

Abstract
Predicting clinical outcomes in cancer using neoantigen burden is imperfect because current algorithms use only the binding affinity of putative neoantigens to HLA. A new study models pancreatic tumour response through a deeper understanding of tumour immunology, providing new tools for identifying neoantigens and characteristics that define their quality.
The field of cancer immunology hypothesizes that tumours with the greatest numbers of mutations respond best to immunotherapy1. Nowhere is this phenomenon clearer than in the differences observed in response rates between patients with DNA mismatch repair deficiency and those with intact DNA repair mechanisms when treated with checkpoint inhibitors2. A greater mutation burden typically results in increased neoantigen expression by tumour cells, which provides a greater pool of potential epitopes (the part ofthe antigen recognized by the immune system) to be displayed on the tumour cell surface by HLA molecules. It is assumed that presentation of more neoantigens leads to better antitumour immune responses; however, this is not the whole story. Not all neoepitopes predicted to bind to the HLA molecule ultimately lead to any immune response, and only a minority of these epitopes seem capable of inducing tumour-killing CD8+ T cells3.
In a study published in Nature, Balachandran and colleagues4 describe a new effort to model tumour response to immunotherapy by identifying a subset of high-quality neoantigens present in tumours of patients with pancreatic cancer. As with previous efforts, the authors began by performing exome sequencing on pancreatic tumours to identify somatic coding mutations and their resulting putative neoantigens. Using existing in silico tools, each peptide in this pool was then scored for its HLA binding potential relative to the wild-type peptide, assuring that the resulting candidate neoantigens were sufficiently different from self-antigens. Next, the authors developed and tested two competing methods for predicting tumour response that they term the quantity and quality models. In both models, the tumour response is represented as a phylogenetic tree in which the survival probability of each clone is determined by its immunogenicity. In the quantity model, immunogenicity is treated as a function of the number of neoantigens present (defined as candidates exceeding an HLA binding threshold), whereas the quality model treats immunogenicity as a function of the quality score of the best predicted antigen, calculated using its physical similarity to pathogen-derived antigens known to elicit T cell responses (FIG. 1). The quality model was better able to predict overall survival than the quantity model in two separate cohorts of patients (n = 58 and n = 166 patients). The authors identify two potential explanations for this performance disparity: in an example of molecular mimicry, T cell receptors (TCRs) that recognize pathogen-derived antigens also recognize physically similar cancer neoantigens; or scoring peptides in this manner simply selects neoantigens with less resemblance to self-peptides.
Figure 1 |. Tumour evolution in response to immune pressure in pancreatic cancer.
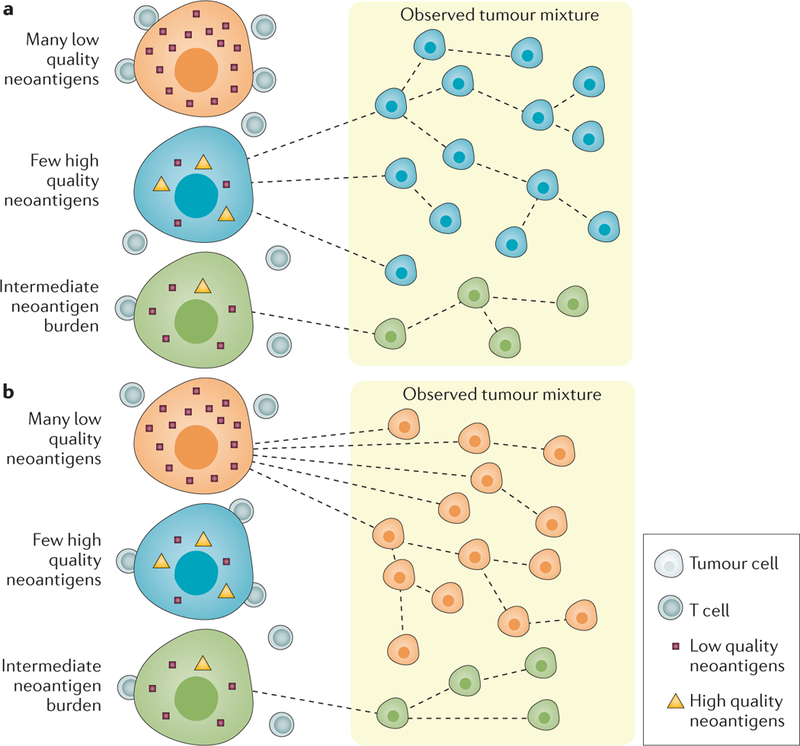
Tumour evolution is modelled using the quantity model (part a), in which survival is a function of the number of HLA binding neoantigens in each tumour clone, or the quality model (part b), in which the survival of a clone depends on high-quality neoantigens, identified by their ability to bind HLA as well as their similarity to known pathogen-derived antigens.
If neoantigens identified using the quality model are important for an antitumour immune response, the authors hypothesized that an immunoediting process, in which clones presenting high-quality neoantigens are killed by CD8+ T cells, should result in selection of clones with reduced antigen expression. This hypothesis was confirmed in a representative patient in which all three high-quality antigens identified in the primary tumour were undetectable in metastases. Additionally, T cells responding to neoantigens defined using the quality model were identified in peripheral blood and archival tumours from the majority of a cohort of seven very long-term survivors of pancreatic cancer (median survival 10.5 years), providing further evidence that the quality model reveals important immune targets. In all seven patients studied, identical TCR clones were shown to expand in response to both tumour-derived high-quality neoantigens and physically similar pathogen-derived antigens, providing evidence for the molecular mimicry hypothesis. This theory, which states that TCRs specific for pathogen-derived antigens are also able to recognize cancer-derived neoantigens, has been proposed as a potential mechanism for development of antitumour T cells5, but has never been demonstrated.
“The quality model was better able to predict overall survival than the quantity model... ”
Finally, the authors implicated the MUC16 gene, previously shown in a preclinical model of ovarian cancer to harbour targetable neoantigens6, as a hotspot of neoantigen-generating mutations in the long-term survivor cohort. High-quality MUC16-derived neoantigens identified in primary tumours were undetectable in metastases, whereas low-quality candidates remained unchanged or increased in abundance, providing further evidence of a survival advantage for clones with a reduced burden of high-quality neoantigens.
Balachandran et al. create a new framework for conceptualizing tumour growth in the context of immune suppression by modelling not only the neoantigen-HLA interaction, but also the often-neglected TCR recognition interaction. The addition of a quality score arising from pathogen-derived antigen similarity was shown to improve the predictive power of the model, demonstrating that current methods relying solely on HLA binding algorithms for neoantigen prediction have room for improvement. Additionally, the inclusion of the allele frequency of each neoantigen in the framework improves accuracy by acknowledging the importance of tumour heterogeneity. The authors show both the predictive power of this model in individuals who survived pancreatic cancer, and its ability to identify biologically relevant neoantigens. In the process, the authors provide evidence that molecular mimicry might be not only possible, but more common than originally thought. By shifting the conversation away from the quantity of neoantigens and towards the quality of expressed neoantigens, the authors highlight the importance of immunotherapy in diseases like pancreatic cancer, which typically harbour very few mutations7.
In summary, Balachandran and colleagues have contributed to the field of cancer immunotherapy by enhancing existing immunogenicity models, while also providing valuable insights into clonal evolution and the factors that make a successful tumour antigen. Improvements such as these to neoantigen discovery methods could dramatically improve vaccine design efforts. Furthermore, because their novel methods rely on in silico techniques, these findings are also easily translatable to the clinic.
Footnotes
Competing interests statement
E.J. has the potential to receive royalties from Aduro Biotech for vaccines developed in her laboratory.
References
- 1.Mehnert JM et al. The challenge for development of valuable immuno-oncology biomarkers. Clin. Cancer Res. 23, 4970–4979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le DT et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahin U et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Balachandran VP et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 10.1038/nature24462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zitvogel L, Ayyoub M, Routy B & Kroemer G Microbiome and anticancer immunosurveillance. Cell 165, 276–287 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Chekmasova AA et al. Successful eradication of established peritoneal ovarian tumors in SCID-beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin. Cancer Res. 16, 3594–3606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalmers ZR et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]