a ‘collaborative attack’ to kill neurons, attempts at neuroprotection should adopt a ‘multi‐target approach’ or involve the use of combined drug therapy. Cannabinoids fit perfectly with this strategy, through the use of either a broad‐spectrum cannabinoid or a mixture of two or more cannabinoids or, even, in combination with other therapeutic agents. The advantage of cannabinoids is that the cellular and molecular mechanisms mediating their neuroprotective effects are quite diverse and often complementary. For example, a single cannabinoid or a combination of cannabinoids acting in conjunction might reduce excitotoxicity, oxidative stress and glial activation or promote autophagy, eliminate protein aggregates and enhance metabolic and neurotrophic support. Cannabinoids can also activate NPC proliferation, maturation and differentiation, which may facilitate not only neuroprotection but also cell replacement, a critical issue in neurodegenerative disorders that are currently diagnosed when an important amount of neuronal damage/loss has already occurred. Such a combination of neuroprotective and neuroreparative properties is aided by two important characteristics of these compounds: (i) their natural pharmacological targets within the endocannabinoid signalling system can be found in neuromodulatory structures, offering endogenous protection and (ii) these endocannabinoid targets are located in cell substrates and CNS structures that are crucial for the preservation, rescue, repair and replacement of neurons. It is important to note that the intense preclinical work carried out in the last 15 years on cannabinoid‐based therapies for these diseases has provided solid evidence to justify further efforts aimed at developing these molecules (or combinations thereof) from their current preclinical state to a true clinical application. This is the challenge for the forthcoming years, to initiate clinical trials aimed at validating in patients the promise and expectations generated by cannabinoids in cell and animal models of different neurodegenerative disorders.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c, 2017d, 2017e).
Conflict of interest
The author declares no conflicts of interest.
Acknowledgements
This work was supported by grants from CIBERNED (CB06/05/0089) and MINECO (SAF2015‐68580‐C2‐1‐R). The author is indebted to all those who participated in the studies carried out by his group that were mentioned in this review and to Yolanda García‐Movellán for administrative assistance. He also wants to acknowledge EPHAR for allowing him to deliver a conference at the recent 8th European Workshop on Cannabinoid Research on which this review is based.
Fernández‐Ruiz, J. (2019) The biomedical challenge of neurodegenerative disorders: an opportunity for cannabinoid‐based therapies to improve on the poor current therapeutic outcomes. British Journal of Pharmacology, 176: 1370–1383. 10.1111/bph.14382.
This review article was prepared from a conference delivered by the author at the 8th European Workshop on Cannabinoid Research held in Roehamptom, UK (31 August to 2 September 2017), and which was nominated as the EPHAR lecture. The author wants to acknowledge EPHAR for such appointment.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017e). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arévalo‐Martín A, García‐Ovejero D, Gómez O, Rubio‐Araiz A, Navarro‐Galve B, Guaza C et al (2008). CB2 cannabinoid receptors as an emerging target for demyelinating diseases: from neuroimmune interactions to cell replacement strategies. Br J Pharmacol 153: 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athauda D, Foltynie T (2015). The ongoing pursuit of neuroprotective therapies in Parkinson's disease. Nat Rev Neurol 11: 25–40. [DOI] [PubMed] [Google Scholar]
- Baune BT (2015). Inflammation and neurodegenerative disorders: is there still hope for therapeutic intervention? Curr Opin Psychiatry 28: 148–154. [DOI] [PubMed] [Google Scholar]
- Benito C, Nuñez E, Tolon RM, Carrier EJ, Rábano A, Hillard CJ et al (2003). Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque‐associated glia in Alzheimer's disease brains. J Neurosci 23: 11136–11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk C, Paul G, Sabbagh M (2014). Investigational drugs in Alzheimer's disease: current progress. Expert Opin Investig Drugs 23: 837–846. [DOI] [PubMed] [Google Scholar]
- Choi IY, Ju C, Anthony Jalin AM, Lee DI, Prather PL, Kim WK (2013). Activation of cannabinoid CB2 receptor‐mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol 182: 928–939. [DOI] [PubMed] [Google Scholar]
- Coates JR, Wininger FA (2010). Canine degenerative myelopathy. Vet Clin North Am Small Anim Pract 40: 929–950. [DOI] [PubMed] [Google Scholar]
- Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K (2017). Alzheimer's disease drug development pipeline: 2017. Alzheimers Dement 3: 367–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delenclos M, Jones DR, McLean PJ, Uitti RJ (2016). Biomarkers in Parkinson's disease: advances and strategies. Parkinsonism Relat Disord 22: S106–S110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuth HU, Dijkhuizen RM, Farr TD, Gelderblom M, Horsburgh K, Iadecola C et al (2017). Recent progress in translational research on neurovascular and neurodegenerative disorders. Restor Neurol Neurosci 35: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divers TJ, Mohammed HO, Cummings JF (1997). Equine motor neuron disease. Vet Clin North Am Equine Pract 13: 97–105. [DOI] [PubMed] [Google Scholar]
- Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K et al (2007). Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68: 384–386. [DOI] [PubMed] [Google Scholar]
- Duarte JM, Ferreira SG, Carvalho RA, Cunha RA, Köfalvi A (2012). CB₁ receptor activation inhibits neuronal and astrocytic intermediary metabolism in the rat hippocampus. Neurochem Int 60: 1–8. [DOI] [PubMed] [Google Scholar]
- Eaton SL, Wishart TM (2017). Bridging the gap: large animal models in neurodegenerative research. Mamm Genome 28: 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan SG, Campbell VA (2014). The influence of cannabinoids on generic traits of neurodegeneration. Br J Pharmacol 171: 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzanehfar P (2016). Towards a better treatment option for Parkinson's disease: a review of adult neurogenesis. Neurochem Res 41: 3161–3170. [DOI] [PubMed] [Google Scholar]
- Fernández‐Ruiz J, Romero J, Ramos JA (2015a). Endocannabinoids and neurodegenerative disorders: Parkinson's disease, Huntington's chorea, Alzheimer's disease, and others. Handb Exp Pharmacol 231: 233–259. [DOI] [PubMed] [Google Scholar]
- Fernández‐Ruiz J, Moro MA, Martínez‐Orgado J (2015b). Cannabinoids in neurodegenerative disorders and stroke/brain trauma: from preclinical models to clinical applications. Neurotherapeutics 12: 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Ruiz J, Gómez‐Ruiz M, García C, Hernández M, Ramos JA (2017). Modeling neurodegenerative disorders for developing cannabinoid‐based neuroprotective therapies. Methods Enzymol 593: 175–198. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Boccardi M, Barkhof F, Blennow K, Cappa S, Chiotis K et al (2017). Strategic roadmap for an early diagnosis of Alzheimer's disease based on biomarkers. Lancet Neurol 16: 661–676. [DOI] [PubMed] [Google Scholar]
- Fujii M, Sherchan P, Krafft PR, Rolland WB, Soejima Y, Zhang JH (2014). Cannabinoid type 2 receptor stimulation attenuates brain edema by reducing cerebral leukocyte infiltration following subarachnoid hemorrhage in rats. J Neurol Sci 342: 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galve‐Roperh I, Chiurchiù V, Díaz‐Alonso J, Bari M, Guzmán M, Maccarrone M (2013). Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res 52: 633–650. [DOI] [PubMed] [Google Scholar]
- Geldenhuys WJ, Van der Schyf CJ (2013). Rationally designed multi‐targeted agents against neurodegenerative diseases. Curr Med Chem 20: 1662–1672. [DOI] [PubMed] [Google Scholar]
- Gordon PH, Meininger V (2011). How can we improve clinical trials in amyotrophic lateral sclerosis? Nat Rev Neurol 7: 650–654. [DOI] [PubMed] [Google Scholar]
- Grimm A, Eckert A (2017). Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem 143: 418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson AJ, Grimaldi M, Axelrod J, Wink D (1998). Cannabidiol and (−)∆9‐tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A 95: 8268–8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm IE, Alstrup AK, Luo Y (2016). Genetically modified pig models for neurodegenerative disorders. J Pathol 238: 267–287. [DOI] [PubMed] [Google Scholar]
- Hoque A, Hossain MI, Ameen SS, Ang CS, Williamson N, Ng DC et al (2016). A beacon of hope in stroke therapy – blockade of pathologically activated cellular events in excitotoxic neuronal death as potential neuroprotective strategies. Pharmacol Ther 160: 159–179. [DOI] [PubMed] [Google Scholar]
- Hwang J, Adamson C, Butler D, Janero DR, Makriyannis A, Bahr BA (2010). Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: a neuroprotective therapeutic modality. Life Sci 86: 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iuvone T, Esposito G, De Filippis D, Scuderi C, Steardo L (2009). Cannabidiol: a promising drug for neurodegenerative disorders? CNS Neurosci Ther 15: 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG, Mendell LM (1999). Assessing the decade of the brain. Science 284: 739. [DOI] [PubMed] [Google Scholar]
- Khan TA, Hassan I, Ahmad A, Perveen A, Aman S, Quddusi S et al (2016). Recent updates on the dynamic association between oxidative stress and neurodegenerative disorders. CNS Neurol Disord Drug Targets 15: 310–320. [DOI] [PubMed] [Google Scholar]
- Koprich JB, Kalia LV, Brotchie JM (2017). Animal models of α‐synucleinopathy for Parkinson disease drug development. Nat Rev Neurosci 18: 515–529. [DOI] [PubMed] [Google Scholar]
- Kowal SL, Dall TM, Chakrabarti R, Storm MV, Jain A (2013). The current and projected economic burden of Parkinson's disease in the United States. Mov Disord 28: 311–318. [DOI] [PubMed] [Google Scholar]
- Kudelova J, Fleischmannova J, Adamova E, Matalova E (2015). Pharmacological caspase inhibitors: research towards therapeutic perspectives. J Physiol Pharmacol 66: 473–482. [PubMed] [Google Scholar]
- Maas AI, Murray G, Henney H 3rd, Kassem N, Legrand V, Mangelus M et al (2006). Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo‐controlled, clinical trial. Lancet Neurol 5: 38–45. [DOI] [PubMed] [Google Scholar]
- Mancuso R, Navarro X (2015). Amyotrophic lateral sclerosis: current perspectives from basic research to the clinic. Prog Neurobiol 133: 1–26. [DOI] [PubMed] [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M (2010). Alzheimer's disease: clinical trials and drug development. Lancet Neurol 9: 702–716. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C (2002). Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1 . J Neurochem 80: 448–456. [DOI] [PubMed] [Google Scholar]
- Mazucanti CH, Cabral‐Costa JV, Vasconcelos AR, Andreotti DZ, Scavone C, Kawamoto EM (2015). Longevity pathways (mTOR, SIRT, insulin/IGF‐1) as key modulatory targets on aging and neurodegeneration. Curr Top Med Chem 15: 2116–2138. [DOI] [PubMed] [Google Scholar]
- McLellan MA, Rosenthal NA, Pinto AR (2017). Cre‐loxP‐mediated recombination: general principles and experimental considerations. Curr Protoc Mouse Biol 7: 1–12. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Shohami E (2007). Endocannabinoids and traumatic brain injury. Mol Neurobiol 36: 68–74. [DOI] [PubMed] [Google Scholar]
- Meldolesi J (2017). Neurotrophin receptors in the pathogenesis, diagnosis and therapy of neurodegenerative diseases. Pharmacol Res 121: 129–137. [DOI] [PubMed] [Google Scholar]
- Menalled L, Brunner D (2014). Animal models of Huntington's disease for translation to the clinic: best practices. Mov Disord 29: 1375–1390. [DOI] [PubMed] [Google Scholar]
- Molina‐Holgado F, Pinteaux E, Moore JD, Molina‐Holgado E, Guaza C, Gibson RM et al (2003). Endogenous interleukin‐1 receptor antagonist mediates anti‐inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J Neurosci 23: 6470–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Howland DS (2013). Large genetic animal models of Huntington's disease. J Huntingtons Dis 2: 3–19. [DOI] [PubMed] [Google Scholar]
- OECD (2017). Health at a glance. OECD. [Google Scholar]
- Olanow CW, Kieburtz K, Katz R (2017). Clinical approaches to the development of a neuroprotective therapy for PD. Exp Neurol 298: 246–251. [DOI] [PubMed] [Google Scholar]
- Pacher P, Mechoulam R (2011). Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50: 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov D, Mansfield C, Moussy A, Hermine O (2017). ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front Aging Neurosci 9: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop E (2000). Dexanabinol Pharmos. Curr Opin Investig Drugs 1: 494–503. [PubMed] [Google Scholar]
- Rodrigues FB, Wild EJ (2018). Huntington's disease clinical trials corner: February 2018. J Huntingtons Dis 7: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Cueto C, Benito C, Romero J, Hernández‐Gálvez M, Gómez‐Ruiz M, Fernández‐Ruiz J (2014). Endocannabinoid‐hydrolysing enzymes in the post‐mortem cerebellum of humans affected by hereditary autosomal dominant ataxias. Pathobiology 81: 149–159. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Cueto C, Hernández‐Gálvez M, Hillard CJ, Maciel P, García‐García L, Valdeolivas S et al (2016). Dysregulation of the endocannabinoid signaling system in the cerebellum and brainstem in a transgenic mouse model of spinocerebellar ataxia type‐3. Neuroscience 339: 191–209. [DOI] [PubMed] [Google Scholar]
- Rosser A, Svendsen CN (2014). Stem cells for cell replacement therapy: a therapeutic strategy for HD? Mov Disord 29: 1446–1454. [DOI] [PubMed] [Google Scholar]
- Saba L (2015). Imaging in Neurodegenerative Disorders. Oxford University Press: Oxford, UK. [Google Scholar]
- Salat DH (2011). The declining infrastructure of the aging brain. Brain Connect 1: 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio C, Borowsky B, Reilmann R (2014). Clinical trials in Huntington's disease: interventions in early clinical development and newer methodological approaches. Mov Disord 29: 1419–1428. [DOI] [PubMed] [Google Scholar]
- Sardi SP, Cedarbaum JM, Brundin P (2018). Targeted therapies for Parkinson's disease: from genetics to the clinic. Mov Disord, in press 33: 684–696. 10.1002/mds.27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S (2013). Regulation of autophagy by mTOR‐dependent and mTOR‐independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 41: 1103–1130. [DOI] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B et al (2017). APP mouse models for Alzheimer's disease preclinical studies. EMBO J 36: 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N (2010). Cannabinoid and cannabinoid‐like receptors in microglia, astrocytes, and astrocytomas. Glia 58: 1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker TB, Blair NF, Barker RA (2017). Neural grafting for Parkinson's disease: challenges and prospects. Neural Regen Res 12: 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP (2012). Recruiting adaptive cellular stress responses for successful brain ageing. Nat Rev Neurosci 13: 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United Nations (2015). World population prospects: the 2015 revision. UN
- Vendel E, de Lange EC (2014). Functions of the CB1 and CB2 receptors in neuroprotection at the level of the blood–brain barrier. Neuromolecular Med 16: 620–642. [DOI] [PubMed] [Google Scholar]
- Whitelaw CB, Sheets TP, Lillico SG, Telugu BP (2016). Engineering large animal models of human disease. J Pathol 238: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2016). Life expectancy increased by 5 years since 2000, but health inequalities persist. WHO. [Google Scholar]
- Yang W, Tu Z, Sun Q, Li XJ (2016). CRISPR/Cas9: implications for modeling and therapy of neurodegenerative diseases. Front Mol Neurosci 9: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef SA, Capucchio MT, Rofina JE, Chambers JK, Uchida K, Nakayama H et al (2016). Pathology of the aging brain in domestic and laboratory animals, and animal models of human neurodegenerative diseases. Vet Pathol 53: 327–348. [DOI] [PubMed] [Google Scholar]
- Ziemka‐Nałęcz M, Zalewska T (2012). Endogenous neurogenesis induced by ischemic brain injury or neurodegenerative diseases in adults. Acta Neurobiol Exp 72: 309–324. [DOI] [PubMed] [Google Scholar]