

Abstract
The molecular pathogenesis of Alzheimer’s disease (AD) is complex and sparsely understood. The relationship between AD’s amyloid β (Aβ) peptides and neuronal membranes is central to Aβ’s cytotoxicity, and is directly modulated by the composition of the lipid headgroups. Molecular studies of the insertion of model Aβ40 protofilaments in lipid bilayers revealed strong interactions that can affect the structural integrity of both the membranes and the ordered amyloid aggregates. In particular, electrostatics plays a crucial role in the interaction between Aβ protofilaments and palmytoil-oleoyl-phosphatidylethanolamine (POPE) lipids, a common component of neuronal plasma membranes. Here, we use all-atom molecular dynamics and steered molecular dynamics simulations to systematically compare the effects that POPE and palmytoil-oleoyl-phosphatidylcholine (POPC) headgroups have on the Aβ-lipid interactions. We find that Aβ protofilaments exhibit weaker electrostatic interactions with POPC headgroups, and establish significantly shorter-lived contacts with the POPC bilayer. This illustrates the crucial yet complex role of electrostatic and hydrogen bonding interactions in modulating the anchoring and insertion of Aβ into lipid bilayers. Our study reveals the atomistic details behind the barrier created by the lipid headgroup region in impeding solution-aggregated fibrillar oligomers to spontaneously insert into POPC bilayers, in contrast to the POPE case. While the biological reality is notoriously more complex (e.g., including other factors such as cholesterol), our results evidence a simple experimentally and computationally testable case for probing the factors that control the insertion of Aβ oligomeric aggregates in neuronal cell membranes - a process central to their neurotoxicity.
Keywords: Alzheimer’s disease, amyloid β-peptide fibrils, Aβ fibrillar oligomers, molecular dynamics simulations, toxic amyloid channels, amyloid peptide-lipid membrane interactions
Graphical Abstract
Molecular interactions of Alzheimer’s amyloid fibrils with lipid membranes.
1. Introduction
Amyloid β (Aβ) peptides are cleavage products of β- and γ-secretases upon the transmembrane amyloid precursor proteins. They have an amphipathic nature; their N-terminus is part of the extracellular part of the precursor protein, whereas the more hydrophobic C-terminus is part of the membrane-spanning domain.(1) After its secretion, depending on its environmental conditions, monomeric Aβ adopts structures that are disordered(2) or partially folded.(3, 4) The structure of aggregated Aβ in solution has been shown in vitro to be different from aggregates formed in the presence of membranes.(5, 6) The role of electrostatic interactions in anchoring of amyloid proteins into membranes leading to their subsequent aggregation has been long established.(7–9) Recent experimental studies suggest that even at physiological concentrations, electrostatic interactions between Aβ40 and lipids can lead to peptides accumulating on the membrane surface at higher concentrations than in bulk solution, thus enhancing aggregation. At increased concentrations, this effect is more intense.(5, 10, 11) Upon aggregation, Aβ adopts a β-sheet-rich structure, which disrupts lipid membranes.(12–16) Experimental studies have also shown that oligomers of Aβ and human islet amyloid polypeptide (hIAPP) cause membrane permeation for charged, as well as for neutral lipids.(17, 18) Both aggregated Aβ and IAPP were shown to have the tendency to bind irreversibly to both zwitterionic and anionic bilayers;(19, 20)
In this study, we report strong differences in the molecular interaction mechanism between Aβ fibrillar oligomers and palmytoil-oleoyl-phosphatidylethanolamine (POPE) and palmytoil-oleoyl-phosphatidylcholine (POPC) zwitterionic lipids with headgroups that are electrostatically neutral yet have different morphologies (Fig. 1(a)). In a previous study,(21) we employed all-atom molecular dynamics (MD) simulations to analyse the mechanism of interaction between solid state NMR-based models of Aβ fibrillar octamers(22–24) and a model membrane composed of zwitterionic POPE lipid molecules. The POPE lipids are a major component of plasma membranes (i.e., composing only about 25% of phospholipids in all living cells, but up to 45% in human neurons).(25) We identified the electrostatic attraction between the proximal charged residues in the oligomer and lipid headgroups as the driving force that causes perturbations in the membrane, followed by a further insertion of the Aβ C-termini into the membrane hydrophobic core. The lipid tails in the upper (proximal) leaflet can reorient themselves towards the membrane surface and interact with the C-termini, which leads to a local membrane-thinning effect. This finding is in agreement with experimental studies evidencing that it is the oligomeric aggregates rather than the monomeric Aβ (19, 26) or the mature fibrils(27) that induce neuronal dysfunction.
Figure 1:

(a) Morphological differences between POPE and POPC headgroups. The nitrogen atom in the POPE headgroup is surrounded by three hydrogens, whereas in POPC the nitrogen is surrounded by three methyl groups. (b) A cross-section through the Aβ9–40 fibril, showing a two-peptide layer. Each peptide consists of an N-terminus β-strand (red), a turn region (green) and a C-terminus β-strand (blue). In the N-terminus and turn regions there are both positive (H13, K16, K28; blue) and negative (E11, E22, D23; red) residues. Polar sidechains are represented in green and the hydrophobic in grey, respectively.
2. Results and discussion
The Aβ protofilament structures (Fig. 1(b)) are based on solid-state NMR data,(22–24) and described in detail in our previous study.(21) While carefully avoiding steric clashes, the fibrillar oligomer was placed atop the membrane as close as possible to the lipids.
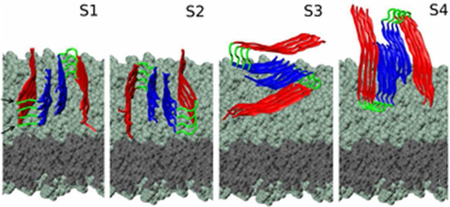
Based on our previous observations regarding the symmetry properties of Aβ protofilaments(21, 28), we generated four main types of initial conditions (denoted S1 to S4, described in our previous work, depicted in Fig. 2, and summarised in Table S1), that capture the most different possible fibril-membrane interaction modes. Each initial conformation is consisting of an Aβ fibrillar octamer and a POPE or POPC lipid bilayer. Additional MD runs for systems with the same initial conformation i, with i=1, 4, were labeled as Sia, Sib, etc. (see Table S1).
Figure 2:

Initial conditions for S1-S4 systems. The plane of the two-peptide layers (numbered 1 to 4 from the membrane upwards) is oriented either parallel or perpendicular with respect to the plane of the membrane. The N-terminus region is represented in red, turn is in green and C-terminus is in blue. The heavy atoms in the lipid headgroups (teal) and tail (gray) regions are represented as spheres.
Our MD trajectories with explicit water molecules showed that the protofilament lost contact with the POPC membrane in S1a, S3a and S4a systems. In contrast, Aβ in the S2a system maintained contact with the membrane throughout the trajectory. The S2-type conformation also exhibited the strongest interaction with a POPE bilayer in our previous study.(21) In the S2-type system, the two-peptide plane is parallel to the membrane, thus our current results are consistent with the theory that fibrillar oligomers are more cytotoxic than fibrils, due to displaying a higher area of reactive ends.(29) This preliminary result lead us to infer that the interaction was conformation-dependent and that Aβ in the S2 conformation was the most prone to maintaining contact with the lipids. We tested this observation by simulating systems in all S1-S4 conformations with carefully constructed and equilibrated systems (see Table S1). The subsequent simulations showed consistent behavior for the S1-, S3- and S4-type systems. In contrast to S2a, in the S2b and the S2c systems our fibrillar oligomer lost contact with the membrane, subsequent to a short interaction.
2.1. Changes in the structure of the protofilament during MD
The secondary structure of Aβ amyloid fibrils consists mostly of β-sheets(22), which are stabilized by backbone hydrogen bonds (HBs) between the layers in the fibril. Thus, the NHB between the layers is closely related to the secondary structure of the fibril; the more numerous the HBs, the higher the β-sheet content. We analyzed the effect of the lipid interaction on the secondary structure of the Aβ protofilament. Fig. 3 shows the percentage of residues that are part of β-sheets. According to experimental studies, β-sheets are formed for the F10-E22 and A30-V40 residues,(22, 23) which represents 84% of the total number of amino acids.
Figure 3:

Secondary structure- the variation of the β-sheet content during the simulations. Compared to Aβ-POPE simulations, in Aβ-POPC simulations the fibril preserves a higher portion of its intrinsic β-sheet structure. The β-sheet content is maintained at 50% for S1 (panel a) and S2 (b) systems, and at 70% for the S3 (c) and S4 (d) systems
The β-sheet content decreased from 80% to 50% in the case of S1- and S2-type conformations and to 75% in the case of S3- and S4-type conformations. By comparison, the β-sheet structure was preserved better than during Aβ-POPE simulations(21), which can be explained by the briefer interaction between the oligomer and the lipids. We observed no helical structure. The charged residues in either the N-terminus or turn region in S3-type and S4-type conformations initially in contact with the membrane, exhibited the shortest interaction with the lipids, therefore preserving the highest β-sheet structure. As other indicators of the overall change of structure, we analyzed the root mean square deviation (RMSD) for the Cα atoms with respect to the initial structure (RMSD0) plots and the number of the inter-strand contacts (Ncontacts) variation, shown in Figs. S1 and S2, respectively. The lowerer RMSD0 values and the high Ncontacts values compared to Aβ-POPE systems are correlated with the higher preservation of β-sheet structure. Due to the interaction with the membrane, the RMSD0 values for either Aβ-POPE or Aβ-POPC systems are higher than the RMSD0 registered in previous simulations of fibrillar Aβ in solution.(24, 28)
2.2. Analysis of the Aβ-POPC interaction
POPE headgroups are well known for forming HBs acting as both donors and an acceptors.(30) HBs between POPC-POPC headgroups or between POPC lipids and transmembrane proteins have been observed in previous computational studies.(31, 32) However, compared to POPE, POPC lipids show a decreased tendency to form HBs with the peptides, due to the fact that POPC has a more voluminous headgroup than POPE and it restricts the interaction between Aβ and the phosphate and carbonyl moieties. Similarly to the analysis of Aβ-POPE simulations, we compared the NHB between layer 1 and layer 2 with the NHB between layer 1 and the lipids for the S1 and S2 conformations (see Fig. S3). Due to the geometry of the S3 and S4 conformations- the β-sheet planes parallel to the membrane plane, the possibility of creating HBs between the protofilament and the lipids is greatly reduced and thus disregarded. S1-type and S2-type systems exhibit the strongest peptide-lipid interaction, due to the positioning of the plane of the Aβ layers parallel to the plane of the membrane. This correlates well with previous theories stating that, subsequent to an initial electrostatic interaction between Aβ and the membrane, there is a peptide-lipid alignment such that the hydrophobic residues are oriented towards the core of the membrane.(33) But, unlike the sustained Aβ-POPE interaction, in the Aβ-POPC systems (except for S2a system), the HBs between the fibril and the lipids are relatively short-lived. The only case where the HBs are maintained is S2a; the increase in the NHB layer 1 in the protofilament and the lipids (represented by a black line in Fig. S3) leads to a decline in the NHB between layer 1 and layer 2 (red line in Fig. S3). This effect is noticed in the secondary structure plots. We chose S1b as a representative case of the interaction between Aβ oligomers and POPC bilayers, as it is a striking example of the reduced degree of interaction between Aβ and lipids. Throughout the simulation, as depicted in Fig. 4(a), the NHB between layer 1 and layer 2 fluctuates between 40 and 60, whereas the initial 20-30 HBs between the protofilament and the lipids, decreases to zero. Nevertheless, both in S2a and in S1b systems, we observed a similar anti-correlation of the NHB due to the β-sheet preservation/lipid interaction interplay as in the Aβ-POPE simulations, albeit greatly reduced.
Figure 4:

In system S1b, Aβ interacted with the membrane only transiently. (a) The number of hydrogen bonds, NHB, established between layer 1 and layer 2 (red) compared to the NHB between layer 1 and the lipids (black), and (b) the electrostatic energy (Ue) between D23 and K28 in layer 1 (green) and between D23, K28 in layer 1 and the lipids (black).
The electrostatic energy (Ue) values (see Fig. 4(b) for system S1b and Fig. S4 for all systems) provide a quantitative measure of the effect the attraction between the charged residues and the lipid headgroups has upon the salt bridge formed in the initially ordered protofilament. The charged residues are E11, H13, K16 in the N-terminus and E22, D23, K28 in the turn region. It has been suggested that the N-terminus plays a very important role in mediating the Aβ-lipid interaction(34), modulated by the electrostatic charges. Also, Aβ absorption to hydrophilic surfaces may also be mainly driven by electrostatic interactions.(35) K28 has been shown to act as an anchor in POPC bilayers interacting with lipid headgroup and superficial waters(36) and to be part of a HB network that promotes Aβ oligomerization.(37) The salt bridge between D23 and K28 is an important structural element of Aβ fibrils, as demonstrated by several experimental and computational studies and supports the stability of the fibril.(20, 38, 39) We have previously shown that the D23 – K28 salt bridge competes with the interaction between D23, K28 and the POPE lipids. Not only does the latter interaction break the salt bridge, it also disrupts the membrane, as shown in a previous computational study.(20)
An overview of the variation of the electrostatic energy Ue for all simulated systems is presented in Fig. S4. The lowest energy for the two stable D23-K28 salt bridges in layer 1 in the protofilament is −180 kcal/mol. In S1a the salt bridges remained fully formed for the entire 60-ns simulation. As a result of the weak electrostatic interaction between Aβ and POPC, the protofilament drifted away from the membrane and lost contact with the lipids, as shown in Fig. 5(b). We constructed S1b by placing the protofilament closer to the membrane and as a result it remained in the proximity of the membrane for longer due to a stronger electrostatic interaction between the charged residues and the headgroups, as shown in Fig. 4(b). This clearly destabilized the salt bridge as energy values increased to −90 kcal/mol, indicating that one of the salt bridges broke. But, 20 ns into the simulation, due to the instability of the HBs between Aβ and the lipids, as shown by Fig. 4(a), and to the water inserting at the fibril/membrane interface which led to a screening of the charges, the Ue value for the D23, K28 and the lipids interaction increases to zero. The subsequent effect was that the salt bridge regenerates and Ue values return to −180 kcal/mol (Fig. 4(b)).
Figure 5:

Relative z-axis position for selected regions in layer 1 of Aβ in the S1-type conformation interacting with (a) a POPE bilayer, and (b) a POPC bilayer (system S1a). The COM positions of charged residues in the N-terminus region, of charged residues in the turn region and of the C-termini are depicted in red, green and blue, respectively. To distinguish between the two strands in the fibril we used a solid smooth line for one and a solid line with a circular markers for the other. The horizontal black line represents the phosporus atoms plane. The gray shaded area is occupied by the headgroups.
A detailed analysis of water dynamics at the Aβ-lipid interface in representative system S1b-POPC (Fig. S8) shows that in only about 38 ns the water molecules accumulate between the fibril and the lipids such that the fibrillar oligomer becomes effectively solvated, as shown at the end of the simulation (55ns, Fig. S8c).
In the case of S2-type systems, the electrostatic interaction between the fibril and lipids is stronger and the disturbance of the salt bridge more pronounced than for the S1-type systems, as shown in Fig. S5. Although the salt bridges in system S2a were disturbed, the connection between D23 and K28 was not fully broken and the energy occasionally reached values of −90 kcal/mol or −180 kcal/mol even towards the end of the simulation. Positioning the protofilament closer to the lipids led to an initial decrease in the values for the charged residues-lipids interaction energy in the first 5 ns of the simulations of S2b and S2c. During these simulations the interaction between the salt bridge residues was also disturbed by the lipids, as indicated by the fluctuations of the D23-K28 Ue values. The peptide-lipid attraction tended to be unstable, as the energy increased towards zero or positive values, with rare events of negative energy values. In S2b both salt bridges break and reform throughout the simulation, whereas in S2c, after 10 ns, one of the salt bridges breaks and remains so for the rest of the simulation. For the systems in the S3 and S4 conformations we plotted the values of Ue between lipids and the charged residues in all four layers in one N-terminus in S3 and in the N-terminus and turn regions in S4 (see conformation geometry in Fig. 2) and compared it to the interaction energy between the closest D23-K28 pairs to the membrane (black line and green line, respectively, in Fig. S4). Thus, each green line represents the Ue values for salt bridges in the four layers, the lowest value being −700 kcal/mol when all were stably formed. As shown in previous studies of Aβ protofilaments in solution (24, 28), there are both intramolecular and intermolecular salt bridges between adjacent peptide layers (i.e. the residues in layer i can also interact with the residues in i–1, and i+1 layers), thus the total energy is lower than for four isolated layers as the interaction network makes the salt bridges more stable. In S3b and S3c, Ue values for the D23-K28 interaction increased from −700 kcal/mol to −490 kcal/mol, occasionally in discrete step-like jumps between the values which indicated that certain salt bridges are broken, which correlated with decreases in the Ue between D23, K28 and the lipids.
2.3. Representative differences between Aβ-POPE versus Aβ-POPC systems
The electrostatic interaction and the hydrogen bonding between Aβ and the lipids decided whether the protofilament remained in the proximity of the membrane. In our previous Aβ-POPE simulations (21) we identified the electrostatic attraction as being the initial step of interaction between the fibril and the headgroups, followed by the hydrophobic effect between the C-termini and the lipid tails. We observed a reorganization of the headgroups(40) around the protofilament, whilst the C-terminus inserts into the lipid tail region. Fig. 5(a) depicts a clear interaction between the POPE lipids and the charged residues in the N-termini and turn regions, as the COM of these regions are just above the headgroup area (red lines and green lines, respectively). One of the C-termini gets inserted into the membrane core, as indicated by the negative values of the COM for this region (smooth blue line in Fig. 5(a)). In the current study we observed that a strong peptide-lipid interaction occurred only during the S2a simulation.
By contrast, in most Aβ-POPC systems, the protofilament lost contact with the membrane. As a clear example, we show the dynamics of system S1a (see Fig. 5(b). The fibril started at similar position as in the S1 Aβ-POPE system, about 5 Å above the phosphorus atoms plane. At around 25 ns, the system starts to drift away from the membrane into the bulk solution and towards the end of the simulation the COM of one of the N-termini reaches a z-value of over 45 Å; due to the twisting motion of the fibril, the opposed N-terminus was the last part to lose contact with lipids. This effect of the twisting motion occurred in all other Aβ-POPC systems (except for S2a and S3a).
2.4. Possible lipid tail extraction from the membrane in the S2a Aβ-POPC system
S2a was the only molecular system in which Aβ peptides maintained contact with the membrane and the C-termini interacted with the lipid tails. Initial visual inspection of the trajectory revealed that while the C-terminus was slightly protruding into the hydrophobic membrane core, two of the POPC lipid tails changed their downwards orientation and ascended above the membrane plane to interact with the octamer. One oleoyl tail and one palmytoil tail from different lipids began to change their orientation and fully interacted with the protofilament after 10 ns of simulation. Fig. 6(a) shows the z-axis coordinates for the end carbon atoms of the palmitoyl and oleoyl tails of the upper lipid leaflet.
Figure 6:

a) z-axis coordinates versus time for the end carbon atoms in the lipid tails, oleoyl is shown in light grey, palmitoyl in dark grey, the dotted black line represents the phosphorus atoms plane. (b) z-axis coordinates for the COM of charged residues in the N-terminus, E11, H13, K16 (red lines), in the turn region, E22, D23, K28 (bright-green lines) and of the C-terminus (blue lines). The dark green zone represents the headgroup region and the grey region the hydrophobic core of the membrane. (c) The palmitoyl tail (left) and the oleoyl tail (right) interact with the hydrophobic residues in the protofilament. The former interacts with the C-termini in the first two layers in the octamer, whereas the latter has the ability to interact with hydrophobic regions in all four layers.
The palmitoyl tail positioned itself between the two strands in the C-termini region as shown in Fig. 6(c) and interacted chiefly with the hydrophobic M35 and the small polar G37 residue. After approximately 50 ns, probably due to the fact that it was not sterically feasible to maintain its position between the strands and to the high energy penalty for crossing the headgroup region, it regained its initial orientation. This protrusion between the Aβ strands affected the inter-strand contacts between the C-termini in the protofilament. As shown in Fig. S2, the Ncontacts decreased from 50 to 20, but After 80 ns, the contacts were reestablished, due to the palmitoyl tail retreating below the headgroup plane. The oleoyl tail started its upwards movement at the beginning of the simulation, but reached the maximum stretch at 40 ns, with the end carbon atom 20 Å above the phosphorus plane. At this point, the tail stretched along all four layers of the protofilament, maximizing the hydrophobic interaction. This tail interacted with a different part of the protofilament, namely the inner region of one of the hairpin-like cross-β unit, mainly with L17VFFA21 in the N-terminus on one side and residues G29AIIGLM35 in the C-terminus on the other. The residues in these Aβ segments are mainly hydrophobic, such that the penalty for leaving the membrane core was balanced by the hydrophobic interactions with these regions. The upward movement of the tails and their subsequent interaction with different parts of the protofilament had a stabilizing effect on the octamer, maintaining it atop the membrane. At the same time, it prevented the C-termini from interacting with the lipid tails below the headgroup region. As shown in Fig. 6(b), the COM of the C-termini in both strands were above the headgroup region until 50 ns into the simulation, when one of them started to protrude through the headgroups to interact with the tails beneath them. Fig. 6(c) is a representative snapshot of the interaction between different parts of the protofilament and the palmitoyl and oleoyl tails, from the beginning of the simulation until the palmitoyl tail retraction below the headgroup region. When the palmitoyl tail reassumed its initial orientation in the membrane core, it pulled down one of the C-termini in layer 1, indicated by the corroboration between the first two panels in Fig. 6. The C-terminus remains in contact with the lipids for the remainder of the simulation, as inferred from the low value of its z-axis coordinate in Fig. 6(b). These data demonstrate that the protrusion of lipid tails above the hydrophilic region of the membrane and their subsequent hydrophobic interaction with the C-termini could enhance their protrusion through the headgroups, once the tails reassumed their initial downward orientation.
2.5. Dynamics of the fibril structure during the SMD simulations
We performed SMD simulations on Aβ-POPE and Aβ-POPC systems in the S2 conformation (henceforth S2-POPE and S2-POPC). Pulling started After the equilibration stage. During the simulations the protofilament was pressed through the lipid headgroup region and we observed the subsequent Aβ-lipid interaction. The details of the approach are described in the Experimental section. We carefully monitored the structure of the fibril throughout the simulations. The RMSD0 plot for the Cα atoms (Fig. S6), indicates that pulling restricts the overall movement of the protofilament, and thus it takes longer for the fibril to reach an equilibrated structure. The maximum RMSD0 value of 8 Å during the S2-POPE SMD simulation is similar to the RMSD0 obtained in the MD simulations, whereas in the case of S2-POPC, the RMSD0 = 8 Å is higher than the registered value of 7 Å during the MD simulations. The increase in the RMSD0 translates in a variation of the β-sheet content registered in each case (as shown in Fig. S7). The β-sheet content of S2-POPE decreases from 80% to 50% for the first 35 ns of the simulation. The pulling speed was initially set to 1 Å/ns for the first 4 ns for the fibril to travel to the surface of the membrane and then to 0.1 Å/ns while pulling on the Cα atoms of E11, N27 and G38. The percentage of β-sheet increases when pulling speed is further reduced to 0.01 Å/ns and all the Cα atoms are selected. For the S2-POPC system we used a higher velocity than for S2-POPE in order to overcome the fibril drifting into bulk water, which in return hindered the equilibration and dynamics of the lipids around the protofilament, which would permit the lipids to reorganize, leading to more stable contacts between the protofilament and the lipids, as indicated by experiments.(19) During the simulation of S2-POPC, the content of β-sheet first dropped to 45% during the first 5 ns of pulling on the Cα atoms of E11, N27 and G38 with a speed of v = 1 Å/ns. It further decreased until 15 ns, when selecting only the same atoms only in layer 3 and layer 4 and decreasing the speed to v=0.1 Å/ns. When selecting all the Cα atoms again at a speed of 0.1 Å/ns, the percentage increased to 50%. In both S2-POPE and S2-POPC we observed a small percentage of helical structure. In S2-POPE it involved residues A21-S26 in the N-terminus and turn regions. In the S2-POPC simulation, the helix was formed in the turn and C-terminus regions, between residues G25-A30, similar to the helix observed in our previous MD simulations of Aβ-POPE bilayers.(21) The turn region has the lowest content of β-sheet followed by the C-terminus and both preserve relatively the same percentage of residues in this conformation.
2.6. Aβ-lipid interaction during SMD
The set goal of performing the SMD simulations is for the protofilament to pass the hydrophilic barrier of the headgroups and for the C-termini to interact with the lipid tails. The process is described in Fig. 7. We analyzed whether the decrease in the relative z-axis distance between the COM of the C-termini and that of the phosphorus (P) atoms plane correlated with the Ncontacts made between the C-termini and the tails. The plot of distance versus Ncontacts in Fig. 8 reveals that when z-distance values decreased, Ncontacts values increased. In the S2-POPE system, the protofilament passes through the headgroups more easily and thus most of the data is registered for distances between 0-2 Å. The lipids reoriented themselves and the tails hydrophobically interacted with the C-termini. In S2-POPC the protofilament spent more time just above the P atoms plane making few contacts with the lipids, following which it passed the barrier and protrudes deeper into the core of the membrane reaching 5 Å below the P atoms plane.
Figure 7:

Left column: representation of the SMD simulation at constant speed, resulting in the fibril insertion into the lipid bilayer. Right column: final conformations for the S2-POPE and S2-POPC systems, respectively.
Figure 8:

Upper panels: Contour plots for the distance versus Ncontacts histograms, showing maximum counts between 0-2 Å for S2-POPE (left) and between 5-10 Å for S2-POPC (right) simulations. Ncontacts during SMD simulations is evaluated between the C-termini in layer 1 of the fibril and the lipid tails, with a cutoff of 2.7 Å between heavy atoms. Lower panels: z-axis coordinates for charged residues in the N-terminus and turn region and for the C-termini versus time for S2-POPE (left) and S2-POPC (right) following the SMD simulations.
2.7. Aβ-lipid interaction subsequent to SMD
After performing the SMD simulations we ran short MD simulations for each system in order to assess if the interaction with the membrane is maintained. The MD runs were 14-ns and 26-ns long for S2-POPE and S2-POPC, respectively. As shown in Fig. 8 (second row), in the S2-POPE system the Aβ C-termini continue their interaction with the lipid tails, similarly to simulations in our previous study. However, in the S2-POPC case, the C-termini are expelled from the lipid tail region and move rapidly (10 ns) towards the surface of the membrane. Our SMD results offer a qualitative description of the behaviour of the protofilament interacting with the lipid bilayer hydrophilic barrier and highlight the difficulty of transferring a solution-formed β-sheet rich Aβ protofilament segment through the lipid headgroup region.
2.8. Conclusions
We studied molecular systems consisting of Aβ9–40 fibrillar octamers, constructed using atomistic constraints from solid-state NMR experiments,(22–24, 28) and POPE and POPC model lipid bilayers under a variety of initial conformations and relative orientations. We investigated how the different chemical composition and morphology of POPE and POPC headgroups influences the interaction between the protofilament and the lipids. In contrast to the strong, favorable electrostatic interaction between Aβ and POPE lipids observed in Ref. (21), our present study of the Aβ-POPC systems shows a significantly weaker affinity between the peptide protofilaments and the membrane. These results are illustrated clearly in the two animation movies provided in the SI. Interestingly, we find the S2-type conformation as the most prone to interacting directly with lipids, in agreement with our previous Aβ-POPE interaction studies.(21, 40) This is supported by the analysis of the number of hydrogen bonds, side chain contacts, hydration dynamics (Fig. S8) and interaction energies between the protofilament and the lipid headgroups for the S2 conformation. However, while the protofilament-membrane interactions are the strongest during the S2a simulation, this system is the only one in which we observed the initiation of the Aβ C-termini protrusion through the hydrophilic headgroups barrier and their direct interaction with the lipid tails. The primarily hydrophobic association between the C-termini and the lipid tails stems from the upwards movement of the latter, rather than from the insertion of the C-termini into the membrane core (as previously observed for POPE lipids (21)). This process was triggered by a POPC lipid tail regaining its normal orientation in the membrane, After a direct interaction with the C-terminus of an Aβ peptide. The fact that it has been previously shown that hydrophobic exposure is required for cytotoxicity (41) is strenghtening our conclusion that the S2 conformation- exhibiting the hydrophobic C-termini towards the membrane- is the ideal orientation for initiation of the interaction with the membrane. The upwards movement of the POPC lipid tails in the presence of the protofilament could be related to the detergent-like effect on membrane observed for amyloid-forming peptides, such as the hIAPP amyloid peptides,(42) which were shown to have the ability to extract lipids from membranes; this behaviour has been proposed also for Aβ.(43, 44) Our study brings the first computational evidence that Aβ peptides could also behave in such a manner when interacting with POPC lipids.
To further probe the weak affinity between Aβ protofilaments and the POPC membrane, we performed additional constant velocity pulling SMD simulations. The SMD runs were used to facilitate the protofilament insertion into the membranes consisting of both types of lipids. Once again, we used the conformation that exhibited the strongest interactions with two types of lipids: S2-POPC and S2-POPE. Although initial contacts had been established between Aβ peptides and the lipid tails, After SMD, the POPC lipids appear to expel the Aβ protofilament from the membrane, once again bringing into focus the importance of the electrostatic interactions. The analysis of the SMD runs showed that, in both systems, the fibrils can make contact with the lipid tails when being actively pulled along the −z direction (i.e., towards the membrane core). However, in the MD runs following SMD, the contacts are maintained only for the S2-POPE system. In the latter, following the insertion of the Aβ protofilament into the bilayer during SMD, the headgroups reorganize themselves around the peptides to maximize the electrostatic interaction, while lipid tails make favourable contacts with their C-termini. This finding indicates clearly that the POPC headgroups impose a significantly higher energy barrier to the insertion of Aβ protofilaments as compared to POPE lipids.
We note that Aβ peptides aggregated in solution could present different structures and toxicity than when formed on or near membranes.(45) Experimentally observed amyloid channels (46, 47) could thus be formed through different pathways than by insertion of solution-formed oligomers into the membrane. Cellular membranes are complex heterogeneous systems, also involving many types of molecules other than the model phospholipids used in our simulations. Other studies summarized the key membrane characteristics involved in membrane-mediated aggregation, stressing the importance of the presence of cholesterol and gangliosides in rafts in neuronal cells, which interact with Aβ and, even preceding this, with the amyloid precursor protein,(48) as the C99 structure depends on the lipid bilayer composition.(49) The importance of cholesterol and sphingolipids in binding and oligomerization of amyloidogenic proteins has been stressed before(50) and strengthened by newer studies on the role cholesterol plays in amyloid binding to membranes(51) and in amyloid channel formation.(37) Also, gangliosides in lipid rafts exert a chaperone effect on amyloidogenic proteins.(16, 52) Our studies show that spontaneous insertion of the Aβ C-termini into a lipid membrane core may occur when the energetic barrier imposed by the hydrophilic headgroups is overcome by one of two possible mechanisms: (a) in the case of Aβ-POPE systems, the headgroups are sufficiently small such that the strong electrostatic attraction between the charged residues in the protofilament and the lipids overcomes the headgroup barrier to membrane insertion,(21) and (b) in the case of the more voluminous POPC lipids, through upwards movement of the lipid tail and direct interaction with the Aβ and the subsequent lipid retraction/Aβ insertion. Since in our simulations the later mechanism involving lipid tails changing their orientation and traversing the hydrophilic headgroups barrier to interact with the hydrophobic regions of Aβ was a rare event, its likelihood of occurrence can only be assessed in future studies. Nevertheless, our results using solution-stable Aβ fibrillar oligomers and simple models of lipid membranes demonstrate that even in the same class of phospholipids, relatively minor chemical differences in the headgroups molecular architecture can lead to striking differences in the lipid membrane interactions with amyloidogenic peptide aggregates such as amyloid protofilaments. While biological membranes are notoriously more complex (e.g., including other factors such as cholesterol, and cell cycle-dependent lipid composition(53)), our results evidence a simple experimentally and computationally testable case for probing the factors that control the insertion of Aβ protofilaments in neuronal cell membranes. These findings could advance the understanding of molecular mechanisms underlying membrane-mediated fibril nucleation or Aβ channel formation processes, which play central roles in AD, type II diabetes and other amyloid-related diseases.
3. Experimental
We carried out the simulations using the NAMD(54) program with the CHARMM27 force field.(55) Each Aβ-lipids system consisted of between 74,000 and 89,000 atoms (see Table S1). Simulations were performed in an NPT ensemble, at constant number of particles, constant pressure and constant temperature of 310 K, close to the physiological temperature. The pressure was maintained constant at 1 atm using the Langevin piston method, and the temperature was controlled with Langevin dynamics.(55, 56) The integration step was 1 fs. We used the TIP3P explicit solvent model.(57) Visual Molecular Dynamics(58) was employed for generating the initial conditions, for monitoring the simulations and for partly analyzing the obtained data.(21, 40)
3.1. MD simulations of model lipid bilayers
In this study we used model POPE and pure POPC lipid bilayers. Details on the construction of the POPE bilayers can be found in our previous work(21), and for the POPC bilayer in Table S1. We built the bilayer by arranging 9 × 9 POPC lipids per leaflet and adding water 40 Å above (enough for later accommodating the fibril) and 10Å below the bilayer. To ensure their stability, the POPC bilayers were minimised (with fixed phosphorus atoms of the lipid headgroups), gradually heated, equilibrated (using restraints for the phosphorus atoms) and simulated in an NPT ensemble.
3.2. MD simulations of Aβ-POPC membrane systems
We used Aβ9–40 to model Aβ1–40, as experimental studies showed that the first eight residues in the peptide do not contribute to stabilizing the fibril structure. We placed the equilibrated fibrillar amyloid protofilament to the equilibrated lipid bilayers and removed the water molecules within 2.5 Å of the amyloid. We then minimized thoroughly the fibril-membrane systems (i.e., using initially a fixed backbone and fixed lipid phosphorus atoms), and we gradually heated and equilibrated (using initially restraints on the Cα and lipid phosphorus atoms) and simulated the system in the NPT ensemble. After constructing and equilibrating our molecular systems using an system preparation steps similar to our previous MD studies of amyloid aggregates(21, 40, 59), we generated long all-atom molecular dynamics (MD) trajectories ranging from 25 ns to 150 ns (see Table S1), for a total of almost 500 ns. For most cases, simulations were stopped when the oligomer lost contact with the membrane.
3.3. SMD simulations of fibril-lipid membrane systems
Testing the insertion of a large molecular ensemble into a membrane with voluminous head-groups would require a time scale that is not affordable by carrying out direct MD simulations. A method used to accelerate such processes is Steered Molecular Dynamics (SMD)(60, 61), that can be performed using the NAMD(54) package. SMD allows the application of controlled forces to selected atoms to drive them along a predefined direction, and has been use in other previous studies of amyloid fibrils.(62) We conducted SMD simulations on two systems in the S2 conformation, one containing a POPE and the other a POPC bilayer (henceforth referred to as S2-POPE and S2-POPC, respectively). Following heating and equilibration, we applied SMD on selected protofilament atoms at constant velocities in the −z direction (towards the bilayer) to explore the possible permeation pathways of the oligomer through the headgroup region. We used the same bilayers in the same NPT conditions as in our preceding MD simulations of the S2 systems. The details of the simulations are given in Table S2. We ran 10-ns and 20-ns segments of simulations at pulling speeds between 0.01 Å/ns and 1 Å/ns on selected Cα atoms. The speed was altered depending on the result of the previous simulation segment: it was increased if in the previous simulation segment the oligomer was drifting away from the membrane; it was decreased, if the oligomer pressed against the bilayer. To prevent the lipid bilayer from drifting under the force applied by the pressing protofilament, the phosphorus atoms of the lipids in the lower leaflet are constrained in the (x,y) plane.
3.4. Analysis
The secondary structure was analysed using STRIDE.(63) In the analysis of the number of hydrogen bonds (NHB), either within the fibril or between the fibril and the lipids, we used the same donor-acceptor distance and angle cutoff as for the Aβ-POPE simulations.(21) For plotting the Ue, we employed VMD and calculated the energy in the same conditions as in our simulations (PME, 8.5 Å switching distance and 10 Å cut-off distance).
Supplementary Material
Acknowledgement
The authors thank the DJEI/DES/SFI/HEA Irish Centre for High-End Computing (ICHEC), and the Biowulf Linux cluster at the National Institutes of Health, USA (http://biowulf.nih.gov) for the provision of computational facilities and support. F.T. and B.R.B. were supported by the intramural research program of the National Heart, Lung and Blood Institute, National Institutes of Health. F.T. and N.V.B are also grateful for financial support from the Irish Research Council.
Footnotes
Supporting Information Available: This article contains supporting information (SI) including Tables S1 and S2, Figs. S1-S8, and two animations (movies S1 and S2) illustrating representative trajectories of Aβ-POPE and Aβ-POPC membrane interactions, respectively, as described in the SI document. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Sawamura N, Morishima-Kawashima M, Waki H, Kobayashi K, Kuramochi T, Frosch MP, Ding K, Ito M, Kim TW, Tanzi RE, Oyama F, Tabira T, Ando S, and Ihara Y (2000) Mutant presenilin 2 transgenic mice: A large increase in the levels of Aβ 42 is presumably associated with the low density membrane domain that contains decreased levels of glycerophospholipids and sphingomyelin. J. Biol. Chem. 275, 27901–27908. [DOI] [PubMed] [Google Scholar]
- 2.Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, Lu Y, Felix AM, Maggio JE, and Lee JP (2000) The Alzheimer’s peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 130, 130–141. [DOI] [PubMed] [Google Scholar]
- 3.Vivekanandan S, Brender JR, Lee SY, and Ramamoorthy A (2011) A partially folded structure of amyloid-β(1-40) in an aqueous environment. Biochem. Biophys. Res. Commun. 411, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danielsson J, Jarvet J, Damberg P, and Graslund A (2005) The Alzheimer β-peptide shows temperature-dependent transitions between left-handed 3(1)-helix, beta-strand and random coil secondary structures. FEBS J. 272, 3938–3949. [DOI] [PubMed] [Google Scholar]
- 5.Niu Z, Zhao W, Zhang Z, Xiao F, Tang X, and Yang J (2014) The Molecular Structure of Alzheimer β-Amyloid Fibrils Formed in the Presence of Phospholipid Vesicles. Angew. Chem., Int. Ed. 53, 9294–9297. [DOI] [PubMed] [Google Scholar]
- 6.Morriss-Andrews A, Brown FLH, and Shea J-E (2014) A Coarse-Grained Model for Peptide Aggregation on a Membrane Surface. J. Mol. Biol. 118, 8420–8432. [DOI] [PubMed] [Google Scholar]
- 7.Hertel C, Terzi E, Hauser N, JakobRotne R, Seelig J, and Kemp JA (1997) Inhibition of the electrostatic interaction between β-amyloid peptide and membranes prevents beta-amyloid-induced toxicity. Proc. Natl. Acad. Sci. U. S. A. 94, 9412–9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao HX, Tuominen EKJ, and Kinnunen PKJ (2004) Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 43, 10302–10307. [DOI] [PubMed] [Google Scholar]
- 9.Kubo S, Nemani VM, Chalkley RJ, Anthony MD, Hattori N, Mizuno Y, Edwards RH, and Fortin DL (2005) A combinatorial code for the interaction of alpha-synuclein with membranes. J. Biol. Chem. 280, 31664–31672. [DOI] [PubMed] [Google Scholar]
- 10.Bokvist M, Lindstrom F, Watts A, and Grobner G (2004) Two types of Alzheimer’s β-amyloid (1-40) peptide membrane interactions: Aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 335, 1039–1049. [DOI] [PubMed] [Google Scholar]
- 11.Aisenbrey C, Borowik T, Bystrom R, Bokvist M, Lindstrom F, Misiak H, Sani MA, and Grobner G (2008) How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur. Biophys. J. 37, 247–255. [DOI] [PubMed] [Google Scholar]
- 12.Lau TL, Ambroggio EE, Tew DJ, Cappai R, Masters CL, Fidelio GD, Barnham KJ, and Separovic F (2006) Amyloid-β peptide disruption of lipid membranes and the effect of metal ions. J. Mol. Biol. 356, 759–770. [DOI] [PubMed] [Google Scholar]
- 13.de Planque MRR, Raussens V, Contera SA, Rijkers DTS, Liskamp RMJ, Ruysschaert JM, Ryan JF, Separovic F, and Watts A (2007) β-sheet structured β-amyloid(1-40) perturbs phosphatidylcholine model membranes. J. Mol. Biol. 368, 982–997. [DOI] [PubMed] [Google Scholar]
- 14.Wong PT, Schauerte JA, Wisser KC, Ding H, Lee EL, Steel DG, and Gafni A (2009) Amyloid-β Membrane Binding and Permeabilization are Distinct Processes Influenced Separately by Membrane Charge and Fluidity. J. Mol. Biol. 386, 81–96. [DOI] [PubMed] [Google Scholar]
- 15.Hane F, Drolle E, Gaikwad R, Faught E, and Leonenko Z (2011) Amyloid-β Aggregation on Model Lipid Membranes: An Atomic Force Microscopy Study. J. Alzheimer’s Dis. 26, 485–494. [DOI] [PubMed] [Google Scholar]
- 16.Sasahara K, Morigaki K, and Shinya K (2013) Effects of membrane interaction and aggregation of amyloid β-peptide on lipid mobility and membrane domain structure. Phys. Chem. Chem. Phys. 15, 8929–8939. [DOI] [PubMed] [Google Scholar]
- 17.Engel MFM, Khemtemourian L, Kleijer CC, Meeldijk HJD, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, and Hoppener JWM (2008) Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. U. S. A. 105, 6033–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams TL, Day IJ, and Serpell LC (2010) The Effect of Alzheimer’s Aβ Aggregation State on the Permeation of Biomimetic Lipid Vesicles. Langmuir 26, 17260–17268. [DOI] [PubMed] [Google Scholar]
- 19.Kremer JJ, and Murphy RM (2003) Kinetics of adsorption of β-amyloid peptide Aβ(1-40) to lipid bilayers. J. Biochem. Biophys. Methods 57, 159–69. [DOI] [PubMed] [Google Scholar]
- 20.Poojari C, and Strodel B (2013) Stability of Transmembrane Amyloid β-Peptide and Membrane Integrity Tested by Molecular Modeling of Site-Specific Aβ 42 Mutations. PLoS One 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tofoleanu F, and Buchete N-V (2012) Molecular Interactions of Alzheimer’s A Protofilaments with Lipid Membranes. J. Mol. Biol. 421, 572–586. [DOI] [PubMed] [Google Scholar]
- 22.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, and Tycko R (2002) A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U. S. A. 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petkova AT, Yau WM, and Tycko R (2006) Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchete N-V, Tycko R, and Hummer G (2005) Molecular Dynamics Simulations of Alzheimer’s β-Amyloid Protofilaments. J. Mol. Biol. 353, 804–821. [DOI] [PubMed] [Google Scholar]
- 25.Vance JE, and Tasseva G (2013) Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta 1831, 543–54. [DOI] [PubMed] [Google Scholar]
- 26.Haass C, and Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. [DOI] [PubMed] [Google Scholar]
- 27.Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, and Chiti F (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147. [DOI] [PubMed] [Google Scholar]
- 28.Buchete NV, and Hummer G (2007) Structure and dynamics of parallel β-sheets, hydrophobic core, and loops in Alzheimer’s Aβ fibrils. Biophys. J. 92, 3032–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glabe CG (2009) Amyloid Oligomer Structures and Toxicity. Open Biol. J. 2, 222–227. [Google Scholar]
- 30.Murzyn K, Rog T, and Pasenkiewicz-Gierula M (2005) Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 88, 1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasenkiewicz-Gierula M, Murzyn K, Rog T, and Czaplewski C (2000) Molecular dynamics simulation studies of lipid bilayer systems. Acta Biochim. Pol. 47, 601–11. [PubMed] [Google Scholar]
- 32.Kandasamy SK, and Larson RG (2007) Binding modes of protegrin-1, a β-strand antimicrobial peptide, in lipid bilayers. Mol. Simul. 33, 799–807. [Google Scholar]
- 33.Shai Y (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta, Biomembr. 1462, 55–70. [DOI] [PubMed] [Google Scholar]
- 34.Urbanc B, Betnel M, Cruz L, Li H, Fradinger EA, Monien BH, and Bitan G (2011) Structural Basis for Aβ(1-42) Toxicity Inhibition by Aβ C-Terminal Fragments: Discrete Molecular Dynamics Study. J. Mol. Biol. 410, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giacomelli CE, and Norde W (2005) Conformational changes of the amyloid β-peptide (1-40) adsorbed on solid surfaces. Macromol. Biosci. 5, 401–7. [DOI] [PubMed] [Google Scholar]
- 36.Dominguez L, Meredith SC, Straub JE, and Thirumalai D (2014) Transmembrane Fragment Structures of Amyloid Precursor Protein Depend on Membrane Surface Curvature. J. Am. Chem. Soc. 136, 854–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Scala C, Troadec JD, Lelievre C, Garmy N, Fantini J, and Chahinian H (2014) Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer β-amyloid peptide. J. Neurochem. 128, 186–95. [DOI] [PubMed] [Google Scholar]
- 38.Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, and Meredith SC (2005) Aβ40-Lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry 44, 6003–14. [DOI] [PubMed] [Google Scholar]
- 39.Reddy G, Straub JE, and Thirumalai D (2009) Influence of preformed Asp23-Lys28 salt bridge on the conformational fluctuations of monomers and dimers of Aβ peptides with implications for rates of fibril formation. J. Phys. Chem. B 113, 1162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tofoleanu F, and Buchete NV (2012) Alzheimer A beta peptide interactions with lipid membranes Fibrils, oligomers and polymorphic amyloid channels. Prion 6, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ladiwala AR, Litt J, Kane R, Aucoin D, Smith S, Ranjan S, Davis J, Nostrand W, and Tessier P (2012) Conformational Differences between Two Amyloid β Oligomers of Similar Size and Dissimilar Toxicity. J. Biol. Chem. 287, 24765–24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sparr E, Engel MFM, Sakharov DV, Sprong M, Jacobs J, de Kruijff B, Hoppener JWM, and Killian JA (2004) Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 577, 117–120. [DOI] [PubMed] [Google Scholar]
- 43.Butterfield SM, and Lashuel HA (2010) Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem., Int. Ed. 49, 5628–54. [DOI] [PubMed] [Google Scholar]
- 44.Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R, and Kapurniotu A (2010) Identification of Hot Regions of the Aβ-IAPP Interaction Interface as High-Affinity Binding Sites in both Cross- and Self-Association. Angew. Chem., Int. Ed. 49, 3081–3085. [DOI] [PubMed] [Google Scholar]
- 45.Kotler SA, Walsh P, Brender JR, and Ramamoorthy A (2014) Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 43, 6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jang H, Connelly L, Teran Arce F, Ramachandran S, Kagan BL, Lal R, and Nussinov R (2013) Mechanisms for the Insertion of Toxic, Fibril-like β-Amyloid Oligomers into the Membrane. J. Chem. Theory Comput. 9, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jang H, Arce F, Ramachandran S, Kagan B, Lal R, and Nussinov R (2013) Conformational Differences between Two Amyloid β Oligomers of Similar Size and Dissimilar Toxicity. J. Phys. Chem. B 117, 11518–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Straub JE, and Thirumalai D (2014) Membrane-Protein Interactions Are Key to Understanding Amyloid Formation. J. Phys. Chem. Lett. 5, 633–635. [DOI] [PubMed] [Google Scholar]
- 49.Song Y, Mittendorf KF, Lu Z, and Sanders CR (2014) Impact of bilayer lipid composition on the structure and topology of the transmembrane amyloid precursor c99 protein. J. Am. Chem. Soc 136, 4093–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fantini J, and Yahi N (2010) Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: common mechanisms in neurodegenerative diseases. Expert. Rev. Mol. Med. 12, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fantini J, Yahi N, and Garmy N (2013) Cholesterol accelerates the binding of Alzheimer’s β-amyloid peptide to ganglio-side GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 4, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, and Matsuzaki K (2002) Interactions of amyloid β-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 41, 7385–90. [DOI] [PubMed] [Google Scholar]
- 53.Atilla-Gokcumen G, Muro E, Relat-Goberna J, Sasse S, Bedigian A, Coughlin M, Garcia-Manyes S, and Eggert U (2014) Dividing Cells Regulate Their Lipid Composition and Localization. Cell 156, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, and Schulten K (1999) NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312. [Google Scholar]
- 55.MacKerell AD et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 56.Feller SE, Zhang YH, Pastor RW, and Brooks BR (1995) Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J. Chem. Phys. 103, 4613–4621. [Google Scholar]
- 57.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983) Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- 58.Humphrey W, Dalke A, and Schulten K (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–&. [DOI] [PubMed] [Google Scholar]
- 59.Kelly CM, Northey T, Ryan K, Brooks BR, Kholkin AL, Rodriguez BJ, and Buchete N-V (2015) Conformational dynamics and aggregation behavior of piezoelectric diphenylalanine peptides in an external electric field. Biophys. Chem. 196, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grubmüller H, Heymann B, and Tavan P (1996) Ligand Binding: Molecular Mechanics Calculation of the Streptavidin-Biotin Rupture Force. Science 271, 997–999. [DOI] [PubMed] [Google Scholar]
- 61.Izrailev S, Stepaniants S, Balsera M, Oono Y, and Schulten K (1997) Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72, 1568–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raman EP, Takeda T, Barsegov V, and Klimov DK (2007) Mechanical Unbinding of A Peptides from Amyloid Fibrils. J. Mol. Biol. 373, 785–800. [DOI] [PubMed] [Google Scholar]
- 63.Heinig M, and Frishman D (2004) STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 32, W500–W502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.