

Abstract
Background and Purpose
Non‐invasive in vivo imaging of cannabinoid CB2 receptors using PET is pursued to study neuroinflammation. The purpose of this study is to evaluate the in vivo binding specificity of [18F]MA3, a CB2 receptor agonist, in a rat model with local overexpression of human (h) CB2 receptors.
Methods
[18F]MA3 was produced with good radiochemical yield and radiochemical purity. The radiotracer was evaluated in rats with local overexpression of hCB2 receptors and in a healthy non‐human primate using PET.
Key Results
Ex vivo autoradiography demonstrated CB2‐specific binding of [18F]MA3 in rat hCB2 receptor vector injected striatum. In a PET study, increased tracer binding in the hCB2 receptor vector‐injected striatum compared to the contralateral control vector‐injected striatum was observed. Binding in hCB2 receptor vector‐injected striatum was blocked with a structurally non‐related CB2 receptor inverse agonist, and a displacement study confirmed the reversibility of tracer binding. This study identified the utility of mutated inactive vector model for evaluation of CB2 receptor agonist PET tracers. [18F]MA3 PET scans in the non‐human primate showed good uptake and fast washout from brain, but no CB2 receptor‐specific binding was observed.
Conclusion and Implications
Evaluation of [18F]MA3 in a rat model with local overexpression of hCB2 receptors showed CB2 receptor‐specific and reversible tracer binding. [18F]MA3 showed good brain uptake and subsequent washout in a healthy non‐human primate, but no specific binding was observed. Further clinical evaluation of [18F]MA3 in patients with neuroinflammation is warranted.
Linked Articles
This article is part of a themed section on 8th European Workshop on Cannabinoid Research. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.10/issuetoc
Abbreviations
- 18F‐FDG
2‐[18F]fluoro‐2‐deoxy‐D‐glucose
- AAV
adeno‐associated virus
- BPND
binding potential
- NaI (Tl)
thallium‐doped sodium iodide
- SRTM
simplified reference tissue model
- SUV
standardized uptake value
- TAC
time‐activity curve
Introduction
The endocannabinoid system comprises the two cannabinoid receptors, CB1 and CB2, alonmg with the endogenous ligands, 2‐arachidonoyl glycerol and N‐arachidonoyl ethanolamine, and other proteins for their biosynthesis and degradation (Matsuda et al., 1990). The CB1 and CB2 receptors belong to the G‐protein coupled receptor family, and they share 44% overall homology (Felder et al., 1995). The CB1 receptors are mainly expressed in the CNS with a high level of receptor expression observed in the globus pallidus, entopenducular nucleus, substantia nigra pars reticulata and cerebellum (Freund et al., 2003). The expression of CB1 receptors has been imaged using [18F]MK‐9470 PET in monkey and human brains (Burns et al., 2007). The CB2 receptors, on the other hand, are principally expressed in organs with immune functions, and high expression is found in spleen, tonsils and leucocytes (Lynn and Herkenham, 1994). In the brain, CB2 receptor expression is very low under physiological conditions and is mainly detected in Purkinje cells of the cerebellum, hippocampal neurons and brain stem (Atwood and Mackie, 2010). The expression of CB2 receptors is up‐regulated during pathological conditions such as (neuro) inflammation and is predominantly observed in the activated microglia of the brain (Moldovan et al., 2016). The CB2 receptors play both neuroprotective and immunomodulatory roles. Furthermore, CB2 receptor agonists proved to be effective in neuropathic pain which makes these receptors a promising therapeutic target (Guindon and Hohmann, 2008; Atwood et al., 2012). Finally, activation of CB2 receptors does not have the psychoactive side effects which are common for CB1 receptor agonists (Spinelli et al., 2018).
Non‐invasive imaging of CB2 receptors with PET has a promising role in identifying neuropathological changes. Hence, it might be beneficial for diagnosis and treatment follow‐up of neurodegenerative disorders. such as Alzheimer's, Parkinson's and Huntington's disease, multiple sclerosis and amyotrophic lateral sclerosis, in which overexpression of CB2 receptors has been reported (Ashton and Glass, 2007; Cassano et al., 2017).
Several CB2 receptor selective agents and PET tracers have been developed in the past, but for some compounds, it is not clear whether they are agonist, antagonist or inverse agonist at these receptors (Evens et al., 2008; Evens et al., 2009; Horti et al., 2010; Evens et al., 2011; Turkman et al., 2011; Evens et al., 2012; Turkman et al., 2012; Moldovan et al., 2016; Caillé et al., 2017). A detailed overview of previously developed PET tracers for CB2 receptor imaging was recently discussed by Spinelli et al. (2018). Horti et al. (2010) developed and evaluated [11C]A‐836339 (agonist, Ki hCB2 = 0.64 nM) as a CB2 receptor‐specific tracer that was evaluated in an LPS‐induced neuroinflammation mouse model and in an Alzheimer's disease mouse model (APPswe/PS1dE9 mice). Similarly, Turkman et al. (2012) developed two [18F] labelled 2‐oxoquinolines ([18F]‐14, Ki hCB2 = 2.8 nM) and examined these in in vitro receptor binding and ex vivo autoradiography studies. However, no in vivo data were reported. Moldovan et al. (2016) developed several PET tracers for CB2 receptors, and further evaluation was carried out with [18F]29, a derivative of A‐836339 (agonist, Ki hCB2 = 4 nM) which showed specific binding in the brain with LPS‐induced neuroinflammation in mouse models. However, rapid tracer metabolism limited further evaluation. More recently, [11C]RS‐016 (Ki hCB2 = 0.7 nM) emerged as a specific PET tracer for detection of atherosclerotic plaques, although it was unsuccessful in distinguishing stable from vulnerable plaques (Meletta et al., 2017). Our group has previously described [11C]NE40 (inverse agonist, Ki hCB2 = 9.6 nM) (Figure 1) as a PET tracer specific for CB2 receptors. Uptake of [11C]NE40 was observed in the spleen of rodents, organs with known high expression of CB2 receptors and in a rat model with local overexpression of human (h) CB2 receptors in brain (Evens et al., 2009; Evens et al., 2012). [11C]NE40 was further evaluated for its safety and tolerability in healthy human male subjects in biodistribution and dosimetry studies (Ahmad et al., 2013).
Figure 1.
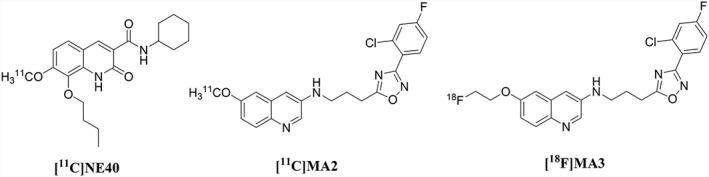
Chemical structures of [11C]NE40, [11C]MA2 and [18F]MA3, PET radiotracers for in vivo CB2 receptor imaging.
In 2016, we reported the synthesis, in vivo biodistribution and in vitro binding affinity data of two brain penetrant, high‐affinity CB2 receptor agonist PET tracers [11C]MA2 and [18F]MA3 (Ahamed et al., 2016; Figure 1). MA3 has an almost 100 times higher affinity for hCB2 receptors (agonist, Ki hCB2 = 0.8 nM) compared with MA2 (agonist, Ki hCB2 = 87 nM).
Agonists only bind to the (functionally) active state of CB2 receptors (Gullapalli et al., 2010), whereas inverse agonists like NE40 bind to both (functionally) active and inactive states. PET imaging with an agonist thus would provide quantification of the active state fraction of CB2 receptors which may be more relevant to the study of the role of these receptors in various pathologies. MA3 is an agonist that has proven brain exposure and hence can be used in pharmacological experiments aiming for the activation of CB2 receptors in vivo. In this study, we further focused on [18F]MA3 and evaluated its potential as specific CB2 receptor tracer. We conducted PET imaging in a rat model with local overexpression of hCB2 receptors combined with ex vivo autoradiography. In the same animal model, we compared the binding potential (BPND) with that of [11C]NE40 and finally performed a PET study in a rhesus monkey.
Methods
Synthesis of compounds
All the chemicals employed in the (radio) synthesis were purchased from commercial suppliers Aldrich (Overijse, Belgium), TCI Europe (Zwijndrecht, Belgium), Acros (Merelbeke, Belgium) and used without further purification unless stated. The CB2 receptor‐selective inverse agonist NE40 (8‐butoxy‐N‐cyclohexyl‐1,2‐dihydro‐7‐methoxy‐2‐oxoquinoline‐3‐carboxamide) was synthesized as described previously (Evens et al., 2009). The synthesis of the precursor MA1 (3‐(3‐(3‐(2‐chloro‐4‐fluorophenyl)‐1,2,4‐oxadiazol‐5‐yl)propylamino)quinolin‐6‐ol) used for radiosynthesis of [18F]MA3 and the synthesis of the non‐radioactive reference material MA3 (6‐(2‐fluoroethoxy)‐N‐(3‐(3‐(2‐chloro‐4‐fluorophenyl)‐1,2,4‐oxadiazol‐5‐yl)propyl)quinolin‐3‐amine) were described in our previous publication (Ahamed et al., 2016).
HPLC was performed using a Shimadzu LC‐2010A HT system connected to a UV spectrometer. Radioactivity in the column eluent was monitored using a 3 in thallium‐doped sodium iodide [NaI (Tl)] scintillation detector connected to a single channel analyser (Gabi box, Raytest, Straubenhardt, Germany). Radioactivity in HPLC eluent fractions of samples for radiometabolite analysis was quantified with an automated γ counter, equipped with a 3 in NaI (Tl) well crystal coupled to a multichannel analyser (Wallac 1480 Wizard, Wallac, Turku, Finland). The results were corrected for background radiation, detector dead time and physical decay during counting.
Animals
All animal care and experimental procedures were approved by the local Animal Ethics Committee of the University of Leuven and were in accordance with European Ethics Committee guidelines (decree 86/609/EEC). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010). Female Wistar rats (RjHan, RRID:RGD_13508588 from Janvier labs, Le Genest‐Saint‐Isle, France) were housed in individually ventilated cages in a thermoregulated (22°C) and humidity‐controlled environment under a 12 h/12 h day/night cycle with free access to food and water. The body mass of the rats ranged from 223 to 233 g.
Experimental design
Our experiments are exploratory studies and/or proof‐of‐principle studies, where we check whether the developed tracer meets certain requirements for its potential clinical suitability. No statistical analysis was performed on the results acquired from our animal experiments because sample size did not reach minimum n = 5, according to ARRIVE guidelines. The in vivo microPET studies were performed in three conditions, namely, baseline, block and displacement study with three rats with local overexpression of the hCB2 receptor. Additional blocking studies were performed with the CB1 receptor selective compound, rimonabant, in baseline and block with three control rats. As microPET is a non‐invasive imaging method, the rats served as their own control. The ex vivo autoradiography study was performed on nine brain sections from a single rat. The non‐human primate, Rhesus macaque (Macaca mulatta, NCBI:taxid9544), study (n = 1) was performed as a proof‐of‐concept study to qualitatively evaluate the tracer brain kinetics in order to predict the clinical suitability of the developed radiotracer. The vehicle and NE40 used in the studies were blinded to the operator and prepared and numbered by the designer.
Rat model with local overexpression of hCB2 receptors
The preparation and validation of the rat model with local overexpression of hCB2 receptors in brain was described by Vandeputte et al. (2011). Briefly, four female Wistar rats (RjHan, RRID:RGD_13508588 from Janvier Labs, Le Genest‐Saint‐Isle, France) were stereotactically injected with an adeno‐associated viral (AAV) vector serotype 2/7 encoding hCB2 (D80N), in the right striatum (relative to Bregma: anteroposterior 0 mm, lateral −2.8 mm and dorsoventral −5.5 mm to −4.5 mm) under control of a CaMKII promoter. A control AAV2/7 vector expressing the eGFP under control of a CaMKII promoter was injected in the left striatum at the contralateral side (anteroposterior 0 mm, lateral 2.8 mm and dorsal −5.5 mm to −4.5 mm). Each animal was injected with 4 μL vector with comparable vector titres (4.8 E+12 GC mL−1 and 5.7 E+12 GC mL−1 respectively).
In vivo PET study in a rat model with local hCB2 receptor overexpression
PET imaging experiments were performed on a Focus™ 220 PET scanner (Concorde Microsystems, Knoxville, TN) using three rats with local overexpression of hCB2 receptors. The first scan was performed with [18F]MA3, 11 weeks after the surgery: rat n°1 was scanned baseline, rat n°2 was scanned after pretreatment (block) and rat n°3 was scanned in a displacement study. In the second scan 12 weeks after surgery, rat n°1 was scanned after blocking, rat n°2 in a displacement and rat n°3 for baseline. Finally in the last scan 14 weeks after surgery, rat n°1 was scanned in a displacement, rat n°2 for baseline and rat n°3 after blocking. Using this protocol, each rat served as its own control.
During PET imaging, animals were kept under anaesthesia (2.5% isoflurane in O2 at 1 L min−1 flow rate). Upon i.v. injection with about 30 MBq tracer, a 90 min ([11C]NE40) or 120 min ([18F]MA3) dynamic PET scan was started. All scans were acquired in list mode. Acquisition data were Fourier rebinned in 24 time frames (4 × 15 s, 4 × 60 s, 5 × 180 s, 8 × 300 s, 3 × 600 s) for 90 min acquisitions and 27 time frames (4 × 15 s, 4 × 60 s, 5 × 180 s, 8 × 300 s, 6 × 600 s) for 120 min acquisitions and reconstructed using maximum a posteriori iterative reconstruction. A summed image of the reconstructed data was spatially normalized to an in‐house created 18F‐FDG template of the rat brain in Paxinos coordinates (Casteels et al., 2006) using an affine transformation. The latter was then used to normalize all time frames of the dynamic PET data set to allow automated and bilateral volumes of interest (VOIs) analyses. Time‐activity curves (TACs) were generated using PMOD software (PMOD software, Ver# 3.2, RRID:SCR_016547).
The rats in the baseline (only tracer injection) and pretreatment study were anaesthetized with 2.5% isoflurane in O2 at 1 L min−1 flow rate and injected i.p. with vehicle (see below) or NE40 (1 mg kg−1) 30 min prior to tracer injection respectively. For the displacement study, NE40 (1 mg kg−1) was administered i.v. 20 min after tracer injection. NE40 was dissolved in a vehicle containing 20% DMSO, 40% 2‐hydroxypropyl β‐cyclodextrin and 5% tween 80 in water for injection at a concentration of 1 mg mL−1. BPND was calculated using simplified reference tissue model (SRTM) and the left striatum as a reference region.
MicroPET experiments with CB1 receptor blockade were performed with healthy rats in baseline and pretreatment conditions, using the procedure described above. Vehicle or rimonabant (1 mg kg−1) was injected i.v. 15 min before tracer injection. Rimonabant was dissolved in vehicle consisting of 5% DMSO, 8% PEG 400 and 1% Tween 80 in water for injection.
Ex vivo autoradiography in a rat model with local hCB2 receptor overexpression
A rat with local overexpression of hCB2 receptors (n = 1) was injected via the tail vein with about 48 MBq [18F]MA3 under anaesthesia (2.5% isoflurane in O2 at 1 L min−1 flow rate). The rat was killed by decapitation 30 min after tracer administration. The brain was removed, rapidly frozen in 2‐methylbutane cooled to −40°C. Transverse sections (20 μm) from the brain were obtained using a cryotome (Shandon cryotome FSE, Thermo Fisher, Waltham, MA; n = 9) and were exposed to a high performance phosphor storage screen for 1 h (Super resolution screen; Perkin Elmer, Waltham, MA). The screens were read using a Cyclone Plus system (Perkin Elmer), and data were analysed using Optiquant software (Perkin Elmer, RRID_SCR_016769).
In vivo PET study in a non‐human primate
Dynamic 120 min [18F]MA3 PET brain scans were acquired with a Focus™ 220 PET scanner in a female rhesus monkey (M. mulatta, 5.2 kg). Sedation was performed by i.m. injection of a combination of 0.3 mL Rompun (xylazine 2% solution) and 0.35 mL Nimatek (ketamine 100 mg mL−1). About 60 min after the first injection, the monkey received an additional dose of 0.15 mL Rompun and 0.175 mL Nimatek via i.v. injection. During the last part of the scan, dosing was done less frequently based on the heartbeat frequency. The O2 and CO2 saturation in the blood and the heartbeat were constantly monitored. The body temperature was regulated via a heating pad. The breathing frequency and the eye response were checked visually. The head of the animal was placed centrally in the field of view of the PET scanner. The monkey was injected with about 170 MBq of [18F]MA3 via the saphenous vein. Scans were acquired in list mode and Fourier rebinned in 27 time frames (4 × 15 s, 4 × 60 s, 5 × 180 s, 8 × 300 s, 6 × 600 s). Data were reconstructed using a 3D maximum a posteriori iterative reconstruction. TACs of the whole brain and several brain regions were generated using VOIs with PMOD software, and radioactivity concentration in the brain was expressed as standardized uptake value (SUV) as a function of time after tracer injection. For the pretreatment study, NE40 was dissolved in a vehicle containing 20% DMSO and 40% 2‐hydroxypropyl β‐cyclodextrin in water for injection. Pretreatment consisted of i.v. injection of NE40 at a dose of 1 mg kg−1 10 min before tracer injection.
Non‐human primate plasma radiometabolite analysis
During the baseline and pretreatment scan, blood samples were collected at 10, 30 and 60 min post tracer injection (p.i.) via the contralateral saphenous vein in EDTA‐containing tubes (4 mL tubes; BD vacutainer, BD, Franklin Lakes, NJ) and stored on ice. The blood samples were weighed, and activity was counted in the automated γ counter. Next, the blood was centrifuged for 10 min at 2330× g to separate the plasma. About 0.5 mL of plasma was weighed and counted for radioactivity in an automated γ counter. The plasma was then diluted with an equal volume of acetonitrile and centrifuged for 5 min at 2330× g to precipitate the proteins. Next, a volume of 0.5 mL of the supernatant was filtered through a 0.22 μm filter (0.2 μm nylon filter, Acrodisc 13, PALL Life Sciences, Billerica, MA, USA) containing authentic non‐radioactive compound MA3 (20 μL of 1 mg mL−1 solution in H2O/CH3CN 50:50 v/v) and counted for radioactivity in the automated γ counter. The solution was then injected onto an HPLC system consisting of an analytical XBridge column (C18, 3.0 μm, 3 mm × 100 mm, Waters, Milford, Connecticut, USA) eluted with a mixture of 0.05 M sodium acetate buffer (pH 5.5) and acetonitrile (55:45 v/v) at a flow rate of 0.6 mL min−1. After passing through the UV detector (254 nm) and the 3 in NaI (Tl) scintillation detector, the HPLC eluate was collected as 1 mL fractions using an automatic fraction collector, and the radioactivity in these fractions was measured using an automated γ counter.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Quantitative data are expressed as mean ± SD. Error bars correspond to SD. TAC data were generated using PMOD software, and data were plotted using GraphPad Prism version 5.00 for Windows (GraphPad Prism, RRID:SCR_002798). Chemical structures were drawn using ChemDraw Ultra 8.0 (Perkin Elmer RRID:SCR_016768).
Materials
Rimonabant (5‐(4‐chlorophenyl)‐1‐(2,4‐dichlorophenyl)‐4‐methyl‐N‐(piperidin‐1‐yl)‐1H‐pyrazole‐3‐carboxamide) was kindly provided by Professor Bertrand Kuhnast, CEA, France.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Radiosynthesis of [11C]NE40 and [18F]MA3
[11C]NE40 synthesis was carried out according to the protocol described by Evens et al. (2009). The alkylation yield for [11C]NE40 was 45% (based on the AUC of the radiometric channel of the preparative HPLC), and [11C]NE40 was obtained with a radiochemical purity of more than 99% and a molar activity of 30 GBq μmol−1 (n = 1) at the end of synthesis (EOS). [18F]MA3 was synthesized by alkylation of the phenol precursor using the secondary labelling agent 1‐bromo‐2‐[18F] fluoroethane (18FEtBr) as described previously (Ahamed et al., 2016). [18F]MA3 was produced with alkylation yield of 19% (based on the AUC of the radiometric channel of the preparative HPLC), a radiochemical purity in the range of 96–99% and a molar activity in the range of 50–240 GBq μmol−1 (n = 7) at EOS.
Validation of the developed rat model using [11C]NE40
To validate the model of local overexpression of hCB2 receptors in rats, a 90 min baseline dynamic PET scan (n = 3) with the previously reported specific CB2 receptor PET tracer [11C]NE40 was performed. Figure 2 shows a high‐intensity signal in the hCB2 receptor vector‐injected right striatum. This was also reflected in the baseline TACs in which accumulation of radioactivity was observed in the right striatum with faster washout from the left striatum. Uptake in the right striatum (hCB2 receptor vector) was twofold higher than that in the left striatum (eGFP control). SRTM BPND using the left striatum as a reference region was 2.1 ± 0.2.
Figure 2.
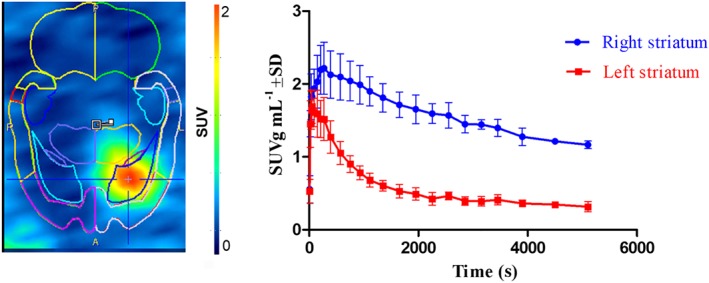
PET study with [11C]NE40 in rat model with local overexpression of hCB2 receptors. Left, summed images of 0–90 min after injection; right, corresponding TACs of the right and left striatum. Data are expressed as SUV. Error bars correspond to SD.
In vivo PET study of [18F]MA3 in a rat model with local hCB2 receptor overexpression
In baseline conditions (Figure 3A; n = 3), a high‐intensity signal was observed in the right striatum which had been injected with the hCB2 receptor vector and only background radioactivity in the eGFP control vector‐injected left striatum. This was also reflected in the baseline TACs in which accumulation of radioactivity was observed in the right striatum with washout from the left striatum. Uptake in the right striatum (hCB2 receptor vector) was higher than that in the left striatum (control).
Figure 3.
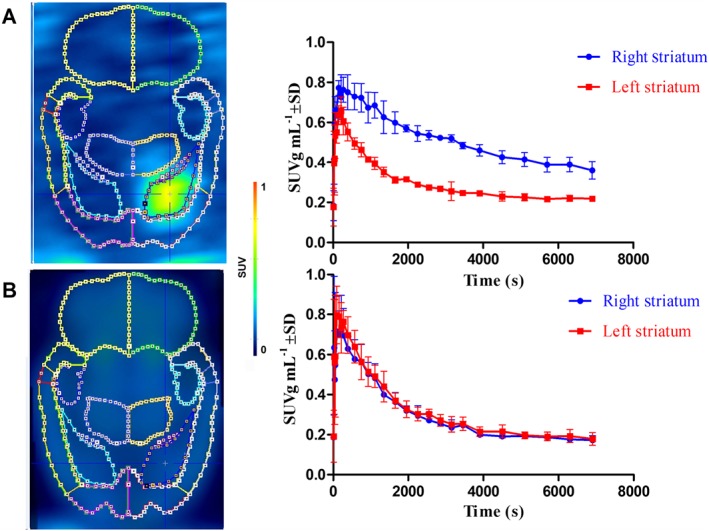
PET data of [18F]MA3 in an hCB2 receptor rat model: the right striatum was stereotactically injected with AAV‐hCB2 receptor; and the left striatum was injected with control vector AAV‐eGFP. (A) Baseline study (n = 3): summed image of 0–120 min post‐injection of [18F]MA3 with corresponding TACs of the right and left striatum. (B) Pretreatment study with NE40 (n = 3): summed image of 0–120 min post‐injection of [18F]MA3 with corresponding TACs of the right and left striatum. NE40 (in vehicle) was injected i.p. at a dose of 1 mg kg−1 30 min before tracer injection. In the baseline scan, only vehicle was administered i.p. 30 min before tracer injection. Data are expressed as SUV. Error bars correspond to SD.
Pretreatment by i.p. injection of NE40 (1 mg kg−1) 30 min before tracer injection resulted in a decrease of the radioactivity concentration in the right striatum, expressing hCB2 receptors (Figure 3B; n = 3). After blocking, no significant difference in tracer binding was observed between the right and left striatum. SRTM BPND using the left striatum as a reference region was 2.0 ± 0.2.
Figure 4A (n = 3) represents the summed image of 0–20 min of the PET scan before NE40 displacement injection, demonstrating higher tracer binding in the right striatum, with average SUV values (0–20 min) of 0.7 ± 0.1, comparable with the baseline scans. After i.v. injection of NE40 (1 mg kg−1), 20 min after injection, the TAC of the right striatum dropped to the level of the left striatum [average SUV values (20–120 min) 0.2 ± 0.0] (Figure 4B).
Figure 4.
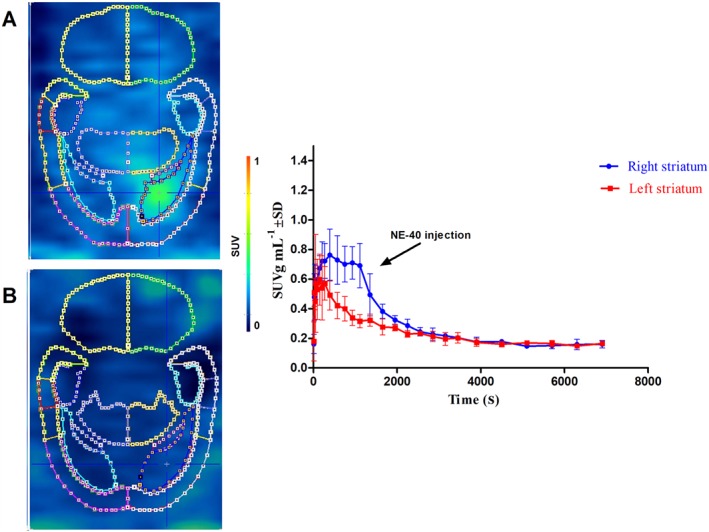
PET data of [18F]MA3 displacement study in a hCB2 receptor rat model. The right striatum was stereotactically injected with AAV‐hCB2 receptor; and the left striatum was injected with control vector AAV‐eGFP. NE40 (1 mg kg−1) was injected i.v. (arrow) 20 min after tracer injection. Images (from transverse sections) corresponding to the chase experiment: summed image (0–20 min after tracer injection) before chase injection (A) and summed image (20–120 min after tracer injection) after chase injection (B), with corresponding TACs of the right and left striatum. Data are expressed as SUV. Error bars correspond to SD.
In vivo PET study of [18F]MA3 in a rat with the CB1 receptor selective antagonist rimonabant
The results from the study with the CB1 receptor selective antagonist rimonabant are shown in Figure 5. To investigate the specificity of [18F]MA3 for CB2 over CB1 receptors, we have performed additional microPET blocking experiments with the CB1 receptor‐specific antagonist rimonabant. The [18F]MA3 brain TACs analysis showed rapid entry with slightly lower brain retention values from 30 min after injection, in the blocking conditions.
Figure 5.
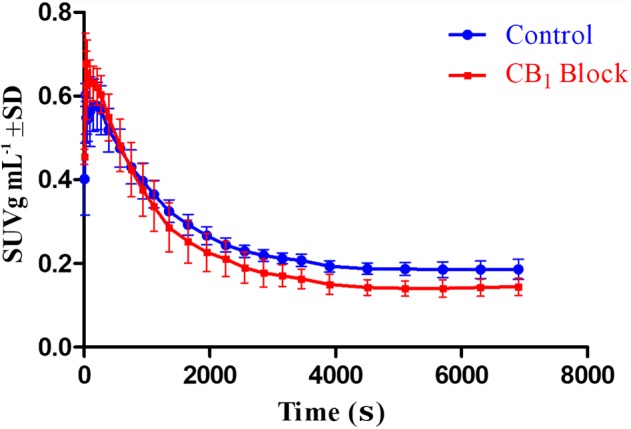
PET data of study with rimonabant blockade of CB1 receptors. (A) Baseline study (n = 3) of 0–120 min post‐injection of [18F]MA3 TACs of whole brains and pretreatment study with rimonabant (n = 3) of 0–120 min after injection of [18F]MA3 TACs of whole brains. Rimonabant (1 mg kg−1 in vehicle) was injected i.v. 15 min before tracer injection. In the baseline scan, only vehicle was administered i.v. 15 min before tracer injection. Data are expressed as SUV. Error bars correspond to SD.
Ex vivo autoradiography of [18F]MA3 in rat model with local hCB2 receptor overexpression
Figure 6 shows the autoradiogram developed from cryosections of rat brain obtained 30 min after i.v. injection of [18F]MA3. This study confirmed that the radiotracer binds to the hCB2 receptor viral vector‐injected site, whereas the binding to the rest of the brain was low. Tracer binding was quantified and expressed as digital light units per mm2. The ratio of right to left striatum was 2.7.
Figure 6.
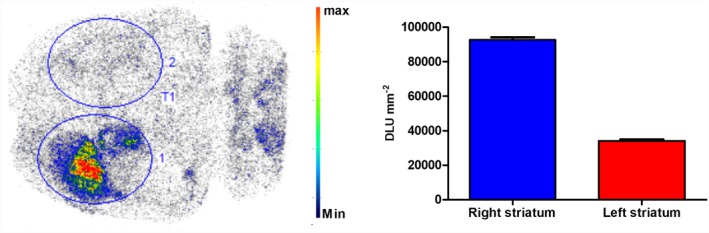
Transverse section of an ex vivo autoradiography study performed in a rat model with local overexpression of hCB2 receptors in the right striatum (n = 1). [18F]MA3 was injected i.v. via the tail vein, and the rat was killed at 30 min after injection. The autoradiogram shows specific binding of [18F]MA3 in the right striatum. Binding is expressed as digital light units (DLU) mm−2 of the right and left striatum. Max, maximum; min, minimum.
In vivo PET study of [18F]MA3 in a non‐human primate
The results of the 120 min baseline and pretreatment PET scans of [18F]MA3 in monkey brain are shown in Figure 7. The TAC of the baseline scan showed a fast, initial brain uptake (SUV 1.4 total brain). The uptake was homogeneous throughout the brain (Figure 7A). Regional brain concentrations were highest in caudate (average SUV20–120 min = 0.7 ± 0.1), followed by thalamus (average SUV20–120 min = 0.6 ± 0.1), hippocampus (average SUV20–120 min = 0.6 ± 0.1), frontal cortex (average SUV20–120 min = 0.6 ± 0.1) and cerebellum (average SUV20–120 min = 0.6 ± 0.1) (TACs of different brain regions not shown). After 40 min, the concentration of radioactivity in the brain reached a steady state (average SUV20–120 min 0.6 ± 0.1 total brain). Higher brain uptake (peak SUV 1.8 total brain) was observed after pretreatment with NE40 (1 mg kg−1) for the total duration (0–120 min) of the scan (average SUV20–120 min 0.8 ± 0.2 total brain).
Figure 7.
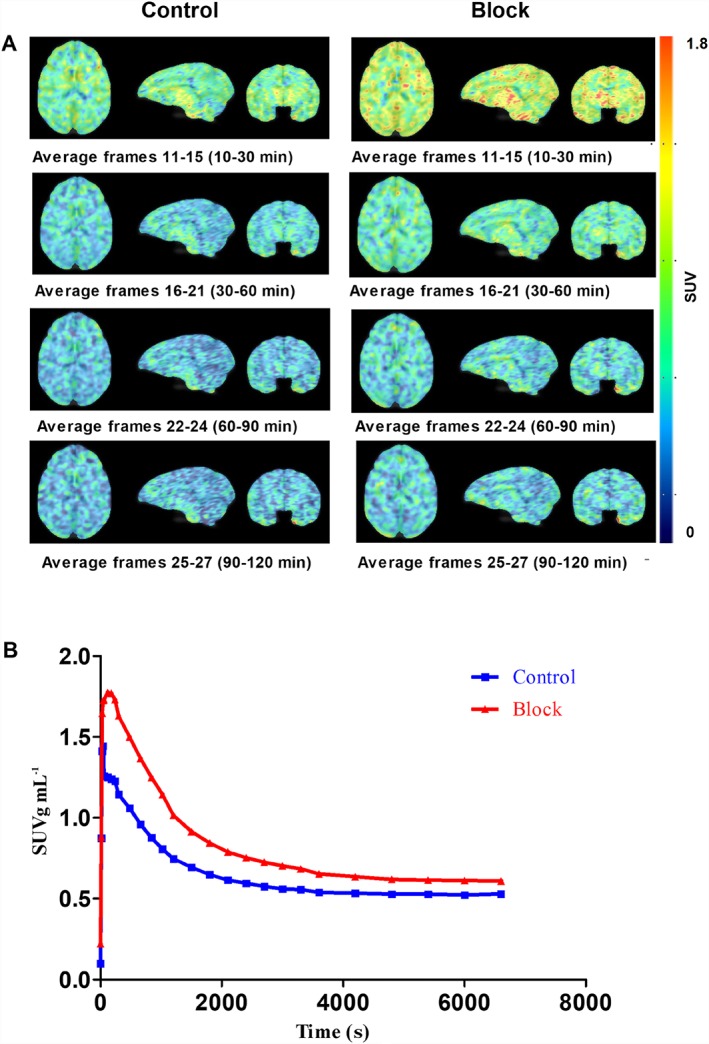
PET data of [18F]MA3 in Rhesus monkey. (A) Transverse, sagittal and frontal PET sections of monkey brain. Summed images of 10–30, 30–60, 60–90 and 90–120 min after tracer injection for the baseline study (left images) and for the pretreatment study with NE40 (1 mg kg−1; i.v.) 10 min before tracer injection (right images). (B) Corresponding TACs of total monkey brain at baseline (control) and after pretreatment (block).
Plasma radio‐metabolite analysis in non‐human primate
The reconstructed radio‐chromatograms of monkey plasma analysis at 10, 30 and 60 min after [18F]MA3 injection showed two peaks (graphs not shown) with the most apolar peak corresponding to the parent tracer. At 10 min post‐injection of the radiotracer, 16% of the recovered radioactivity in plasma corresponded to intact tracer, and this percentage decreased to 12% at 30 min and to 7% at 60 min after injection. The percentage of intact tracer in plasma was slightly higher in blocking conditions with 21% at 10 min, 13% at 30 min and 9% at 60 min after injection. The recovery of the HPLC‐injected radioactivity was in the range of 86–99%. The plasma SUV values of parent [18F]MA3 in baseline and blocking conditions are shown in Figure 8. Brain‐to‐plasma SUV ratios in baseline conditions were 1.2, 1.2 and 1.9, and after pretreatment with NE40, they were 1.4, 1.4 and 1.7 at 10, 30 and 60 min after injection respectively.
Figure 8.
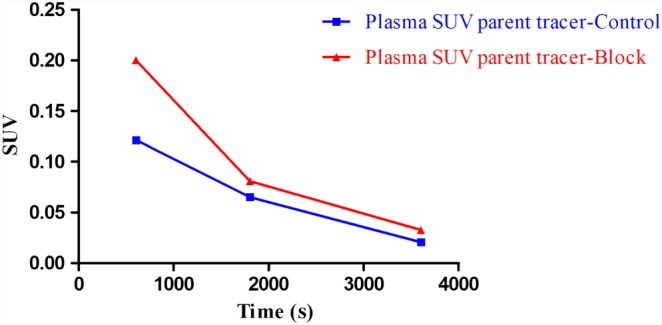
Monkey plasma radiometabolite analysis and quantification of intact tracer in control and block conditions at 10, 30 and 60 min after tracer injection.
Discussion
In the past, our group has reported the synthesis and preclinical evaluation of the inverse agonist tracer [11C]NE40 in a rat model with local overexpression of hCB2 receptors in the brain. Further, [11C]NE40 was studied in healthy volunteers, and currently, studies were ongoing in patients with neuroinflammatory conditions. [11C]NE40 is an inverse agonist, which thus binds to both the active and inactive fraction of CB2 receptors. The limitation is the short half‐life of the 11C label (20.3 min) which necessitates an onsite cyclotron. In the present report, we describe an agonist PET tracer [18F]MA3 that is expected to bind only to the (functionally) active fraction of CB2 receptors which may be more relevant in the study of the role of CB2 receptors in different pathologies. [18F]MA3 has the further advantage of being labelled with 18F which has a longer half‐life (109.8 min), providing us with the potential opportunity to transport the developed radiotracer to nearby PET centres.
We have also reported the synthesis, in vitro affinity and biodistribution study of two agonist tracers [11C]MA2 and [18F]MA3 in NMRI mice (Ahamed et al., 2016). In the present study, [18F]MA3 the CB2 receptor agonist tracer with the highest affinity (0.8 nM) was further evaluated in a rat model with local over expression of inactive hCB2 receptors. To the best of our knowledge, this is the first report demonstrating that CB2 receptor agonists bind to the mutated inactive hCB2 receptors so that the AAV hCB2 receptor rat model can be used for comparative evaluation of CB2 receptor agonist PET tracers.
Validation of the developed rat model using clinically tested [11C]NE40 showed specific binding of the tracer in the right striatum indicating local overexpression of hCB2 receptors. The results were in line with the previously reported data (Evens et al., 2012). [18F]MA3 was further extensively studied in the validated rat model. TACs obtained from the baseline scan showed [18F]MA3 binding in the striatum injected with the hCB2 receptor vector and a fast washout from the contralateral striatum injected with eGFP vector.The concentration of radioactivity (SUV) in the right striatum was two‐fold higher than that in the left striatum ~15 min after injection of the tracer. This was confirmed by ex vivo autoradiography where a three‐fold higher binding was observed in the right, compared with the left, striatum.
The specificity of binding to hCB2 receptors was demonstrated by blocking experiments with a structurally un‐related CB2 receptor‐specific inverse agonist NE40. After pretreatment, the TAC of [18F]MA3 in the right striatum aligned with the TAC of the left striatum. A displacement study with NE40 demonstrated that binding of [18F]MA3 to hCB2 receptors was reversible. Initially, baseline conditions were observed with higher tracer concentration in the right, compared to the left, striatum. However, after i.v. injection of NE40, the TAC of the right striatum (overexpression of hCB2 receptors) aligned to the TAC of the left (control) striatum demonstrating fast in vivo binding reversibility of [18F]MA3. We performed in vitro autoradiography on spleen sections, but none of a large number of protocols tested was able to sufficiently wash off non‐specific binding. Hence, we did not observe any difference between control and blocking condition and failed to identify CB2 receptor‐specific binding in spleen in contrast to earlier reports with CB2 receptor agonist tracers (Caillé et al., 2017). Two possible reasons for this discrepancy are suboptimal autoradiography protocols and / or a high off‐rate constant (koff) value of [18F]MA3.
To investigate the in vivo specific binding of [18F]MA3 to CB1 receptors, we have performed microPET blocking experiments with the CB1 receptor‐specific antagonist, rimonabant. For the three rats, we consistently observed slightly lower brain retention values from 30 min after injection onwards, suggesting that there may indeed be a small contribution of binding of the tracer to CB1 receptors.
In this report, we did a head‐to‐head comparison study with [11C]NE40 (inverse agonist) and [18F]MA3 (agonist) in a rat model that overexpresses an inactive mutant of hCB2 receptors. Mutation of highly conserved aspartate residues in the second transmembrane domain doesn't produce signal transduction in cannabinoid receptors but retains their ligand recognition properties which are similar to wild type receptors (Tao and Abood, 1998). Interestingly, we found that this rat model can be used to test agonist tracers such as [18F]MA3. In our experiments, we found a similar BPND (Figure 9) for both [11C]NE40 and [18F]MA3 indicating that the mutant CB2 receptor seems to have the active‐like conformation. The developed mutated AAV rat model can therefore be used to evaluate and compare both the agonist and inverse agonist tracers. The combination of the AAV technology with high‐affinity, CB2 receptor agonist PET tracers further allows cell and gene therapy in the brain to be tracked non‐invasively.
Figure 9.
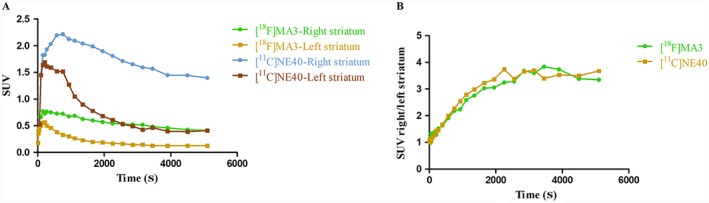
(A) Baseline TACs of [11C]NE40 and [18F]MA3 in the left and right striatum of rats overexpressing hCB2 receptors. (B) Right‐to‐left striatum SUV ratios as a function of time after injection of [11C]NE40 and [18F]MA3.
Preliminary evaluation of [18F]MA3 in a healthy non‐human primate showed moderate brain uptake with homogenous distribution and fast washout. The appearance of radiometabolites in monkey plasma was rather fast. Only polar radiometabolites were detected. Also, bone uptake of [18F]MA3 was low, as previously reported in the biodistribution studies (Ahamed et al., 2016). Identification of radiometabolites presents some difficulties due to very low mass amounts of tracers that are typically injected. We assume that the radiometabolites are not [18F]fluoroethanol or its oxidation products [18F]fluoroacetaldehyde or [18F]fluoroacetic acid as these radiometabolites generate [18F]fluoride, and we did not observe any bone uptake. TAC analysis did not show any increase in brain radioactivity over time indicating that radio‐metabolites were not entering the brain. Species differences in susceptibility to radio‐defluorination is known for many tracers. Although less extensive metabolism is generally expected in higher species, further in vivo evaluation in humans will be necessary to check the extent of defluorination (Celen et al., 2010).
In contrast to what was expected, pretreatment with the inverse agonist NE40 (1 mg kg−1, i.v.) increased the concentration of radioactivity in monkey brain. This could be explained by the combination of low (monkey) CB2 receptor expression in healthy conditions and a peripheral blocking effect. In view of the (albeit limited) blocking effect of rimonabant in rats, it is also possible that the higher CB2 receptor selective blocker NE40 displaced [18F]MA3 from peripheral binding sites increasing the brain exposure and binding to CB1 receptors in the brain, resulting in higher TAC values. We were not able to observe any blocking effect of NE40 pretreatment in monkey brain. Expression of CB2 receptors may be too low in healthy conditions so that further evaluation in humans, in both healthy and disease conditions, are required to further evaluate the potential of [18F]MA3 as a CB2 receptor‐specific agonist PET tracer in humans.
This study also confirmed that MA3 is a specific CB2 receptor agonist with good brain exposure in rats and monkeys and is thus suitable for pharmacological experiments where in vivo activation of CB2 receptors is required.
In conclusion, evaluation of [18F]MA3 in a rat model with local striatal overexpression of hCB2 receptors showed specific and reversible tracer binding in the striatum injected with the hCB2 receptor vector, indicating that this tracer has potential for in vivo imaging of the active state fraction of CB2 receptors with PET. This developed animal model is further useful for studies with both agonist and inverse agonist CB2 receptor PET radiotracers. The longer half‐life of [18F]MA3 offers the opportunity to transport the developed radiotracer to nearby PET centres where no cyclotron is available. Further evaluation of [18F]MA3 in patients with neuroinflammation is warranted.
Author contributions
B.A. and S.C. conducted the radiochemical synthesis and animal experiments. M.A. performed non‐radiochemical synthesis. B.A., S.C. and M.K. performed data analysis. C.V.H. prepared and provided the animal model. B.A., S.C., W.V. and G.B. designed the research, made data interpretation and prepared the manuscript. All the authors reviewed the results and approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Julie Cornelis, Ivan Sannen and Pieter Haspeslagh (Laboratory for Radiopharmaceutical Research, KU Leuven) for their help with the animal studies and Christophe Ulens form the Laboratory of Neuro‐ and Psychophysiology for assisting in the PET monkey studies. This work was supported by IMIR (In Vivo Molecular Imaging Research group), KU Leuven, and by SBO project MIRIAD (Molecular Imaging Research Initiative for Acceleration of Drug Development‐130065) funded by IWT Flanders and by the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. HEALTH‐F2‐2011‐278850 (INMiND) and Research Foundation Flanders (FWO‐Flanders) G0007.12; KU Leuven Programme Financing.
Attili, B. , Celen, S. , Ahamed, M. , Koole, M. , Haute, C. V. D. , Vanduffel, W. , and Bormans, G. (2019) Preclinical evaluation of [18F]MA3: a CB2 receptor agonist radiotracer for PET. British Journal of Pharmacology, 176: 1481–1491. 10.1111/bph.14564.
References
- Ahamed M, van Veghel D, Ullmer C, Van Laere K, Verbruggen A, Bormans G (2016). Synthesis, biodistribution and in vitro evaluation of brain permeable high affinity type 2 cannabinoid receptor agonists [11C]MA2 and [18F]MA3. Front Neurosci 10: 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad R, Koole M, Evens N, Serdons K, Verbruggen A, Bormans G et al (2013). Whole‐body biodistribution and radiation dosimetry of the cannabinoid type 2 receptor ligand [11C]‐NE40 in healthy subjects. Mol Imaging Biol 15: 384–390. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopouloas A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S14–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton JC, Glass M (2007). The cannabinoid CB2 receptor as a target for inflammation‐dependent neurodegeneration. Curr Neuropharmacol 5: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Wager‐Miller J, Haskins C, Straiker A, Mackie K (2012). Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands. Mol Pharmacol 81: 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Mackie K (2010). CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol 160: 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns HD, Van Laere K, Sanabria‐Bohórquez S, Hamill TG, Bormans G, Eng WS et al (2007). [18F]MK‐9470, a positron emission tomography (PET) tracer for in vivo human PET brain imaging of the cannabinoid‐1 receptor. Proc Natl Acad Sci U S A 104: 9800–9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillé F, Cacheux F, Peyronneau MA, Jego B, Jaumain E, Pottier G et al (2017). From structure–activity relationships on thiazole derivatives to the in vivo evaluation of a new radiotracer for cannabinoid subtype 2 PET imaging. Mol Pharm 14: 4064–4078. [DOI] [PubMed] [Google Scholar]
- Cassano T, Calcagnini S, Pace L, De Marco F, Romano A, Gaetani S (2017). Cannabinoid receptor 2 signaling in neurodegenerative disorders: from pathogenesis to a promising therapeutic target. Front Neurosci 11: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteels C, Vermaelen P, Nuyts J, Van Der Linden A, Baekelandt V, Mortelmans L et al (2006). Construction and evaluation of multitracer small‐animal PET probabilistic atlases for voxel‐based functional mapping of the rat brain. J Nucl Med 47: 1858–1866. [PubMed] [Google Scholar]
- Celen S, Koole M, De Angelis M, Sannen I, Chitneni SK, Alcazar J et al (2010). Preclinical evaluation of 18F‐JNJ41510417 as a radioligand for PET imaging of phosphodiesterase‐10A in the brain. J Nucl Med 51: 1584–1591. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evens N, Bosier B, Lavey BJ, Kozlowski JA, Vermaelen P, Baudemprez L et al (2008). Labelling and biological evaluation of [11C]methoxy‐Sch225336: a radioligand for the cannabinoid‐type 2 receptor. Nucl Med Biol 35: 793–800. [DOI] [PubMed] [Google Scholar]
- Evens N, Muccioli GG, Houbrechts N, Lambert DM, Verbruggen AM, Van Laere K et al (2009). Synthesis and biological evaluation of carbon‐11‐ and fluorine‐18‐labeled 2‐oxoquinoline derivatives for type 2 cannabinoid receptor positron emission tomography imaging. Nucl Med Biol 36: 455–465. [DOI] [PubMed] [Google Scholar]
- Evens N, Vandeputte C, Muccioli GG, Lambert DM, Baekelandt V, Verbruggen AM et al (2011). Synthesis, in vitro and in vivo evaluation of fluorine‐18 labelled FE‐GW405833 as a PET tracer for type 2 cannabinoid receptor imaging. Bioorg Med Chem 19: 4499–4505. [DOI] [PubMed] [Google Scholar]
- Evens N, Vandeputte C, Coolen C, Janssen P, Sciot R, Baekelandt V et al (2012). Preclinical evaluation of [11C]NE40, a type 2 cannabinoid receptor PET tracer. Nucl Med Biol 39: 389–399. [DOI] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Brilley EM, Mansouri J, Mackie K, Blond O et al (1995). Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48: 443–450. [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017–1066. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG (2008). Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol 153: 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullapalli S, Amrutkar D, Gupta S, Kandadi MR, Kumar H, Gandhi M et al (2010). Characterization of active and inactive states of CB1 receptor and the differential binding state modulation by cannabinoid agonists, antagonists and inverse agonists. Neuropharmacology 58: 1215–1219. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horti AG, Gao Y, Ravert HT, Finley P, Valentine H, Wong DF et al (2010). Synthesis and biodistribution of [11C]A‐836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2). Bioorg Med Chem 18: 5202–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn AB, Herkenham M (1994). Localization of cannabinoid receptors and nonsaturable high‐density cannabinoid binding sites in peripheral tissues of the rat: implications for receptor‐mediated immune modulation by cannabinoids. J Pharmacol Exp Ther 268: 1612–1623. [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346: 561–564. [DOI] [PubMed] [Google Scholar]
- Meletta R, Slavik R, Mu L, Rancic Z, Borel N, Schibli R et al (2017). Cannabinoid receptor type 2 (CB2) as one of the candidate genes in human carotid plaque imaging: Evalu1ation of the novel radiotracer [11C]RS‐016 targeting CB2 in atherosclerosis. Nucl Med Biol 47: 31–43. [DOI] [PubMed] [Google Scholar]
- Moldovan RP, Teodoro R, Gao Y, Deuther‐Conrad W, Kranz M, Wang Y et al (2016). Development of a high‐affinity PET radioligand for imaging cannabinoid subtype 2 receptor. J Med Chem 59: 7840–7855. [DOI] [PubMed] [Google Scholar]
- Spinelli F, Mu L, Ametamey SM (2018). Radioligands for positron emission tomography imaging of cannabinoid type 2 receptor. J Labelled Comp Radiopharm 61: 299–308. [DOI] [PubMed] [Google Scholar]
- Tao Q, Abood ME (1998). Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G‐protein coupling. J Pharmacol Exp Ther 285: 651–658. [PubMed] [Google Scholar]
- Turkman N, Shavrin A, Ivanov RA, Rabinovich B, Volgin A, Gelovani JG et al (2011). Fluorinated cannabinoid CB2 receptor ligands: synthesis and in vitro binding characteristics of 2‐oxoquinoline derivatives. BioorgMed Chem 19: 5698–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkman N, Shavrin A, Paolillo V, Yeh HH, Flores L, Soghomonian S et al (2012). Synthesis and preliminary evaluation of [18F]‐labeled 2‐oxoquinoline derivatives for PET imaging of cannabinoid CB2 receptor. Nucl Med Biol 39: 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeputte C, Evens N, Toelen J, Deroose CM, Bosier B, Ibrahimi A et al (2011). A PET brain reporter gene system based on type 2 cannabinoid receptors. J Nucl Med 52: 1102–1109. [DOI] [PubMed] [Google Scholar]