

Abstract
The mammalian target of rapamycin (mTOR) inhibiting drug rapamycin (Sirolimus) has severe side effects in patients including hyperlipidemia, an established risk factor for atherosclerosis. Recently, it was shown that rapamycin decreases hepatic LDL receptor (LDL-R) expression, which likely contributes to hypercholesterolemia. Scavenger receptor, class B, type I (SR-BI) is the major HDL receptor and consequently regulating HDL-cholesterol levels and the athero-protective effects of HDL. By using the mTOR inhibitor rapamycin, we show that SR-BI is down-regulated in human umbilical vein endothelial cells (HUVECs). This reduction of SR-BI protein as well as mRNA levels by about 50% did not alter HDL particle uptake or HDL-derived lipid transfer. However, rapamycin reduced HDL-induced activation of eNOS and stimulation of endothelial cell migration. The effects on cell migration could be counteracted by SR-BI overexpression, indicating that decreased SR-BI expression is in part responsible for the rapamycin-induced effects. We demonstrate that inhibition of mTOR leads to endothelial cell dysfunction and decreased SR-BI expression, which may contribute to atherogenesis during rapamycin treatment.
Abbreviations: ABCG1, ATP binding cassette transporter G1; BP-C, Bodipy-cholesterol; BP-CE, Bodipy-cholesteryl oleate; CETP, cholesteryl ester transfer protein; DAF-2 DA, 4,5-diaminofluorescein diacetate; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; FAS, fatty acid synthase; HCAECs, human coronary artery endothelial cells; HUVECs, human umbilical vein endothelial cells; LDL-R, LDL-receptor; LXR, liver X receptor; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; PPARγ, peroxisome proliferator-activated receptor gamma; S6, ribosomal protein S6; S6K1, S6 kinase 1; S1P, sphingosine-1-phosphate; SR-BI, scavenger receptor, class B, type I; SREBP, sterol regulatory element binding protein; RCT, reverse cholesterol transport; TSC2, tuberous sclerosis 2 protein
Keywords: mTOR, SR-BI, Endothelial cell, HDL, Cell migration, eNOS
Highlights
-
•
The mTOR inhibitor rapamycin decreases the expression of the HDL receptor SR-BI in endothelial cells.
-
•
Rapamycin causes endothelial dysfunction by impairing HDL-induced NO production and cell migration.
-
•
Our observations contribute to the understanding of the mechanisms by which rapamycin influences atherogenesis.
1. Introduction
Plasma concentrations of HDL cholesterol exhibit an inverse correlation with the incidence of coronary artery disease [1]. HDL particles possess anti-inflammatory, anti-oxidant, and anti-thrombotic properties and are capable of activating eNOS (reviewed in [2]). The cardio-protective effect of HDL is further ascribed to its ability to transfer lipids from peripheral cells, such as macrophage foam cells residing in the arterial intima, back to the liver for excretion into the bile, a process called reverse cholesterol transport (RCT) [3]. To achieve the removal of excess cholesterol deposited in the arterial intima, HDL must cross the endothelial barrier to come into close proximity to macrophage foam cells found in atherosclerotic plaques. The mechanisms necessary for this transport are not fully understood (reviewed in [4]).
The mammalian target of rapamycin (mTOR) pathway is a central regulator of cellular growth and metabolism in response to nutrients, growth factors, or cellular energy levels (reviewed in [5]). The conserved serine/threonine kinase mTOR forms two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which differ in their input signals and output functions. The mTORC1 substrates S6 kinase 1 (S6K1) and eIF4E binding protein 1 are involved in protein synthesis (reviewed in [6]). mTORC1 is also known to activate gene transcription, e.g. via the sterol regulatory element binding protein (SREBP) pathway, a key transcriptional regulator of cellular cholesterol metabolism, or the peroxisome proliferator-activated receptor gamma (PPARγ) [7], [8], [9]. mTORC2 is involved in cell survival and metabolism and cytoskeletal organization via its substrates, the kinases Akt, serum- and glucocorticoid-induced protein kinase 1 and protein kinase C-α (reviewed in [5]).
Rapamycin (Sirolimus) in complex with FK506-binding-protein binds to the FKBP12–rapamycin-binding domain of mTORC1, thereby inhibiting its kinase activity. Rapamycin was first approved as an immunosuppressant agent to prevent allograft rejection; thus a plethora of clinical data are available from kidney transplant patients. The majority of these patients developed dyslipidemia [10], which contributes to cardiovascular disease, the most common cause of death in kidney-transplant recipients [11]. Although much effort has been made in understanding rapamycin-related dyslipidemia, for most mTOR inhibitors the net effect on atherosclerosis remains elusive. The decrease in LDL receptor (LDL-R) expression may be a reason for hyperlipidemia observed in rapamycin-treated patients [10], [12]. Indeed, it was reported recently that down-regulation of hepatic LDL-R expression is mediated via mTORC1 and leads to increased LDL-cholesterol levels in mice [13]. Interestingly, HDL-cholesterol levels increased in rapamycin-treated mice, indicating a complex mode of action of this drug. Several groups found increased HDL-cholesterol levels upon rapamycin treatment in different mouse models [13], [14], [15], and scavenger receptor, class B, type I (SR-BI) loss of function in humans as well as in mice leads to a malfunction of HDL metabolism and abnormal HDL cholesterol levels (reviewed in [16]).
Both the HDL receptor SR-BI and the ATP binding cassette transporter G1 (ABCG1) participate in endothelial HDL transport [17]. Besides playing a role in lipoprotein uptake, SR-BI mediates signal transduction of HDL (reviewed in [18]). This is of particular importance in endothelial cells, where HDL was shown to induce cell migration and eNOS activation, thereby stimulating NO production, which is essential for maintaining vascular homeostasis [18], [19], [20]. Further studies identified the involvement of eNOS phosphorylation at serin 1177 via Akt, mediated by Src kinases and PI3 kinase, in this process [21]. Defective NO production causes impaired vasodilation, which together with a pro-inflammatory and pro-thrombotic state of endothelial cells results in endothelial dysfunction. The latter represents an early event in the development of atherosclerosis.
In the present study, we have investigated the consequences of mTOR inhibition on SR-BI expression and function in primary endothelial cells. After rapamycin treatment, a time-dependent down-regulation of SR-BI in human umbilical vein endothelial cells (HUVECs) was found. Interestingly, HDL uptake was unaltered under these conditions, while HDL-stimulated endothelial NO production and cell migration were impaired. The contribution of SR-BI to these two hallmarks of endothelial dysfunction was assessed by analyzing the effects of SR-BI knockdown and overexpression. SR-BI knockdown alone could not mimic the rapamycin induced effects on eNOS activation or cell migration, whereas SR-BI overexpression in rapamycin-treated cells could partly attenuate endothelial dysfunction.
2. Material and methods
2.1. Materials
Antibodies to SR-BI (CLA-1) and phospho-eNOS (S1177) were purchased from BD Transduction Laboratories. Antibodies to total mTOR, total TSC2, phospho-Akt (S473), total Akt, phospho-S6 ribosomal protein (S240, 244), total S6 ribosomal protein, GAPDH, and total eNOS were obtained from Cell Signaling Technology. The antibody to ß-actin was purchased from Abcam. HUVECs were incubated with the following inhibitors: 20 nM rapamycin (Calbiochem, Merck), 250 nM Torin 1 (Tocris Bioscience), 10 μM S6K1 inhibitor (PF4708671, Sigma) and 1 mM Akt inhibitor (MK-2206). We further applied 10 μM lovastatin (Sigma) and two PPARγ agonists: 10 μM troglitazone (Merck) and 10 μM pioglitazone (Sigma).
2.2. Cell culture
Human umbilical vein endothelial cells (HUVECs) and human coronary artery endothelial cells (HCAECs) (PromoCell) were cultured in flasks coated with 0.5% gelatin in Endothelial Cell Growth Medium (PromoCell) containing endothelial cell growth supplement, epidermal growth factor, basic fibroblast growth factor, heparin and hydrocortisone, supplemented with 5% fetal calf serum. Passages from 4 to 7 were used for the experiments. HUVECs were maintained in serum-free Endothelial Cell Growth Medium (PromoCell) containing the endothelial cell growth supplement mix. HepG2 and Huh7 cells (ATCC) were maintained in minimal essential medium (MEM; GE Healthcare) supplemented with 1% penicillin/streptomycin, 10% FCS, 1% non-essential amino acids and 1% glutamine. For experiments, FCS was reduced to 1%. HepG2 cells were incubated with 20 to 100 nM rapamycin. Fluorescently labeled HDL uptake experiments were performed as described for HUVECs.
2.3. Lipoprotein isolation and labeling with fluorescent dyes
Plasma was collected from healthy volunteers and HDL was prepared by sequential ultracentrifugation (d = 1.21 g/ml) [22]. The apolipoprotein part of HDL was covalently labeled with fluorescent Alexa Fluor 488 (Molecular Probes) according to the manufacturer's instructions. Loading of HDL with Bodipy-cholesteryl oleate (BP-CE) or Bodipy-cholesterol (BP-C) was performed as described previously [23]. Labeling of HDL with the fluorescent phospholipid DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) was carried out by incubation of HDL in human lipoprotein deficient serum overnight at 37 °C followed by ultracentrifugation [24]. HUVECs were incubated with 50 μg/ml labeled HDL for 60 min. After the cells were fixed in 4% para-formaldehyde for 30 min at 4 °C, samples were mounted and examined using a fluorescence microscope (Axiovert 135, Zeiss).
2.4. [3H-CE-, 125I]-HDL labeling and uptake experiments
HDL was double labeled with [3H]-cholesteryl-oleate (Perkin Elmer) and sodium [125] iodine (Hartmann Analytics) as previously described [25]. For uptake experiments, HUVECs were seeded in 12-well plates. To calculate unspecific binding, a 40-fold excess of unlabeled HDL was added to every fourth well. [3H-CE-, 125I]-HDL was added to each well at a concentration of 10 μg/ml. After 60 min cells were washed twice with cold PBS + BSA (2 mg/ml) and twice with cold PBS. Cells were then lysed with NaOH (0.1 M). [125I]-radioactivity in the lysates was counted using a gamma-counter (COBRAII Auto-gamma; Perkin Elmer). [3H] was analyzed using 15 ml Ready-Safe (Beckman Coulter) and a beta-counter (Tri-Carb 2800TR; Perkin Elmer). Measurements were normalized to protein content, determined by the Bradford protein assay (Biorad).
2.5. Quantitative real-time PCR
RNA was isolated using the RNeasy Plus Kit (Qiagen) and cDNA was synthesized with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). For real-time PCR TaqMan® Assays (Life Technologies) and a StepOne Real Time PCR System (Applied Biosystems) were utilized. The following TaqMan® probes were used: GAPDH (Hs99999905_m1), SCARB1 (Hs00969821_m1), HMGCR (Hs00168352_m1), LDLR (Hs00181192_m1), FASN (Hs01005622_m1), ABCA1 (Hs00194045_m1) and ABCG1 (Hs01555193). Results are shown as relative expression normalized to GAPDH.
2.6. TSC2 knockdown using RNAi
siRNA mediated knockdown in HUVECs was performed using the RNAiFect Transfection Reagent (Qiagen) according to the manufacturer's instructions. HUVECs were transfected for 48 h with scrambled control or TSC2 targeting siRNA using a final siRNA concentration of 30 nM (ON-TARGETplus SMARTpool, Dharmacon, Thermo Scientific). Knockdown efficiency was tested using Western Blot analysis.
2.7. Western Blot analysis
Western Blot analysis was carried out according to standard procedures. Densitometric quantification was performed using the TotalLab Software (TotalLab Ltd).
2.8. SR-BI silencing and overexpression using lentiviral particles
HUVECs were seeded in 24-well plates, and the next day 250 μl of Endothelial Cell Growth Medium was added containing 8 μg/ml of Polypren and 2 ∗ 105 TU of shRNA lentiviral transduction particles targeting SR-BI (SHCLNV, TRCN0000056963, MISSION ® Lentiviral Transduction Particles, Sigma), scrambled control (SHC002V, MISSION® pLKO.1-puro Non-Mammalian shRNA Control Transduction Particles, Sigma) or SCARB1 (LP-G0781-Lv105-0200-S, Gene Copoeia). Cells were centrifuged for 90 min at 30 °C and 1300 g. Two days after transduction cells were changed to selection medium containing 0.5 μg/ml of Puromycin (Life Technologies). Knockdown and overexpression efficiency were tested by Western Blot analysis.
2.9. Cell surface biotinylation
Cell surface biotinylation and cell fractionation were performed using the Cell Surface Protein Isolation Kit (Thermo Scientific) according to the manufacturer's instructions. Cell surface and cytosolic fractions obtained were analyzed by Western Blot. The total amount of a specific protein in each fraction was calculated according to the fraction volume.
2.10. NO quantification using 4,5-diaminofluorescein diacetate (DAF-2 DA)
For the quantification of intracellular NO concentrations, DAF-2 DA (Sigma) was used [26]. HUVECs were seeded into clear-bottom 96-well plates. After the cells were pretreated with or without 20 nM rapamycin for 24 h and washed twice with PBS, we added PBS containing 1 μM DAF-2 DA with or without 100 μg/ml HDL. DAF-2 fluorescence intensity was measured at an excitation wavelength of 488 nm every 2 min for 60 min using a fluorometer (Zenyth 3100, Anthos). The assay was performed at least in triplicate. DAF-2 fluorescence from untreated cells was subtracted from HDL-treated cells.
2.11. Cell migration assay
HUVECs were seeded in 6-well plates. Cell monolayers were scratched using a pipette tip and migration into the wounded area was assessed after 6 and 24 h. Medium was changed immediately after wounding and 20 nM rapamycin and/or 100 μg/ml HDL were added. Cell migration at the 24 h time point was quantified by measuring the scratch area. Results are calculated as % of the initial scratch area; results are expressed as % of control treated cells.
2.12. Filipin staining
Free cholesterol distribution and the amount within the cells were assessed using filipin staining. Cells were fixed in 4% para-formaldehyde for 30 min at 4 °C, washed twice with PBS, and incubated with 1 mg/ml filipin III (Sigma) for 2 h. Samples were washed twice with PBS, mounted and imaged using a fluorescence microscope (Axiovert 135, Zeiss).
2.13. Statistical analysis
Results were expressed as mean ± SD. Data were analyzed using a two-tailed Student's t-test. p-Values below 0.005 were considered as significant.
3. Results
3.1. SR-BI expression is decreased by rapamycin
Deregulation of the mTOR pathway provokes dyslipidemia but its consequences on the interaction of HDL with endothelial cells are still elusive. Therefore, we sought to analyze the effect of the mTOR inhibitor rapamycin on SR-BI in HUVECs. Upon treatment with rapamycin (20 nM) for 24 h SR-BI expression was reduced by about 50% in HUVECs on the protein as well as on the mRNA level (Fig. 1A). As expected, mTORC1 activity was inhibited upon rapamycin treatment as shown by decreased S6 phosphorylation (Fig. 1A). Similar results were found in human coronary artery endothelial cells (HCAECs) (Fig. S1) as well as in the human hepatoma cell lines HepG2 (Fig. S2A) and Huh7 (data not shown). Next, the time dependence of this regulation was analyzed in HUVECs (Fig. 1B). Down-regulation of SR-BI occurred rather late, resulting in a 30% reduction of SR-BI protein after 16 h. Rapamycin strongly inhibited the phosphorylation of the mTORC1 downstream target ribosomal protein S6 (S6) after 1 h, while Akt phosphorylation at serine 473 decreased more slowly. Concomitantly to SR-BI protein, SR-BI mRNA expression was decreased by about 30% after 16 h of incubation with rapamycin (Fig. 1B). These data show that the HDL receptor SR-BI is down-regulated upon inhibition of mTOR by rapamycin, on the protein as well as on the mRNA level.
Fig. 1.
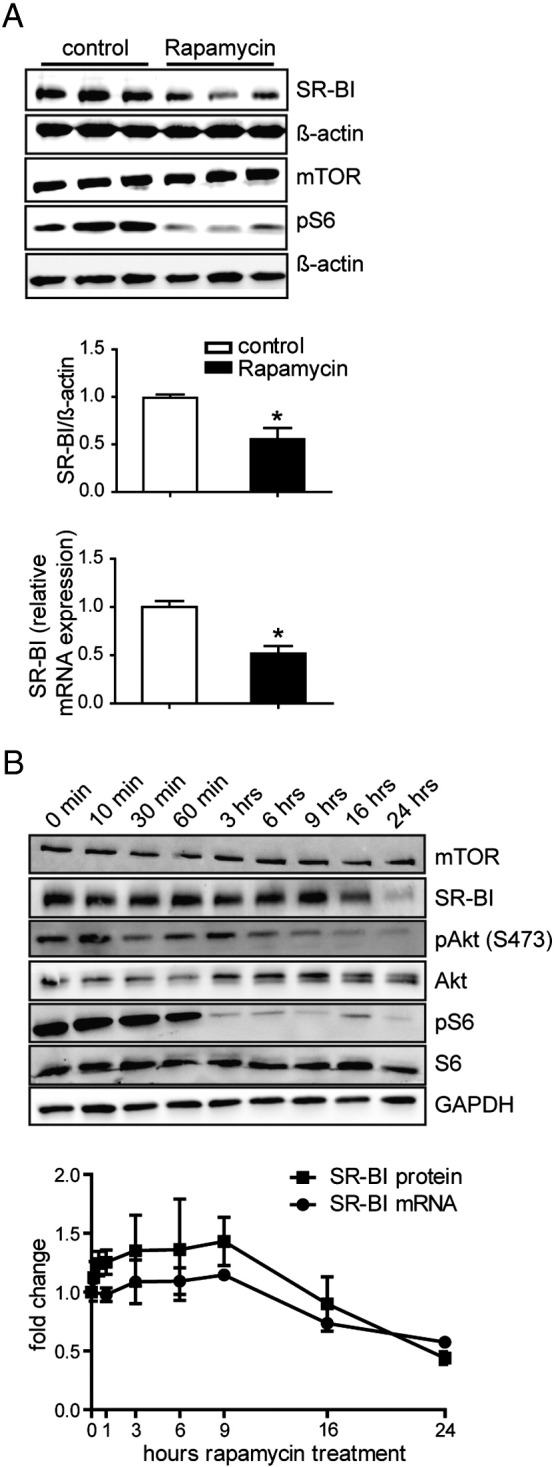
Analysis of SR-BI expression in HUVECs. A, HUVECs were incubated with 20 nM rapamycin for 24 h, and then lysed and Western Blot analysis or quantitative real time PCR was performed. SR-BI protein and mRNA expression upon rapamycin treatment was quantified (n = 6). B, cells were incubated with 20 nM rapamycin and lysed at the indicated time points. Afterwards Western Blot analysis or quantitative real time PCR was performed. SR-BI protein (squares) and mRNA (circles) expression upon rapamycin treatment was quantified (n = 2). A representative Western Blot is shown in A and B. *p < 0.005.
3.2. SR-BI regulation is mediated via mTORC1
Having identified the involvement of mTOR in the regulation of SR-BI, we next aimed to dissect the mTOR signaling network using specific inhibitors and activators to identify mTOR downstream effectors (illustrated in Fig. 2D). We investigated the effect of rapamycin, Torin, S6K1 inhibitor (PF4708671), and Akt inhibitor (MK-2206) on SR-BI expression (Fig. 2A). Rapamycin and Torin blocked both mTOR complexes in HUVECs, as demonstrated by decreased pAkt and pS6 levels (Fig. 2A). MK-2206 significantly blocked Akt phosphorylation, and PF4708671 decreased S6 phosphorylation. SR-BI protein expression was down-regulated by about 50% after 24 h of incubation with rapamycin or Torin, but not upon inhibition of S6K1 or Akt alone (Fig. 2A). A similar expression pattern of SR-BI was seen on the mRNA level (Fig. 2B). This indicates that Akt, a major downstream target of mTORC2, and S6K1 are not involved in the down-regulation of SR-BI.
Fig. 2.

Effect of mTOR pathway modulation on SR-BI expression. A and B, HUVECs were treated for 24 h with either 20 nM rapamycin, 250 nM Torin, 10 μM PF4708671 or 1 μM MK-2206. Cells were lysed and Western Blot analysis (A) or quantitative real time PCR (B) was performed (n = 3). C, RNAi mediated TSC-2 knockdown was performed in HUVECs, cells were lysed and Western Blot analysis was performed. D, simplified illustration of mTOR signaling and the influence of the specific inhibitors used in HUVECs. *p < 0.005.
We next activated mTORC1 signaling by knockdown of the mTORC1 upstream inhibitor tuberous sclerosis 2 protein (TSC2) (Fig. 2C). This knockdown led to an increase of SR-BI protein expression as well as mTORC1 activity, as shown by increased S6 phosphorylation. Taken together, these results suggest an involvement of mTORC1 in the regulation of SR-BI, which seems to be independent of S6K1.
3.3. SR-BI regulation is independent of the SREBP-LDL-R axis
Regulation of cellular lipid metabolism via mTOR is mainly mediated by the transcription factors SREBPs and PPARγ (reviewed in [9], [27]). Thus we analyzed the expression of target genes of these transcription factors after inhibition of mTOR and in combination with specific activators (Fig. 3). After rapamycin treatment, the mRNA expression of the LDL-R and hydroxymethylglutaryl-CoA-reductase (HMG-CoA-R), both targets of SREBP-2, and of fatty acid synthase (FAS) which is a SREBP-1 target, was reduced by about 50% (Fig. 3A). To assess whether this regulation is mediated by an alteration of the cellular cholesterol content, we used lovastatin, an inhibitor of HMG-CoA-R, the rate-limiting enzyme in the cholesterol biosynthetic pathway. Lovastatin decreased the intracellular free cholesterol content, as demonstrated by filipin staining (Fig. 3B). While LDL-R, HMG-CoA-R, and FAS mRNA expressions increased after lovastatin treatment, SR-BI expression remained unchanged. Rapamycin treatment with or without lovastatin resulted in a decrease of SR-BI expression as well as LDL-R, HMG-CoA-R, and FAS expressions (Fig. 3A). In contrast to lovastatin, rapamycin alone did not affect cellular cholesterol content, as demonstrated by filipin staining, but a combination of both had the same effect as lovastatin alone (Fig. 3B). Thus, while the cholesterol responsive genes were reactivated by lovastatin, SR-BI regulation was only influenced by rapamycin treatment.
Fig. 3.

Analysis of SR-BI down-regulation. A, HUVECs were incubated for 24 h with 20 nM rapamycin (R) and/or 10 μM lovastatin (L). Cells were lysed and quantitative real time PCR was performed (n = 3). B, after preincubation with rapamycin and/or lovastatin, cells were fixed, filipin staining was performed, and cells were imaged using a fluorescence microscope. C, HUVECs were incubated for 24 h with 20 nM rapamycin and/or 10 μM troglitazone or 10 μM pioglitazone. Cells were lysed and Western Blot analysis was performed. *p < 0.005.
In contrast to SREBP regulated genes ABCA1 and ABCG1, both liver X receptor (LXR) targets, were decreased by lovastatin but not by rapamycin treatment, arguing against an involvement of LXRs in regulating SR-BI upon mTOR inhibition (Fig. 3A). We further analyzed the role of PPARγ, another important transcription factor controlling cellular lipid metabolism, in the regulation of SR-BI by mTOR. We found that PPARγ mRNA was decreased upon rapamycin treatment (data not shown). However, activation of PPARγ using two different ligands, troglitazone and pioglitazone, did not alter basal SR-BI expression and did not interfere with the rapamycin-mediated down-regulation of SR-BI (Fig. 3C). Our results indicate that these classical transcription factors regulating cellular lipid metabolism are not involved in the down-regulation of SR-BI upon rapamycin treatment.
3.4. Rapamycin treatment does not alter HDL uptake
Next, we investigated functional consequences emerging from the alteration in SR-BI expression levels in HUVECs upon rapamycin treatment. We first analyzed the uptake of fluorescently labeled HDL (Fig. 4A). Reconstituted HDL particles containing the fluorescent cholesterol analogs Bodipy-cholesterol (BP-C) and Bodipy-cholesteryl oleate (BP-CE) were used to follow the uptake of HDL-associated sterols. HDL-Alexa 488 was used to visualize the HDL particle itself and HDL-DiI to follow the fate of HDL-derived phospholipids. BP-C was distributed throughout the cells, with enrichment in the perinuclear area. BP-CE was also enriched in the perinuclear area. HDL-Alexa 488 and HDL-DiI showed a more vesicular staining pattern. After treatment with rapamycin, there was no alteration in the distribution or in the amount of HDL or HDL-derived lipids taken up. Similar results were observed in HepG2 cells (Fig. S2B) and Huh7 cells (data not shown). To quantify this, we used double labeled [3H-CE-, 125I]-HDL (Fig. 4B). No difference was found in specific HDL particle uptake, represented by the [125I]-HDL fraction, or in specific [3H-CE] uptake from HDL particles in HUVECs. To exclude the involvement of cellular redistribution of SR-BI, we analyzed SR-BI distribution by separating plasma membrane and cytosolic proteins using cell surface biotinylation (Fig. 4C). No major shift upon rapamycin treatment was seen. Thus, despite a reduction of SR-BI levels by ~ 50% upon treatment with rapamycin we found no difference in HDL uptake, pointing to a compensatory mechanism.
Fig. 4.

Investigation of HDL uptake and SR-BI cell surface expression. A, after treatment with 20 nM rapamycin for 24 h, cells were incubated with 50 μg/ml HDL-Alexa 488, HDL-BP-CE, HDL-BP-C, or HDL-DiI for 1 h at 37 °C. Cells were then fixed and imaged using a fluorescence microscope. B, HUVECs were incubated with 20 nM rapamycin for 24 h. Cells were then incubated with 10 μg/ml radioactively dual labeled [3H-CE-; 125I]-HDL. After 1 h at 37 °C cells were lysed, and the radioactivity was counted in the cell lysates and normalized to protein content (n = 3). C, cell surface biotinylation was performed, and lysates from each fraction were analyzed by Western Blot and protein expression was quantified by densitometric analysis (n = 3). No significant changes were detected.
3.5. Rapamycin treatment causes endothelial dysfunction
Since an essential function of HDL is to activate cellular signaling pathways in endothelial cells, thereby stimulating NO production and cell migration, we assessed whether rapamycin has any influence on these processes. Upon rapamycin pretreatment, HDL failed to stimulate Akt phosphorylation to the same extent as in control cells (Fig. 5A). In HCAECs a similar result was seen (Fig. S1). Consequently, eNOS phosphorylation at S1177, which reflects enzyme activity, was decreased compared to control cells. To further test whether this results in impaired eNOS activity, we assessed NO production in HUVECs using DAF-2 DA (Fig. 5B). Indeed, following rapamycin pretreatment NO production was not stimulated by HDL (Fig. 5B, gray regression line). In contrast, in control cells addition of HDL increased the amount of intracellular NO in a time-dependent manner (Fig. 5B, black regression line). The underlying cause of decreased eNOS activity upon rapamycin treatment is likely linked to impaired Akt phosphorylation, since treatment with the Akt inhibitor MK-2206 had a similar effect on HDL-induced eNOS phosphorylation as rapamycin treatment (Fig. S3). To further analyze the functional consequences of impaired HDL-induced cellular signaling, we carried out experiments on the stimulation of cell migration by HDL. Rapamycin significantly blocked basal as well as HDL-stimulated cell migration in HUVECs (Fig. 5C). These data indicate that rapamycin causes endothelial dysfunction by impairing NO production and endothelial cell migration.
Fig. 5.

Analysis of HDL-stimulated eNOS activation and cell migration. A, after 20 nM rapamycin pretreatment for 24 h, HUVECs were incubated with 100 μg/ml HDL for the indicated time. Cells were then lysed and Western Blot analysis was performed. B, HUVECs were preincubated with 20 nM rapamycin for 24 h. Cells were then incubated with 1 μM DAF-2 DA in PBS for 60 min with or without 100 μg/ml HDL. Fluorescence intensity was measured every 2 min using a fluorometer. Linear regression of three independent experiments is shown; results are expressed as mean ± SEM. C, HUVEC monolayers were scratched and migration into the wounded area was assessed after 6 and 24 h in the presence of 20 nM rapamycin and/or 100 μg/ml HDL. Cell migration after 24 h was quantified. A representative image is shown. Results are calculated as % of the initial scratch area; results are expressed as % of control (n = 3). *p < 0.005.
3.6. SR-BI knockdown does not mimic the rapamycin induced effects
Having discovered that rapamycin triggers endothelial dysfunction in HUVECs, we aimed to investigate the significance of SR-BI in these processes. We first performed a SR-BI knockdown using a lentiviral system in HUVECs to assess the consequences of SR-BI down-regulation separately, without the influence of rapamycin (Fig. 6). Upon incubation with HDL, Akt as well as eNOS phosphorylation increased in control and to a similar extent in SR-BI knockdown cells (Fig. 6A). No difference in basal or HDL-stimulated cell migration was seen upon SR-BI knockdown compared to control cells (Fig. 6B). These results indicate that the effects of rapamycin on endothelial cell migration and NO production are not mediated by SR-BI alone.
Fig. 6.

Influence of SR-BI knockdown on HDL-stimulated eNOS activation and cell migration. A, after lentiviral knockdown of SR-BI HUVECs were incubated with or without 100 μg/ml HDL for 30 min. Cells were lysed and analyzed by Western Blot (n = 3). A representative Western Blot is shown. B, HUVEC monolayers were scratched and migration into the wounded area was assessed after 6 and 24 h in the presence of 20 nM rapamycin and/or 100 μg/ml HDL. Cell migration after 24 h was quantified. A representative image is shown. Results are calculated as % of the initial scratch area; results are expressed as % of control (n = 3). *p < 0.005.
3.7. SR-BI overexpression partly overcomes the rapamycin induced effects
Finally, we made use of SR-BI overexpression to assess whether unimpaired SR-BI expression can counteract the effects mediated by rapamycin. Therefore, HDL-stimulated signaling and cell migration were assessed in wildtype versus SR-BI overexpressing HUVECs (Fig. 7). Akt and eNOS phosphorylation increased upon incubation with HDL in wildtype as well as SR-BI overexpressing cells (Fig. 7A). This effect was almost abolished upon rapamycin treatment. Even SR-BI overexpression could not attenuate defective stimulation of Akt and eNOS phosphorylation by HDL. In addition, basal phospho-Akt and phospho-S6 levels were similar in wildtype and SR-BI overexpressing HUVECs. Again, we investigated endothelial cell migration upon stimulation with HDL during rapamycin treatment (Fig. 7B). HDL-stimulated cell migration was improved in SR-BI overexpressing cells compared with wildtype cells under control as well as under rapamycin-treated conditions. There was no difference in basal unstimulated cell migration. Taken together, we found that SR-BI overexpression did not influence HDL-stimulated Akt and eNOS phosphorylation but improved HDL-stimulated cell migration, suggesting that SR-BI is in part involved in the rapamycin induced effects contributing to endothelial dysfunction.
Fig. 7.
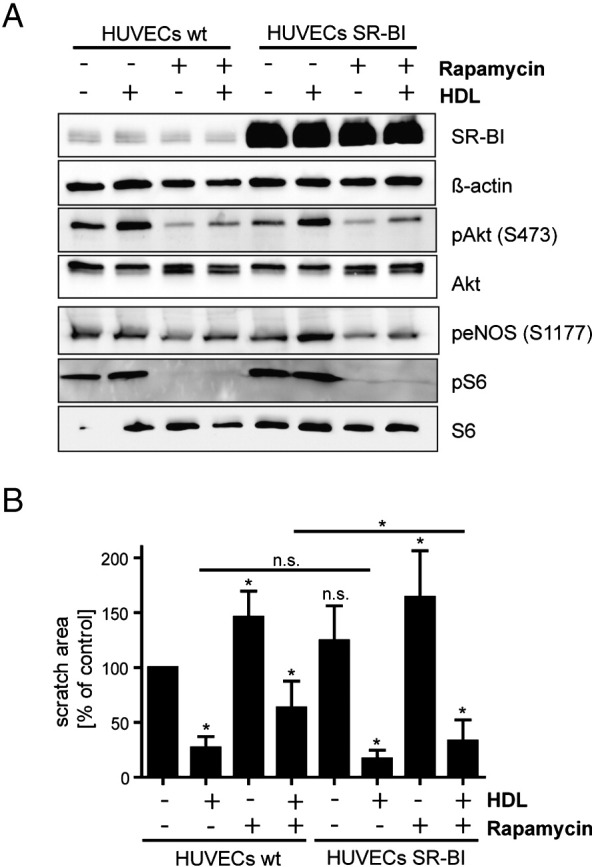
Influence of SR-BI overexpression on HDL-stimulated eNOS activation and cell migration. A, after lentiviral overexpression of SR-BI in HUVECs, wildtype (HUVECs wt) or SR-BI overexpressing (HUVECs SR-BI) cells were preincubated with 20 nM rapamycin for 24 h followed by a stimulation with 100 μg/ml HDL for 30 min. Cells were lysed and analyzed by Western Blot (n = 2). A representative Western Blot is shown. B, HUVEC monolayers were scratched and migration into the wounded area was assessed after 6 and 24 h in the presence of 20 nM rapamycin and/or 100 μg/ml HDL. Cell migration after 24 h was quantified. Results are calculated as % of the initial scratch area; results are expressed as % of control (n = 3). *p < 0.005.
4. Discussion
Here we show that the mTOR inhibitor rapamycin, which is in clinical use as an immunosuppressant, for anti-restenosis, and for the treatment of various cancers, down-regulates SR-BI protein as well as mRNA levels in human endothelial cells and induces endothelial dysfunction.
Acute rapamycin treatment specifically inhibits mTORC1, while long-term treatment was shown to impair the function of mTORC2 in a cell type and tissue specific manner [28]. Rapamycin treatment for 24 h in HUVECs efficiently inhibited the activity of both mTOR complexes as downstream target phosphorylation decreased strongly. HepG2 cells exhibited a similar response. Rapamycin and Torin, which both inhibit mTORC1 and mTORC2, decreased SR-BI protein as well as mRNA levels in HUVECs. Rapamycin also decreased SR-BI expression in HepG2 cells, however a higher concentration of rapamycin was necessary to induce the same effect as in HUVECs. Interestingly, neither inhibition of Akt, a main downstream target of mTORC2, nor inhibition of S6K1, a classical mTORC1 target, had a significant effect on SR-BI expression in HUVECs. Constitutive activation of mTORC1 by TSC2 knockdown led to an increase of SR-BI protein, emphasizing the importance of mTORC1 in this process. mTORC1 exerts its downstream effects by various mechanisms and was previously shown to activate SREBPs in different ways, the induction of SREBP mRNA being independent of S6K1 [29].
Since rapamycin treatment affected SR-BI mRNA and protein levels to the same extent, a regulation of transcription or RNA stability is most likely involved in this regulation. Data on the regulation of endothelial SR-BI expression are limited. Hepatic SR-BI expression is very complex and regulated by multiple factors on the transcriptional as well as on the post-transcriptional level (reviewed in [30]). The actions of rapamycin are diverse and operate on several levels, on the transcriptional, translational, and also post-translational levels, as shown recently for the LDL-R [13]. To assess which factors might be involved in the regulation of SR-BI upon inhibition of mTOR, we analyzed the relevance of transcription factors that are important in lipid and lipoprotein metabolism and also regulate SR-BI expression, at least in the liver [30]. Our data do not suggest an involvement of SREBPs, LXRs, or PPARγ in the regulation of SR-BI by rapamycin. It appears conceivable that a combination of factors is regulating SR-BI transcription upon modulation of mTOR activity. Furthermore, post-transcriptional mechanisms may also play a role in the regulation of SR-BI by rapamycin.
Unexpectedly, we found no change in HDL uptake upon rapamycin treatment. Since SR-BI is not solely responsible for HDL uptake in endothelial cells, this finding may be explained by the contribution of several other proteins. One possible candidate is ABCG1, which was shown to be involved in HDL uptake and transport in bovine aortic endothelial cells [17]. We found in our experiments slightly increased ABCG1 expression upon rapamycin treatment, which might compensate decreased SR-BI expression. The beta-chain of cell surface F(0)F(1) ATPase was found to participate in HDL transcytosis in endothelial cells and in hepatic HDL endocytosis [31], [32]. The regulation of endothelial cholesterol metabolism needs to be tightly controlled, emphasizing the importance of the redundancy of these proteins involved in regulating HDL uptake.
Rapamycin pretreatment strongly impaired HDL-stimulated Akt and eNOS phosphorylation, NO production, and endothelial cell migration. These data are in agreement with previous work showing that rapamycin blocks endothelial cell migration [33]. To test whether these adverse effects leading to endothelial dysfunction are directly related to decreased SR-BI expression or rather evolve from a combination of rapamycin dependent effects, we performed SR-BI knockdown in HUVECs. Lentiviral SR-BI knockdown resulted in a 60 to 70% decrease of SR-BI protein levels in HUVECs, which was considered to be appropriate to mimic down-regulation of SR-BI by about 50% upon rapamycin treatment. Despite this substantial reduction of SR-BI levels, we found no difference in HDL-induced signaling or cell migration compared to control cells. In contrast, in aortas of SR-BI knockout mice HDL no longer induced NO dependent vasorelaxation [19]. This emphasizes the significance of SR-BI in stimulating eNOS activity in vivo. Sphingosine-1-phosphate (S1P) was shown to induce NO production via eNOS in endothelial cells and might fill in for SR-BI [34], [35]. Signaling via SR-BI is important for HDL-induced endothelial cell migration in vitro [36]. In vivo evidence also demonstrates that SR-BI is an important player in this process as re-endothelialization is decreased in SR-BI knockout mice [36]. We did not observe decreased HUVEC migration after rapamycin treatment in SR-BI knockdown cells, again indicating that this effect is not mediated by SR-BI alone.
We finally could demonstrate that HDL-stimulated endothelial cell migration was improved in cells overexpressing SR-BI compared to wildtype cells. Thus, we conclude that decreased SR-BI expression can only in part account for the effects caused by rapamycin in endothelial cells.
Despite increasing clinical applications of mTOR inhibitors, one has to consider their serious systemic side effects, especially alterations of the lipid profile. Hyperlipidemia is observed frequently in rapamycin-treated patients [10], [12] and is presumably connected to decreased LDL-R expression. Via mTORC1 rapamycin down-regulates hepatic LDL-R expression in mice, leading to elevated LDL-cholesterol levels [13]. This, together with our finding that SR-BI expression is decreased in vitro and that rapamycin induces endothelial dysfunction, may contribute to atherogenesis in rapamycin-treated patients. However, atherogenesis is a complex process with multiple players and pro- as well as anti-atherogenic factors and events. Thus, the net effect of rapamycin in humans is not predictable. Several studies have identified rapamycin as a positive as well as a negative regulator of atherogenic events [14], [37], [38]. Although the in vivo contribution of endothelial SR-BI to anti-atherogenic processes is still not known (reviewed in [39]), decreased availability of the receptor due to rapamycin treatment may be considered as pro-atherogenic.
In summary, we reveal that the mTOR inhibitor rapamycin regulates SR-BI expression without affecting HDL uptake in endothelial cells. Rapamycin impaired HDL-mediated endothelial cell migration and eNOS activation, leading to endothelial dysfunction, a hallmark of early atherogenesis.
The following are the supplementary data related to this article.
Consequences of rapamycin treatment on SR-BI expression and HDL-stimulated signaling in HCAECs. After 20 nM rapamycin pretreatment for 24 h, HCAECs were incubated with 100 μg/ml HDL for 30 min. Cells were then lysed and Western Blot analysis was performed.
Consequences of rapamycin treatment on SR-BI expression and HDL uptake in HepG2 cells. A, HepG2 cells were incubated with 20, 50, or 100 nM rapamycin for 24 h, lysed and Western Blot analysis was performed. B, after treatment with 100 nM rapamycin for 24 h, cells were incubated with 50 μg/ml HDL-Alexa 488, HDL-BP-CE, HDL-BP-C, HDL-Dil for 1 h at 37 °C. Cells were then fixed and imaged using a fluorescence microscope. Note the dose-dependent down-regulation of mTOR activity by rapamycin and the concomitant SR-BI decrease.
Mechanisms of rapamycin-induced eNOS inhibition. HUVECs were incubated with 20 nM rapamycin or 1 μM MK-2206 for 24 h and then were incubated with 100 μg/ml HDL for 30 min. Afterwards cells were lysed and Western Blot analysis was performed (n = 2). Note the phosphorylation of eNOS after rapamycin as well as MK-2206 treatment, which was not stimulated by HDL incubation.
Acknowledgements
We thank Jelena Brankovic for excellent technical assistance and Monika Strobl for carefully reading the manuscript. This work was supported by the Austrian Science Fund (FWF) P22838-B13 (to H.S.). Stefanie Fruhwürth is a recipient of a DOC-fFORTE fellowship of the Austrian Academy of Sciences at the Institute of Medical Chemistry, Center for Pathobiochemistry and Genetics, Medical University of Vienna, Vienna, Austria.
References
- 1.Gordon D.J., Rifkind B.M. High-density lipoprotein—the clinical implications of recent studies. N. Engl. J. Med. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 2.Rye K.A., Barter P.J. Cardioprotective functions of HDL. J. Lipid Res. 2014;55(2):168–179. doi: 10.1194/jlr.R039297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rader D.J., Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–913. doi: 10.1038/nature06796. [DOI] [PubMed] [Google Scholar]
- 4.von Eckardstein A., Rohrer L. Transendothelial lipoprotein transport and regulation of endothelial permeability and integrity by lipoproteins. Curr. Opin. Lipidol. 2009;20:197–205. doi: 10.1097/MOL.0b013e32832afd63. [DOI] [PubMed] [Google Scholar]
- 5.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma X.M., Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 7.Lewis C.A., Griffiths B., Santos C.R., Pende M., Schulze A. Regulation of the SREBP transcription factors by mTORC1. Biochem. Soc. Trans. 2011;39:495–499. doi: 10.1042/BST0390495. [DOI] [PubMed] [Google Scholar]
- 8.Brown M.S., Goldstein J.L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 9.Laplante M., Sabatini D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009;19:R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathis A.S., Dave N., Knipp G.T., Friedman G.S. Drug-related dyslipidemia after renal transplantation. Am. J. Health Syst. Pharm. 2004;61:565–585. (quiz 586–567) [PubMed] [Google Scholar]
- 11.Howard R.J., Patton P.R., Reed A.I., Hemming A.W., Van der Werf W.J., Pfaff W.W., Srinivas T.R., Scornik J.C. The changing causes of graft loss and death after kidney transplantation. Transplantation. 2002;73:1923–1928. doi: 10.1097/00007890-200206270-00013. [DOI] [PubMed] [Google Scholar]
- 12.Kasiske B.L., de Mattos A., Flechner S.M., Gallon L., Meier-Kriesche H.U., Weir M.R., Wilkinson A. Mammalian target of rapamycin inhibitor dyslipidemia in kidney transplant recipients. Am. J. Transplant. 2008;8:1384–1392. doi: 10.1111/j.1600-6143.2008.02272.x. [DOI] [PubMed] [Google Scholar]
- 13.Ai D., Chen C., Han S., Ganda A., Murphy A.J., Haeusler R., Thorp E., Accili D., Horton J.D., Tall A.R. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J. Clin. Invest. 2012;122:1262–1270. doi: 10.1172/JCI61919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elloso M.M., Azrolan N., Sehgal S.N., Hsu P.L., Phiel K.L., Kopec C.A., Basso M.D., Adelman S.J. Protective effect of the immunosuppressant sirolimus against aortic atherosclerosis in apo E-deficient mice. Am. J. Transplant. 2003;3:562–569. doi: 10.1034/j.1600-6143.2003.00094.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang N., Su D., Qu S., Tse T., Bottino R., Balamurugan A.N., Xu J., Bromberg J.S., Dong H.H. Sirolimus is associated with reduced islet engraftment and impaired beta-cell function. Diabetes. 2006;55:2429–2436. doi: 10.2337/db06-0173. [DOI] [PubMed] [Google Scholar]
- 16.Mineo C., Shaul P.W. Functions of scavenger receptor class B, type I in atherosclerosis. Curr. Opin. Lipidol. 2012;23:487–493. doi: 10.1097/MOL.0b013e328357ba61. [DOI] [PubMed] [Google Scholar]
- 17.Rohrer L., Ohnsorg P.M., Lehner M., Landolt F., Rinninger F., von Eckardstein A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ. Res. 2009;104:1142–1150. doi: 10.1161/CIRCRESAHA.108.190587. [DOI] [PubMed] [Google Scholar]
- 18.Mineo C., Shaul P.W. Regulation of signal transduction by HDL. J. Lipid Res. 2013;54:2315–2324. doi: 10.1194/jlr.R039479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuhanna I.S., Zhu Y., Cox B.E., Hahner L.D., Osborne-Lawrence S., Lu P., Marcel Y.L., Anderson R.G., Mendelsohn M.E., Hobbs H.H., Shaul P.W. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 2001;7:853–857. doi: 10.1038/89986. [DOI] [PubMed] [Google Scholar]
- 20.Mineo C., Shaul P.W. Role of high-density lipoprotein and scavenger receptor B type I in the promotion of endothelial repair. Trends Cardiovasc. Med. 2007;17:156–161. doi: 10.1016/j.tcm.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Mineo C., Yuhanna I.S., Quon M.J., Shaul P.W. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J. Biol. Chem. 2003;278:9142–9149. doi: 10.1074/jbc.M211394200. [DOI] [PubMed] [Google Scholar]
- 22.Schumaker V.N., Puppione D.L. Sequential flotation ultracentrifugation. Methods Enzymol. 1986;128:155–170. doi: 10.1016/0076-6879(86)28066-0. [DOI] [PubMed] [Google Scholar]
- 23.Rohrl C., Meisslitzer-Ruppitsch C., Bittman R., Li Z., Pabst G., Prassl R., Strobl W., Neumuller J., Ellinger A., Pavelka M., Stangl H. Combined light and electron microscopy using diaminobenzidine photooxidation to monitor trafficking of lipids derived from lipoprotein particles. Curr. Pharm. Biotechnol. 2012;13:331–340. doi: 10.2174/138920112799095338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitas R.E., Innerarity T.L., Weinstein J.N., Mahley R.W. Acetoacetylated lipoproteins used to distinguish fibroblasts from macrophages in vitro by fluorescence microscopy. Arteriosclerosis. 1981;1:177–185. doi: 10.1161/01.atv.1.3.177. [DOI] [PubMed] [Google Scholar]
- 25.Stangl H., Hyatt M., Hobbs H.H. Transport of lipids from high and low density lipoproteins via scavenger receptor-BI. J. Biol. Chem. 1999;274:32692–32698. doi: 10.1074/jbc.274.46.32692. [DOI] [PubMed] [Google Scholar]
- 26.Kojima H., Sakurai K., Kikuchi K., Kawahara S., Kirino Y., Nagoshi H., Hirata Y., Nagano T. Development of a fluorescent indicator for nitric oxide based on the fluorescein chromophore. Pharm. Bull. 1998;46:373–375. doi: 10.1248/cpb.46.373. [DOI] [PubMed] [Google Scholar]
- 27.Lamming D.W., Sabatini D.M. A Central Role for mTOR in Lipid Homeostasis. Cell Metab. 2013;18(4):465–469. doi: 10.1016/j.cmet.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarbassov D.D., Ali S.M., Sengupta S., Sheen J.H., Hsu P.P., Bagley A.F., Markhard A.L., Sabatini D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 29.Owen J.L., Zhang Y., Bae S.H., Farooqi M.S., Liang G., Hammer R.E., Goldstein J.L., Brown M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. U. S. A. 2012;109(40):16184–16189. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leiva A., Verdejo H., Benitez M.L., Martinez A., Busso D., Rigotti A. Mechanisms regulating hepatic SR-BI expression and their impact on HDL metabolism. Atherosclerosis. 2011;217:299–307. doi: 10.1016/j.atherosclerosis.2011.05.036. [DOI] [PubMed] [Google Scholar]
- 31.Cavelier C., Ohnsorg P.M., Rohrer L., von Eckardstein A. The beta-chain of cell surface F(0)F(1) ATPase modulates apoA-I and HDL transcytosis through aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012;32:131–139. doi: 10.1161/ATVBAHA.111.238063. [DOI] [PubMed] [Google Scholar]
- 32.Martinez L.O., Jacquet S., Esteve J.P., Rolland C., Cabezon E., Champagne E., Pineau T., Georgeaud V., Walker J.E., Terce F., Collet X., Perret B., Barbaras R. Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature. 2003;421:75–79. doi: 10.1038/nature01250. [DOI] [PubMed] [Google Scholar]
- 33.Moss S.C., Lightell D.J., Jr., Marx S.O., Marks A.R., Woods T.C. Rapamycin regulates endothelial cell migration through regulation of the cyclin-dependent kinase inhibitor p27Kip1. J. Biol. Chem. 2010;285:11991–11997. doi: 10.1074/jbc.M109.066621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dantas A.P., Igarashi J., Michel T. Sphingosine 1-phosphate and control of vascular tone. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H2045–H2052. doi: 10.1152/ajpheart.01089.2002. [DOI] [PubMed] [Google Scholar]
- 35.Levkau B. Sphingosine-1-phosphate in the regulation of vascular tone: a finely tuned integration system of S1P sources, receptors, and vascular responsiveness. Circ. Res. 2008;103:231–233. doi: 10.1161/CIRCRESAHA.108.181610. [DOI] [PubMed] [Google Scholar]
- 36.Seetharam D., Mineo C., Gormley A.K., Gibson L.L., Vongpatanasin W., Chambliss K.L., Hahner L.D., Cummings M.L., Kitchens R.L., Marcel Y.L., Rader D.J., Shaul P.W. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ. Res. 2006;98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 37.Ouimet M., Franklin V., Mak E., Liao X., Tabas I., Marcel Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao L., Ding T., Cyrus T., Cheng Y., Tian H., Ma M., Falotico R., Pratico D. Low-dose oral sirolimus reduces atherogenesis, vascular inflammation and modulates plaque composition in mice lacking the LDL receptor. Br. J. Pharmacol. 2009;156:774–785. doi: 10.1111/j.1476-5381.2008.00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mineo C., Shaul P.W. Functions of scavenger receptor class B, type I in atherosclerosis. Curr. Opin. Lipidol. 2012;23(5):487–493. doi: 10.1097/MOL.0b013e328357ba61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Consequences of rapamycin treatment on SR-BI expression and HDL-stimulated signaling in HCAECs. After 20 nM rapamycin pretreatment for 24 h, HCAECs were incubated with 100 μg/ml HDL for 30 min. Cells were then lysed and Western Blot analysis was performed.
Consequences of rapamycin treatment on SR-BI expression and HDL uptake in HepG2 cells. A, HepG2 cells were incubated with 20, 50, or 100 nM rapamycin for 24 h, lysed and Western Blot analysis was performed. B, after treatment with 100 nM rapamycin for 24 h, cells were incubated with 50 μg/ml HDL-Alexa 488, HDL-BP-CE, HDL-BP-C, HDL-Dil for 1 h at 37 °C. Cells were then fixed and imaged using a fluorescence microscope. Note the dose-dependent down-regulation of mTOR activity by rapamycin and the concomitant SR-BI decrease.
Mechanisms of rapamycin-induced eNOS inhibition. HUVECs were incubated with 20 nM rapamycin or 1 μM MK-2206 for 24 h and then were incubated with 100 μg/ml HDL for 30 min. Afterwards cells were lysed and Western Blot analysis was performed (n = 2). Note the phosphorylation of eNOS after rapamycin as well as MK-2206 treatment, which was not stimulated by HDL incubation.