

Abstract
One-electron oxidized guanine (G•+) in DNA generates several short-lived intermediate radicals via proton transfer reactions resulting in the formation of neutral guanine radicals (scheme 1). The identification of these radicals in DNA is of fundamental interest to understand the early stages of DNA damage. Herein, we used time-dependent density functional theory (TD-ωB97XD-PCM/6-31G(3df,p)) to calculate the vertical excitation energies of one electron oxidized G and guanine(G)-cytosine(C) base pair in various protonation states: G•+, G(N1-H)• and G(N2-H)•, as well as G•+-C, G(N1-H)•-(H+)C, G(N1-H)•-(N4-H+)C), G(N1-H)•-C and G(N2-H)•-C in aqueous phase. The calculated UV-vis spectra of these radicals are in good agreement with experiment for the G radical species when the calculated values are red-shifted by 40 - 70 nm. The present calculations show that the lowest energy transitions of proton transferred species (G(N1-H)•-(H+)C, G(N1-H)•-(N4-H+)C), and G(N1-H)•-C) are substantially red-shifted in comparison to the spectrum of G•+-C. The calculated spectrum of G(N2-H)•-C shows intense absorption (high oscillator strength) which matches the strong absorption in the experimental spectra of G(N2-H)• at 600 nm. The present calculations predict the lowest charge transfer transition from C→G•+ is π→π* in nature and lies in the UV-region (3.4 – 4.3 eV) with small oscillator strength.
Graphical Abstract
Introduction
The exposure of high-energy or ultraviolet (UV) radiation to cellular DNA excites as well as ionizes DNA bases resulting in a variety of base damages.1–3 In DNA, all nucleobases may potentially be ionized, however, guanine is the most easily ionized as it has the lowest ionization potential compared to other native DNA bases.3–6 For this reason guanine is known as a hole transfer sink in DNA.3,4,7 The individual DNA base cation radicals in nucleosides as well as in DNA have been extensively studied using pulse radiolysis, ESR and photochemistry experiments3,4,6–9 and theory.10–13 Many works have identified the UV-vis spectra of individual nucleoside cation radicals and their neutral deprotonated forms13–18, however, the absorption spectra of oxidized-DNA or base pairs are less well identified and a subject of some controversy.19–23 Their identification is made difficult owing to various factors such as hydrogen-bonding, sequence, stacking interactions, and protonation/deprotonation reactions.19–24 Very recently, using nanosecond transient absorption spectroscopy with 266 nm excitation, Markovitsi and coworkers20 recorded the absorption spectra of one-electron oxidized duplex DNA of ten base pairs having alternate guanine-cytosine sequence. With the aid of time-dependent density functional theory (TD-DFT) calculations the transient absorption spectra was characterized as deprotonated guanine radical (G(N1-H)•. To match the experimental spectra the TD-DFT calculated spectra were red-shifted by 0.6 eV.20
Recently, ESR and UV-visible spectroscopy was used to characterize the radicals generated in one-electron oxidized DNA oligomer d[TGCGCGCA]2 at different pHs.19 The ESR and UV-visible spectra of one-electron oxidized 1-methylguanosine were used as benchmark to identify the radicals in one-electron oxidized d[TGCGCGCA]2. The G(N2-H)• generated from one-electron oxidized 1-methylguanosine has characteristic absorption band around ca. 600 nm.14 At pH ≥ 7 the initial radical in one-electron oxidized d[TGCGCGCA]2 was characterized as G(N1-H)• at 155 K. On further annealing to 175 K, an equilibrium mixture of G(N1-H)• and G(N2-H)• was identified. The ESR hyperfine couplings of N2 (Azz = 16 Gauss) and the N2-H proton coupling 8 Gauss in water were obtained which clearly identified G(N2-H)• in one-electron oxidized d[TGCGCGCA]2.19 The recorded UV-visible spectra of one-electron oxidized d[TGCGCGCA]2 having absorption band around ca. 600 nm also supports G(N2-H)• formation in one-electron oxidized d[TGCGCGCA]2. The relative stability of G(N1-H)•-C and G(N2-H)•-C in the gas phase25 and in solution19 was calculated using the B3LYP method and G(N2-H)•-C was found to be slightly more stable than G(N1-H)•-C by 1.0 kcal/mol (gas phase) and 0.4 kcal/mol (solution).
In this work, we have employed TD-DFT (TD-ωB97XD method) to investigate the vertical transition energies of radicals which are likely generated during one-electron oxidation of G-C base pair in DNA with the goal of finding an appropriate and predictive method to aid the identification of various one electron oxidized G-C species via UV-vis spectroscopy. The suitability of the chosen TD-ωB97XD method is assessed by comparing it and two other methods, TD-ωB97X and M05–2X, with the available equation of motion coupled-cluster (EOM-CCSD(T))26 and coupled cluster singles and doubles (CC2)27 calculated excited state energies of G-C base pair in the gas phase. EOM-CCSD(T) method26 has been used as a benchmark to assess the suitability of the TD-DFT to calculate the excited states of DNA base dimers and tetramers.28 In Scheme 1, we present the structures of different radicals which are considered in this study. The nomenclature of these radicals presented in scheme 1(a) to 1(e) are: 1(a) G•+-C (the initial one-electron oxidized G-C base pair); 1(b) G(N1-H)•-(H+)C, (the intra-base proton transfer species); 1(c) (G(N1-H)•-(N4-H+)C) (the species formed by the deprotonation of 1(b) from N4 of C to solvent)29,30; 1(d) G(N1-H)•-C (the species formed by base shift and proton rearrangement of 1(c) or 1(e)19,25,30; and 1(e) G(N2-H)•-C, (species formed by deprotonation of G•+-C from N2 of G to solvent)19.
Scheme 1.

Molecular structures of different possible radicals produced during one electron oxidation of G-C base pair in DNA. (a) G•+-C, (b) G(N1-H)•-(H+)C, (c) proton transfer from N4 of C to solvent (G(N1-H)•-(N4-H+)C), (d) displaced base pair held by two hydrogen bonds G(N1-H)•-C and (e) deprotonation from N2 of G to solvent G(N2-H)•-C. Pink, blue and green circles highlight the N1(G), N2(G) and N4(C) protons which are involved in proton transfer reactions.
Methods of Calculation
The ground state geometries of radicals shown in scheme 1 are fully optimized using the ωB97XD method and 6–31g(3df,p) basis set. ωB97XD is a long-range corrected hybrid density functional with damped atom–atom dispersion corrections developed by Chai and Head-Gordon.31,32 This functional (ωB97XD) has been found reliable for the calculation of excited states including charge transfer (CT) excited states than results found using earlier density functionals.31–33 For the calculation of vertical excited state transition energies, the time-dependent variant (TD-ωB97XD/6–31G(3df,p)) was used considering the ωB97XD/6–31G(3df,p) optimized ground state geometries. The ground state geometry optimization and excited state calculations were carried out in the aqueous phase via the integral equation formalism of the polarized continuum model (IEF-PCM) of Tomasi et al.34 For IEF-PCM calculation the cavity of the solvent treated as continuum was generated using the default options set into the Gaussian program. The complete methodology for excited state calculation is abbreviated as TD-ωB97XD-PCM/6–31g(3df,p). Total 20 transition energies were calculated for each chosen radicals. All the calculations were carried out using Gaussian 16 suite of programs.35 Gabedit36 software was used to generate data points to plot the calculated absorption spectrum and IQMOL37 molecular modeling programs was used to plot molecular orbitals. In the calculation, the hydrogens at N9 of guanine and at N1 of cytosine in hydrogen-bonded G-C base pair radicals, shown in scheme 1, were substituted by methyl group to mimic the effect of deoxyribose (sugar) ring as these sites are attached to the sugar ring in DNA.
Results and Discussion
(i). Suitability of the TD-ωB97XD method.
To test the suitability of the TD-DFT method for excited state calculations, we considered G-C base pair as a test case and calculated the singlet vertical excitation energies using ωB97X, ωB97XD and M05–2X methods with 6–31G(d), 6–31G(d, p), 6–31++G(d, p), 6–31G(3df, p) and TZVP basis sets. The calculated excitation energies of G-C base pair by these chosen TD-DFT methods are compared with those calculated using ab initio EOM-CCSD(T)26 and CC227 level of theories, see Table 1 and Tables S1 – S4 in the supporting information. In Table 1, we present the vertical excitation energies of G-C base pair calculated by ωB97X, ωB97XD and M05–2X with the 6–31G(3df, p) basis set along with those calculated by EOM-CCSD(T)26 and CC227 methods.
Table 1.
Comparison of methods: vertical singlet excited state energies (ΔE) in eV and oscillator strength (f) of G–C base pair calculated with EOM-CCSD(T), CC2/TZVP, ωb97x/6-31G(3df,p), M05-2x/6-31G(3df,p) and ωb97xd/6-31G(3df,p) in the gas phase. Charge transfer (CT) transitions are shown in bold.
| Guanine (G)-Cytosine (C) base pair | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| State | CC2/TZVPa | EOM-CCSD(T)b | ωb97x/6-31G(3df,p) | M05-2x/6-31G(3df,p) | ωb97xd/6-31G(3df,p) | |||||
| Transition | ΔE (f) | Transition | ΔE (f) | Transition | ΔE (f) | Transition | ΔE (f) | Transition | ΔE (f) | |
| S1 | G(π)→G(π)* | 4.88 (0.061) | G(π)→G(π)* | 4.85 (0.07) | G(π)→G(π)* | 5.19 (0.0666) |
G(π)→C(π)* (CT) |
5.06 (0.0411) | G(π)→G(π)* | 5.04 (0.0682) |
| S2 | C(π)→C(π)* | 5.06 (0.063) | C(π)→C(π)* | 4.92 (0.10) | C(π)→C(π)* | 5.32 (0.1753) | C(π)→C(π)* | 5.31 (0.1738) | C(π)→C(π)* | 5.20 (0.1523) |
| S3 |
G(π)→C(π)* (CT) |
5.23 (0.028) |
G(π)→C(π)* (CT) |
5.36 (0.01) | G(π)→G(π)* | 5.82 (0.4531) | G(π)→C(π)* G(π)→G(π)* |
5.32 (0.0347) |
G(π)→C(π)* (CT) |
5.53 (0.0038) |
| S4 | C(n)→C(π)* | 5.49 (0.001) | G(π)→G(π)* | 5.48 (0.41) | GC(n)→C(π)* G(n)→G(π)* |
5.94 (0.0011) | G(π)→G(π)* | 5.74 (0.4392) | G(π)→G(π)* | 5.62 (0.3981) |
| S5 | C(π)→C(π)* | 5.50 (0.183) | C(n)→C(π)* | 5.65 (0.00) | GC(n)→C(π)* G(n)→G(π)* |
6.02 (0.0000) | G(n)→G(π)* | 5.79 (0.0007) | C(n)→C(π)* G(n)→C(π)* |
5.73 (0.0010) |
| S6 | G(π)→G(π)* | 5.51 (0.425) | G(n)→G(π)* | 5.76 (0.00) | GC(π)→C(π)* | 6.09 (0.1232) | C(n)→C(π)* | 5.89 (0.0004) | GC(π)→C(π)* | 5.83 (0.0956) |
| S7 | C(n)→C(π)* | 5.80 (0.000) |
G(π)→C(π)* (CT) |
6.27 (0.0020) | GC(π)→C(π)* | 6.04 (0.0967) | G(n)→G(π)* | 5.90 (0.0001) | ||
| S8 | G(n)→G(π)* | 5.90 (0.000) | G(n)→G(π)* | 6.54 (0.0022) | G(n)→G(π)* | 6.35 (0.0019) | G(n)→G(π)* | 6.32 (0.0014) | ||
| S9 | C(n)→C(π)* | 6.60 (0.0001) | GC(n)→C(π)* | 6.41 (0.0000) | C(n)→C(π)* | 6.50 (0.0000) | ||||
From Table 1, we see that TD-ωB97XD/6–31G(3df, p) predicts the lowest two transitions (S1 and S2) as local excitations (LE) which are 1ππ* type. The corresponding transition energies are 5.04 eV and 5.20 eV, respectively, with oscillator strength 0.0682 and 0.1523. The third transition (S3) is a charge transfer (CT) in nature 1G(π)→C(π)* and occurs at 5.53 eV with oscillator strength 0.0038. The S4 – S8 transitions are 1ππ*, 1nπ*, 1ππ*, 1nπ* and 1nπ* and have transition energies (oscillator strength) as 5.62 (0.3981), 5.73 (0.0010), 5.83 (0.0956), 5.90 (0.0001) and 6.32 (0.0014), respectively. Szalay et al. calculated the singlet excited states of G-C base pair in the gas phase using the EOM-CCSD(T)/TZVP level of theory. The EOM-CCSD(T)/TZVP calculated S1 – S6 transitions are 1ππ*(LE), 1ππ*(LE), 1ππ*(CT), 1ππ*(LE), 1nπ*(LE) and 1nπ*(LE) types and their corresponding transition energies (oscillator strength) are 4.85 (0.07), 4.92 (0.10), 5.36 (0.01), 5.48 (0.41), 5.65 (0.00) and 5.76 (0.00), respectively. The S6 transition at 5.76 eV predicted by EOM-CCSD(T)/TZVP is 1nπ*(LE), while TD-ωB97XD/6–31G(3df, p) predicts S6 and S7 transitions very close in energy as 1ππ* (5.83 eV), 1nπ* (5.90 eV), respectively. Thus, we found that TD-ωB97XD/6–31G(3df, p) correctly predicts S1 – S5 transitions as obtained by EOM-CCSD(T)/TZVP level of theory. The TD-ωB97XD/6–31G(3df, p) calculated transition energies are, also, in very good agreement with those calculated by the EOM-CCSD(T)/TZVP level of theory having a maximum difference of 0.28 eV with the average only 0.12 eV larger than the EOM-CCSD(T)/TZVP calculated values, see Table 1.
Recently, TD-M05–2X has been suggested to be suitable method for excited state calculations of DNA ion radicals.15,20,38,39 However, we find the TD-M05–2X/6–31G(3df, p) calculation predicts first lowest transition S1 as the charge transfer 1G(π)→C(π)* and S2 and S3 transitions as 1ππ*(LE) and 1ππ*(LE) in nature, see Table 1. Whereas, EOM-CCSD(T)/TZVP predicts S1 – S3 transitions as 1ππ*(LE), 1ππ*(LE) and 1G(π)→C(π)*(CT) types and LE occur on G, C. Thus, TD-M05–2X/6–31G(3df, p) incorrectly predicts the nature of the lowest transition and further transition energies are over estimated (ca. 0.5 eV) in comparison to the EOM-CCSD(T)/TZVP calculated values, see Table 1. From Table 1, we found that TD-ωb97x/6–31G(3df,p) method correctly predicts the nature of the S1 and S2 transitions as obtained by EOM-CCSD(T)/TZVP method but fails to predict the S3 transition as a CT type. On the whole, the vertical excitation energies were systematically overestimated by around 0.4 eV. Thus, from our initial test, we conclude that among TD-M05–2X, TD-ωb97x and TD-ωB97XD methods, the TD-ωB97XD functional is the best choice for present study with an average only 0.12 eV larger than the EOM-CCSD(T)/TZVP values.
(ii). Vertical excited states of one-electron oxidized guanine.
The absorption spectra of one-electron oxidized deoxyguanosine (dGuo) at pHs 3.1 and 6.6 and 1-methylgaunosine (1-metGuo) at pH 7.3 were reported by Candeias and Steenken14 using pulse radiolysis. The spectra of deoxyguanosine at pH 3.1 was characterized as deoxyguanosine radical cation (dGuo•+) and at pH 6.6 was characterized as N1-deprotonated neutral radical (dGuo(N1-H)•). The spectra of 1-metGuo at pH 7.3 was characterized as N2-deprotnated neutral radical (1-met-Guo(N2-H)•). It should be noted that N1 and N2 deprotonation have similar pKa’s with N1 favored slightly. Methyl substation at N1 forces the N2 deprotonation.
The absorption spectrum of dGuo•+ shows absorptions at around 300, 385 nm and a broad band extending from 470 – 550 nm, respectively. The TD-ωb97xd-PCM/6–31G(3df,p) calculated transitions of dGuo•+ were presented in Table S5 in the supporting information. From a comparison between the calculated and experimental transitions we found that the calculated transitions must be red-shifted by ca. 40 nm to match the first two higher energy transitions, see Table S5 in the supporting information. The low energy transition is very broad in the experimental spectrum but it appears that the shift needed would be less than 40 nm. The simulated spectrum of dGuo•+ along with the experimental spectrum is shown in Figure 1(a).
Figure 1.

TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical absorption spectrum of (a) deoxyguanosine radical cation (dGuo•+) (yellow color) and 40 nm red-shifted (red color). (b) deoxyguanosine radical (dGuo(N1-H)•) (yellow color) and 50 nm red-shifted (red color) and (c) 1-methylgaunosine radical (1-met-Guo(N2-H)•) (yellow color) and 70 nm red-shifted (red color). The experimental data points (blue color) in Figures 1(a) and 1(b) were taken from Figure 1 of reference 14, the pulse radiolysis of one-electron oxidized deoxyguanosine monitored at 10 μs after pulse radiolysis. The experimental data points (blue color) in Figure 1(c) were taken from Figure 3 of reference 14, the pulse radiolysis of one-electron oxidized 1-methylguanosine monitored at 100 μs after pulse radiolysis. The simulated spectra of 20 lowest transitions in 1(a), 1(b) and 1(c) were convoluted using a Gaussian function with a half width at half maximum of 30 nm, 25 nm and 35 nm, respectively.
The experimental absorption spectra of dGuo(N1-H)• shows absorption at around 311, 390 nm and a broad band extending from 470 – 550 nm centering at 500 nm, respectively, and has low intensity transitions between 600 – 700 nm.14 From the calculated transitions of dGuo(N1-H)•, presented in Table S6 in the supporting information, we found that a red-shift of 50 nm gives an excellent match with the experiment for all transitions. The simulated spectra of dGuo(N1-H)• with experimental spectrum is shown in Figure 1(b).
Experiment shows that 1-met-Guo(N2-H)• has very intense absorption at around 306 nm and at around 610 nm and a weak shoulder at 380 nm. The intense absorption at around 610 nm is the characteristic of guanine(N2-H)• which is used to distinguish it from guanine(N1-H)•. The TD-ωb97xd-PCM/6–31G(3df,p) calculated transitions of 1-met-Guo(N2-H)• shows intense absorption at 522 and 242 nm and moderate absorption at 343 nm, respectively, see Table S7 in the supporting information.
Thus, for this case the TD-ωb97xd-PCM/6–31G(3df,p) calculated transitions need a red-shift of 70 nm to match the experimental absorption spectra of 1-met-Guo(N2-H)•, see Table S7 and the simulated spectra in Figure 1(c).
Thus, from these calculations we infer that TD-ωb97xd-PCM/6–31G(3df,p) calculated transitions are blue-shifted by 40 – 70 nm in comparison to the experiment. For 40 nm red-shift, the energy shifts needed decrease from ca. 0.6 eV at 310 nm to ca. 0.15 eV at 600 nm. Below we explore sensitivity of the long wavelength transition in G(N2-H)• to the degree of twist of the N2H bond with respect to the molecular plane. The wavelength red shift is found to increase with rotation of the N2H bond (vide infra).
(iii). Vertical excited states of G•+-C base pair.
The twelve lowest vertical transition energies of G•+-C base pair, calculated using the TD-ωB97XD-PCM/6–31G(3df,p) method, are presented in Table 2. The 1st transition (D1) is n→π* type and has energy 2.31 eV and very weak oscillator strength 0.0003. The D2 and D3 transitions are local excitations G(π)→G(π*) and have energies 2.59 eV and 2.94 eV and oscillator strengths 0.0100 and 0.0377, respectively. D4 has energy 3 eV and it is n→π* type. D5 is a CT (C(π)→G(π*)) transition and has energy 3.34 eV and oscillator strength 0.0136. In this transition, electron from π-MO of cytosine is transferred to β-LUMO localized on guanine. D6 transition has energy 3.54 eV and oscillator strength 0.1148. This is π→π* type local excitation occurs on guanine base, see Table 2 and Figure S1 in the supporting information. The D7 is also a CT transition but has lower oscillator strength than D5.
Table 2.
TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical lowest lying transition energies (ΔE) in eV, and oscillator strength (f) of G•+-C base pair. Structure is shown in scheme 1(a). See Figure S1 in the supporting information for MOs involved in transitions.
| State | Transitiona | ΔE (eV) |
λ (nm) |
f | Contribution | Red-shift 40 nm | Exp. (nm)c |
|
|---|---|---|---|---|---|---|---|---|
| ΔE (eV) | λ (nm) | |||||||
| D1 | GC(n)→G(π*)b | 2.31 | 536 | 0.0003 | 97% b | 2.15 | 576 | |
| D2 | G(π)→G(π*) | 2.59 | 479 | 0.0100 | 78% | 2.39 | 519 | |
| D3 | G(π)→G(π*) | 2.94 | 422 | 0.0377 | 65% | 2.68 | 462 | 480, 470–550 |
| D4 | GC(n)→G(π*) b | 3.00 | 413 | 0.0000 | 66% | 2.74 | 453 | |
| D5 | C(π)→G(π*) | 3.34 | 372 | 0.0136 | 94% | 3.01 | 412 | |
| D6 | G(π)→G(π*) | 3.54 | 351 | 0.1148 | 86% | 3.17 | 391 | 400, 385 |
| D7 | C(π)→G(π*) | 3.64 | 341 | 0.0017 | 88% | 3.25 | 381 | |
| D8 | C(π)→GC(π*) GC(π)→GC(π*) b |
3.68 | 337 | 0.0000 | 71% | 3.29 | 377 | |
| D9 | G(n) →G(π*) | 3.83 | 324 | 0.0000 | 75% | 3.41 | 364 | |
| D10 | GC(n) →G(π*) b | 4.29 | 289 | 0.0000 | 88% | 3.77 | 329 | |
| D11 | GC(π)→ G(π*) b | 4.50 | 275 | 0.1272 | 71% | 3.94 | 315 | 300 |
| D12 | C(π)→ GC(π*) b | 4.59 | 270 | 0.0000 | 50% | 4.00 | 310 | |
The transient absorption spectra of G•+ in one-electron oxidized DNA, recorded between 350 – 700 nm using pulse radiolysis by Tagawa and coworkers, showed two absorption bands at around 400 nm (3.1 eV) and 480 nm (2.6 eV), respectively.21,22 The time-resolved spectrum of one-electron oxidized oligonucleotides containing alternating G-C base pair (spectra recorded at 100 μs) shows absorptions at around 300, 400 and a band between 450 – 550 nm, respectively.20 The later values compare well with the absorption spectrum of the free guanine nucleoside cation radical (dGuo•+) recorded between 300 – 700 nm, which show absorptions at around 300, 385 nm and a band extending from 470 – 550 nm, respectively.14 In Figure 2, we have plotted the calculated absorption spectra of G•+-C along with the pulse radiolysis spectrum of dGuo•+ and we found that the calculated spectrum must be red-shifted by 40 nm to match the experiment. Transitions 270 – 536 nm red-shifted by 40 nm involves transition energy changes which vary from 0.6 eV – 0.16 eV, see Table 2.
Figure 2.

TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical absorption spectrum of G•+-C (yellow color) and 40 nm red-shifted (red color). The experimental data points (blue color) were taken from Figure 1 of reference 14, the pulse radiolysis of one-electron oxidized deoxyguanosine at pH 3.1 and monitored at 10 μs after pulse radiolysis. The simulated spectra of 20 lowest calculated transitions were convoluted using a Gaussian function with a half width at half maximum of 30 nm. The red color curve is y-shifted by 0.02.
(iv). Vertical excited states of G(N1-H)•-(H+)C base pair.
Pulse radiolysis21,22, ESR19,40 experiments and theory41 showed that G•+, base paired with cytosine in DNA, transfers its N1 proton to N3 of cytosine, see structure G(N1-H)•-(H+)C in scheme 1(b). From deoxynucleosides in aqueous solution, it is known that the pKa of dG•+ for the N1 proton is 3.93,14 and pKa of N3-protonated dC is 4.3.21,42 This gives an estimate that the transfer from G to C in DNA is slightly favorable. So G(N1-H)•-(H+)C is an important contributor to the UV-vis spectra of ionized DNA.
The twelve (D1 – D12) lowest vertical transition energies of G(N1-H)•-(H+)C are presented in Table 3 and MOs involved in the electronic transitions are shown in Figure S2 in the supporting information. The D1 transition is a G(n)→G(π*) type and has energy 2.07 eV with oscillator strength 0.0003. The D2 and D3 transitions are G(π)→G(π*) in nature and have transition energies 2.27 eV and 2.70 eV and oscillator strengths 0.0165 and 0.0609, respectively. Transitions from the ground state to the D4 and D5 states have vanishingly low oscillator strengths. D6 is G(π)→G(π*) type occurs at 3.51 eV with substantial oscillator strength 0.1933. D8 transition is a CT and π→π* in nature occurs from C to G. It has transition energy 4.27 eV and oscillator strength 0.0019, see Table 3. In the G•+-C base pair the CT transition is at 3.34 eV (see Table 2) which is ca. 1 eV less than the CT transition (4.27 eV) of G(N1-H)•-(H+)C. This is in accord with a recent theoretical study12 that showed for G(N1-H)•-(H+)C, the HOMO localized on C is as expected lies below the SOMO (singly occupied molecular orbital) localized on G, whereas, in G•+-C, SOMO-HOMO level switching occurs and the HOMO localized on C lies above the SOMO on G. D9 – D12 transitions are local transitions lying between 4.44 – 4.88 eV, respectively.
Table 3.
TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical lowest lying transition energies (ΔE) in eV, and oscillator strength (f) of G(N1-H)•-(+H+)C. Structure is shown in scheme 1(b). See Figure S2 in the supporting information for MOs involved in transitions.
| State | Transitiona | ΔE (eV) |
λ (nm) |
f | Contribution | Red-shift 40 nm | Exp. (nm)c |
|
|---|---|---|---|---|---|---|---|---|
| ΔE (eV) | λ (nm) | |||||||
| D1 | G(n)→G(π*) | 2.07 | 598 | 0.0003 | 90% | 1.94 | 638 | |
| D2 | G(π)→G(π*) | 2.27 | 547 | 0.0165 | 83% | 2.11 | 587 | 550 - 650, 600 - 700 |
| D3 | G(π)→G(π*) | 2.70 | 460 | 0.0609 | 83% | 2.48 | 500 | 500, 470 – 540 |
| D4 | G(n)→G(π*) | 2.91 | 426 | 0.0000 | 86% | 2.66 | 466 | |
| D5 | C(π)→C(π*) | 3.46 | 358 | 0.0001 | 47% | 3.12 | 398 | |
| D6 | G(π)→G(π*) | 3.51 | 353 | 0.1933 | 92% | 3.16 | 393 | 380, 390 |
| D7 | GC(n)→G(π*)b | 3.55 | 349 | 0.0000 | 57% | 3.19 | 389 | |
| D8 | C(π)→G(π*) | 4.27 | 290 | 0.0019 | 98% | 3.76 | 330 | |
| D9 | G(n) )→G(π*) | 4.44 | 280 | 0.0002 | 81% | 3.88 | 320 | |
| D10 | G(π)→G(π*) | 4.45 | 278 | 0.2302 | 86% | 3.90 | 318 | 311 |
| D11 | C(π)→C(π*) | 4.76 | 260 | 0.0000 | 52% | 4.13 | 300 | |
| D12 | C(π)→C(π*) | 4.88 | 254 | 0.3608 | 95% | 4.22 | 294 | |
Using nanosecond pulse radiolysis, the transient absorption spectra of deprotonation of G•+ in oligonucleotides were measured by Kobayashi et al.21,22 over the range 350 – 700 nm and it was proposed that the pulse radiolysis spectrum of one-electron oxidized oligonucleotide monitored at 250 ns after the pulse21 was due to G(N1-H)•. The spectrum has absorption at 380 nm and 500 nm and some characteristic shoulder in the spectral range of 550 – 650 nm. The absorption spectra of dGuo(N1-H)• shows absorption at around 311, 390 nm and a broad band extending from 470 – 540 nm centering at 500 nm, respectively, and has low intensity transitions between 600 – 700 nm.14 The absorption spectrum of G(N1-H)•-(+H+)C (yellow curve) calculated using TD-ωb97xd-PCM/6–31G(3df,p) is shown in Figure S3 in the supporting information along with experimental spectrum (blue curve) of dGuo(N1-H)• produced due to one-electron oxidation of deoxyguanosine at pH 6.6 and monitored at 10 μs after pulse radiolysis by Candeias and Steenken.14 The calculated spectrum has absorption at 353 nm and 460 nm and has some absorption at around 550 nm range (yellow curve) and it is blue-shifted with respect to the experimental spectrum (blue curve), see Figure S3 in the supporting information. To match with experiment, the calculated spectrum of G(N1-H)•-(+H+)C was red-shifted by 40 nm (see yellow curve in Figure 3) and an excellent match was obtained. The Shifted spectrum has absorptions at 318, 393, 500 nm and some low intensity absorption at around 587 nm, respectively, see Figure 3 and Table 3. Since G(N1-H)•-(+H+)C, G(N1-H)•-(N4-H+)C and G(N1-H)•-C (see structures in scheme 1(b) – 1(d)) are produced due to N1 deprotonation of G•+, we speculate that these species contribute/influence the spectrum of G(N1-H)• in DNA.14,21,22 Thus, for easy comparison, we presented the calculated spectra of G(N1-H)•-(N4-H+)C (green curve) and G(N1-H)•-C (red curve) in Figure S3 in the supporting information and their corresponding red-shifted spectra in Figure 3.
Figure 3.

TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical absorption spectra of G(N1-H)•-(+H+)C (yellow color), G(N1-H)•-(N4-H+)C (green color) and G(N1-H)•-C (red color) red-shifted by 40 nm are shown. The experimental data points (blue color) were taken form Figure 1 of reference 14, the pulse radiolysis of one-electron oxidized deoxyguanosine at pH 6.6 and monitored at 10 μs after pulse radiolysis. The simulated spectra of 20 lowest transitions were convoluted using a Gaussian function with a half width at half maximum of 30 nm. For clarity in presentation spectra in blue, yellow, green and red are y-shifted by 0.12, 0.09, 0.05 and 0.01, respectively.
(v). Vertical excited states of G(N1-H)•-(N4-H+)C base pair.
The structure of G(N1-H)•-(N4-H+)C is shown in Scheme 1(c). A likely mechanism for the formation of this species in DNA is as follows. The initially formed G•+-C (Scheme 1(a)) in DNA undergoes proton transfer to G(N1-H)•-(H+)C40 (Scheme 1(b)) and subsequently (H+)C deprotonates from its N4 site to solvent to produce G(N1-H)•-(N4-H+)C (Scheme 1(c)).21,22 The lowest vertical transitions (D1 – D12) of G(N1-H)•-(H+)C, calculated using TD-ωb97xd-PCM/6–31G(3df,p) method, are presented in Table 4 and MOs involved in the transitions are shown in Figure S4 in the supporting information. The calculated transitions for G(N1-H)•-(N4-H+)C are red-shifted in comparison to those of G•+-C and G(N1-H)•-(H+)C, see Tables 2 and 3, respectively. The D1 transition has energy 1.95 eV and oscillator strength 0.0002. This transition is n→π* type and has some CT character mixed with local excitation. D2 – D5 transitions are local excitations and are G(π)→G(π*), G(n)→G(π*), G(π)→G(π*) and C(π)→C(π*) types, respectively. The transition energies of these transitions (D2 – D5) are 2.18, 2.68, 2.75 and 3.24 eV and their oscillator strengths are 0.0104, 0.0001, 0.0280 and 0.0001, respectively. D6 is a pure CT in nature occurring from C(π)→G(π*). The energy of this transition is 3.26 eV with oscillator strength 0.0029. D8 and D12 transitions are local transitions occurring from G(π)→G(π*) and have high oscillator strengths (0.1158 and 0.1703). D10 is CT in nature and has very low oscillator strength than D6.
Table 4.
TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical lowest lying transition energies (ΔE) in eV, and oscillator strength (f) of G(N1-H)•-(N4-H+)C. Structure is shown in scheme 1(c). See Figure S4 in the supporting information for MOs involved in transitions.
| State | Transitiona | ΔE (eV) |
λ (nm) |
f | Contribution | Red-shift 40 nm | Exp. (nm)c |
|
|---|---|---|---|---|---|---|---|---|
| ΔE (eV) | λ (nm) | |||||||
| D1 | GC(n)→G(π*)b | 1.95 | 637 | 0.0002 | 92% | 1.83 | 677 | |
| D2 | G(π)→G(π*) | 2.18 | 569 | 0.0104 | 84% | 2.04 | 609 | 550 – 650 |
| D3 | G(n)→G(π*) | 2.68 | 462 | 0.0001 | 73% | 2.47 | 502 | |
| D4 | G(π)→G(π*) | 2.75 | 450 | 0.0280 | 87% | 2.53 | 490 | 500 |
| D5 | C(π)→C(π*) | 3.24 | 383 | 0.0001 | 88% | 2.93 | 423 | |
| D6 | C(π)→G(π*) | 3.26 | 380 | 0.0029 | 92% | 2.95 | 420 | |
| D7 | GC(n)→C(π*)b | 3.47 | 357 | 0.0000 | 91% | 3.12 | 397 | |
| D8 | G(π)→G(π*) | 3.63 | 342 | 0.1158 | 89% | 3.25 | 382 | 380 |
| D9 | G(n)→G(π*) | 4.36 | 285 | 0.0001 | 71% | 3.82 | 325 | |
| D10 | C(π)→G(π*) | 4.39 | 282 | 0.0005 | 91% | 3.85 | 322 | |
| D11 | GC(n)→G(π*) | 4.53 | 274 | 0.0000 | 97% | 3.95 | 314 | |
| D12 | G(π)→G(π*) | 4.55 | 273 | 0.1703 | 79% | 3.96 | 313 | |
Nature (D1= LE, CT; D2= LE; D3=LE; D4=LE; D5=LE; D6=CT; D7=LE, CT; D8= LE; D9= LE; D10= CT; D11= LE, CT; D12= LE). LE = Local excitation; CT = charge transfer.
Delocalized on G and C.
See figure 5 in reference 21.
The UV-vis difference spectra of one-electron oxidized oligonucleotide monitored at 250 ns after the pulse by Kobayashi and Tagawa21 was suggested by them to be accounted for G(N1-H)•-(N4-H+)C. The spectrum recorded between 350 – 700 nm and monitored at 250 ns has absorptions at around 380 nm (3.3 eV), 500 nm (2.5 eV) and a broad band extending from ca. 550 – 650 nm (2.3 – 1.9 eV) with gradually decreasing intensity, respectively. The calculated spectrum (green color) after a 40 nm red-shift is shown in Figure 3. The shifted spectrum (green curve in Figure 3 is very similar to the experimental G(N1-H)• (blue curve) and calculated G(N1-H)•-(+H+)C (yellow curve). We notice that the lowest energy transition for the deprotonated radicals to solvent from N4 of cytosine G(N1-H)•-(N4-H+)C (green curve) and G(N1-H)•-C (red curve, discussed below) are red-shifted in comparison to the inter-base proton transfer (G(N1-H)•-(+H+)C (yellow curve in Figure 3).
(vi). Vertical excited states of G(N1-H)•-C base pair.
The structure of G(N1-H)•-C is shown in scheme 1(d). This structure (G(N1-H)•-C) can result from deprotonation of G•+-C to solvent by several paths (scheme 1). The formation of G(N1-H)•-C in one-electron oxidized d[TGCGCGCA]2 at pH ≥ 7 was confirmed by ESR experiment by annealing the sample at 155 K.19 Our calculations using ωb97xd-PCM/6–31G(3df,p) method showed that G(N1-H)•-C (scheme 1(d)) is ca. 6 kcal/mol more stable than G(N1-H)•-(N4-H+)C which is in agreement with an earlier theoretical study.30 The TD-ωb97xd-PCM/6–31G(3df,p) calculated D1 – D12 transition energies of G(N1-H)•-C are presented in Table 5 and MOs involved in the transitions are shown in Figure S5 in the supporting information. Among D1 – D12, the most intense transitions are D2 (GC(π)→G(π*)), D4 (G(π)→G(π*)), D6 (G(π)→G(π*)) and D11 and energies (oscillator strength) of these transitions are 2.09 eV (0.0266), 2.75 eV (0.0576), 3.57 eV (0.1849) and 4.50 eV (0.2149), respectively, see Table 5.
Table 5.
TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical lowest lying transition energies (ΔE) in eV, and oscillator strength (f) of G(N1-H)•-C. Structure is shown in scheme 1(d). See Figure S5 in the supporting information for MOs involved in transitions.
| State | Transitiona | ΔE (eV) |
λ (nm) |
f | Contribution | Red-shift 40 nm | |
|---|---|---|---|---|---|---|---|
| ΔE (eV) | λ (nm) | ||||||
| D1 | G(n)→G(π*)b | 1.92 | 644 | 0.0003 | 90% | 1.81 | 684 |
| D2 | GC(π)→G(π*)c | 2.09 | 592 | 0.0266 | 91% | 1.96 | 632 |
| D3 | GC(n)→G(π*)c | 2.65 | 469 | 0.0001 | 88% | 2.44 | 509 |
| D4 | G(π)→G(π*) | 2.73 | 455 | 0.0576 | 91% | 2.51 | 495 |
| D5 | GC(n)→G(π*)b | 3.43 | 362 | 0.0000 | 80% | 3.08 | 402 |
| D6 | G(π)→G(π*) | 3.57 | 347 | 0.1849 | 91% | 3.20 | 387 |
| D7 | C(π)→C(π*) | 3.65 | 340 | 0.0000 | 90% | 3.26 | 380 |
| D8 | C(π)→G(π*) | 3.84 | 323 | 0.0004 | 75% | 3.42 | 363 |
| D9 | GC(π)→G(π*)c | 4.04 | 307 | 0.0003 | 99% | 3.57 | 347 |
| D10 | G(n)→G(π*) | 4.30 | 288 | 0.0002 | 70% | 3.78 | 328 |
| D11 | G(π)→G(π*) | 4.50 | 276 | 0.2149 | 83% | 3.92 | 316 |
| D12 | C(π)→C(π*) | 4.52 | 274 | 0.0003 | 61% | 3.95 | 314 |
Nature (D1= LE; D2= LE, CT; D3=LE, CT; D4=LE; D5=LE; D6=LE; D7=LE; D8= CT; D9= LE, CT; D10= LE; D11= LE; D12= LE). LE = Local excitation; CT = charge transfer.
Very small mixing with cytosine.
Delocalized on G and C.
The TD-ωb97xd-PCM/6–31G(3df,p) calculated absorption spectrum of G(N1-H)•-C after 40 nm red-shift (red curve) is shown in Figure 3. The shifted spectrum is similar to the experimental G(N1-H)• (blue curve) and has absorption around 630 nm. From a comparison of the absorption spectra of G•+-C, G(N1-H)•-(+H+)C, G(N1-H)•-(N4-H+)C and G(N1-H)•-C, it is evident that spectra of these species are similar but it is noticeable that as proton transfers from N1 of guanine to cytosine and finally to solvent to ultimately produce the more stable species G(N1-H)•-C (scheme 1(d)), the outer band is progressively red-shifted.
(vii). Vertical excited states of G(N2-H)•-C base pair.
The structure of G(N2-H)•-C is shown in Scheme 1(e). Our calculation using ωb97xd-PCM/6–31G(3df,p) method shows that G(N2-H)•-C is nearly isoenergetic with G(N1-H)•-C with G(N2-H)•-C favored by 0.19 kcal/mol. In an earlier theoretical study19, the structures of G(N1-H)•-C and G(N2-H)•-C surrounded by 11 water molecules were optimized by B3LYP/6–31+G** method and G(N2-H)•-C was found ca. 0.4 kcal/mol more stable than G(N1-H)•-C. The TD-ωb97xd-PCM/6–31G(3df,p) calculated transition energies of G(N2-H)•-C are presented in Table 6 and MOs involved in the transitions are shown in Figure S6 in the supporting information. The D1 transition has mainly local excitation occurring on G but with a minor mixing with C and it is π→π* in nature, see Table 6 and Figure S6 in the supporting information. This transition (D1) has energy 2.3 eV and largest oscillator strength (0.1903) of all the other transitions (D2 – D12). D2 and D3 transitions are n→π* and have energies 2.62 eV and 2.70 eV, respectively. D4 and D7 are the π→π* type local excitation occurring on G. These transitions have energies 3.03 eV and 3.86 eV, respectively, and corresponding oscillator strengths are 0.0178 and 0.0672, respectively. The D8 transition is CT type (C(π)→G(π*)) and has energy 4.03 eV and very small oscillator strength 0.0020. D10 is G(π)→GC(π*) in nature has large oscillator strength (0.1315) and transition energy 4.34 eV. D14 is C(π)→C(π*) and has a slightly greater oscillator strength than D1.
Table 6.
TD-ωb97xd-PCM/6-31G(3df,p) calculated vertical lowest lying transition energies (ΔE) in eV, and oscillator strength (f) of G(N2-H)•-C. Structure is shown in scheme 1(e). See Figure S6 in the supporting information for MOs involved in transitions.
| State | Transitiona | ΔE (eV) |
λ (nm) |
f | Contribution | Red-shift 70 nm | Exp. (nm)d |
|
|---|---|---|---|---|---|---|---|---|
| ΔE (eV) | λ (nm) | |||||||
| D1 | GC(π)→G(π*)b | 2.30 | 540 | 0.1903 | 83%a | 2.03 | 610 | 610 |
| D2 | GC(n)→G(π*)b | 2.62 | 473 | 0.0008 | 80%a | 2.28 | 543 | |
| D3 | GC(n)→G(π*)b | 2.70 | 459 | 0.0009 | 66%a | 2.34 | 529 | |
| D4 | G(π)→G(π*) | 3.03 | 409 | 0.0178 | 93% | 2.59 | 479 | |
| D5 | C(π)→C(π*) | 3.67 | 338 | 0.0000 | 87% | 3.04 | 408 | |
| D6 | GC(n)→G(π*)b | 3.69 | 336 | 0.0001 | 87%a | 3.05 | 406 | |
| D7 | G(π)→G(π*) | 3.86 | 321 | 0.0672 | 86% | 3.17 | 391 | 380 |
| D8 | C(π)→G(π*) | 4.03 | 307 | 0.0020 | 84% | 3.29 | 377 | |
| D9 | GC(π)→G(π*)b | 4.28 | 290 | 0.0002 | 72% | 3.44 | 360 | |
| D10 | G(π)→G(π*)c | 4.34 | 286 | 0.1315 | 71% | 3.48 | 356 | |
| D11 | G(n)→G(π*) | 4.36 | 284 | 0.0000 | 75% | 3.50 | 354 | |
| D12 | C(π)→C(π*)c | 4.55 | 273 | 0.0000 | 63% | 3.62 | 343 | |
| D13 | GC(n)→G(π*)b | 4.82 | 257 | 0.0001 | 56% | 3.79 | 327 | |
| D14 | C(π)→C(π*) | 5.04 | 246 | 0.1953 | 86% | 3.92 | 316 | 306 |
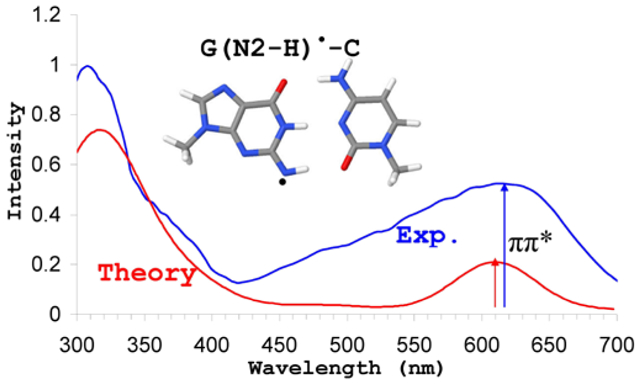
The absorption spectra of G(N2-H)• generated from one-electron oxidized 1-methylguanosine has characteristic broad absorption band at around 610 nm (2.0 eV).14,19 The absorption spectra of one-electron oxidized d[TGCGCGCA]2 at pH = 9 at 180 K showed absorption band around 620 nm which characterizes the formation of G(N2-H)•-C in one-electron oxidized d[TGCGCGCA]219. Kobayashi et al.22 studied the UV-vis spectra of one-electron oxidized oligonucleotide (sequence G1AA) monitored at 500 ns after pulse radiolysis between 350 – 700 nm. The spectra shows absorption at around 350 nm, a weak shoulder at 400 nm and broad band extending from 550 – 650 nm, respectively. Our calculated spectrum of G(N2-H)•-C has absorption at 540 nm (2.3 eV) (Table 6) which is ca. 70 nm blue-shifted with respect to experiment.14,19
In Figure 4, we presented the calculated (yellow curve) and 70 nm red-shifted (red curve) spectra along with four experimental spectra: (a) the pulse radiolysis of one-electron oxidized 1-met-Guo(N2-H)• at pH 7.3 and monitored at 100 μs after pulse radiolysis.14 (b) the UV-visible absorption spectra of 1-met-Guo(N2-H)• recorded at 77 K in 7.5 M LiCl glass/D2O at pD ca. 8.5.19 (c) The UV-visible absorption spectra of one-electron oxidized d[TGCGCGCA]2 in glassy 7.5 M LiCl in H2O at pH 9 recorded at 180 K.19 and (d) The pulse radiolysis of one-electron oxidized oligonucleotide monitored at 500 ns after pulse radiolysis.22 These experimental spectra show absorption at around 610 nm which is the characteristic of G(N2-H)•.14 The UV-vis spectrum of 1-met-Guo(N2-H)• shows intense absorptions at around 310 nm and 610 nm, respectively, see blue and green curves in Figure 4. Our TD-ωb97xd-PCM/6–31G(3df,p) calculated spectrum of G(N2-H)•-C shows four intense absorptions at 246, 286, 321 and 540 nm, respectively, (Table 6) and the shape of the spectra also matched very well with the experiment. When calculated spectrum is red-shifted by 70 nm (red curve), an excellent agreement between theory and experiment is achieved, see Figure 4. We also observe that in comparison to the other radicals shown in scheme 1(a) – 1 (d), the first lowest transition (D1) of this (G(N2-H)•-C) has the highest oscillator strength (0.1903) and experiment also shows an intense corresponding peak at 610 nm, see Figure 4.
Figure 4.

TD-ωb97xd-PCM/6–31G(3df,p) calculated vertical absorption spectrum of G(N2-H)•-C (yellow color) and 70 nm red-shifted (red color) are shown. Experimental spectra are as follows: Blue color- The experimental data points were taken from Figure 3 of reference 14, the pulse radiolysis of one-electron oxidized 1-methylguanosine at pH 7.3 and monitored at 100 μs after pulse radiolysis. Green color- The data points were taken from the UV-visible absorption spectra of 1-Methyl-G(N2–H)• recorded at 77 K in 7.5 M LiCl glass/D2O at pD ca. 8.5, see Figure 1 of reference 19. Black color- The data points were taken form Figure 5 of reference 22, the pulse radiolysis of one-electron oxidized oligonucleotide (sequence G1AA) monitored at 500 ns after pulse radiolysis. Pink color- The data points were taken from the UV-visible absorption spectra of one-electron oxidized d[TGCGCGCA]2 in glassy 7.5 M LiCl in H2O at pH 9 recorded at 180 K, see Figure 6 of reference 19. The simulated spectra of 20 lowest transitions were convoluted using a Gaussian function with a half width at half maximum of 35 nm. For clarity in presentation the experimental spectra in blue, green, black and pink are y-scaled by 2.5, 1.6, 17 and 4.5, respectively, while calculated spectra in yellow and red colors are y-shifted by 0.02.
The calculated spectrum of G(N2-H)•, shown in Figure 1(c)) and Figure 4, needs 20 – 30 nm more red-shift than the other radicals (scheme 1(a) – 1(d)) to match the experimental spectrum. The out of plane rotation of the N2-H group is a possible factor which may account for the larger red shift needed to match the the experimental spectrum of G(N2-H)·. Thus, in view of this, we rotated the N2-H group from 0 – 60 degree with respect to purine ring in the step size of 10 degree and refined 20 – 40 degree range by step size of 2 degrees and calculated the vertical transition energies. The calculated first lowest transition is presented in Table S8 in the supporting information. The calculation shows that the first lowest transition of G(N2-H)• is clearly influenced by the non-planarity of the N2-H group in guanine. In the range of 0 – 40 degree the TD-ωb97xd-PCM/6–31G(3df,p) calculated first lowest transition varies from 500 – 640 nm and the corresponding oscillator strength varies from 0.1056 – 0.0800, see Table S8 in the supporting information. We note that at only 24 degrees there is a red shift of over 40 nm at an energy cost of only 0.1 eV which is significantly less than a single hydrogen bond. The non-planarity of amino group in isolated DNA bases and in base pairs is an intrinsic property and in base pair occurs during propeller twisting of purine and pyrimidine rings.43
We note this does not account for the overall redshift needed in all our calculations. This is a common issue in DFT calculations of these systems and is also reported in previous work.20 The choice of the TD-ωb97xd method reduced the shift needed significantly from 0.6 eV in this earlier work20 to 0.2 eV in the present work.
Conclusions
DNA on one-electron oxidation transiently forms G•+ which undergoes proton transfer reactions internally to C forming several short-lived intermediate G-C cationic radicals (see scheme 1). These species ultimately deprotonate to solvent resulting in the formation of neutral guanine radicals (G(N1-H)• or G(N2-H)•). These radicals have been suggested from their UV-vis spectra after pulse radiolysis and several confirmed by ESR experiments.3,4,6,13,19,21,22,40 In this work, we calculated vertical transition energies for the various one-electron oxidized G-C species to better understand the nature of the transitions involved in the UV-vis spectra of these intermediates. Our chosen method (TD-ωb97xd-PCM/6–31G(3df,p)) was tested by comparison to high level methods (EOM-CCSD(T)26) for the neutral G-C base pair and found to give excellent results. This gave us confidence in the usefulness of the method for other G-C species investigated in this work. From a comparison of our TD-ωb97xd-PCM/6–31G(3df,p) calculated transition energies of one-electron oxidized guanine radical (G•+, G(N1-H)• and G(N2-H)•) and G-C base pair with those available from pulse radiolysis experiments,14,19,21,22 we found that the calculated spectra are blue-shifted by 40 – 70 nm with respect to experiment. Thus, to match with experiment, the calculated spectra were red-shifted by of 40 – 70 nm. We note that for 40 nm red-shift, the energy shifts decrease from ca. 0.6 eV at 310 nm to ca. 0.15 eV at 600 nm. The present calculations show that the spectra of G•+-C, G(N1-H)•-(+H+)C, G(N1-H)•-(N4-H+)C and G(N1-H)•-C are quite similar and not likely to be distinguishable by experiment from their UV-vis spectra which are poorly resolved. However, the present calculation predicts that as proton transfers from N1 of guanine to cytosine and finally to solvent to form the more stable species G(N1-H)•-C (scheme 1(d)), the outer band is progressively red-shifted, see Figure 3 and Figure S3 in the supporting information. This shift is not seen in the experiment owing to the low intensity and poor resolution of the longest wavelength transition.
For G(N2-H)•-C our calculations predict an intense absorption around 540 nm (2.3 eV) and the corresponding oscillator strength is 0.1903. The experimental absorption spectra of G(N2-H)• have a characteristic absorption band with high intensity at around 610 nm (2.0 eV).14,19 Our TD-ωb97xd-PCM/6–31G(3df,p) calculated transition (540 nm) of G(N2-H)•-C is therefore red-shifted by 70 nm to give an overall match, see Figure 4. We also note that G(N2-H)•-C is a more stable structure than G(N1-H)•-(N4-H+)C and favored slightly over G(N1-H)•-C. Our calculations also show that the lowest energy transitions in all the radicals considered in this study (Scheme 1) are local excitations taking place from G→G and they are either π→π* or n→π* in nature, see Tables 2 - 6. The lowest energy pure CT transitions have low oscillator strength and occur from C→G•+ and are π→π* in nature with transition energies lying between 3.36 – 4.27 eV.
Supplementary Material
ACKNOWLEDGMENTS
Authors thank the National Cancer Institute of the National Institutes of Health (Grant R01CA045424) for support.
Footnotes
Supporting Information
Tables of vertical singlet excited state energies of G–C base pair calculated by CC2, EOM-CCSD(T), TD-ωb97x, TD-M05–2x and TD-ωb97xd methods. Tables of vertical excited state and of dGuo•+, dGuo(N1-H)• and 1-methylG(N2-H)•. Transition energies plots with MOs of G•+-C, G(N1-H)•-(H+)C, G(N1-H)•-(N4-H+)C), G(N1-H)•-C and G(N2-H)•-C.
The authors declare no competing financial interest.
References
- (1).Cadet J; Douki T; Ravanat J-L Oxidatively Generated Damage to Cellular DNA by UVB and UVA Radiation. Photochem. Photobiol 2015, 91 (1), 140–155. [DOI] [PubMed] [Google Scholar]
- (2).Markovitsi D UV-Induced DNA Damage: The Role of Electronic Excited States. Photochem. Photobiol 2016, 92 (1), 45–51. [DOI] [PubMed] [Google Scholar]
- (3).Steenken S Purine Bases, Nucleosides, and Nucleotides: Aqueous Solution Redox Chemistry and Transformation Reactions of Their Radical Cations and e- and OH Adducts. Chem. Rev 1989, 89 (3), 503–520. [Google Scholar]
- (4).Kumar A; Sevilla MD Proton-Coupled Electron Transfer in DNA on Formation of Radiation-Produced Ion Radicals. Chem. Rev. 2010, 110 (12), 7002–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li X; Sevilla MD DFT Treatment of Radiation Produced Radicals in DNA Model Systems In Advances in Quantum Chemistry; Brändas JRS and E., Ed.; Academic Press, 2007; Vol. 52, pp 59–87. [Google Scholar]
- (6).Kumar A; Sevilla MD Theoretical Modeling of Radiation-Induced DNA Damage In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; Greenberg MM, Ed.; John Wiley & Sons, Inc., 2009; pp 1–40. [Google Scholar]
- (7).Harris MA; Mishra AK; Young RM; Brown KE; Wasielewski MR; Lewis FD Direct Observation of the Hole Carriers in DNA Photoinduced Charge Transport. J. Am. Chem. Soc 2016, 138 (17), 5491–5494. [DOI] [PubMed] [Google Scholar]
- (8).Giese B; Amaudrut J; Köhler A-K; Spormann M; Wessely S Direct Observation of Hole Transfer through DNA by Hopping between Adenine Bases and by Tunnelling. Nature 2001, 412 (6844), 318–320. [DOI] [PubMed] [Google Scholar]
- (9).Lewis FD; Wu T; Liu X; Letsinger RL; Greenfield SR; Miller SE; Wasielewski MR Dynamics of Photoinduced Charge Separation and Charge Recombination in Synthetic DNA Hairpins with Stilbenedicarboxamide Linkers. J. Am. Chem. Soc 2000, 122 (12), 2889–2902. [Google Scholar]
- (10).Kumar A; Sevilla MD Density Functional Theory Studies of the Extent of Hole Delocalization in One-Electron Oxidized Adenine and Guanine Base Stacks. J. Phys. Chem. B 2011, 115 (17), 4990–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Barnett RN; Cleveland CL; Joy A; Landman U; Schuster GB Charge Migration in DNA: Ion-Gated Transport. Science 2001, 294 (5542), 567–571. [DOI] [PubMed] [Google Scholar]
- (12).Kumar A; Sevilla MD Proton Transfer Induced SOMO-to-HOMO Level Switching in One-Electron Oxidized A-T and G-C Base Pairs: A Density Functional Theory Study. J. Phys. Chem. B 2014, 118 (20), 5453–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Adhikary A; Kumar A; Becker D; Sevilla MD The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J. Phys. Chem. B 2006, 110 (47), 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Candeias LP; Steenken S Structure and Acid-Base Properties of One-Electron-Oxidized Deoxyguanosine, Guanosine, and 1-Methylguanosine. J. Am. Chem. Soc 1989, 111 (3), 1094–1099. [Google Scholar]
- (15).Banyasz A; Ketola T-M; Muñoz-Losa A; Rishi S; Adhikary A; Sevilla MD; Martinez-Fernandez L; Improta R; Markovitsi D UV-Induced Adenine Radicals Induced in DNA A-Tracts: Spectral and Dynamical Characterization. J. Phys. Chem. Lett 2016, 7 (19), 3949–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Candeias LP; Steenken S Ionization of Purine Nucleosides and Nucleotides and Their Components by 193-Nm Laser Photolysis in Aqueous Solution: Model Studies for Oxidative Damage of DNA. J. Am. Chem. Soc 1992, 114 (2), 699–704. [Google Scholar]
- (17).Candeias LP; Steenken S Electron Transfer in Di (Deoxy) Nucleoside Phosphates in Aqueous Solution: Rapid Migration of Oxidative Damage (via Adenine) to Guanine. J. Am. Chem. Soc 1993, 115 (6), 2437–2440. [Google Scholar]
- (18).Kumar A; Pottiboyina V; Sevilla MD Hydroxyl Radical (OH • ) Reaction with Guanine in an Aqueous Environment: A DFT Study. J. Phys. Chem. B 2011, 115 (50), 15129–15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Adhikary A; Kumar A; Munafo SA; Khanduri D; Sevilla MD Prototropic Equilibria in DNA Containing One-Electron Oxidized GC: Intra-Duplex vs. Duplex to Solvent Deprotonation. Phys. Chem. Chem. Phys 2010, 12 (20), 5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Banyasz A; Martínez-Fernández L; Improta R; Ketola T-M; Balty C; Markovitsi D Radicals Generated in Alternating Guanine–Cytosine Duplexes by Direct Absorption of Low-Energy UV Radiation. Phys. Chem. Chem. Phys 2018, 20 (33), 21381–21389. [DOI] [PubMed] [Google Scholar]
- (21).Kobayashi K; Tagawa S Direct Observation of Guanine Radical Cation Deprotonation in Duplex DNA Using Pulse Radiolysis. J. Am. Chem. Soc 2003, 125 (34), 10213–10218. [DOI] [PubMed] [Google Scholar]
- (22).Kobayashi K; Yamagami R; Tagawa S Effect of Base Sequence and Deprotonation of Guanine Cation Radical in DNA. J. Phys. Chem. B 2008, 112 (34), 10752–10757. [DOI] [PubMed] [Google Scholar]
- (23).Rokhlenko Y; Cadet J; Geacintov NE; Shafirovich V Mechanistic Aspects of Hydration of Guanine Radical Cations in DNA. J. Am. Chem. Soc 2014, 136 (16), 5956–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kumar A; Sevilla MD Photoexcitation of Dinucleoside Radical Cations: A Time-Dependent Density Functional Study. J. Phys. Chem. B 2006, 110 (47), 24181–24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bera PP; Schaefer HF (G–H)•–C and G–(C–H)• Radicals Derived from the Guanine·cytosine Base Pair Cause DNA Subunit Lesions. Proc. Natl. Acad. Sci 2005, 102 (19), 6698–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Szalay PG; Watson T; Perera A; Lotrich V; Bartlett RJ Benchmark Studies on the Building Blocks of DNA. 3. Watson–Crick and Stacked Base Pairs. J. Phys. Chem. A 2013, 117 (15), 3149–3157. [DOI] [PubMed] [Google Scholar]
- (27).Yamazaki S; Taketsugu T Photoreaction Channels of the Guanine–Cytosine Base Pair Explored by Long-Range Corrected TDDFT Calculations. Phys. Chem. Chem. Phys 2012, 14 (25), 8866–8877. [DOI] [PubMed] [Google Scholar]
- (28).Sun H; Zhang S; Zhong C; Sun Z Theoretical Study of Excited States of DNA Base Dimers and Tetramers Using Optimally Tuned Range-Separated Density Functional Theory. J. Comput. Chem 2016, 37 (7), 684–693. [DOI] [PubMed] [Google Scholar]
- (29).Anderson RF; Shinde SS; Maroz A Cytosine-Gated Hole Creation and Transfer in DNA in Aqueous Solution. J. Am. Chem. Soc 2006, 128 (50), 15966–15967. [DOI] [PubMed] [Google Scholar]
- (30).Adhikary A; Kumar A; Bishop CT; Wiegand TJ; Hindi RM; Adhikary A; Sevilla MD π-Radical to σ-Radical Tautomerization in One-Electron-Oxidized 1-Methylcytosine and Its Analogs. J. Phys. Chem. B 2015, 119 (35), 11496–11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chai J-D; Head-Gordon M Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10 (44), 6615–6620. [DOI] [PubMed] [Google Scholar]
- (32).Chai J-D; Head-Gordon M Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys 2008, 128 (8), 084106. [DOI] [PubMed] [Google Scholar]
- (33).Kumar A; Sevilla MD Excited State Proton-Coupled Electron Transfer in 8-OxoG–C and 8-OxoG–A Base Pairs: A Time Dependent Density Functional Theory (TD-DFT) Study. Photochem. Photobiol. Sci 2013, 12 (8), 1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tomasi J; Mennucci B; Cammi R Quantum Mechanical Continuum Solvation Models. Chem. Rev 2005, 105 (8), 2999–3094. [DOI] [PubMed] [Google Scholar]
- (35).Frisch MJ; Trucks GW; Cheeseman JR; Scalmani G; Caricato M; Hratchian HP; Li X; Barone V; Bloino J; Zheng G; et al. Gaussian 09; Gaussian Inc; Wallingford CT: 2009. [Google Scholar]
- (36).Allouche A-R Gabedit—A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem 2011, 32 (1), 174–182. [DOI] [PubMed] [Google Scholar]
- (37).IQmol Molecular Viewer http://iqmol.org/ (accessed Oct 2, 2018).
- (38).Banyasz A; Ketola T; Martínez-Fernández L; Improta R; Markovitsi D Adenine Radicals Generated in Alternating AT Duplexes by Direct Absorption of Low-Energy UV Radiation. Faraday Discuss. 2018, 207, 181–197. [DOI] [PubMed] [Google Scholar]
- (39).Bhat V; Cogdell R; Crespo-Hernández CE; Datta A; De A; Haacke S; Helliwell J; Improta R; Joseph J; Karsili T; et al. Photocrosslinking between Nucleic Acids and Proteins: General Discussion. Faraday Discuss. 2018, 207, 283–306. [DOI] [PubMed] [Google Scholar]
- (40).Adhikary A; Khanduri D; Sevilla MD Direct Observation of the Hole Protonation State and Hole Localization Site in DNA-Oligomers. J. Am. Chem. Soc 2009, 131 (24), 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kumar A; Sevilla MD Influence of Hydration on Proton Transfer in the Guanine–Cytosine Radical Cation (G •+ −C) Base Pair: A Density Functional Theory Study. J. Phys. Chem. B 2009, 113 (33), 11359–11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ts’o POP Bases, Nucleosides, and Nucleotides In Basic Principles in Nucleic Acid Chemistry; Ts’o POP, Ed.; Academic Press: New York, 1974; Vol. 1, pp 454–584. [Google Scholar]
- (43).Komarov VM; Polozov RV; Konoplev GG Non-Planar Structure of Nitrous Bases and Non-Coplanarity of Watson-Crick Pairs. J. Theor. Biol 1992, 155 (3), 281–294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.