

Abstract
The endocannabinoid system has emerged as an important target for the treatment of many diverse diseases. In addition to the well‐established palliative effects of cannabinoids in cancer therapy, phytocannabinoids, synthetic cannabinoid compounds and inhibitors of endocannabinoid degradation have attracted attention as possible systemic anticancer drugs. Results emerging from preclinical studies suggest cannabinoids elicit effects at different levels of cancer progression, including inhibition of proliferation, neovascularization, invasion and chemoresistance, induction of apoptosis and autophagy as well as enhancement of tumour immune surveillance. Although the clinical use of cannabinoid receptor ligands is limited by their psychoactivity, non‐psychoactive compounds, such as cannabidiol, have gained attention due to preclinically established anticancer properties and a favourable risk‐to‐benefit profile. Thus, cannabinoids may complement the currently used collection of chemotherapeutic agents, as a broadly diversified option for cancer treatment, while counteracting some of their severe side effects.
Linked Articles
This article is part of a themed section on 8th European Workshop on Cannabinoid Research. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.10/issuetoc
Abbreviations
- 2‐AG
2‐arachidonoylglycerol
- AA‐5HT
N‐arachidonoyl serotonin
- AEA
N‐arachidonoylethanolamine, arachidonoylethanolamide, anandamide
- ATF
activating transcription factor
- Bcl‐2
B‐cell lymphoma 2
- CBD
cannabidiol
- Cdc
cell division cycle
- CDK
cyclin‐dependent kinase
- DAGL
DAG lipase
- EMT
epithelial‐to‐mesenchymal transition
- FAAH
fatty acid amide hydrolase
- ICAM‐1
intercellular adhesion molecule 1
- Id
inhibitor of DNA binding
- MAGL
monoacylglycerol lipase
- Met‐F‐AEA
2‐methyl‐2′‐F‐anandamide
- mTOR
mechanistic target of rapamycin
- mTORC1
mechanistic target of rapamycin complex 1
- NAPE‐PLD
N‐acyl‐phosphatidylethanolamine‐selective PLD
- OEA
N‐oleoylethanolamine, oleoylethanolamide
- PEA
N‐palmitoylethanolamine, palmitoylethanolamide
- Rb
retinoblastoma‐associated protein
- Sox
sex‐determining region Y‐box
- TRB
tribbles homologue
- xCT
cystineglutamate transporter
The endocannabinoid system
The components of the ‘classical’ endocannabinoid system have been intensively investigated and reviewed during the last decades. According to its initial definition, the endocannabinoid system comprised the Pertussis toxin sensitive, Gi/o‐coupled, cannabinoid CB1 and CB2 receptors (Matsuda et al., 1990; Munro et al., 1993), as well as their endogenous ligands N‐arachidonoylethanolamine (anandamide, AEA) and 2‐arachidonoylglycerol (2‐AG) (Devane et al., 1992; Mechoulam et al., 1995).
The endocannabinoid system further covers other cannabinoid receptor ligands, such as 2‐arachidonyl glyceryl ether (noladin ether, 2‐AGE) (Hanus et al., 2001), N‐arachidonoyldopamine (NADA) (Bisogno et al., 2000) and O‐arachidonoylethanolamine (virodhamine) (Porter et al., 2002). In addition, the fatty acid amides N‐homo‐y‐linolenylethanolamine and N‐docosatetra‐7,10,13,16‐enoylethanolamine were reported to exhibit cannabinoid receptor binding properties (Hanus et al., 1993).
Synthesizing and degrading enzymes of AEA and 2‐AG comprise a group of proteins that have been investigated intensively following the discovery of endocannabinoids (see Di Marzo, 2009). AEA and other N‐acylethanolamines are endogenously synthesized from membrane phospholipids by the enzyme N‐acylphosphatidylethanolamine‐PLD (NAPE‐PLD) and via alternative biosynthetic pathways. 2‐AG can be generated via phospholipase C or by turnover of DAG via DAG lipase (DAGL) α and β. The intracellular degradation of endocannabinoids is catabolized by the serine hydrolase fatty acid amide hydrolase (FAAH) (Deutsch and Chin, 1993) and, in terms of 2‐AG, by the monoacylglycerol lipase (MAGL) (Blankman et al., 2007).
Following the discovery of the two cannabinoid receptors, the cation channel TRPV1 has been described as an additional receptor, activated by AEA (Zygmunt et al., 1999) and cannabidiol (CBD), a non‐psychoactive phytocannabinoid (Bisogno et al., 2001). Moreover, recent investigations revealed TRPV2 channels to be involved in the modulation of cell fate by CBD (Nabissi et al., 2013, 2015).
In addition to these ionotropic cannabinoid receptors, several GPCRs were deorphanized as cannabinoid‐triggered targets. Thus, GPR55 was antagonized by CBD and activated by a panel of cannabinoid compounds (Ryberg et al., 2007). Accordingly, the synthetic cannabinoid GP55940 was found to activate GPR55, whereas WIN 55,212‐2 did not bind or activate GPR55. The latter study further reported the endocannabinoid‐like substance palmitoylethanolamide (PEA), as well as 2‐AG and virodhamine, to show a significantly more potent action through GPR55 than through either CB1 or CB2 receptors, whereas AEA was equally active on CB receptors and on GPR55.
Finally, several N‐acylethanolamines, including AEA as well as the endocannabinoid‐like substances PEA, oleoylethanolamide (OEA), stearoylethanolamide and linoleoylethanolamide, were revealed as activators of PPARα with OEA exerting the highest efficacy (Artmann et al., 2008). Furthermore, recent investigations suggest cannabinoid compounds can enhance PPARγ expression and activation (Ramer et al., 2013; Vara et al., 2013).
The following sections focus on the different levels of anti‐tumour effects of cannabinoids and the role of components of the endocannabinoid system in this process. An overview of selected pathways involved in mediating the anticancerogenic effect of cannabinoids is provided in Figure 1.
Figure 1.
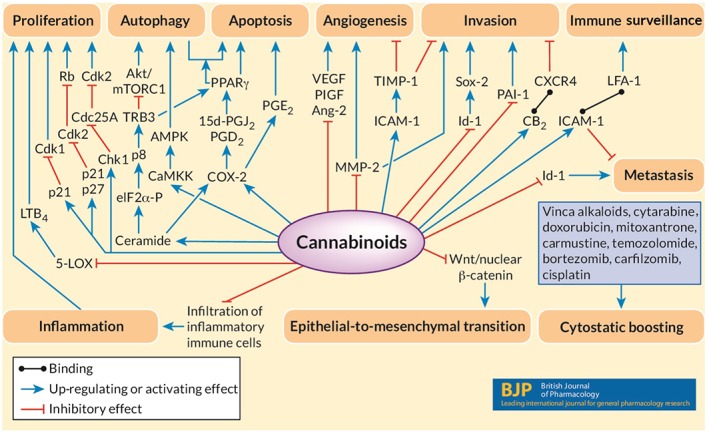
Selected pathways involved in anticancerogenic effects of cannabinoids. The diagram shows selected pro‐cancerogenic and anti‐cancerogenic pathways in cancer cells. Coloured arrows indicate inhibitory (red) and stimulatory (blue) effects of cannabinoids on these processes, finally leading to inhibition of cancer cell growth and spreading. Lines with dots at the end indicate binding between two factors.
Regulation of the endocannabinoid system in cancer tissues
A large number of investigations provided proof for up‐regulation of components of the endocannabinoid system in malignant tissue and an association with adverse patient outcome. In this context, poor prognoses for cancer patients were associated with high expression of CB1 receptors in malignant tissue, such as pancreatic cancer (Michalski et al., 2008). Up‐regulation was likewise observed for CB2 receptors in a variety of cancer types (see Ramer and Hinz, 2017). Endogenous ligands at these receptors were similarly elevated, as has been reported for 2‐AG, for example, in prostate cancer (Nithipatikom et al., 2004). In human meningiomas, AEA, but not 2‐AG, was up‐regulated (Maccarrone et al., 2001), whereas a converse regulation with decreased AEA and increased 2‐AG was observed in blood analyses of circulating endocannabinoids from patients suffering from different kinds of cancers (Sailler et al., 2014) as well as in glioma tissues (Wu et al., 2012). The latter investigation further reported down‐regulation of both expression and activity for NAPE‐PLD, FAAH and MAGL, whereas the expression of DAGL remained unchanged. In colon tumour patients, MAGL expression was likewise down‐regulated in tumour tissue, compared with the neighbouring healthy tissue and was either absent or reduced in the majority of primary colorectal cancer cases (Sun et al., 2013). This study also reported reduced MAGL expression associated with lung, breast, stomach and ovary cancers. Analyses of biopsies obtained from prostate cancer patients further revealed higher expression levels of FAAH in cancer tissue (Endsley et al., 2008). However, the association between levels of the endocannabinoid‐degrading enzymes and prognosis for cancer patients was less clear‐cut. Thus, high levels of FAAH and MAGL positively correlated with the prognosis of patients with pancreatic ductal adenocarcinomas (Michalski et al., 2008), while an increase of MAGL expression was reported for highly malignant tissues, such as high‐grade primary ovary tumours (Nomura et al., 2010).
The functional implication of increased cannabinoid receptor expression and increased endocannabinoid levels in tumour tissue is currently a matter of active debate (see Ramer and Hinz, 2017). Moreover, components of the endocannabinoid system obviously do not possess useful uniform marker properties for reliable cancer prognosis. On the other hand, the overall view of the endocannabinoid system as an anticancer system is supported by numerous reports on anticancerogenic properties of cannabinoid compounds and inhibitors of endocannabinoid turnover, as discussed in the following sections.
Anti‐tumour actions of cannabinoids
Tumour cell growth and viability
The first study monitoring the effects of phytocannabinoids on cancer regression in animal experiments was published by Munson et al. (1975), who showed suppression of tumour growth by Δ8‐tetrahydrocannabinol, Δ9‐tetrahydrocannabinol (THC) and cannabinol, long before the discovery of cannabinoid receptors and endocannabinoids. Meanwhile, cannabinoid compounds were revealed as potent inhibitors of cancer progression on different levels of cancer cell growth and spreading in numerous preclinical investigations.
During the last few decades, a large body of evidence has accumulated to suggest endocannabinoids, phytocannabinoids and synthetic cannabinoids exert an inhibitory effect on cancer growth via blockade of cell proliferation and induction of apoptosis. In the late 1990s, De Petrocellis et al. (1998), addressing the role of cannabinoid receptors in the growth inhibitory action of several cannabinoid compounds, reported AEA to reduce the proliferation of breast cancer cell lines at IC50 values of 0.5 μM (MCF‐7 cells) and 1.5 μM (EFM‐19 cells) via a CB1 receptor‐dependent mechanism. In their study, the synthetic cannabinoid HU‐210, likewise, elicited a comparable antiproliferative action on EFM‐19 cells but with less potency. Notably, although the authors did not mention the IC50 levels for the antiproliferative effects of HU‐210, half maximal inhibition of proliferation can be estimated to be above 5 μM. Two years later, the first study demonstrating that THC and the synthetic cannabinoid WIN 55,212‐2 caused regression of glioma growth in Wistar rats and in Rag2−/− mice was published (Galve‐Roperh et al., 2000). In that study, rats, treated with THC or WIN 55,212‐2, were shown to live significantly longer than vehicle‐treated animals. These experiments were supplemented by in vitro investigations showing that THC, as well as the synthetic cannabinoids WIN 55,212‐2, CP55940 and HU‐210 inhibited glioma cell growth, associated with CB1‐dependent and CB2‐dependent sustained ceramide accumulation and p42/44 MAPK activation. Subsequently, the selective CB2 receptor agonist JWH‐133 also inhibited growth of glioma cells (Sánchez et al., 2001). Following these and other pioneering studies (see Caffarel et al., 2012), a large number of investigations on anticancerogenic cannabinoid actions have been published.
In terms of the intracellular mechanisms of the antiproliferative actions of cannabinoids, several studies have demonstrated cannabinoid compounds to modulate cell cycle checkpoints. As such, cannabinoid receptor activation induced melanoma cell cycle arrest via inhibition of Akt and hypophosphorylation of the retinoblastoma‐associated protein (Rb) (Blázquez et al., 2006). While the latter and other studies reported cannabinoids to confer cell cycle arrest at the G1–S transition, another study using breast cancer cells showed that THC arrested the G2–M transition, via down‐regulation of cyclin‐dependent kinase 1 [CDK1/cell division cycle (Cdc)2] and induction of p21, a CDK inhibitor that suppresses Cdc2–cyclin B activation (Caffarel et al., 2006). A further investigation addressing the antiproliferative impact of a stable AEA analogue, 2‐methyl‐2′‐F‐anandamide (Met‐F‐AEA), on breast cancer cells, found a transient and delayed cell cycle checkpoint response (Laezza et al., 2006). Accordingly, Met‐F‐AEA caused an increase of p21waf and p27kip1 and a decrease of cyclins A and E. As upstream events of cyclin degradation, the authors demonstrated a rapid activation of checkpoint kinase 1 that induced downstream degradation of Cdc 25 homologue A and Cdk2 inactivation. As a delayed response, Met‐F‐AEA activated the p21waf cascade that additionally resulted in Cdk2 inhibition. These effects were further associated with a reduction of Rb activity, a prominent target of Cdk2 activity.
As a mechanism of glioma cell death in response to THC treatment, a CB2 receptor‐mediated up‐regulation of the stress‐associated transcriptional co‐activator p8 mediated a proapoptotic action via up‐regulation of the endoplasmic reticulum stress‐related genes activating transcription factor (ATF)‐4 and the pseudo‐kinase tribbles homolog (TRB)3 (Carracedo et al., 2006). A contribution of p8 induction to cannabinoid‐induced apoptosis was later substantiated for THC and HU‐210 in rhabdomyosarcoma cells. In that study, cannabinoid‐induced apoptosis was associated with inhibition of Akt signalling and, as shown for HU‐210, restored by a CB1 receptor antagonist (Oesch et al., 2009). Another investigation reported THC and the selective CB2 receptor agonist, JWH‐133, to inhibit the growth of highly aggressive ErbB2‐positive breast cancers, associated with inhibition of the pro‐tumourigenic Akt pathway (Caffarel et al., 2010).
A panel of investigations reported cannabinoid‐induced impaired cancer cell viability via mechanisms bypassing activation of cannabinoid receptors. For example, CP55940, JW015 and the FAAH inhibitor, N‐arachidonoyl serotonin (AA‐5HT), inhibited proliferation of rat glioma cells independently of both CB receptors and TRPV1 channel activation (Jacobsson et al., 2001). In the same investigation, however, AEA and 2‐AG exerted antiproliferative actions via cannabinoid receptor‐dependent and TRPV1‐dependent oxidative stress and calpain activation. Furthermore, R(+)‐methanandamide induced a cannabinoid receptor‐ and TRPV1‐independent apoptosis in human neuroglioma cells by de novo synthesis of ceramide (Hinz et al., 2004). In the latter type of cells, the proapoptotic mechanism of R(+)‐methanandamide was based on a ceramide‐dependent up‐regulation of COX‐2 expression (Ramer et al., 2003) and increased synthesis of proapoptotic PGE2 (Hinz et al., 2004).
Notably, reports on anticancer effects of CBD, a non‐psychoactive cannabinoid with low affinity to CB receptors, revealed proapoptotic effects, without CB receptor activation. Thus, CBD suppressed proliferation of glioma cells via decreased activity and content of 5‐lipoxygenase and of its end product LTB4 (Massi et al., 2008). Another study found CBD to induce PPARγ‐dependent toxicity by upstream induction of COX‐2‐dependent PGs of the D and J series in lung cancer cells (Ramer et al., 2013). A further investigation addressing inhibition of glioma stem cell growth demonstrated CBD as a potential ‘redox therapeutic’ that inhibited glioma stem cell survival, associated with Akt phosphorylation via activation of the p38 MAPK pathway, as well as down‐regulation of stem cell marker proteins such as sex‐determining region Y‐box (Sox)2 and inhibitor of DNA binding (Id)‐1, an inhibitor of basic helix–loop–helix transcription factors (Singer et al., 2015). Here, growth inhibition by CBD was mediated via production of ROS that induced a regrowth via a counter‐regulated induction of the antioxidant protein SLC7A11 (xCT, cystine‐glutamate transporter). The antineoplastic action of CBD was further substantiated by McAllister et al. (2011), who demonstrated inhibition of growth and spread of breast cancer cells via mitochondrial damage, increased levels of ROS and down‐regulation of Id‐1. In breast cancer cells, CBD induced intrinsic apoptosis associated with autophagic pathways as indicated by decreased levels of a phosphorylated mechanistic target of rapamycin (mTOR), eukaryotic translation initiation factor (eIF) 4E‐binding protein 1 and cyclin D1 (Shrivastava et al., 2011).
Recent research concerning induction of cancer cell death has focused on autophagy as an underlying mechanism of cannabinoid‐induced antineoplastic action. In this context, THC induces ceramide accumulation, leading to downstream phosphorylation of eIF2α and subsequent endoplasmic reticulum stress associated with autophagy in glioma cells (Salazar et al., 2009). This autophagic effect was, likewise, associated with up‐regulation of ATF‐4 and TRB3, conferring downstream inhibition of the prosurvival kinase Akt with subsequent inhibition of the mechanistic target of rapamycin complex 1 (mTORC1) and thus induction of autophagic cell death. A contribution of the Akt/mTORC1 axis to a CB2 receptor‐dependent autophagy by THC and the CB2 receptor agonist JWH015 was further confirmed in experiments using hepatocellular carcinoma in vitro and in vivo (Vara et al., 2011). In addition to the death‐inducing Akt/mTORC1 modulation by cannabinoids, the latter investigation revealed a separate pathway leading to autophagy that encompasses a calmodulin‐activated kinase kinase subtype, which subsequently phosphorylates AMP‐activated kinase, thereby conferring autophagy as a response to cannabinoid treatment. Another study was able to demonstrate that cannabinoid‐induced TRB3 expression was associated with an up‐regulation of PPARγ that appeared essential for an appropriate autophagosome operation, thereby serving as a causal link to cannabinoid‐induced autophagic cell death (Vara et al., 2013). Noteworthy, results from these studies suggested autophagy, as a response to cannabinoid treatment, to be involved in the upstream activation of apoptosis rather than acting as apoptosis‐alternative pathway leading to cell death (Salazar et al., 2009; Vara et al., 2011). In this context, a recent investigation revealed TRPV2 as a cannabinoid target, involved in CBD‐induced autophagy (Nabissi et al., 2015).
Within the past years, several cannabinoids were found to modulate GPR55, another important key player of cancer progression. GPR55 was found to promote carcinogenesis and was up‐regulated in human carcinomas compared with corresponding healthy tissues (Pérez‐Gómez et al., 2013). Although the complex network of cannabinoid action on GPR55 signalling requires more data, a recent investigation led authors to pursue the hypothesis of a negative crosstalk between GPR55 and CB2 receptors and a bidirectional cross‐antagonism between both receptors (Moreno et al., 2014).
Besides these antineoplastic effects of exogenously added cannabinoid compounds, a large body of evidence suggests endocannabinoids are similarly able to inhibit cancer cell growth (see Ramer and Hinz, 2016). In agreement with the proposed role of endocannabinoids as cancer‐repressive substances, a panel of investigations provided evidence for inhibitors of endocannabinoid turnover to exert comparable effects on cancer cells. In the first publication that revealed a contribution of cannabinoid receptors to the antiproliferative action of AEA, arachidonoyl‐trifluoromethylketone, an inhibitor of FAAH, enhanced the antiproliferative effects of exogenously added AEA, while arachidonoyl‐trifluoromethylketone alone did not affect the proliferation of breast carcinoma cells (De Petrocellis et al., 1998). The concept of inhibition of FAAH as an anticancer strategy was later supported by reports of the FAAH inhibitor, AA‐5HT, inhibiting the growth of xenografts generated from thyroid cancer cells (Bifulco et al., 2004) and reducing colonic carcinogenesis (Izzo et al., 2008). Furthermore, targeting the 2‐AG‐degrading MAGL has revealed a possible option for suppression of cancer progression. Knockdown of MAGL, using small hairpin RNA and pharmacological inhibition of MAGL with the MAGL inhibitor JZL184, suppressed the growth of prostate carcinoma in vivo via partial involvement of CB1 receptors (Nomura et al., 2011). Using breast, ovarian and melanoma cancer cells, another study found MAGL inhibition suppressed cancer aggressiveness, without cannabinoid receptor activation, through a decrease of free fatty acids resulting in less cancer‐promoting PGs and lysophosphatidic acid (Nomura et al., 2010). In the latter study, inhibition of cancer growth by MAGL inhibition was reversed by adding back free fatty acids.
Inhibition of cancer growth in a murine xenograft model has further been demonstrated for colorectal carcinoma cells (Ye et al., 2011). Here, xenograft growth was inhibited in mice that received colorectal cancer cells transfected with MAGL siRNA or treatment with JZL184. In vivo experiments of this investigation revealed knockdown of MAGL, as well as treatment with JZL184 inhibiting cancer cell proliferation and invasion associated with down‐regulation of cyclin D1 and B‐cell lymphoma 2 (Bcl‐2). Similar results using colorectal carcinoma cells were reported by Ma et al. (2016). The latter study found an apoptosis‐related decrease of Bcl‐2 and increase of Bcl‐2‐associated X protein as a response to treatment with JZL184. However, these studies did not evaluate the contribution of cannabinoid‐activated receptors to the observed antiproliferative effects. In another study, lentivirus‐mediated MAGL knockdown in HT29 colon cancer cells and MDA‐MB‐231 breast cancer cells was associated with increased Akt phosphorylation, thereby exhibiting growth inhibition (Sun et al., 2013). Again, in this study, phosphorylation of Akt was constitutively suppressed by MAGL, but the contribution of cannabinoid receptors to this phenomenon was not addressed.
Invasion and metastasis
In addition to the numerous investigations demonstrating cannabinoids to elicit antiproliferative and proapoptotic effects on cancer cells, the first report on anti‐invasive effects of cannabinoids found CB1 receptor activation by 2‐AG inhibited prostate cancer cell invasion (Nithipatikom et al., 2004). Down‐regulation of Id‐1, a hallmark of cannabinoid‐attenuated cancer cell invasion, was observed in CBD‐treated breast (McAllister et al., 2007) and brain cancer cells (Soroceanu et al., 2013). The latter work further confirmed down‐regulation of Sox‐2, a downstream target of Id‐1. Additionally, Id‐1 down‐regulation, associated with inhibition of cancer cell invasion, was confirmed for the CB2 receptor agonist O‐1663 (Murase et al., 2014).
A reduction of cancer cell aggressiveness by Met‐F‐AEA additionally involved the Wnt‐1 signalling pathway (Laezza et al., 2012). Accordingly, the authors showed that treatment of human breast cancer cells with Met‐F‐AEA decreased nuclear accumulation of β‐catenin in a CB1 receptor‐dependent manner and was associated with suppression of β‐catenin‐triggered target oncogenes, such as cyclin D1, c‐myc and MMP‐2. Furthermore, the authors observed suppression of mesenchymal markers, such as vimentin, N‐cadherin and fibronectin, as well as down‐regulation of markers of epithelial‐to‐mesenchymal transition (EMT), such as Snail1, Slug and Twist. Migration inhibition associated with regulation of the EMT markers, such as down‐regulation of vimentin and Snail, as well as up‐regulation of E‐cadherin, was likewise observed in colorectal cancer cells treated with JZL184 (Ma et al., 2016).
Contributions of both CB receptors to the anti‐invasive action of THC and, in terms of R(+)‐methanandamide, an additional involvement of TRPV1 channels, were demonstrated using cervical and lung cancer cells (Ramer and Hinz, 2008). In the latter report, up‐regulation of tissue inhibitor of matrix metalloproteinases (TIMP)‐1 was causally linked to the anti‐invasive potential of both cannabinoids and was later confirmed for the anti‐invasive impact of CBD on cervical and lung cancer cells (Ramer et al., 2010a; Ramer et al., 2012). In terms of cannabinoid‐induced inhibition of lung cancer cell invasion, TIMP‐1 up‐regulation occurred via upstream induction of the intercellular adhesion molecule 1 (ICAM‐1) (Ramer et al., 2012). In addition to the TIMP‐1‐dependent anti‐invasive properties of phytocannabinoids and R(+)‐methanandamide, a recent publication found a causal link between TIMP‐1 induction by the FAAH inhibitors, AA‐5HT and URB597, FAAH siRNA, as well as AEA and OEA and invasion inhibition in lung cancer cells (Winkler et al., 2016). Inhibitor experiments pointed towards a role of CB2 receptors and TRPV1 channels in the anti‐invasive effects of FAAH inhibitors and FAAH siRNA. In the same study, both FAAH inhibitors investigated, endocannabinoids (AEA and 2‐AG) and endocannabinoid‐like substances (PEA and OEA) elicited antimetastatic effects in nude mice.
In other reports, MMP‐2 down‐regulation was associated with decreased invasion of THC‐treated glioma cells (Blázquez et al., 2008). Down‐regulation of the plasminogen activator inhibitor‐1 via activation of CB receptors and TRPV1 channels was found using lung cancer cells and is an additional pathway involving CBD's anti‐invasive action (Ramer et al., 2010b). A recent study further postulated heterodimerization of the chemokine receptor CXCR4 with the CB2 receptor, contributing to attenuation of breast cancer cell invasion (Coke et al., 2016). Taken together, cannabinoids, as well as inhibitors of endocannabinoid degradation, by virtue of their abilities to modulate regulation of MMPs and to down‐regulate Id‐1, may present options to specifically suppress the spread of cancer cells.
Angiogenesis
Tumour neovascularization is another important hallmark of cancer progression that was observed to be suppressed by cannabinoid treatment. In this context, data obtained from animal experiments clearly suggest that the antiangiogenic impact of cannabinoids is a general antineoplastic mechanism of this group of substances (see Ramer and Hinz, 2016). Notably, in vivo results published by Casanova et al. (2003) showed that cannabinoid‐induced inhibition of tumour vascularization was associated with down‐regulation of the proangiogenic factors: VEGF, placental growth factor and angiopoietin‐2. Several other investigations further provided evidence for the inhibition of angiogenic features of endothelial cells following cannabinoid treatment. Antiproliferative and/or antimigratory effects towards HUVEC were reported for WIN 55,212‐2 and JWH‐133, which were associated with down‐regulation of VEGF and MMP‐2 (Blazquez et al., 2003). A contribution of MMP‐2 down‐regulation to antiangiogenic effects was further confirmed for Met‐F‐AEA (Pisanti et al., 2007) and CBD (Solinas et al., 2012).
In contrast to the consistent data from animal experiments, some in vitro studies presented ambiguous cannabinoid effects towards endothelial cells. Accordingly, Kogan et al. (2006) reported no antiangiogenic impacts of endocannabinoids on HUVEC proliferation and even proangiogenic effects of low concentrations of THC and CBD. Another study revealed that nanomolar concentrations of AEA stimulated basic fibroblast growth factor‐induced proliferation of endothelial cells, without affecting proliferation of quiescent endothelial cells (Pisanti et al., 2011). A tendency towards enhanced angiogenic capacities of HUVEC was later confirmed for CBD, THC, R(+)‐methanandamide and JWH‐133 (Ramer et al., 2014). Thus, some cannabinoids may induce, rather than inhibit, angiogenesis at lower, pharmacologically relevant, concentrations through direct interaction with endothelial cells. This notion, however, contrasts with numerous studies that uniformly reported inhibition of tumour vascularization by cannabinoid compounds in vivo. Recent investigations suggest that cannabinoid compounds specifically inhibit angiogenic processes at microenvironmental sites of malignant tissue. In agreement with this assumption, conditioned media from AEA‐treated breast cancer cells were found to inhibit endothelial cell proliferation linked to down‐regulation of angiogenesis‐related factors, such as VEGF, leptin, interferon‐γ and thrombopoietin (Picardi et al., 2014). Another study demonstrated that enhanced levels of TIMP‐1 in conditioned media of CBD‐treated, THC‐treated, R(+)‐methanandamide‐treated and JWH‐133‐treated lung cancer cells suppressed the angiogenic capacities of endothelial cells (Ramer et al., 2014).
Tumour–immune interactions
Some studies support the hypothesis that cannabinoids may enhance immune responses against the progressive growth and spread of tumours. In vivo analyses of melanoma xenograft growth revealed that WIN 55,212‐2 produced a more efficient tumour‐regressive action in immunocompetent, versus immunodeficient, mice (Blázquez et al., 2006). Another study found CBD, THC and R(+)‐methanandamide enhanced the susceptibility of lung cancer cells towards lysis by lymphokine‐activated killer cells (Haustein et al., 2014). The underlying mechanism involved a cannabinoid‐induced up‐regulation of ICAM‐1 on the tumour cell surface and a subsequent crosslink with lymphocyte function‐associated antigen‐1 on the surface of killer cells. In addition, data from a recent investigation suggested that a reduced infiltration of experimental skin cancer with macrophages and neutrophils in THC‐treated animals was associated with tumour regression (Glodde et al., 2015). Thus, cannabinoids may elicit anti‐tumour immune responses via differential mechanisms, enabling a more effective action of immune cells to combat cancer or by favouring conditions that result in the local reduction of immune cells that cause an inflammatory pro‐cancerogenic microenvironment within the cancer tissue. However, one study reported THC increased growth and spreading of mammary carcinoma cells, as a result of inhibition of anti‐tumour immune response (McKallip et al., 2005). Therefore, more investigations are necessary to evaluate the effects of cannabinoids on tumour‐immune interactions.
Clinical data
Currently, there are no data from large, multicentre, double‐blinded, placebo‐controlled trials available concerning the systemic anticancer effect of cannabinoids. One clinical pilot study demonstrated that intracranially administered THC was safe in glioblastoma patients (Guzmán et al., 2006). A recently announced exploratory, phase II, randomized, placebo‐controlled clinical trial with recurrent glioblastoma multiforme patients provides a signal for the potential efficacy of cannabinoids as add‐on anticancer drugs. In this study involving 21 patients, 12 patients were randomized to a combination of THC and CBD in addition to dose‐intensive temozolomide, whereas the remaining nine patients were randomized to placebo plus standard of care. The proof‐of‐concept study showed a significantly higher 1 year survival rate in the cannabinoid group, (83 vs. 53% in the placebo cohort). Moreover, the median survival for the cannabinoid group was greater than 550 days compared with 369 days in the group randomized to placebo (GW Pharmaceuticals, 2017 press release).
As a matter of principle, however, the clinical use of cannabinoids may be limited by their psychoactive effects (Wang et al., 2008), as well as by a probable risk for the development of liver fibrosis that has been ascribed to activation of CB1 receptors (Teixeira‐Clerc et al., 2006). Therefore, non‐psychoactive substances, such as CBD that exert highly efficient anticancer properties and a considerable safety profile in preclinical experiments, have attracted scientific interest. Notably, a recent study on CBD treatment for Dravet syndrome revealed that a CBD dose of 20 mg per kilogram of body weight per day was associated with somnolence and elevation of liver enzyme levels, when compared with the control group (Devinsky et al., 2017). As the placebo and the CBD treatment groups received several additional antiepileptic drugs, adverse interactions with CBD, rather than a toxicity of CBD per se, were discussed by the authors as a possible cause of the observed adverse effects. Furthermore, CB2 receptor agonists as non‐psychoactive cannabinoids have gained scientific interest. In this context, it is noteworthy that, in contrast to the profibrotic CB1 receptor activation (Teixeira‐Clerc et al., 2006), CB2 receptor agonists have been found to exert antifibrotic effects on the liver (Julien et al., 2005).
Combinational effects with chemotherapeutic agents
Taking into account that future clinical studies addressing systemic anticancer effects of cannabinoids will most likely not be conducted as monotherapy, but in combination with established chemotherapeutics, recent research concerning probable interactions between cannabinoids and currently used cytostatic drugs suggests cannabinoids as notable additives to a number of chemotherapeutic effects. Figure 2 provides an overview on this subject. As recently summarized, CBD and THC enhance the cytotoxic effects of several chemotherapeutic agents, including vinca alkaloids, cytarabine, doxorubicin, mitoxantrone, carmustine, temozolomide, bortezomib, carfilzomib and cisplatin (Ramer and Hinz, 2017). Thus, THC and CBD have both been shown to enhance the cytostatic effect of vinblastine in resistant leukaemia cells via down‐regulation of P‐glycoprotein (Holland et al., 2006) and of mitoxantrone in embryonic fibroblasts through inhibition of ABCG2 (Holland et al., 2007). An enhancement of the cytostatic properties of cytarabine, doxorubicin and vincristine has, likewise, been demonstrated for THC in leukaemia cells (Liu et al., 2008). The sensitization of cells appeared to be dependent on decreased p42/44 MAPK activity in response to sublethal concentrations of THC. In addition, a decreased chemoresistance of myeloma cells towards the proteasome inhibitor bortezomib was observed when combined with CBD (Morelli et al., 2014). The toxic effect of CBD alone, or in combination with bortezomib, was found to exert higher efficacy in multiple myeloma cells that express TRPV2 channels, when compared with TRPV2‐deficient cells. A crucial role for TRPV2 channels in the reduction of chemoresistance has further been confirmed for carmustine, doxorubicin and temozolomide in CBD‐challenged glioma cells (Nabissi et al., 2013), as well as for doxorubicin in triple negative breast cancer cells treated with CBD (Elbaz et al., 2016). THC and CBD, or a combination of both, were further demonstrated to enhance the toxic impact of temozolomide on glioma cells in vitro and in vivo via a mechanism that involves autophagy (Torres et al., 2011). Notably, data from this investigation were one basis for the aforementioned placebo‐controlled, phase II study demonstrating that a combination of THC and CBD produced a prolonged survival when combined with temozolomide in glioblastoma patients. Synergistic actions were further reported for the effect of a CBD/THC combination added to multiple myeloma cells in the presence of carfilzomib. The cannabinoids here were shown to overcome resistance to carfilzomib by reducing the proteasome β5i subunit at both the transcriptional and translational levels (Nabissi et al., 2016). The susceptibility of glioblastoma cells to the cytotoxic action of cisplatin was further found to be enhanced by CBD (Deng et al., 2017).
Figure 2.
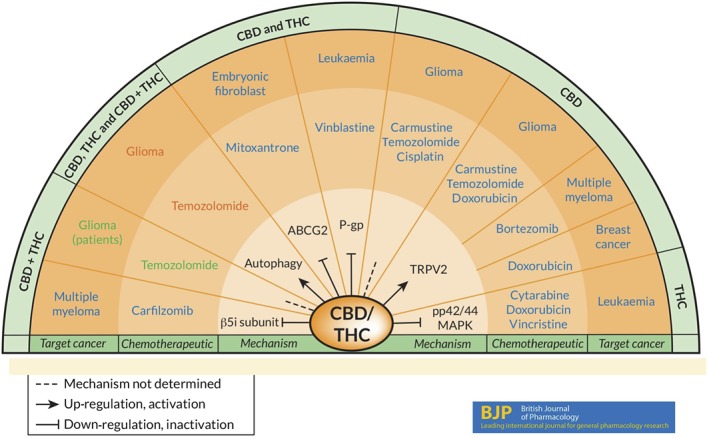
Enhancement of the effects of chemotherapeutic agents on cancer cells or tissue by CBD and/or THC. Arrows in the inner layer specify cannabinoid‐induced up‐regulation/activation or down‐regulation/inactivation of the indicated parameters or mechanisms involved in the anti‐tumour effects of the combination with the indicated anticancer drug in the second layer, respectively. The third layer specifies the cancer type, and the outer layer shows the respective treatment. Dashed lines indicate investigations where no mechanism was assessed. Drugs and cancer types in blue letters indicate cell culture studies that addressed cannabinoid‐induced enhanced sensitivity of cancer cells towards the cytotoxic action of the corresponding chemotherapeutic agents. Red letters indicate a study that demonstrated an enhancement of the cancer‐regressive effect of temozolomide by THC, CBD and a THC/CBD combination and further included in vivo experiments using a murine xenograft model. Details of these preclinical evaluations and references are indicated under the section Combinational effects with chemotherapeutic agents. Green letters specify temozolomide effects boosted by combination with CBD/THC assessed in a phase II, randomized, placebo‐controlled clinical trial with recurrent glioblastoma multiforme patients (GW Pharmaceuticals, 2017 press release). Details concerning this clinical study are described in the section on Clinical data.
Notably, investigations concerning the beneficial interactions between cannabinoids and chemotherapeutics were not confined to synergistic anticancer actions and also addressed probable suppression of adverse chemotherapeutic effects by cannabinoids. Thus, benefits for some cancer patients with chemotherapy‐induced peripheral neuropathy (Gingerich et al., 2009) have been reported under treatment with cannabinoids. Moreover, cannabinoids such as CBD (Pan et al., 2009) and the peripherally restricted cannabinoid CB2 receptor agonist LEI‐101 (Mukhopadhyay et al., 2016) attenuated cisplatin‐induced nephrotoxicity in mice.
Outlook
Taken together, pharmacological use of cannabinoid receptors and other components of the endocannabinoid system, in the broader sense, represents an attractive option for anticancer therapy, at least at the preclinical level. As a matter of fact, the endocannabinoid system offers a broad spectrum of targets that influence cancer cell fate. Particular scientific interest has been attracted by non‐psychoactive cannabinoids such as CBD, which has been demonstrated to exert a broad array of anticarcinogenic properties, such as antiproliferative action towards cancer cells (Ligresti et al., 2006), anti‐invasive and antimetastatic effects (Ramer et al., 2010a, 2012) as well as induction of apoptosis (Massi et al., 2004) and autophagy (Shrivastava et al., 2011; Nabissi et al., 2015). The spectrum of the cancer‐regressive action of CBD is further complemented by its property to enhance the susceptibility of resistant cancer cells towards chemotherapeutic agents (Holland et al., 2006) and to inhibit angiogenesis (Solinas et al., 2012). As a further treatment option for systemic cancer therapy, modulation of endocannabinoid‐degrading enzymes has gained considerable interest. There is a consistent line of evidence suggesting that inhibitors of FAAH exert cancer‐suppressive actions, while sparing psychoactive effects. For instance, FAAH inhibitors have been found to lack hypomotility (Schlosburg et al., 2009). Another investigation found dual inhibition of FAAH and MAGL mimicked the effects of direct CB1 receptor agonists, such as THC (Long et al., 2009). The latter study further reported that the MAGL inhibitor JZL184 induced THC‐like drug discrimination responses, which may reflect a contribution of the MAGL/2‐AG pathway to mimic THC‐like psychoactive effects.
When a high percentage of fatal cancer progressions results from intravasation and extravasation of cancer cells, it highlights the importance of new drugs that efficiently block metastasis of malignant neoplastic cells. In view of the large number of animal studies that demonstrate cannabinoid compounds to exert antimetastatic effects, these substances may serve as feasible add‐on options for the currently used cytostatics. This view is further substantiated by a number of preclinical investigations that provide evidence for the beneficial synergistic action of cannabinoids combined with chemotherapeutic drugs. However, potential adverse drug interactions must be carefully evaluated in future studies. Additionally, cannabinoids may provide a pharmacotherapeutic option as low molecular weight inhibitors of tumour neovascularization in addition to the currently used TK inhibitors sunitinib, pazopanib, sorafenib, axitinib and bevacizumab that improve overall survival of patients with different cancer types, such as renal cell carcinoma.
Future clinical studies that permit reliable conclusions from successful bench‐to‐bedside translation are urgently needed, particularly, since the passage of new laws allowing patient access to cannabinoids and the growing market of cannabinoid‐based nutraceuticals. Scientists must quickly gather facts concerning the risks and benefits of cannabinoid compounds for cancer patients, or else the non‐scientific media will create its own ‘facts’.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c, 2017d).
Conflict of interest
The authors declare no conflicts of interest.
Hinz, B. , and Ramer, R. (2019) Anti‐tumour actions of cannabinoids. British Journal of Pharmacology, 176: 1384–1394. 10.1111/bph.14426.
References
- Alexander SP, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174 (Suppl 1): S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174 (Suppl 1): S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017d). The Concise Guide to Pharmacology 2017/18: Transporters. Br J Pharmacol 174 (Suppl 1): S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artmann A, Petersen G, Hellgren LI, Boberg J, Skonberg C, Nellemann C et al (2008). Influence of dietary fatty acids on endocannabinoid and N‐acylethanolamine levels in rat brain, liver and small intestine. Biochim Biophys Acta 1781: 200–212. [DOI] [PubMed] [Google Scholar]
- Bifulco M, Laezza C, Valenti M, Ligresti A, Portella G, Di Marzo V (2004). A new strategy to block tumor growth by inhibiting endocannabinoid inactivation. FASEB J 18: 1606–1608. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Melck D, Bobrov MY, Gretskaya NM, Bezuglov VV, De Petrocellis L et al (2000). N‐acyl‐dopamines: novel synthetic CB1 cannabinoid‐receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem J 351 (Pt 3): 817–824. [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I et al (2001). Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol 134: 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF (2007). A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2‐arachidonoylglycerol. Chem Biol 14: 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazquez C, Casanova ML, Planas A, Gomez Del Pulgar T, Villanueva C, Fernandez‐Acenero MJ et al (2003). Inhibition of tumor angiogenesis by cannabinoids. FASEB J 17: 529–531. [DOI] [PubMed] [Google Scholar]
- Blázquez C, Carracedo A, Barrado L, Real PJ, Fernández‐Luna JL, Velasco G et al (2006). Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J 20: 2633–2635. [DOI] [PubMed] [Google Scholar]
- Blázquez C, Salazar M, Carracedo A, Lorente M, Egia A, González‐Feria L et al (2008). Cannabinoids inhibit glioma cell invasion by down‐regulating matrix metalloproteinase‐2 expression. Cancer Res 68: 1945–1952. [DOI] [PubMed] [Google Scholar]
- Caffarel MM, Sarrió D, Palacios J, Guzmán M, Sánchez C (2006). Δ9‐Tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res 66: 6615–6621. [DOI] [PubMed] [Google Scholar]
- Caffarel M, Andradas C, Mira E, Pérez‐Gómez E, Cerutti C, Moreno‐Bueno G et al (2010). Cannabinoids reduce ErbB2‐driven breast cancer progression through Akt inhibition. Mol Cancer 9: 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffarel MM, Andradas C, Pérez‐Gómez E, Guzmán M, Sánchez C (2012). Cannabinoids: a new hope for breast cancer therapy? Cancer Treat Rev 38: 911–918. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Gironella M, Lorente M, Garcia S, Guzmán M, Velasco G et al (2006). Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress‐related genes. Cancer Res 66: 6748–6755. [DOI] [PubMed] [Google Scholar]
- Casanova ML, Blázquez C, Martínez‐Palacio J, Villanueva C, Fernández‐Aceñero MJ, Huffman JW et al (2003). Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J Clin Invest 111: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coke CJ, Scarlett KA, Chetram MA, Jones KJ, Sandifer BJ, Davis AS et al (2016). Simultaneous activation of induced heterodimerization between CXCR4 chemokine receptor and cannabinoid receptor 2 (CB2) reveals a mechanism for regulation of tumor progression. J Biol Chem 291: 9991–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Ng L, Ozawa T, Stella N (2017). Quantitative analyses of synergistic responses between cannabidiol and DNA‐damaging agents on the proliferation and viability of glioblastoma and neural progenitor cells in culture. J Pharmacol Exp Ther 360: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Melck D, Palmisano A, Bisogno T, Laezza C, Bifulco M et al (1998). The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci U S A 95: 8375–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch DG, Chin SA (1993). Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 46: 791–796. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G et al (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946–1949. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R et al (2017). Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med 376: 2011–2020. [DOI] [PubMed] [Google Scholar]
- Di Marzo V (2009). The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol Res 60: 77–84. [DOI] [PubMed] [Google Scholar]
- Elbaz M, Ahirwar D, Xiaoli Z, Zhou X, Lustberg M, Nasser MW et al (2016). TRPV2 is a novel biomarker and therapeutic target in triple negative breast cancer. Oncotarget . 10.18632/oncotarget.9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endsley MP, Thill R, Choudhry I, Williams CL, Kajdacsy‐Balla A, Campbell WB et al (2008). Expression and function of fatty acid amide hydrolase in prostate cancer. Int J Cancer 123: 1318–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galve‐Roperh I, Sánchez C, Cortés ML, Gómez del Pulgar T, Izquierdo M, Guzmán M (2000). Anti‐tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal‐regulated kinase activation. Nat Med 6: 313–319. [DOI] [PubMed] [Google Scholar]
- Gingerich J, Wadhwa D, Lemanski L, Krahn M, Daeninck PJ (2009). The use of cannabinoids (CBs) for the treatment of chemotherapy‐induced peripheral neuropathy (CIPN): a retrospective review. J Clin Oncol 27: e20743. [Google Scholar]
- Glodde N, Jakobs M, Bald T, Tüting T, Gaffal E (2015). Differential role of cannabinoids in the pathogenesis of skin cancer. Life Sci 138: 35–40. [DOI] [PubMed] [Google Scholar]
- Guzmán M, Duarte MJ, Blazquez C, Ravina J, Rosa MC, Galve‐Roperh I et al (2006). A pilot study of Δ9‐tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer 95: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GW Pharmaceuticals. GW Pharmaceuticals achieves positive results in phase 2 proof of concept study in glioma. Feb 07, 2017. Available at: https://www.gwpharm.com/about‐us/news/gw‐pharmaceuticals‐achieves‐positive‐results‐phase‐2‐proof‐concept‐study‐glioma
- Hanus L, Gopher A, Almog S, Mechoulam R (1993). Two new unsaturated fatty acid ethanolamides in brain that bind to the cannabinoid receptor. J Med Chem 36: 3032–3034. [DOI] [PubMed] [Google Scholar]
- Hanus L, Abu‐Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE et al (2001). 2‐Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci U S A 98: 3662–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haustein M, Ramer R, Linnebacher M, Manda K, Hinz B (2014). Cannabinoids increase lung cancer cell lysis by lymphokine‐activated killer cells via upregulation of ICAM‐1. Biochem Pharmacol 92: 312–325. [DOI] [PubMed] [Google Scholar]
- Hinz B, Ramer R, Eichele K, Weinzierl U, Brune K (2004). Up‐regulation of cyclooxygenase‐2 expression is involved in R(+)‐methanandamide‐induced apoptotic death of human neuroglioma cells. Mol Pharmacol 66: 1643–1651. [DOI] [PubMed] [Google Scholar]
- Holland ML, Panetta JA, Hoskins JM, Bebawy M, Roufogalis BD, Allen JD et al (2006). The effects of cannabinoids on P‐glycoprotein transport and expression in multidrug resistant cells. Biochem Pharmacol 71: 1146–1154. [DOI] [PubMed] [Google Scholar]
- Holland ML, Lau DT, Allen JD, Arnold JC (2007). The multidrug transporter ABCG2 (BCRP) is inhibited by plant‐derived cannabinoids. Br J Pharmacol 152: 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Aviello G, Petrosino S, Orlando P, Marsicano G, Lutz B et al (2008). Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J Mol Med (Berl) 86: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsson SO, Wallin T, Fowler CJ (2001). Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J Pharmacol Exp Ther 299: 951–959. [PubMed] [Google Scholar]
- Julien B, Grenard P, Teixeira‐Clerc F, Van Nhieu JT, Li L, Karsak M et al (2005). Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 128: 742–755. [DOI] [PubMed] [Google Scholar]
- Kogan NM, Blazquez C, Alvarez L, Gallily R, Schlesinger M, Guzmán M et al (2006). A cannabinoid quinone inhibits angiogenesis by targeting vascular endothelial cells. Mol Pharmacol 70: 51–59. [DOI] [PubMed] [Google Scholar]
- Laezza C, Pisanti S, Crescenzi E, Bifulco M (2006). Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett 580: 6076–6082. [DOI] [PubMed] [Google Scholar]
- Laezza C, d'Alessandro A, Malfitano AM, Maria Malfitano A, Chiara Proto M, Gazzerro P et al (2012). Anandamide inhibits the Wnt/β‐catenin signalling pathway in human breast cancer MDA‐MB 231 cells. Eur J Cancer 48: 3112–3122. [DOI] [PubMed] [Google Scholar]
- Ligresti A, Moriello AS, Starowicz K, Matias I, Pisanti S, De Petrocellis L et al (2006). Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther 318: 1375–1387. [DOI] [PubMed] [Google Scholar]
- Liu WM, Scott KA, Shamash J, Joel S, Powles TB (2008). Enhancing the in vitro cytotoxic activity of Δ9‐tetrahydrocannabinol in leukemic cells through a combinatorial approach. Leuk Lymphoma 49: 1800–1809. [DOI] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X et al (2009). Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A 106: 20270–20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Bai J, Ling Y, Chang W, Xie G, Li R et al (2016). Monoacylglycerol lipase inhibitor JZL184 regulates apoptosis and migration of colorectal cancer cells. Mol Med Rep 13: 2850–2856. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Attinà M, Cartoni A, Bari M, Finazzi‐Agrò A (2001). Gas chromatography‐mass spectrometry analysis of endogenous cannabinoids in healthy and tumoral human brain and human cells in culture. J Neurochem 76: 594–601. [DOI] [PubMed] [Google Scholar]
- Massi P, Vaccani A, Ceruti S, Colombo A, Abbracchio MP, Parolaro D (2004). Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J Pharmacol Exp Ther 308: 838–845. [DOI] [PubMed] [Google Scholar]
- Massi P, Valenti M, Vaccani A, Gasperi V, Perletti G, Marras E et al (2008). 5‐Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor activity of cannabidiol, a non‐psychoactive cannabinoid. J Neurochem 104: 1091–1100. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner T (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346: 561–564. [DOI] [PubMed] [Google Scholar]
- McAllister SD, Christian RT, Horowitz MP, Garcia A, Desprez PY (2007). Cannabidiol as a novel inhibitor of Id‐1 gene expression in aggressive breast cancer cells. Mol Cancer Ther 6: 2921–2927. [DOI] [PubMed] [Google Scholar]
- McAllister SD, Murase R, Christian RT, Lau D, Zielinski AJ, Allison J et al (2011). Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res Treat 129: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKallip RJ, Nagarkatti M, Nagarkatti PS (2005). Δ‐9‐Tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J Immunol 174: 3281–3289. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben‐Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR et al (1995). Identification of an endogenous 2‐monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50: 83–90. [DOI] [PubMed] [Google Scholar]
- Michalski CW, Oti FE, Erkan M, Sauliunaite D, Bergmann F, Pacher P et al (2008). Cannabinoids in pancreatic cancer: correlation with survival and pain. Int J Cancer 122: 742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli MB, Offidani M, Alesiani F, Discepoli G, Liberati S, Olivieri A et al (2014). The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type‐2. Int J Cancer 134: 2534–2546. [DOI] [PubMed] [Google Scholar]
- Moreno E, Andradas C, Medrano M, Caffarel MM, Pérez‐Gómez E, Blasco‐Benito S et al (2014). Targeting CB2–GPR55 receptor heteromers modulates cancer cell signaling. J Biol Chem 289: 21960–21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Baggelaar M, Erdelyi K, Cao Z, Cinar R, Fezza F et al (2016). The novel, orally available and peripherally restricted selective cannabinoid CB2 receptor agonist LEI‐101 prevents cisplatin‐induced nephrotoxicity. Br J Pharmacol 173: 446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu‐Shaar M (1993). Molecular characterization of a peripheral receptor for cannabinoids. Nature 365: 61–65. [DOI] [PubMed] [Google Scholar]
- Munson AE, Harris LS, Friedman MA, Dewey WL, Carchman RA (1975). Antineoplastic activity of cannabinoids. J Natl Cancer Inst 55: 597–602. [DOI] [PubMed] [Google Scholar]
- Murase R, Kawamura R, Singer E, Pakdel A, Sarma P, Judkins J et al (2014). Targeting multiple cannabinoid antitumor pathways with a resorcinol derivate leads to inhibition of advanced stages of breast cancer. Br J Pharmacol 171: 4464–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabissi M, Morelli MB, Santoni M, Santoni G (2013). Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 34: 48–57. [DOI] [PubMed] [Google Scholar]
- Nabissi M, Morelli MB, Amantini C, Liberati S, Santoni M, Ricci‐Vitiani L et al (2015). Cannabidiol stimulates Aml‐1a‐dependent glial differentiation and inhibits glioma stem‐like cells proliferation by inducing autophagy in a TRPV2‐dependent manner. Int J Cancer 137: 1855–1869. [DOI] [PubMed] [Google Scholar]
- Nabissi M, Morelli MB, Offidani M, Amantini C, Gentili S, Soriani A et al (2016). Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 7: 77543–77557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithipatikom K, Endsley MP, Isbell MA, Falck JR, Iwamoto Y, Hillard CJ et al (2004). 2‐Arachidonoylglycerol: a novel inhibitor of androgen‐independent prostate cancer cell invasion. Cancer Res 64: 8826–8830. [DOI] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF (2010). Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ et al (2011). Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol 18: 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch S, Walter D, Wachtel M, Pretre K, Salazar M, Guzmán M et al (2009). Cannabinoid receptor 1 is a potential drug target for treatment of translocation‐positive rhabdomyosarcoma. Mol Cancer Ther 8: 1838–1845. [DOI] [PubMed] [Google Scholar]
- Pan H, Mukhopadhyay P, Rajesh M, Patel V, Mukhopadhyay B, Gao B et al (2009). Cannabidiol attenuates cisplatin‐induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J Pharmacol Exp Ther 328: 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Gómez E, Andradas C, Flores JM, Quintanilla M, Paramio JM, Guzmán M et al (2013). The orphan receptor GPR55 drives skin carcinogenesis and is upregulated in human squamous cell carcinomas. Oncogene 32: 2534–2542. [DOI] [PubMed] [Google Scholar]
- Picardi P, Ciaglia E, Proto M, Pisanti S (2014). Anandamide inhibits breast tumor‐induced angiogenesis. Transl Med UniSa 10: 8–12. [PMC free article] [PubMed] [Google Scholar]
- Pisanti S, Borselli C, Oliviero O, Laezza C, Gazzerro P, Bifulco M (2007). Antiangiogenic activity of the endocannabinoid anandamide: correlation to its tumor‐suppressor efficacy. J Cell Physiol 211: 495–503. [DOI] [PubMed] [Google Scholar]
- Pisanti S, Picardi P, Prota L, Proto MC, Laezza C, McGuire PG et al (2011). Genetic and pharmacologic inactivation of cannabinoid CB1 receptor inhibits angiogenesis. Blood 117: 5541–5550. [DOI] [PubMed] [Google Scholar]
- Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J et al (2002). Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther 301: 1020–1024. [DOI] [PubMed] [Google Scholar]
- Ramer R, Weinzierl U, Schwind B, Brune K, Hinz B (2003). Ceramide is involved in r(+)‐methanandamide‐induced cyclooxygenase‐2 expression in human neuroglioma cells. Mol Pharmacol 64: 1189–1198. [DOI] [PubMed] [Google Scholar]
- Ramer R, Hinz B (2008). Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases‐1. J Natl Cancer Inst 100: 59–69. [DOI] [PubMed] [Google Scholar]
- Ramer R, Merkord J, Rohde H, Hinz B (2010a). Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases‐1. Biochem Pharmacol 79: 955–966. [DOI] [PubMed] [Google Scholar]
- Ramer R, Rohde A, Merkord J, Rohde H, Hinz B (2010b). Decrease of plasminogen activator inhibitor‐1 may contribute to the anti‐invasive action of cannabidiol on human lung cancer cells. Pharm Res 27: 2162–2174. [DOI] [PubMed] [Google Scholar]
- Ramer R, Bublitz K, Freimuth N, Merkord J, Rohde H, Haustein M et al (2012). Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule‐1. FASEB J 26: 1535–1548. [DOI] [PubMed] [Google Scholar]
- Ramer R, Heinemann K, Merkord J, Rohde H, Salamon A, Linnebacher M et al (2013). COX‐2 and PPAR‐γ confer cannabidiol‐induced apoptosis of human lung cancer cells. Mol Cancer Ther 12: 69–82. [DOI] [PubMed] [Google Scholar]
- Ramer R, Fischer S, Haustein M, Manda K, Hinz B (2014). Cannabinoid inhibit angiogenic capacities of endothelial cells via release of tissue inhibitor of matrix metalloproteinases‐1 from lung cancer cells. Biochem Pharmacol 91: 202–216. [DOI] [PubMed] [Google Scholar]
- Ramer R, Hinz B (2016). Antitumorigenic targets of cannabinoids – current status and implications. Expert Opin Ther Targets 20: 1219–1235. [DOI] [PubMed] [Google Scholar]
- Ramer R, Hinz B (2017). Cannabinoids as anticancer drugs. Adv Pharmacol 80: 397–436. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J et al (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152: 1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailler S, Schmitz K, Jäger E, Ferreiros N, Wicker S, Zschiebsch K et al (2014). Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 1: 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar M, Carracedo A, Salanueva IJ, Hernández‐Tiedra S, Lorente M, Egia A et al (2009). Cannabinoid action induces autophagy‐mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 119: 1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez C, de Ceballos ML, Gomez del Pulgar T, Rueda D, Corbacho C, Velasco G et al (2001). Inhibition of glioma growth in vivo by selective activation of the CB(2) cannabinoid receptor. Cancer Res 61: 5784–5789. [PubMed] [Google Scholar]
- Schlosburg JE, Boger DL, Cravatt BF, Lichtman AH (2009). Endocannabinoid modulation of scratching response in an acute allergenic model: a new prospective neural therapeutic target for pruritus. J Pharmacol Exp Ther 329: 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A, Kuzontkoski PM, Groopman JE, Prasad A (2011). Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross‐talk between apoptosis and autophagy. Mol Cancer Ther 10: 1161–1172. [DOI] [PubMed] [Google Scholar]
- Singer E, Judkins J, Salomonis N, Matlaf L, Soteropoulos P, McAllister S et al (2015). Reactive oxygen species‐mediated therapeutic response and resistance in glioblastoma. Cell Death Dis 6: e1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas M, Massi P, Cantelmo AR, Cattaneo MG, Cammarota R, Bartolini D et al (2012). Cannabidiol inhibits angiogenesis by multiple mechanisms. Br J Pharmacol 167: 1218–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroceanu L, Murase R, Limbad C, Singer E, Allison J, Adrados I et al (2013). Id‐1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res 73: 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Jiang L, Luo X, Jin W, He Q, An J et al (2013). Potential tumor‐suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene 32: 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres S, Lorente M, Rodríguez‐Fornés F, Hernández‐Tiedra S, Salazar M, García‐Taboada E et al (2011). A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther 10: 90–103. [DOI] [PubMed] [Google Scholar]
- Teixeira‐Clerc F, Julien B, Grenard P, Tran Van Nhieu J, Deveaux V, Li L et al (2006). CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med 12: 671–676. [DOI] [PubMed] [Google Scholar]
- Vara D, Salazar M, Olea‐Herrero N, Guzmán M, Velasco G, Díaz‐Laviada I (2011). Anti‐tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK‐dependent activation of autophagy. Cell Death Differ 18: 1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara D, Morell C, Rodríguez‐Henche N, Diaz‐Laviada I (2013). Involvement of PPARγ in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis 4: e618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Collet JP, Shapiro S, Ware MA (2008). Adverse effects of medical cannabinoids: a systematic review. CMAJ 178: 1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler K, Ramer R, Dithmer S, Ivanov I, Merkord J, Hinz B (2016). Fatty acid amide hydrolase inhibitors confer anti‐invasive and antimetastatic effects on lung cancer cells. Oncotarget 7: 15047–15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Han L, Zhang X, Li L, Jiang C, Qiu Y et al (2012). Alteration of endocannabinoid system in human gliomas. J Neurochem 120: 842–849. [DOI] [PubMed] [Google Scholar]
- Ye L, Zhang B, Seviour EG, Tao KX, Liu XH, Ling Y et al (2011). Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett 307: 6–17. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V et al (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452–457. [DOI] [PubMed] [Google Scholar]