

Abstract
Background and Purpose
Cannabinoid CB2 receptors are up‐regulated in reactive microglia in the spinal cord of TDP‐43 (A315T) transgenic mice, an experimental model of amyotrophic lateral sclerosis. To determine whether this up‐regulation can be exploited pharmacologically, we investigated the effects of different treatments that affect CB2 receptor function.
Experimental Approach
We treated TDP‐43 (A315T) transgenic mice with the non‐selective agonist WIN55,212‐2, alone or combined with selective CB1 or CB2 antagonists, as well as with the selective CB2 agonist HU‐308, and evaluated their effects on the pathological phenotype.
Key Results
WIN55,212‐2 had modest beneficial effects in the rotarod test, Nissl staining of motor neurons, and GFAP and Iba‐1 immunostainings in the spinal cord, which were mediated in part by CB2 receptor activation. HU‐308 significantly improved the rotarod performance of the transgenic mice, with complete preservation of Nissl‐stained motor neurons in the ventral horn. Reactive astrogliosis labelled with GFAP was also attenuated by HU‐308 in the dorsal and ventral horns, in which CB2 receptors colocalize with this astroglial marker. Furthermore, HU‐308 reduced the elevated Iba‐1 immunostaining in the ventral horn of TDP‐43 transgenic mice, but did not affect this immunoreactivity in white matter, in which CB2 receptors also colocalize with this microglial marker.
Conclusions and Implications
Our study shows an important role for glial CB2 receptors in limiting the progression of the pathological phenotype in TDP‐43 (A315T) transgenic mice. Such benefits appear to derive from the activation of CB2 receptors concentrated in astrocytes and reactive microglia located in spinal dorsal and ventral horns.
Linked Articles
This article is part of a themed section on 8th European Workshop on Cannabinoid Research. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.10/issuetoc
Abbreviations
- ALS
amyotrophic lateral sclerosis
- CB1
cannabinoid receptor type‐1
- CB2
cannabinoid receptor type‐2
- FAAH
fatty acid amide hydrolase
- FTD
frontotemporal dementia
- FUS
fused in sarcoma
- GFAP
glial fibrillary acidic protein
- GLAST
glutamate‐aspartate transporter
- GLT‐1
glutamate transporter type‐1
- TDP‐43
TAR‐DNA binding protein‐43
Introduction
Amyotrophic lateral sclerosis (ALS) is a chronic neurodegenerative disorder characterized by the progressive damage in the upper and lower motor neurons, which results in muscle denervation, atrophy and paralysis (Hardiman et al., 2011; Van Damme et al., 2017). The combination of several cytotoxic events, for example, excessive glutamate levels, oxidative stress, protein aggregation and chronic inflammation, are responsible for this neuronal damage (Foran and Trotti, 2009; Ferraiuolo et al., 2011; Renton et al., 2014). The disease is frequently sporadic (Al‐Chalabi and Hardiman, 2013), but there is also familial cases, in general associated with mutations in genes encoding, among others, for SOD1, TAR‐DNA binding protein‐43 (TDP‐43), FUS (fused in sarcoma) and C9orf72 (Hardiman et al., 2011; Renton et al., 2014). In the case of mutations in TDP‐43, FUS, C90rf72 and other recent genes discovered over the past decade, the disease also presents features of frontotemporal lobar dementia (FTD), which supports the idea that, rather than being one disorder, ALS belongs to a spectrum of disorders having motor but also cognitive deficits (Cruts et al., 2013). One of these dual genes is TARDBP encoding TDP‐43. This protein is involved in pre‐mRNA splicing, transport and/or stability (Buratti and Baralle, 2010; Lagier‐Tourenne et al., 2010), and its alterations represent a new type of proteinopathy characterized by the accumulation of TDP‐43 in the cytosol in the form of protein aggregates (Janssens and Van Broeckhoven, 2013). Different transgenic models for TDP‐43 have been developed (reviewed in Tsao et al., 2012; Van Damme et al., 2017), which represent important tools for the study of ALS and also FTD, being the most used alternative to the classic mutant SOD1 mice generated in the 1990s (Ripps et al., 1995).
Despite the efforts addressed to develop effective treatments able to alleviate specific symptoms or to delay disease progression in ALS, this disorder still lacks an effective therapy, the anti‐excitotoxic agent riluzole (Rilutek®) and the antioxidant compound edaravone (Radicava®), being the only approved medicines, but having only modest effects (Habib and Mitsumoto, 2011; Bova and Kinney, 2013; Rothstein, 2017). Recent preclinical studies have demonstrated neuroprotective effects with cannabinoids (reviewed by de Lago et al., 2015), including the phytocannabinoids Δ9‐tetrahydrocannabinol (Raman et al., 2004) and cannabinol (Weydt et al., 2005), and, to a lesser extent, the phytocannabinoid‐based formulation Sativex® (Moreno‐Martet et al., 2014). Beneficial effects were also obtained with synthetic cannabinoids such as WIN55,212‐2 (Bilsland et al., 2006), the selective cannabinoid receptor type‐2 (CB2 receptor) agonist AM1241 (Kim et al., 2006; Shoemaker et al., 2007) and fatty acid amide hydrolase (FAAH) inhibitors (Bilsland et al., 2006). In this last case, the efficacy of the cannabinoid treatments is likely related to the changes observed in those elements of the endocannabinoid signalling that serve as pharmacological targets for the cannabinoids being investigated. This is the case for the CB2 receptor, which has been found to be overexpressed in microglial cells in ALS patients (Yiangou et al., 2006), or the FAAH enzyme, whose inhibition explains the elevated levels of endocannabinoids found in the spinal cord in ALS (Witting et al., 2004; Bilsland et al., 2006).
So far, all studies that have investigated the neuroprotective potential of cannabinoids in ALS have been conducted in the mutant SOD1 mouse model (Ripps et al., 1995), which was the only experimental model available for this type of study for several years following the discovery of SOD1, the first gene identified to be associated with the disease (Rosen et al., 1993). The recent discovery of new ALS‐related genes has allowed the development of new experimental models, for example, transgenic mouse models for TDP‐43 (Wegorzewska et al., 2009). We recently published the first study that investigated the involvement of cannabinoids in the TDP‐43 (A315T) transgenic mouse model, in which we recorded the damage to motor neurons in the spinal cord in association with possible changes in endocannabinoid ligands, receptors and enzymes (Espejo‐Porras et al., 2015). We used 70–80 day‐old animals to represent an early symptomatic stage, and those aged 100–110 days for the post‐symptomatic stage. Unfortunately, it was not possible to work with older animals, as these TDP‐43 (A315T) transgenic mice develop an intestinal obstruction after the appearance of ALS symptoms, which causes premature death at the age of 120 days after birth (Guo et al., 2012; Esmaeili et al., 2013). In our study, the most important observation was that CB2 receptors are up‐regulated in reactive microglial cells located in the spinal ventral horn of TDP‐43 (A315T) transgenic mice (Espejo‐Porras et al., 2015). This response has been currently found to occur in most neurodegenerative disorders (reviewed in Fernández‐Ruiz et al., 2007; 2015), including ALS, for example, in postmortem ALS tissues (Yiangou et al., 2006). Furthermore, an up‐regulation of CB2 receptors has also been found in reactive astrocytes in mutant SOD1 mice (Espejo‐Porras et al., unpublished results) and in canine ALS, so‐called degenerative myelopathy, which is also dependent on mutant SOD1 (Fernández‐Trapero et al., 2017). This up‐regulation of glial CB2 receptors appears to be of great interest from a pharmacological point of view. Therefore, our objective in this follow‐up study conducted in TDP‐43 (A315T) transgenic mice was to investigate the role played by the activation of CB2 receptors identified in microglial cells in these mice (Espejo‐Porras et al., 2015). To this end, we used different treatments to modulate CB2 receptor function, either alone or in combination with CB1 receptors. We also extended our studies to identify additional cellular substrates expressing CB2 receptors in this experimental ALS model.
Methods
Animals, treatments and sampling
All experiments were conducted with Prp‐hTDP‐43 (A315T) transgenic and non‐transgenic littermate sibling mice bred in our animal facilities from initial breeders purchased from Jackson Laboratories (Bar Harbor, ME, USA) and subjected to genotyping for identifying the presence or absence of the transgene containing the TDP‐43 (A315T) mutation (Wegorzewska et al., 2009). All animals were housed in a room with controlled lighting (08:00–20:00 lights on) and temperature (22 ± 1°C) with free access to standard food and water. All experiments were conducted according to local and European rules (directive 2010/63/EU). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). They were approved by the ethical committees of our university and the regulatory institution (ref. PROEX 059/16). In addition, experiments were conducted in compliance with the journal guidelines concerning design and analysis. Then, once genotyped, wild‐type and transgenic mice were identified by numbered ear marks, prior to the start of the different experiments, they were randomly allocated to the different treatment groups. For data collection, in all behavioural and histological analyses, researchers were blinded to the animal treatment, whereas for the biochemical analyses, due to the form in which the data are collected, blinding was not considered to be necessary.
In the first experiment, we treated non‐transgenic and Prp‐hTDP‐43 (A315T) transgenic male mice with the non‐selective agonist WIN55,212‐2 (Sigma‐Aldrich, Madrid, Spain) at the dose of 5 mg·kg−1 or vehicle (Tween 80‐saline), both administered i.p. The treatment was initiated when animals were 65 days old and prolonged daily up to the age of 90 days. During this period, we recorded the animals’ weight gain and their response in the rotarod test to detect muscle weakness. In the second experiment, Prp‐hTDP‐43 (A315T) transgenic male mice and their corresponding wild‐type controls were treated again with the non‐selective agonist WIN55,212‐2 at the dose of 5 mg·kg−1, in the presence or absence of selective antagonists for the two cannabinoid receptor types. We used rimonabant (provided by Sanofi‐Aventis, Montpellier, France) at the dose of 5 mg·kg−1 to selectively block CB1 receptors and AM630 (Tocris Bioscience, Bristol, UK) at the dose of 5 mg·kg−1 to selectively block CB2 receptors. These doses were selected from previous experiments conducted with the same antagonists and also against WIN55,212‐2 in the context of anti‐inflammatory effects in neurodegenerative disorders (de Lago et al., 2012). All compounds were prepared in Tween 80‐saline as vehicle and were administered i.p. The treatment was initiated again when animals were 65 days old and prolonged daily up to the age of 90 days. During this period, we also recorded the animals’ weight gain (data not shown) and their response in the rotarod test. Lastly, in the third experiment, we treated non‐transgenic and Prp‐hTDP‐43 (A315T) transgenic male mice with the selective CB2 agonist HU‐308 (Tocris Bioscience, Bristol, UK) at the dose of 5 mg·kg−1 or vehicle (Tween 80‐saline), both administered i.p. Again, the treatment was initiated when animals were 65 days old and prolonged daily up to the age of 90 days. During this period, we recorded the animals’ weight gain and their response in the rotarod test.
In the three pharmacological experiments, immediately after the last behavioural recording, animals were killed by rapid decapitation and their spinal cords were dissected and removed. The spinal samples (lumbar area) to be used for histology were fixed for 1 day at 4°C in fresh 4% paraformaldehyde prepared in 0.1 M PBS, pH 7.4. Samples were then cryoprotected by immersion in a 30% sucrose solution for a further day, and finally stored at −80°C for Nissl staining and immunohistochemical analysis. The spinal samples (also lumbar area) to be used for biochemistry were collected and maintained for 24 h at 4°C in RNAlater (Thermo‐Fisher Scientific, Waltham, MA, USA) before being frozen and stored at −80°C for analysis of different markers by qPCR and for glutamate content by using a commercial kit. In the three experiments, we examined the intestinal tract of each animal to exclude those cases in which a premature intestinal lesion might have influenced our results. However, we did not find any intestinal lesions in animals younger than 110 days after birth, according to the data published by Esmaeili et al. (2013). In all experiments, at least 10 animals were used per experimental group, except a few analyses included fewer numbers, but always greater than five animals per group (indicated for each parameter in the legends to figures).
Behavioural recording
TDP‐43 (A315T) transgenic and wild‐type mice were evaluated for possible motor weakness using the rotarod test, using a LE8200 device (Panlab, Barcelona, Spain). After a period of acclimation and training (first session: 0 r.p.m. for 30 s; second and third sessions: 4 r.p.m. for 60 s, with periods of 10 min between sessions) conducted 30 min before, animals were tested with an acceleration from 4 to 40 r.p.m. over a period of 300 s. Mice were tested for three consecutive trials with a rest period of approximately 15 min between trials and the mean of the three trials was calculated.
Real time qRT‐PCR analysis
Total RNA was extracted from tissues using Trizol (Life Technologies, Alcobendas, Spain) and purified using PureLink® RNA Mini Kit RNATidy reagent (Life Technologies, Alcobendas, Spain). The total amount of RNA extracted was quantified by spectrometry at 260 nm, and its purity was evaluated by the ratio between the absorbance values at 260 and 280 nm. Its integrity was confirmed in agarose gels. To prevent genomic DNA contamination, DNA was removed and single‐stranded cDNA was synthesized from 1 μg (or less) of total RNA using a commercial kit (Rneasy Mini Quantitect Reverse Transcription, Qiagen, Izasa, Madrid, Spain). The reaction mixture was kept frozen at −20°C until enzymatic amplification. Quantitative real‐time PCR assays were performed using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) to quantify mRNA levels for the CB2 receptor (Mm00438286_m1), PPAR‐γ (Mm01184322_m1), TNF‐α (Mm99999068_m1), IL‐1β (Mm00434228_m1), glutamate transporter type‐1 (GLT‐1) (Mm00441457_m1) and glutamate‐aspartate transporter (GLAST) (Mm00600697_m1) using GAPDH expression (Mm99999915_g1) as an endogenous control gene for normalization. The PCR assay was performed using the 7300 Fast Real‐Time PCR System (Applied Biosystems, Foster City, CA, USA), and the threshold cycle (Ct) was calculated by the instrument's software. Expression levels were calculated using the 2−ΔΔCt method, but, for presentation, data were transformed to the percentage over the mean obtained in the wild‐type group for each parameter.
Analysis of glutamate tissue contents
Spinal cords were homogenized in 150 mM potassium phosphate buffer, pH 6.8 and then used for the analysis of glutamate contents using a commercial Glutamate Assay Kit (#K629‐100, BioVision, Mountain View, CA, USA), following the instructions provided by the manufacturer. Values were calculated as μg·mg−1 of protein (measured by the Lowry method).
Histological procedures
Tissue slicing
Fixed spinal cords were sliced with a cryostat at the lumbar level (L4‐L6) to obtain coronal sections (20 μm thick) that were collected on gelatin‐coated slides. These sections were then used for the Nissl‐staining and immunofluorescence.
Nissl staining
Slices were used for Nissl staining using cresyl violet, as previously described (Alvarez et al., 2008), which permitted us to determine the effects of particular treatments on cell number. A Leica DMRB microscope (Leica, Wetzlar, Germany) and a DFC300Fx camera (Leica) were used to study and photograph the tissue respectively. To count the number of Nissl‐stained motor neurons (>400 μm2) in the ventral horn, high‐resolution photomicrographs were taken with a 10× objective under the same conditions of light, brightness and contrast. Counting was carried out with the ImageJ software (U.S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2012). Four images from at least three sections per animal were analysed to establish the mean of all animals studied in each group.
Immunofluorescence
Slices were used for detection and quantification of glial fibrillary acidic protein (GFAP) or Iba‐1 immunofluorescence or for double‐labelling analyses with the CB2 receptor. After preincubation for 1 h with Tris‐buffered saline with 1% Triton X‐100 (pH 7.5), sections were sequentially incubated overnight at 4°C with a polyclonal anti‐Iba‐1 (1:500; Wako Chemicals, Richmond, VI, USA) or polyclonal anti‐GFAP (1:200; Dako Cytomation, Glostrup, Denmark), followed by washing in Tris‐buffered saline and incubation (at 37°C for 2 h) with an anti‐rabbit secondary antibody made in donkey (1:200; Biolegend, San Diego, CA, USA), conjugated with Alexa 488 (Invitrogen, Carlsbad, CA, USA) rendering green fluorescence for anti‐Iba‐1, or conjugated with Alexa 546 (Invitrogen, Carlsbad, CA, USA) rendering red fluorescence for anti‐GFAP (except in the double‐labelling analysis and the immunostaining for exploring the activated state of GFAP‐positive cells, which were carried out with green fluorescence as for Iba‐1). For quantification, high‐resolution digital microphotographs were taken with the 10× objective under the same conditions of light and brightness/contrast. They were used to measure the mean density of labelling in a selected area, using the ImageJ software (U.S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2012). All data were expressed in arbitrary units. For quantification of the glial activation state, we classified the GFAP‐ or Iba‐1‐positive cells as resting or activated based on morphological criteria published previously (Guida et al., 2015) or by calculating the ratio between cell body area and cell process length following the procedure published by Ceprián et al. (2017). Both procedures led to equivalent results. For double‐labelling analyses, sections were then washed again and incubated overnight at 4°C with a polyclonal anti‐CB2 receptor (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA). This was followed by washing in Tris‐buffered saline and a further incubation (at room temperature for 2 h) with a biotin‐conjugated anti‐goat (1:200; Vector Laboratories, Burlingame, CA, USA) secondary antibody, followed by another wash and an incubation (at 37°C for 2 h) with red streptavidin (Vector Laboratories, Burlingame, CA, USA) rendering red fluorescence for CB2 receptor. Sections were counter‐stained with the nuclear stain TOPRO‐3‐iodide (Molecular Probes, Eugene, OR, USA) to visualize cell nuclei. A SP5 Leica confocal microscopy was used for slide observation and photography.
Statistics
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Briefly, data were assessed using one‐way or two‐way (with repeated measures) ANOVA followed by the Student–Newman–Keuls test or the Bonferroni test, as required. A P value lower than 0.05 was used as the limit for statistical significance. The sample sizes in the different experimental groups were always ≥5.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, in the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Results
Treatment with WIN55,212‐2 in TDP‐43 (A315T) transgenic mice
Our first experiment consisted of a chronic treatment of TDP‐43 (A315T) transgenic mice (from the age of 65 days up to 90 days) with the non‐selective cannabinoid agonist WIN55,212‐2 or vehicle, using wild‐type animals also treated with vehicle as controls. We found a progressive weight loss in TDP‐43 (A315T) transgenic mice that reached statistical significance compared with wild‐type animals from day 80 (Figure 1A) and this was partially attenuated by the treatment with WIN55,212‐2, although without reaching statistically significant differences compared with TDP‐43 (A315T) transgenic mice treated with vehicle (Figure 1A). A similar pattern was found for the rotarod response with TDP‐43 (A315T) transgenic mice, which showed a deteriorated response from week 10 that worsened at week 13, and the treatment with WIN55,212‐2 improved the response, but no statistically significant differences (due to high variation in this group) were reached when compared with TDP‐43 (A315T) transgenic mice treated with vehicle (Figure 1B). This was paralleled by a certain trend towards a recovery after the treatment with WIN55,212‐2 of the number of Nissl‐stained motor neurons measured in the spinal ventral horn of TDP‐43 (A315T) transgenic mice (Figure 1C, D).
Figure 1.
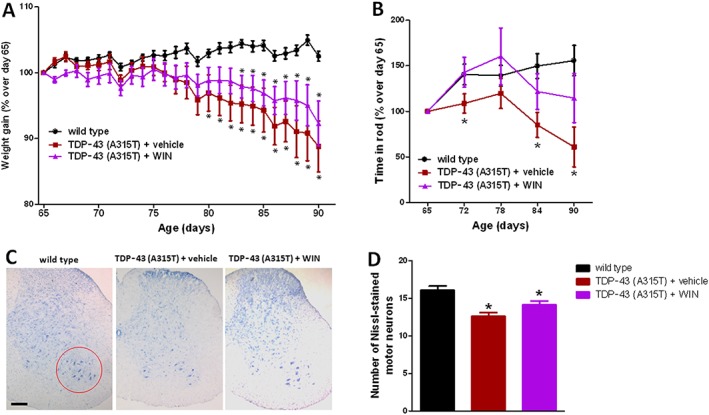
Animal weight (panel A) and rotarod performance (panel B) of TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with WIN55,212‐2 (5 mg·kg−1) or vehicle from day 65 up to day 90 after birth. Quantification of Nissl‐stained motor neurons (panel D), including representative images, in which the area analysed is marked by a circle (panel C; scale bar = 300 μm), in the lumbar ventral horn of the spinal cord in the three experimental groups once animals were killed 24 h after the last injection. In all cases, values are means ± SEM of 12 animals per group. Data were assessed by one‐way (Nissl staining) and repeated measures two‐way (weight and rotarod) ANOVA followed by the Bonferroni test (*P < 0.05 vs. wild type).
We also measured the levels of some inflammation‐related factors by qRT‐PCR in the lumbar spinal cord of TDP‐43 (A315T) transgenic mice and found the CB2 receptor, IL‐1β and TNF‐α levels were all elevated in these mice (Figure 2), but not PPAR‐γ (Figure 2). None of these changes was reversed by the treatment with WIN55,212‐2, rather the elevation in CB2 receptors and, to a lesser extent, in TNF‐α were enhanced by the treatment with this cannabinoid (Figure 2). The contents of glutamate and the expression of its glial transporters GLT‐1 and GLAST were also analysed in the spinal cord, but no changes were found in TDP‐43 (A315T) transgenic mice compared with wild‐type, or after the treatment with WIN55,212‐2 (Figure 2).
Figure 2.
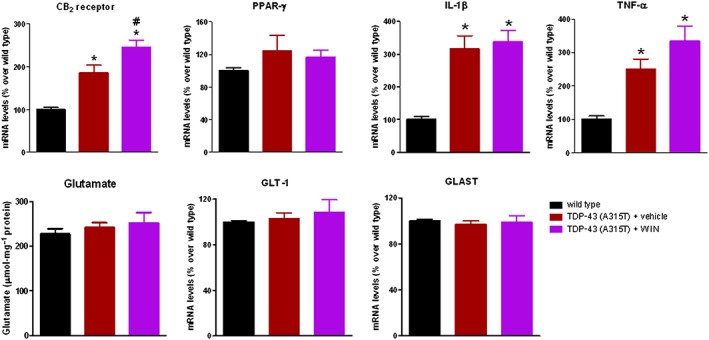
Gene expression for CB2 receptor, PPAR‐γ, IL‐1β, TNF‐α, GLT‐1 and GLAST glutamate transporters measured by qRT‐PCR, and glutamate contents measured with a commercial kit, in the lumbar spinal cord of male TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with WIN55,212‐2 (5 mg·kg−1) or vehicle from day 65 up to day 90 after birth. Values correspond to the percentage over the mean obtained in the wild‐type group for each parameter and are expressed as means ± SEM of 12 animals per group, except for glutamate contents and GLT‐1 and GLAST gene expression that corresponded to five animals per group. Data were assessed by one‐way ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle].
GFAP immunoreactivity was also found to be elevated, reflecting reactive astrogliosis, in the dorsal and ventral horns of the spinal cord of TDP‐43 (A315T) transgenic mice (Figure 3A, B), as previously found in postmortem tissues from ALS patients (Schiffer et al., 1996). The treatment with WIN55,212‐2 attenuated this increase in GFAP immunoreactivity, particularly in the dorsal horn (Figure 3A, B). An elevated Iba‐1 immunostaining, reflecting reactive microgliosis, was also found in numerous grey and white matter areas of the lumbar spinal cord in TDP‐43 (A315T) transgenic mice (Figure 3C–F). In our previous study (Espejo‐Porras et al., 2015), we concentrated on studying changes in the ventral horn, but here, we also investigated changes in the dorsal (ascending somatosensory tracts) and ventral (descending motor tracts) white matter areas. We found elevated Iba‐1 immunostaining in both spinal areas of TDP‐43 (A315T) transgenic mice: dorsal (Figure 3C, E) and ventral white matter (Figure 3D, E). However, the treatment with WIN55,212‐2 was not able to reduce these responses, it even elevated the Iba‐1 immunoreactivity in the case of the ventral white matter (Figure 3C–E). By contrast, the elevated Iba‐1 immunoreactivity detected in the ventral horn of TDP‐43 (A315T) transgenic mice, already found in our previous study (Espejo‐Porras et al., 2015), was attenuated (loss of statistical significance compared with wild‐type animals) after WIN55,212‐2 treatment (Figure 3F).
Figure 3.
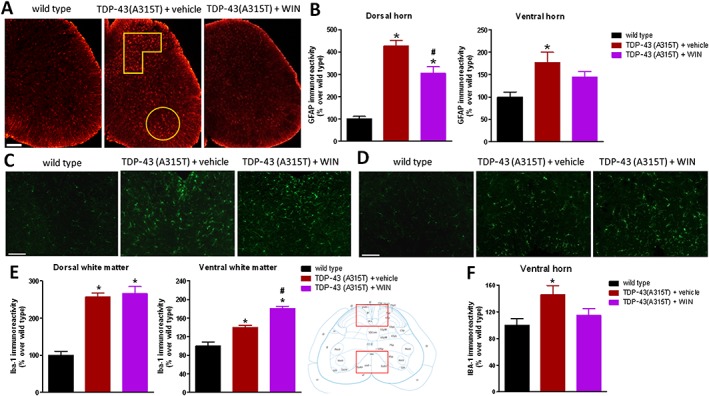
GFAP (panels A and B) and Iba‐1 (panels C–F) immunofluorescence analysis, including representative images (scale bar = 300 μm for GFAP and 200 μm for Iba‐1), in the dorsal and ventral horns for GFAP (analysed areas are marked in the images), and in the dorsal and ventral white matter (indicated in the graphical schemes with representative images in panels C and D) and in the ventral horn for Iba‐1 of the lumbar spinal cord of male TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with WIN55,212‐2 (5 mg·kg−1) or vehicle from day 65 up to day 90 after birth. Values are means ± SEM of 12 animals per group. Data were assessed by one‐way ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle].
Blockade of WIN55,212‐2 effect with selective CB1 and CB2 antagonists
Given that WIN55,212‐2 had effects on rotarod performance, number of Nissl‐stained spinal motor neurons, and GFAP and Iba‐1 immunostaining, we conducted a second experiment aimed at identifying whether these effects of WIN55,212‐2, which is a non‐selective CB receptor agonist, were mediated by the activation of CB1 and/or CB2 receptors, using selective antagonists for these receptors, rimonabant and AM630, respectively, administered at active doses, according to previous studies (de Lago et al., 2012), in combination with WIN55,212‐2 and following the same protocol as in the first experiment. Our data demonstrated a modest reversal (simply a loss of statistical significance compared with TDP‐43 (A315T) transgenic mice treated with vehicle) of the positive effects of WIN55,212‐2 on the deteriorated rotarod performance shown by TDP‐43 (A315T) transgenic mice, with rimonabant and, to a greater extent, with AM630 (Figure 4A), but both effects were just modest. The involvement of CB2 receptors was most clear in the case of the positive effects of WIN55,212‐2 on the number of Nissl‐stained motor neurons in the spinal ventral horn, which were diminished in TDP‐43 (A315T) transgenic mice treated with WIN55,212‐2 and AM630 down to the same values as in TDP‐43 (A315T) transgenic mice treated with vehicle (Figure 4B, C), an effect that did not happen with mice treated with WIN55,212‐2 combined with rimonabant (Figure 4B, C).
Figure 4.
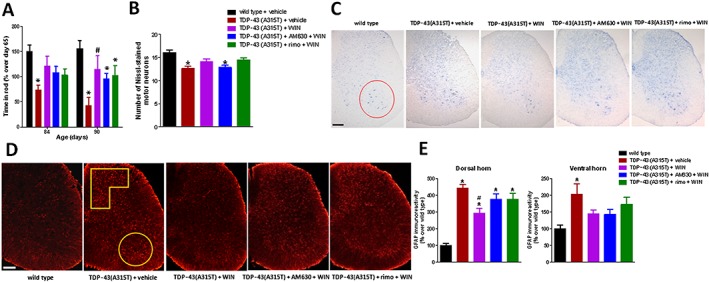
Rotarod performance (panel A) of TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with WIN55,212‐2 (5 mg·kg−1), in the absence or presence of the selective CB1 receptor antagonist rimonabant (5 mg·kg−1) or the selective CB2 receptor antagonist AM630 (5 mg·kg−1), or vehicle, from day 65 up to day 90 after birth. Quantification of Nissl‐stained motor neurons (panel B), including representative images, in which the area analysed in the ventral horn is marked by a circle (panel C; scale bar = 300 μm), and GFAP immunofluorescence analysis (panel E), including representative images (panel D; scale bar = 300 μm), in the dorsal and ventral horns (analysed areas are marked in the images), in the lumbar spinal cord in the five experimental groups once animals were killed 24 h after the last injection. In all cases, values are means ± SEM of 10 animals per group. Data were assessed by one‐way (Nissl staining and GFAP immunofluorescence) and repeated measures two‐way (rotarod) ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild‐type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle].
CB1 receptors, however, appeared to be involved in the reduction in reactive astrogliosis in the spinal dorsal horn mediated by WIN55,212‐2, as the combination of WIN55,212‐2 and rimonabant elevated GFAP immunoreactivity compared with the animals treated with WIN55,212‐2 alone (simply a loss of statistical significance compared with TDP‐43 (A315T) transgenic mice treated with vehicle), and the same happened when CB2 receptors were blocked with AM630 (Figure 4D, E). By contrast, the data in the ventral horn were less clear, with no reversal of WIN55,212‐2's effects after the blockade of CB2 receptors with AM630 and only a trend after the blockade of CB1 receptors with rimonabant (Figure 4D, E).
As regards Iba‐1 immunostaining in the ventral horn, again we found an increase in TDP‐43 (A315T) transgenic mice that was apparently diminished by the treatment with WIN55,212‐2 (loss of statistical significance compared with wild‐type animals; Figure 5A, B). This response was reversed by the blockade of CB1 receptors with rimonabant and, in particular, of CB2 receptors with AM630, which even elevated Iba‐1 levels up to values higher than those seen in TDP‐43 (A315T) transgenic mice treated with vehicle (Figure 5A, B). Lastly, Iba‐1 immunostaining in the spinal white matter areas showed again that WIN55,212‐2 was unable to reverse the elevated Iba‐1 immunoreactivity found in TDP‐43 (A315T) transgenic mice in dorsal white matter (Figure 6A, B), it even elevated the values found in the ventral white matter (Figure 6C, D). The blockade of CB1 receptors with rimonabant did not alter these levels, which remained similar to animals treated with WIN55,212‐2 alone (Figure 6A–D), whereas the same happened with the blockade of CB2 receptors with AM630, but, in this case, the values of Iba‐1 immunoreactivity were even higher when compared with animals treated with WIN55,212‐2 alone (Figure 6A–D), indicating an exacerbated microglia activation after this treatment.
Figure 5.

Iba‐1 immunofluorescence analysis (panel A), including representative images (panel B; scale bar = 200 μm), in the ventral horn (indicated in the graphical scheme) of the lumbar spinal cord of male TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with WIN55,212‐2 (5 mg·kg−1), in the absence or presence of the selective CB1 receptor antagonist rimonabant (5 mg·kg−1) or the selective CB2 receptor antagonist AM630 (5 mg·kg−1), or vehicle, from day 65 up to day 90 after birth. Values are means ± SEM of 10 animals per group. Data were assessed by one‐way ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild type; @P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with WIN55,212‐2 and AM630].
Figure 6.
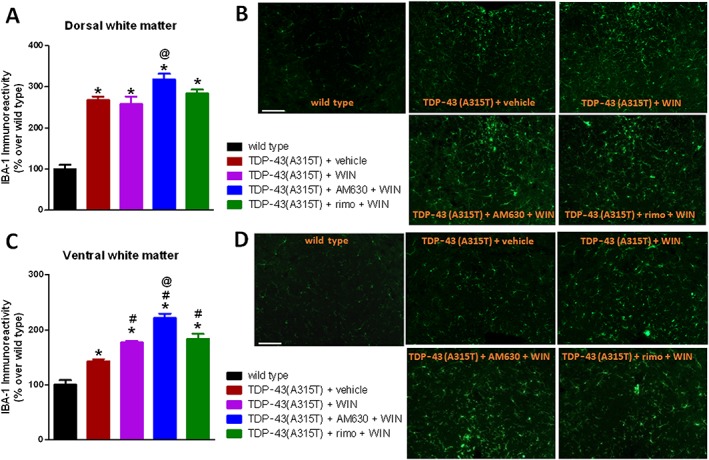
Iba‐1 immunofluorescence analysis (panels A and C), including representative images (panels B and D; scale bar = 200 μm), in the dorsal and ventral white matter areas (indicated in the graphical schemes in Figure 3) of the lumbar spinal cord of male TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with WIN55,212‐2 (5 mg·kg−1), in the absence or presence of the selective CB1 receptor antagonist rimonabant (5 mg·kg−1) or the selective CB2 receptor antagonist AM630 (5 mg·kg−1), or vehicle, from day 65 up to day 90 after birth. Values are means ± SEM of 10 animals per group. Data were assessed by one‐way ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle; @P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with WIN55,212‐2].
Treatment with HU‐308 in TDP‐43 (A315T) transgenic mice
In the third experiment, we wanted to further explore more specifically the role of CB2 receptors in TDP‐43 (A315T) transgenic mice by targeting these receptors with the selective agonist HU‐308, following the same protocol as in the previous experiments. We found that the progressive loss of weight observed in TDP‐43 (A315T) transgenic mice was significantly attenuated by the treatment with HU‐308, in particular in the second part of the period analysed (Figure 7A). The same effect was found with regard to the deteriorated rotarod response observed in TDP‐43 (A315T) transgenic mice when these mice were treated with HU‐308, in particular at week 13 (Figure 7B). The treatment with HU‐308 was also able to completely preserve the Nissl‐stained motor neurons in the spinal ventral horn, with values similar to those found in wild‐type mice and much higher than those found in TDP‐43 (A315T) transgenic mice treated with vehicle (Figure 7C, D).
Figure 7.
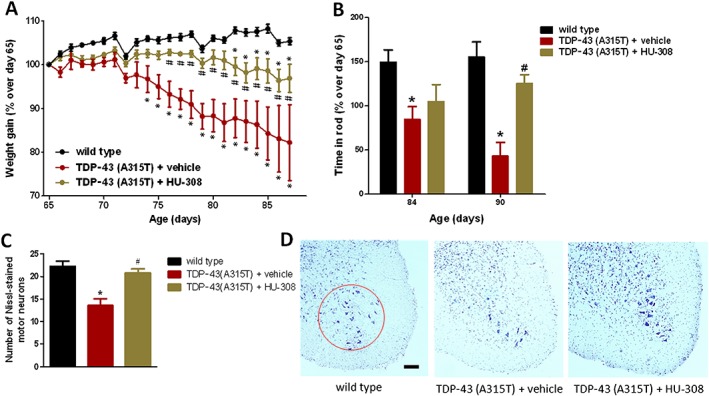
Animal weight (panel A) and rotarod performance (panel B) of TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with HU‐308 (5 mg·kg−1) or vehicle, from day 65 up to day 90 after birth. Quantification of Nissl‐stained motor neurons (panel C), including representative images, in which the area analysed is marked by a circle (panel D; scale bar = 300 μm), in the ventral horn of the lumbar spinal cord in the three experimental groups once animals were killed 24 h after the last injection. In all cases, values are means ± SEM of 10 animals per group. Data were assessed by one‐way (Nissl staining) and repeated measures two‐way (weight and rotarod) ANOVA followed by the Bonferroni test (*P < 0.05 vs. wild type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle].
Reactive astrogliosis, labelled with GFAP immunostaining, was also attenuated in the dorsal and, to a lesser extent, in the ventral horns by the treatment with HU‐308 (Figure 8A, B). This effect was particularly evident with regard to the number of GFAP‐positive cells showing morphological characteristics to be activated, something that was determined using the procedure described by Guida et al. (2015), which, in general, consisted of classifying the cells according to their size (higher cell volume in activated cells) and shape (reduced number and length of processes in activated cells). Using this procedure, the total number of GFAP‐positive cells did not vary among wild type, TDP‐43 (A315T) transgenic treated with vehicle, or treated with HU‐308 (Figure 8C), but the changes were significant when only the number of activated GFAP‐positive cells was quantified, with a significant elevation in TDP‐43 (A315T) transgenic mice (cells with great volume and reduced processes; see Figure 8D) that returned close to the levels of wild‐type animals after the treatment with HU‐308 (Figure 8C).
Figure 8.
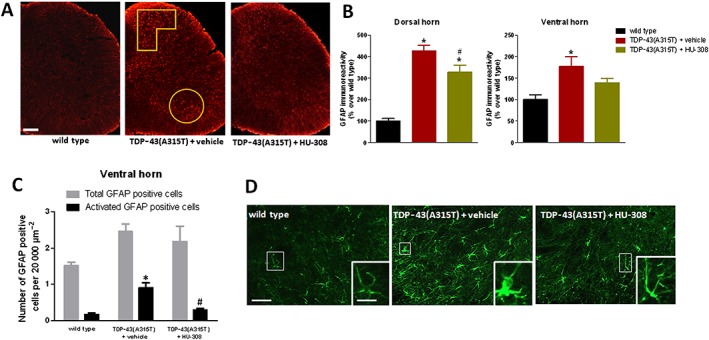
GFAP immunofluorescence analysis (panel B), including representative images (panel A; scale bar = 300 μm), in the dorsal and ventral horns (analysed areas are marked in the images) of the lumbar spinal cord of male TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with HU‐308 (5 mg·kg−1) or vehicle from day 65 up to day 90 after birth. Number of total or activated GFAP positive cells (panel C) identified by their morphological characteristics [see details in the text; representative images are included in panel D; scale bar = 50 and 20 μm (enlargement)] in the ventral horn of the lumbar spinal cord in the three experimental groups. Values are means ± SEM of 10 animals per group. Data were assessed by one‐way ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle].
HU‐308 also reduced the elevated Iba‐1 immunostaining found in the ventral horn of TDP‐43 (A315T) transgenic mice to the levels found in wild‐type animals (Figure 9A, B). As occurred with GFAP immunostaining, this reduction was particularly evident when the number of activated Iba‐1 positive cells was quantified; these were identified by measuring the ratio between the cell area and the length of cell processes, following the method described by Ceprián et al. (2017). Using this method, the number of activated Iba‐1 positive cells was elevated in TDP‐43 (A315T) transgenic mice, but this elevation returned to levels of wild‐type animals when TDP‐43 (A315T) transgenic mice were treated with HU‐308 (Figure 9C, E). The same pattern was seen when the number of activated Iba‐1 positive cells was measured by using the procedure described for GFAP immunostaining (Figure 9D, E). By contrast, the treatment with HU‐308 did not affect, as happened with WIN55,212‐2, the elevated immunoreactivity for Iba‐1 found in the dorsal white matter of TDP‐43 (A315T) transgenic mice (Figure 10A, B), and this immunoreactivity was even exacerbated by HU‐308 in the ventral white matter (Figure 10C, D), suggesting the possible existence of different roles for the CB2 receptors located in the different microglial cell subpopulations labelled in this study.
Figure 9.
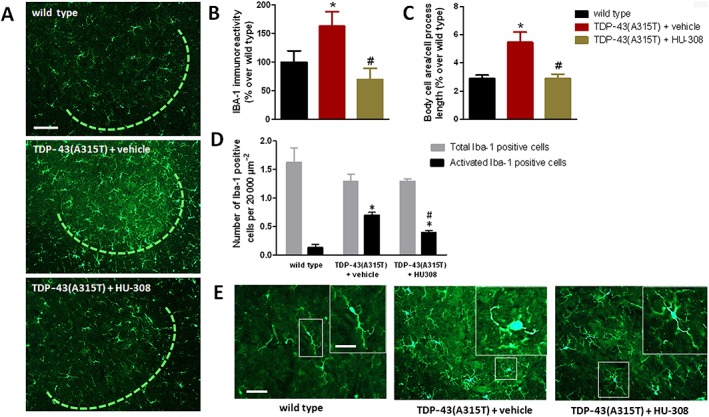
Iba‐1 immunofluorescence analysis (panel B), including representative images, in which the area analysed is marked by a circle (panel A; scale bar = 100 μm), in the ventral horn of the lumbar spinal cord of male TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with HU‐308 (5 mg·kg−1) or vehicle from day 65 up to day 90 after birth. Values for the ratio between body cell area and cell process length (panel C) and number of total or activated Iba‐1 positive cells (panel D) identified by their morphological characteristics [see details in the text; representative images are included in panel E; scale bar = 50 and 20 μm (enlargement)], in the ventral horn of the lumbar spinal cord in the three experimental groups. Values are means ± SEM of eight animals per group. Data were assessed by one‐way ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle].
Figure 10.
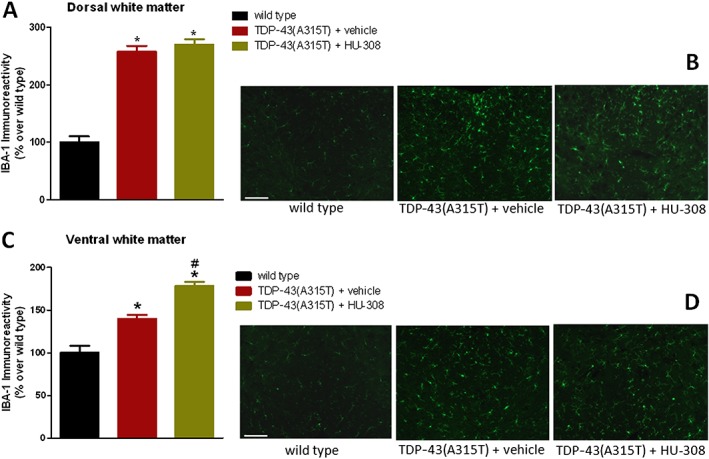
Iba‐1 immunofluorescence analysis (panels A and C), including representative images (panels B and D; scale bar = 200 μm), in the dorsal and ventral white matter areas (indicated in the graphical schemes in Figure 3) of the lumbar spinal cord of male TDP‐43 (A315T) transgenic and wild‐type male mice after the daily treatment with HU‐308 (5 mg·kg−1) or vehicle from day 65 up to day 90 after birth. Values are means ± SEM of 10 animals per group. Data were assessed by one‐way ANOVA followed by the Bonferroni test [*P < 0.05 vs. wild type; #P < 0.05 vs. TDP‐43 (A315T) transgenic mice treated with vehicle].
Double‐labelling analysis to identify cell substrates for the CB2 receptor in TDP‐43 (A315T) transgenic mice
The above pharmacological experiments demonstrated that the most active treatment corresponded to the administration of the selective CB2 receptor agonist. In addition, in our previously published article (Espejo‐Porras et al., 2015), we identified CB2 receptor immunofluorescence only in reactive microglial cells located in the ventral horn of TDP‐43 (A315T) transgenic mice, but we also demonstrated the presence of CB2 receptor‐positive cells in the ventral horn of these animals, whose phenotype was not elucidated in the study (Espejo‐Porras et al., 2015). Therefore, our next experiment consisted of conducting double‐staining analyses for the CB2 receptor and Iba‐1 or GFAP in different areas of the spinal cord altered in TDP‐43 (A315T) transgenic mice. Our analyses indicated that the presence of this receptor was increased in the periphery of astrocytes that were completely labelled with GFAP in the spinal grey matter of TDP‐43 (A315T) transgenic mice, an effect found only marginally in wild‐type mice (Figure 11A). The different subcellular location of both proteins may be because the CB2 receptor is a membrane protein, whereas GFAP is located in the cytosol (Figure 11A). In additional double‐labelling analyses, we also found CB2 receptors located in Iba‐1 positive cells (microglial cells) in the spinal white matter, both in the dorsal and ventral areas, of TDP‐43 (A315T) transgenic mice compared with wild‐type animals in which the signal was residual (Figure 11B). However, the activation of these receptors with HU‐308 did not appear to reduce the microglial reactivity detected with Iba‐1 immunostaining (Figure 10A–D), being that, as indicated previously, the CB2 receptor is located in microglial cells in spinal grey matter areas (Espejo‐Porras et al., 2015) the most probable cellular target for the benefits found after selective activation of these receptors (Figure 9A–E).
Figure 11.
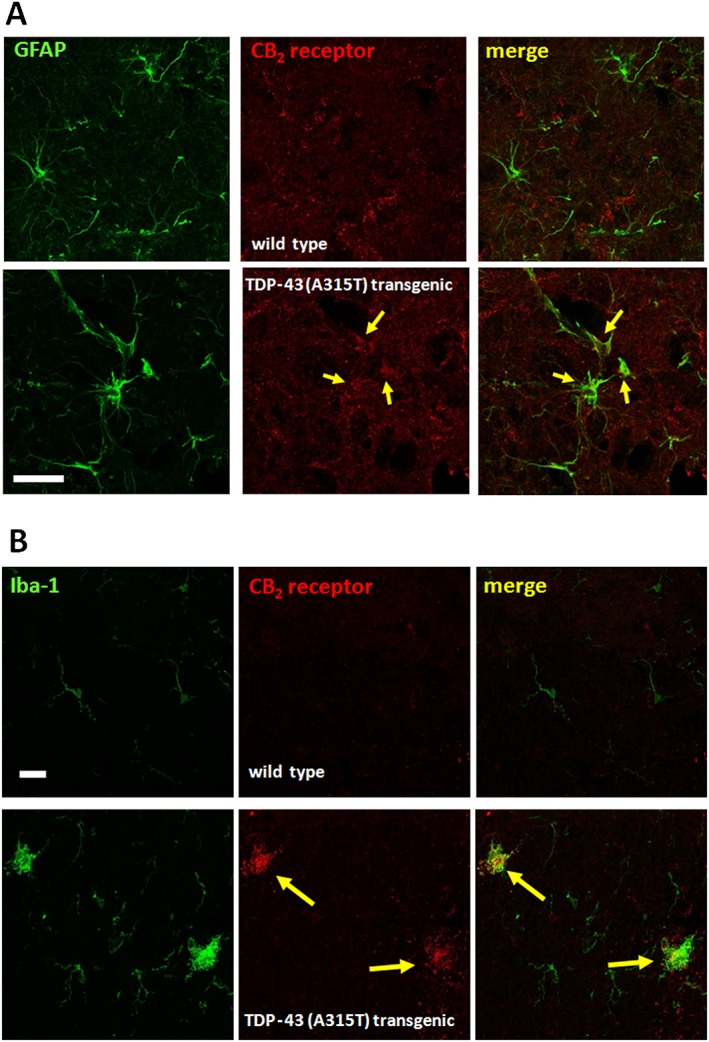
Representative double‐immunofluorescence images for CB2 receptors and GFAP (dorsal horn; see panel A) or Iba‐1 (white matter; see panel B) in the spinal cord of TDP‐43 (A315T) transgenic and wild‐type male mice at 90 days after birth. Immunostainings were repeated in five animals per group. Cells positive for GFAP or Iba‐1 and CB2 receptors are indicated with arrows (scale bar = 25 μm).
Discussion
Our study is the first one to investigate whether cannabinoid treatments have potential as neuroprotective agents in an experimental model of ALS other than the classic mutant SOD1 mice. In our previous study (Espejo‐Porras et al., 2015), we demonstrated the involvement of CB2 receptors in TDP‐43 (A315T) transgenic mice, which became up‐regulated in reactive microglial cells located in the spinal grey matter, so that their manipulation may serve to attenuate the consequences of microglial reactivity and to preserve spinal motor neurons. This has been investigated in this follow‐up study using different pharmacological strategies. Firstly, we used a non‐selective CB1/CB2 receptor agonist, WIN55,212‐2, which showed beneficial effects by improving the deteriorated rotarod response in TDP‐43 (A315T) transgenic mice. We assume that these benefits are not simply the consequence of an acute effect of this cannabinoid agonist increasing motor activity, as the dose of WIN55,212‐2 used is associated with decreased rather than increased motor activity. Conversely, these benefits appear to be more likely related to preservation of spinal motor neurons and the reduction in astroglial and microglial reactivity labelled with GFAP and Iba‐1, respectively, in spinal grey matter areas. It is true that the effects were modest, not particularly elevated, and that they were not exclusively mediated by the activation of CB2 receptors, as a subsequent experiment using WIN55,212‐2 in combination with selective CB1 or CB2 antagonists has proved. However, we also conducted experiments here using a selective CB2 receptor agonist, which does not activate CB1 or other endocannabinoid‐related targets, and, in this case, we obtained a greater improvement in the deteriorated rotarod performance, a complete preservation of spinal motor neurons and a strong attenuation in the activation of astrocytes and microglial cells in the dorsal and ventral horns, in which cell bodies of ascending and descending spinal neurons are located (reviewed in Diaz and Morales, 2016). These effects appear to be genuine and support the therapeutic value of the activation of CB2 receptors in this disease, a finding also observed in other experimental models of ALS (Kim et al., 2006; Shoemaker et al., 2007).
Given the predominant presence of these receptors in glial elements in the spinal cord of TDP‐43 (A315T) transgenic mice, the CB2 receptor‐dependent benefits are likely to be mediated through control of the toxic effects of these cells on neurons, a fact largely investigated in ALS and other neurodegenerative disorders (reviewed in Fernández‐Ruiz et al., 2007, 2015; de Lago et al., 2015). In our previous study (Espejo‐Porras et al., 2015), we demonstrated the presence of CB2 receptors in reactive microglial cells located in the spinal ventral horn, which is the area in which the cell bodies of the damaged spinal motor neurons are located. In the present study, we also detected immunoreactivity for the CB2 receptors in other cells that were not immunolabelled with the Iba‐1 antibody and that were not identified previously (Espejo‐Porras et al., 2015). We have confirmed now that these cells are positive for GFAP, so they are astrocytes located in the vicinity of spinal motor neurons in the ventral horn, but we found that this colocalization was also evident in the spinal dorsal horn. The dorsal horn is the area in which cell bodies of ascending somatosensory pathways are located (for a recent review, see Diaz and Morales, 2016). They receive innervation directly from the skin, and generate nerve tracts that ascend through the dorsal or the lateral white matter areas to reach the encephalic nuclei. For example, the gracilis or the cunneatus fasciculi, which mediate touch and vibration senses, ascend through the dorsal white matter, whereas the spinothalamic tracts, which are involved in mediating pain and thermal sensitivity, ascend through the lateral white matter (reviewed in Diaz and Morales, 2016). The interest of the dorsal white matter in our study is that we detected elevated Iba‐1 immunoreactivity in this area, as well as in the ventral white matter area, in TDP‐43 (A315T) transgenic mice, and further double‐labelling analyses also conducted in the present study detected CB2 receptors in these microglial cells in both areas. This is an important observation of our present study that adds to the identification of CB2 receptors in the spinal ventral horn of TDP‐43 (A315T) transgenic mice in our previous study (Espejo‐Porras et al., 2015). In the present study, we identified CB2 receptors in microglial cells (labelled with Iba‐1) in TDP‐43 (A315T) transgenic mice located in areas of the spinal cord associated with axonal tracts (spinal white matter), not only in areas associated with neuronal cell bodies (spinal horns). As mentioned above, the dorsal white matter area corresponds to somatosensory tracts ascending (afferent) to the encephalic zones, whereas the ventral white matter is the area of descending (efferent) motor tracts (e.g. anterior corticospinal tracts, reticulospinal tracts; reviewed in Diaz and Morales, 2016). In both cases, microglial cells containing CB2 receptors are recruited to these two white matter areas in TDP‐43 (A315T) transgenic mice. However, by contrast with the case of CB2 receptors located in these animals in astrocytes and microglial cells in the dorsal (only astrocytes) and ventral (both astrocytes and microglial cells) horns, whose activation with HU‐308 (and also with WIN55,212‐2, although to a lesser extent and also involving CB1 receptors) reduced the levels of GFAP or Iba‐1, respectively, the activation of microglial CB2 receptors located in white matter areas did not alter the microglial reactivity detected with Iba‐1 immunostaining, even provoked an elevation in the ventral white matter.
Therefore, these results suggest that glial CB2 receptors up‐regulated in TDP‐43 (A315T) transgenic mice may play different functions depending on the type of glial cells (astrocytes vs. microglial cells) or the spinal area (grey vs. white matters, dorsal vs. ventral zones) in which they are located. This is an interesting hypothesis that requires further research. Such a hypothesis is related to the non‐cell autonomous toxicity exerted by both deteriorated astrocytes and microglial cells on neurons, which is well‐known to contribute to ALS progression (Yamanaka et al., 2008; Ilieva et al., 2009; Haidet‐Phillips et al., 2011; Lee et al., 2016), and to the way by which the activation of glial CB2 receptors may mitigate such toxicity. Based on work conducted in other neurodegenerative disorders, the activation of CB2 receptors in astrocytes may enhance the metabolic support exerted by these cells for neurons, as well as facilitating their key role in glutamate clearance in the vicinity of glutamatergic synapses limiting excitotoxic damage (reviewed in Fernández‐Ruiz et al., 2015; de Lago et al., 2015). In the case of microglial CB2 receptors, their activation may reduce the generation of pro‐inflammatory factors and promote the shift of macrophages from M1 to M2 phenotypes with evident benefits for neuronal survival (reviewed in Fernández‐Ruiz et al., 2015; de Lago et al., 2015).
In summary, our study shows an important role for glial CB2 receptors, together with a certain contribution of CB1 receptors, in limiting the progression of the pathological phenotype in TDP‐43 (A315T) transgenic mice. Such benefits apparently derive from the activation of those CB2 receptors concentrated in astrocytes and reactive microglial cells located in spinal dorsal and ventral horns. However, we also detected the presence of microglial CB2 receptors in association with ascending (dorsal white matter area) and descending (ventral white matter area) tracts, whose function and therapeutic potential in this pathological context remain to be determined.
Author contributions
The study design, coordination and supervision were undertaken by J.F.R. and E.d.L. F.E.P. and L.G.T. worked on pharmacological treatments in TDP‐43 (A315T) transgenic mice and on behavioural analysis. F.E.P., L.G.T. and I.S.G. worked on qPCR analysis. F.E.P. and E.d.L. worked on glutamate analysis. F.E.P., L.G.T. and C.A.R.C. performed the histological procedures. J.F.R. with F.E.P., L.G.T., C.A.R.C. and I.S.G. worked on statistical analysis of the data. J.F.R. wrote the manuscript with the revision and approval of all authors.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work has been supported by grants from CIBERNED (CB06/05/0089), MINECO (SAF2012/39173 and SAF2015‐68580‐C2‐1‐R) and GW Pharmaceuticals Ltd. These agencies had no further role in study design, the collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication. Francisco Espejo‐Porras and Laura García‐Toscano are predoctoral fellows supported by the MINECO (FPI Program), whereas Irene García‐Santos is a predoctoral fellow supported by the iPFIS program (Instituto de Salud Carlos III).
The authors are indebted to Yolanda García‐Movellán for administrative assistance and to Henar Suárez for her support and advice with confocal microscopy.
Espejo‐Porras, F. , García‐Toscano, L. , Rodríguez‐Cueto, C. , Santos‐García, I. , de Lago, E. , and Fernandez‐Ruiz, J. (2019) Targeting glial cannabinoid CB2 receptors to delay the progression of the pathological phenotype in TDP‐43 (A315T) transgenic mice, a model of amyotrophic lateral sclerosis. British Journal of Pharmacology, 176: 1585–1600. 10.1111/bph.14216.
Contributor Information
Eva de Lago, Email: elagofem@med.ucm.es.
Javier Fernandez‐Ruiz, Email: jjfr@med.ucm.es.
References
- Al‐Chalabi A, Hardiman O (2013). The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9: 617–628. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Lafuente H, Rey‐Santano MC, Mielgo VE, Gastiasoro E, Rueda M et al (2008). Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic‐ischemic newborn piglets. Pediatr Res 64: 653–658. [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Dick JR, Pryce G, Petrosino S, Di Marzo V, Baker D et al (2006). Increasing cannabinoid levels by pharmacological and genetic manipulation delay disease progression in SOD1 mice. FASEB J 20: 1003–1005. [DOI] [PubMed] [Google Scholar]
- Bova MP, Kinney GG (2013). Emerging drug targets in amyotrophic lateral sclerosis. Expert Opin Orphan Drugs 1: 5–20. [Google Scholar]
- Buratti E, Baralle FE (2010). The multiple roles of TDP‐43 in pre‐mRNA processing and gene expression regulation. RNA Biol 7: 420–429. [DOI] [PubMed] [Google Scholar]
- Ceprián M, Jiménez‐Sánchez L, Vargas C, Barata L, Hind W, Martínez‐Orgado J (2017). Cannabidiol reduces brain damage and improves functional recovery in a neonatal rat model of arterial ischemic stroke. Neuropharmacology 116: 151–159. [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, Van Langenhove T, van der Zee J, Van Broeckhoven C (2013). Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci 36: 450–459. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lago E, Moreno‐Martet M, Cabranes A, Ramos JA, Fernández‐Ruiz J (2012). Cannabinoids ameliorate disease progression in a model of multiple sclerosis in mice, acting preferentially through CB1 receptor‐mediated anti‐inflammatory effects. Neuropharmacology 62: 2299–2308. [DOI] [PubMed] [Google Scholar]
- de Lago E, Moreno‐Martet M, Espejo‐Porras F, Fernández‐Ruiz J (2015). Endocannabinoids and amyotrophic lateral sclerosis In: Fattore L. (ed). Cannabinoids in Neurologic and Mental Disease. Elsevier: The Netherlands, pp. 99–124. [Google Scholar]
- Diaz E, Morales H (2016). Spinal cord anatomy and clinical syndromes. Semin Ultrasound CT MR 37: 360–371. [DOI] [PubMed] [Google Scholar]
- Esmaeili MA, Panahi M, Yadav S, Hennings L, Kiaei M (2013). Premature death of TDP‐43 (A315T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int J Exp Pathol 94: 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejo‐Porras F, Piscitelli F, Verde R, Ramos JA, Di Marzo V, de Lago E et al (2015). Changes in the endocannabinoid signaling system in CNS structures of TDP‐43 transgenic mice: relevance for a neuroprotective therapy in TDP‐43‐related disorders. J Neuroimmune Pharmacol 10: 233–244. [DOI] [PubMed] [Google Scholar]
- Fernández‐Ruiz J, Romero J, Velasco G, Tolón RM, Ramos JA, Guzmán M (2007). Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol Sci 28: 39–45. [DOI] [PubMed] [Google Scholar]
- Fernández‐Ruiz J, Moro MA, Martínez‐Orgado J (2015). Cannabinoids in neurodegenerative disorders and stroke/brain trauma: from preclinical models to clinical applications. Neurotherapeutics 12: 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Trapero M, Espejo‐Porras F, Rodríguez‐Cueto C, Coates JR, Pérez‐Díaz C, de Lago E et al (2017). Upregulation of CB2 receptors in reactive astrocytes in canine degenerative myelopathy, a disease model of amyotrophic lateral sclerosis. Dis Model Mech 10: 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7: 616–630. [DOI] [PubMed] [Google Scholar]
- Foran E, Trotti D (2009). Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal 11: 1587–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guida F, Lattanzi R, Boccella S, Maftei D, Romano R, Marconi V et al (2015). PC1, a non‐peptide PKR1‐preferring antagonist, reduces pain behavior and spinal neuronal sensitization in neuropathic mice. Pharmacol Res 91: 36–46. [DOI] [PubMed] [Google Scholar]
- Guo Y, Wang Q, Zhang K, An T, Shi P, Li Z et al (2012). HO‐1 induction in motor cortex and intestinal dysfunction in TDP‐43 A315T transgenic mice. Brain Res 1460: 88–95. [DOI] [PubMed] [Google Scholar]
- Habib AA, Mitsumoto H (2011). Emerging drugs for amyotrophic lateral sclerosis. Expert Opin Emerg Drugs 16: 537–558. [DOI] [PubMed] [Google Scholar]
- Haidet‐Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A et al (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29: 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardiman O, van den Berg LH, Kiernan MC (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 7: 639–649. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW (2009). Non‐cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187: 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens J, Van Broeckhoven C (2013). Pathological mechanisms underlying TDP‐43 driven neurodegeneration in FTLD‐ALS spectrum disorders. Hum Mol Genet 22: R77–R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Moore DH, Makriyannis A, Abood ME (2006). AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol 542: 100–105. [DOI] [PubMed] [Google Scholar]
- Lagier‐Tourenne C, Polymenidou M, Cleveland DW (2010). TDP‐43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19: R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hyeon SJ, Im H, Ryu H, Kim Y, Ryu H (2016). Astrocytes and microglia as non‐cell autonomous players in the pathogenesis of ALS. Exp Neurobiol 25: 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Martet M, Espejo‐Porras F, Fernández‐Ruiz J, de Lago E (2014). Changes in endocannabinoid receptors and enzymes in the spinal cord of SOD1(G93A) transgenic mice and evaluation of a Sativex®‐like combination of phytocannabinoids: interest for future therapies in amyotrophic lateral sclerosis. CNS Neurosci Ther 20: 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman C, McAllister SD, Rizvi G, Patel SG, Moore DH, Abood ME (2004). Amyotrophic lateral sclerosis: delayed disease progression in mice by treatment with a cannabinoid. Amyotroph Lateral Scler Other Motor Neuron Disord 5: 33–39. [DOI] [PubMed] [Google Scholar]
- Renton AE, Chiò A, Traynor BJ (2014). State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW (1995). Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 92: 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD (2017). Edaravone: a new drug approved for ALS. Cell 171: 725. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A et al (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59–62. [DOI] [PubMed] [Google Scholar]
- Schiffer D, Cordera S, Cavalla P, Migheli A (1996). Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 139 (Suppl): 27–33. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL (2007). The CB2 cannabinoid agonist AM‐1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem 101: 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao W, Jeong YH, Lin S, Ling J, Price DL, Chiang PM et al (2012). Rodent models of TDP‐43: recent advances. Brain Res 1462: 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W, Van Den Bosch L (2017). Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech 10: 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH (2009). TDP‐43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 106: 18809–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Hong S, Witting A, Möller T, Stella N, Kliot M (2005). Cannabinol delays symptom onset in SOD1 (G93A) transgenic mice without affecting survival. Amyotroph Lateral Scler Other Motor Neuron Disord 6: 182–184. [DOI] [PubMed] [Google Scholar]
- Witting A, Weydt P, Hong S, Kliot M, Möller T, Stella N (2004). Endocannabinoids accumulate in spinal cord of SOD1 G93A transgenic mice. J Neurochem 89: 1555–1557. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori‐Tonou N, Yamashita H, Gutmann DH et al (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11: 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C et al (2006). COX‐2, CB2 and P2X7‐immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol 6: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]