

Abstract
Neuroinflammation is implicated in the progression of numerous disease states of the CNS, but early inflammatory signaling events in glial cells that may predispose neurons to injury are not easily characterized in vivo. To address this question, we exposed transgenic mice expressing a nuclear factor-κB (NF-κB)-driven enhanced green fluorescent protein (EGFP) reporter construct to low doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and examined inflammatory activation of astrocytes in relation to neurobehavioral and neuropathological outcomes. The highest dose of MPTP (60 mg/kg total dose) caused a decrease in locomotor activity and a reduction in stride length. No significant loss of dopaminergic neurons in the substantia nigra was apparent at any dose. In contrast, expression of tyrosine hydroxylase in striatal fibers was reduced at 60 mg/kg MPTP, as were levels of dopamine and DOPAC. Colocalized expression of EGFP and inducible nitric oxide synthase (NOS2) occurred in astrocytes at 30 and 60 mg/kg MPTP and was associated with increased protein nitration in nigral dopaminergic neurons. Inhibition of NF-κB in primary astrocytes by expression of mutant IκBα suppressed expression of NOS2 and protected cocultured neurons from astrocyte-mediated apoptosis. These data indicate that inflammatory activation of astrocytes and enhanced nitrosative stress occurs at low doses of MPTP prior to loss of dopaminergic neurons. NF-κB-mediated expression of NOS2 appears to be a sensitive indicator of neuroinflammation that correlates with MPTP-induced neurochemical and neurobehavioral deficits prior to loss of dopaminergic neurons in the subtantia nigra.
Keywords: astrocyte, neuroinflammation, neurodegeneration, NF-κB, MPTP
Neuroinflammation is an important feature in the progression of neurodegenerative disease (Hald et al., 2007; Whitton, 2007). In addition, CNS-related effects are a major reason for safety-related attrition in drug development (Redfern et al., 2010). Increased levels of the proinflammatory cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-1, and IL-6 have been reported in the substantia nigra of post-mortem Parkinson’s disease (PD) brains and in animal models of PD (Nagatsu et al., 2000). Additionally, mice lacking TNF-α receptors (Sririam et al., 2002) and inducible nitric oxide synthase (iNos/NOS2; Liberatore et al., 1999) are protected against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Epidemological studies also suggest that use of nonsteroidal antiinflammatory drugs reduces the risk for both PD (Chen et al., 2003) and Alzheimer’s disease (Szekely and Zandi, 2010). Collectively, these data support a role for inflammatory signaling in neurodegeneration, but the ability to detect early activation of neuroinflammatory pathways with spatial and temporal fidelity has been limited.
Activated microglia and astrocytes have been noted in areas surrounding dying neurons in post-mortem brains of PD patients and in MPTP-treated animals (McGeer et al., 1988; Kurkowska-Jastrzebska et al., 1999). Activated glial cells release numerous neurotoxic substances, including proinflammatory cytokines and reactive oxygen/nitrogen species (Hirsch and Hunot, 2000). Whether this glial reaction is merely a response to degenerating neurons or is a pathogenic cause of neuronal loss is still debated. Regardless, sustained glial activation is detrimental to neuronal survival, and signaling pathways regulating the expression of neuroinflammatory genes in glial cells have been proposed as a promising therapeutic target for PD and Alzheimer’s disease (Camandola and Mattson, 2007; Wilms et al., 2007; Kalinin et al., 2009; Saijo et al., 2009). However, detecting early activation of neuroinflammatory pathways in glial cells in vivo has been problematic both in models of neurodegenerative disease and in development of new therapeutics.
Expression of neuroinflammatory genes such as NOS2 and TNF-α is principally regulated by nuclear factor-κB (NF-κB; Collart et al., 1990; Xie et al., 1993), a Rel protein-family transcription factor involved in inflammation, apoptosis, and immune responses (Karin and Ben-Neriah, 2000; Perkins, 2000). NF-κB is activated through phosphorylation of the inhibitory IκBα subunit by the IκB kinase complex, which facilitates degradation of IκBα and nuclear translocation of p65/RelA (Nakano et al., 1998; Vermeulen et al., 2002). Activation of NF-κB may be an early event in cellular stress responses in glia. Recent data from transgenic animals with microglial-specific (Cho et al., 2008) and astrocyte-specific (Brambilla et al., 2009) gene deletion of NF-κB support the neuroprotective efficacy of blocking this signaling pathway in models of inflammatory neurodegeneration. It has been suggested that NF-κB is an important point of convergence for multiple cellular stress pathways underlying neuroinflammatory injury (Camandola and Mattson, 2007). Detection of NF-κB activation in vivo could therefore allow the identification of endogenous or exogenous stressors with the potential to elicit neuroinflammatory responses.
To understand better the role of NF-κB in glial-mediated inflammatory responses, we postulated that low doses of MPTP would stimulate astrocyte-specific NF-κB activity in transgenic reporter mice prior to loss of dopaminergic neurons. We demonstrate here that doses of MPTP lower than typically used activate NF-κB and induce expression of NOS2 in astrocytes and increase protein nitration in dopaminergic neurons, indicating development of a neuroinflammatory phenotype. Expression of the NF-κB-EGFP reporter occurs concomitantly with changes in neurobehavioral parameters but prior to loss of dopaminergic neurons, suggesting that neuroinflammatory activation of astrocytes may directly contribute to neuronal degeneration during intoxication with MPTP.
MATERIALS AND METHODS
Animals and Treatment
A transgenic fluorescent reporter mouse harboring an EGFP reporter construct containing three NF-κB cis elements (cis-NF-κBEGFP; Magness et al., 2004; generously provided by Dr. Christian Jobin, University of North Carolina at Chapel Hill) was employed in this study. Male mice, 12 weeks of age, were randomly divided into four groups (n = 10) as follows: 1) saline, 2) 15 mg/kg MPTP, 3) 30 mg/kg MPTP, and 4) 60 mg/kg MPTP. Doses of MPTP (prepared in saline as free base; Sigma, St. Louis, MO) were delivered by subcutaneous injection bid 12 hr apart. All animal procedures were performed in accordance with NIH guidelines for the care and use of laboratory animals and were approved by the Colorado State University Institutional Animal Care and Use Committee. Every effort was made to minimize pain and discomfort, and all terminal procedures were performed under deep isofluorane anesthesia.
Neurobehavioral and Locomotor Assessment
Spontaneous locomotor activity.
Ambulatory locomotion was evaluated by using VersaMax open-field activity chambers (Accuscan Instruments, Inc., Columbus, OH). All mice used in the study were monitored over a 10-min period on day 0 to establish baseline and then on days 1, 4, and 6 posttreatment. Monitoring occurred at the same time of day, under lowered ambient light, and in the presence of white noise. VersaDat software (Accuscan Instruments, Inc.) was used to analyze multiple parameters, including total distance traveled, total movement time, number of rearing movements, and margin time.
Hind-limb stride length measurements.
Abnormal gait features, including reduction in stride length, are routinely encountered in disease states involving the basal ganglia such as PD. Stride length in MPTP-treated and control animals were determined 1 day prior to treatment (day 0) and on day 6 posttreatment through manual measurements between two hind-limb paw prints. The hind-paw of each mouse was inked using commercially available nontoxic finger paint. The mice were then placed in a paper-lined runway leading to their home cage. At least four paw prints produced during continuous movement were measured. The initial prints as well as those made immediately adjacent to the cage opening were omitted. The percentage change from the initial measurement to final measurement of each animal was calculated, and the mean percentage change between dose groups was compared.
Tissue Processing and Sectioning
Seven days after treatment, animals were terminally anesthetized with isoflurane and transcardially perfused with 20 mM sodium cacodylate in phosphate-buffered saline (cacodylate-PBS) containing 10 U/ml heparin, followed by 4% paraformaldehyde in cacodylate-PBS. After perfusion fixation, the brains were carefully removed from the skull and immersion fixed in the same fixative at 4°C for 3 hr. The brains were then cryoprotected in cacodylate-PBS containing 15% sucrose overnight, followed by 30% sucrose. The tissue was then embedded in optimal cutting temperature (OCT) compound and frozen in liquid nitrogen-cooled isopentane and stored –80°C until sectioning. Serial 40-µm coronal sections through the midbrain and representative 25-µm sections through striatum were cut at –19°C on a cryostat microtome. Sections were stored at –20°C, free floating, in cryoprotectant (30% w/v sucrose, 30% v/v ethylene glycol; 0.5 M phosphate buffer, pH 7.2) until staining.
Tyrosine Hydroxylase Immunofluorescence and Stereological Counts
Free-floating serial sections used for tyrosine hydroxylase (TH) staining were obtained using systematic random sampling from all sections encompassing the entire length of the SNpc with a section fraction of one-third, where section 1 of the series was randomly selected from the first three sections. The sampled sections were rinsed in 0.05 M Tris-buffered saline (TBS; pH 7.2), followed by incubation for 1 hr in blocking buffer consisting of 1% goat and 1% donkey serum in TBS. Sections were then incubated with primary antibody rabbit anti-TH (1:500 in blocking buffer containing 0.5% Triton X-100; Chemicon, Temecula, CA) overnight at 4°C. Sections were washed with TBS, followed by incubation with Alexafluor conjugated anti-rabbit secondary antibody (1:500 in blocking buffer; Invitrogen, Carlsbad, CA) for 3 hr at room temperature. Tissue sections were then rinsed three times and mounted in medium containing 4ʹ,6-diamidino-2-phenylindole (DAPI) to visualize cell nuclei. Slides were stored at 4°C until imaged for stereological counting.
Stereological counts of TH-positive cells were performed using Slidebook (Intelligent Imaging Innovations) employing the optical fractionator method (West and Gunderson, 1990). Images were captured using a Zeiss Axiovert 200M inverted fluorescence microscope equipped with a Hammatsu ORCA-ER-cooled charge-coupled device camera (Hammamatsu Photonics, Hamamatsu City, Japan). The boundary of the SNpc was determined by low-magnification (10×) montage imaging. Total numbers of TH-positive cells were obtained through imaging (40×) uniform randomly placed counting frames (100 × 100 µm) using an optical dissector of 30 µm with 5 µm upper and lower guard zones.
Representative images of TH expression in both substantia nigra and the striatum were captured using an Olympus FluoView (FV-10i; Olympus Optical Co. Ltd., Tokyo, Japan) laser scanning confocal microscope equipped with water immersion lenses. Montage images were obtained using a 10× objective, and representative high-magnification images were obtained using a 60× objective.
Striatal Dopamine and Dopamine Metabolite Content
Dopamine and DOPAC concentrations in the striatum were quantified by high-performance liquid chromatography (HPLC) coupled with electrochemical detection as described previously (Champney et al, 1992). Briefly, animals were anesthetized with isoflurane and immediately decapitated. Brains were quickly removed from the skull, and the striatal region was isolated out through referencing with a mouse brain matrix block. Striatal brain tissues were flash frozen in liquid N2 and stored at –80°C until analysis. Prior to HPLC quantitation, catecholamines were extracted through sonication in 0.2 M perchloric acid solution containing 1.5 mM EDTA, and 6 M sodium thiosulfate on ice for 2 sec. The samples were cleared by centrifugation at 14,000 rpm for 10 min at 4°C. Protein-free extracts were injected onto an Allure PFP propyl column (2.1 × 250 mm, 5 m), and components were separated by using a gradient elution with the following mobile phases. Mobile phase A consisted of the following (mM): citric acid (15) sodium phosphate (7), EDTA (0.6), and sodium octyl sulfate (0.9). Mobile phase B was 100% methanol. The gradient elution protocol was performed as follows at a flow rate of 0.3 ml/min: 100% A from 0 to 10 min; 96% A, 4% B from 10 to 25 min; 100% A from 25 to 50 min. Prior to the injection of the subsequent sample, the column was allowed to equilibrate for 10 min at 100% A.
Immunofluorescence and Colocalization With EGFP Expression
Cell-specific expression of EGFP was determined by coimmunofluorescence microscopy. Representative sections from the striatum and SNpc were stained, free floating, with a cell marker specific for astrocytes (glial fibrillary acid protein; GFAP) as well as NOS2. Cryoprotectant was removed by multiple washes in TBS. The sections were blocked in blocking buffer consisting of 1% goat and 1% donkey serum in TBS. Sections were then incubated with the cell-specific antibodies GFAP (1:1,000; Dako, Carpinteria, CA) and NOS2 (1:500; Millipore, Bedford, MA) in blocking buffer overnight at 4°C. After primary antibody incubation, sections were rinsed multiple times, followed by incubation for 3 hr at room temperature with Alexafluor-555 and Alexafluor-647 conjugated anti-rabbit and anti-goat (1:500; Invitrogen) secondary antibodies, respectively. Sections were then rinsed repeatedly and mounted using DAPI mounting media and coverslipped. Both primary and secondary antibody controls were used to determine the extent of nonspecific staining. Instrinsic EGFP fluorescence was visualized by excitation at 490 nm excitation/520 nm emission (10 nm bandpass).
Primary Cell Isolations and Treatments
Cortical astrocytes and striatal neurons were isolated from postnatal day P0–P2 cis-NF-κBEGFP transgenic or C57Bl/6 mouse pups as previously described, with slight modifications (Allen et al., 2000; Carbone et al., 2009). Briefly, mouse pups were euthanized by decapitation, followed by rapid dissection of the cortical or striatal regions and removal of meninges. The tissue was then digested with dispase (1.5 U/ml) with constant stirring. Every 10 min, the dispase solution was replaced with fresh solution for a total of 50 min. The extractions containing dissociated cells were kept on ice and pooled at the end of the digestion. Dissociated astrocytes were plated onto 60-mm tissue culture plates at a cell density of 1 × 104 cells/cm2 and allowed to adhere for 24 hr, followed by a media rinse to remove contaminating cell types. Astrocyte cultures were allowed to grow to approximately 80–90% confluence before treatments. Purity of cultures was determined by immunofluorescence to be greater than 95% using GFAP as an astrocyte marker. Dissociated striatal neurons were seeded on poly-D-lysine-coated 30-mm coverslips at a cell density of 4 × 105 cells/coverslip. Neuronal cultures were maintained for 1 week prior to any treatments. Astrocyte and neuronal cultures were maintained at 37°C in a humidified atmosphere of 5% CO2/95% air (v/v) in either minimum essential media supplemented with 10% fetal bovine serum and 1× antibiotic cocktail (0.001 mg/ml penicillin, 0.001 mg/ml streptomycin, 0.002 mg/ml neomycin) or neurobasal media containing 1× B27 supplement and 1× antibiotic cocktail, respectively.
To investigate the role of astrocyte activation in neuronal degeneration, a coculture system was established using permeable cell culture inserts (BD Biosciences, San Jose, CA; 0.4-µm, six-well inserts) that permit diffusion of small molecules but no direct cell–cell contact. Astrocytes were plated on the inserts and cultured until 90% confluent and were subsequently subject to adenovirally mediated suppression of NF-κB signaling through overexpression of a phosphorylation-deficient mutant IκBα (Jobin et al., 1998). Astrocytes were infected with 2 × 106 viral particles per milliliter of Opti-MEM for 24 hr, rinsed, and allowed to recover for an additional 24 hr in complete culture media. The effectiveness of the suppression was checked using real-time RT-PCR analysis of Nos2 induction as described previously (Carbone et al., 2008). After the recovery period, the astrocytes were challenged with an inflammatory stimulus through simultaneous exposure to 10 µM MPTP plus TNF-α (10 pg/ml) and IF-γ (1 ng/ml) for 8 hr, rinsed three times with PBS and then cultured above the primary striatal neurons in neurobasal media for 6 hr. Neuronal apoptosis was determined using live cell fluorescence microscopy utilizing the cell-permeable pan-caspase substrate rhodamine 110, bis-(L-aspartic acid amide; Invitrogen) as a general indicator of caspase activation. At least eight microscopic fields per treatment group were examined, and the experiment was repeated a total of three times.
Statistical Analysis
Data are presented as mean ± SEM. Comparative analysis of treatments was made by one-way ANOVA, followed by Neuman-Keuls post hoc analysis. All statistical analysis were conducted in Prism software (version 4.0; Graph Pad Software, San Diego, Ca). Means were determined to be significant at P < 0.05. Groups that are significantly different (P < 0.05) from each are designated with different letters.
RESULTS
Locomotion and Gait Analysis
The effects of MPTP treatment on neurobehavioral function were assessed by open-field activity measurements (Fig. 1). Over the 7-day exposure period, significant depression of spontaneous locomotor activity was observed on day 1 posttreatment compared with saline controls, followed by a complete recovery to control levels and in some instances hyperrecovery of function on subsequent days (Fig. 1A). Significant reductions were observed only in the 60 mg/kg treatment group in total distance traveled, total movement time, and number of rearing movements on day 1 posttreatment, although a noticeable dose-dependent trend could be discerned (Fig. 1B). No significant changes were observed in the other parameters tested. In addition to changes in spontaneous locomotion, alterations in gait features have also been observed in MPTP-treated mice as well as other toxicant-induced models of basal ganglia dysfunction (Fernagut et al., 2002; Amende et al., 2005). Abnormal gait properties were assessed through hind-limb stride length measurements. A significant decrease in stride length was observed in the 60 mg/kg dose group on day 7 posttreatment in comparison with initial baseline readings obtained on day 0 prior to treatment (Fig. 1C).
Fig. 1.

Low-dose MPTP induces changes in locomotor activity. A: Locomotor activity over the 7-day treatment period as measured by open-field activity. B: Locomotor activity data for day 1 posttreatment. The highest dose of 60 mg/kg MPTP resulted in significant reduction in three of the four locomotor parameters tested 24 hr after dosing, which completely recovered by 6 days posttreatment. Data are expressed as mean ± SEM (n = 10). C: Stride length measurements 6 days posttreatment expressed as percentage change compared with baseline (day 0).
TH Expression and Dopaminergic Neuron Counts
MPTP-induced neurodegeneration was determined using unbiased systematic stereological counting of dopaminergic neurons in the SNpc. Representative photomicrographs are shown in Figure 2A, depicting fluorescent TH immunostaining of dopamineregic neurons in SNpc from both control and 60 mg/kg dose groups. Exposure to MPTP at all doses did not significantly reduce the total number of TH-positive neurons (Fig. 2B). Although quantification of TH-positive neurons did not reveal significant loss in the number of dopaminergic neurons upon treatment with MPTP, a generalized loss of TH immunofluorescence intensity was observed in the SNpc (Fig. 2A). Similarly, the overall intensity of TH immunostaining in the striatum was significantly reduced during exposure to MPTP at the highest dose of 60 mg/kg, consistent with loss of dopaminergic nerve terminals in the striatum.
Fig. 2.

MPTP induces dose-dependent neuropathological changes in the nigrostriatal system. A: Representative 10× confocal montage images of tyrosine hydroxylase (TH; red) expression in the substantia nigra pars compacta of saline control and 60 mg/kg MPTP-treated animals counterstained with DAPI (blue). Representative 60× confocal images of TH-positive neurons in the SNpc of saline control and 60 mg/kg MPTP-treated animals (inset). B: Estimated number of total TH+ neurons in the SNpc as determined by stereological counting (n = 5). C: Representative 10× confocal montage images of TH immunostaining in the striatum of saline control and 60 mg/kg MPTP-treated animals counterstained with DAPI. D: Quantitation of TH expression in the striatum. Data expressed as mean fluorescence intensity in relative fluorescence units ± SEM (n = 5). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Dopamine and Dopamine Metabolite Levels
MPTP effects on striatal concentrations of dopamine and the dopamine metabolite DOPAC were determine by using HPLC analysis. Administration of MPTP at the highest concentration of 60 mg/kg showed significant reduction in both striatal dopamine and DOPAC levels at day 7 posttreatment (Fig. 3A,B). Neither of the two lower doses (15 mg/kg or 30 mg/kg) was sufficient to alter striatal neurochemistry significantly, although a dose-dependent trend in reduction of DOPAC could be observed. The ratio of DOPAC to dopamine also decreased, suggesting a decrease in dopamine oxidation (Fig. 3C).
Fig. 3.
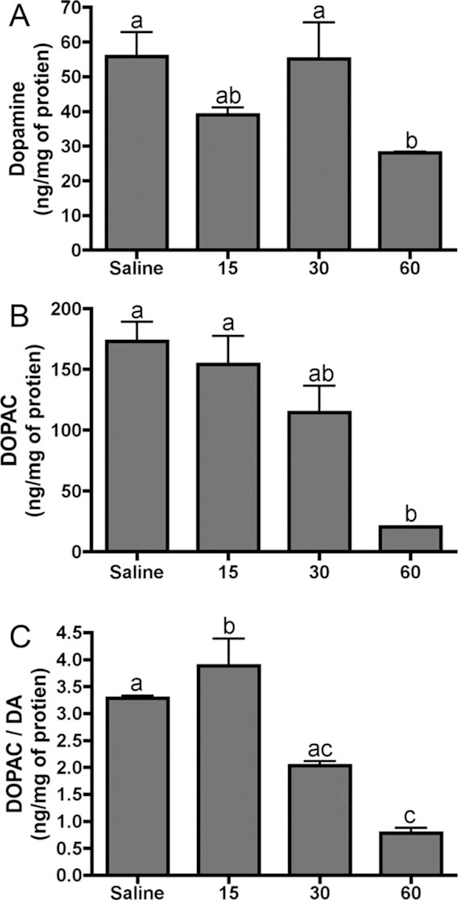
Low-dose MPTP exposure results in decreased striatal dopamine levels. Dopamine (A) and DOPAC (B) concentrations and DOPAC/dopamine ratio (C) as determined by HPLC analysis. Catecholamine concentrations are expressed as ng/mg protein ± SEM (n = 3).
Astrocyte-Specific NF-κB Activation
Astrocyte-specific activation of NF-κB was determined by coimmunoflourescence in cis-NF-κBEGFP reporter mice using the specific marker GFAP. The number of cells in the astrocyte population was assessed for expression of both EGFP and NOS2, a prototypic NF-κB-regulated inflammatory gene. Representaive images showing the colocalization of GFAP astrocyte marker with expression of the GFP reporter and NOS2 are shown in Figure 4A. Exposure to both 30 mg/kg and 60 mg/kg MPTP resulted in significant increases in the number of astrocytes expressing both GFP and inducible NOS2 (Fig. 4B–D).
Fig. 4.

Neuroinflammatory activation of astrocytes occurs in NF-κB reporter mice prior to loss of dopaminergic neurons. A: Representative 40× images of astrocytes (GFAP; purple) expressing intrinsic GFP fluorescence (green), inducible nitric oxide synthase (NOS2; red), and counterstained with DAPI (blue). B: Quantification of the number of GFAP+astrocytes expressing intrinsic GFP. C: Quantification of the number of GFAP+astrocytes expressing NOS2. D: Quantification of the number of GFAP+astrocytes expressing both GFP and NOS2. Data are expressed as percentage ± SEM (n = 9). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Increase Expression of 3-Nitrotyrosine in the SNpc
Induction of NOS2 with subsequent increased production of NO is associated with glial-specific neuroinflammatory responses in PD. Global protein nitration in the SNpc was determined by coimmunofluorescence staining intensity for 3-nitrotyrosine (3-NT) protein adducts in TH-positive neurons (Fig. 5). Mice exposed to the highest dose of 60 mg/kg MPTP showed a significant increase in 3-NT staining, with the most intense staining colocalizing with TH-positive neurons, athough expression is also apparent in non-TH-positve cells.
Fig. 5.
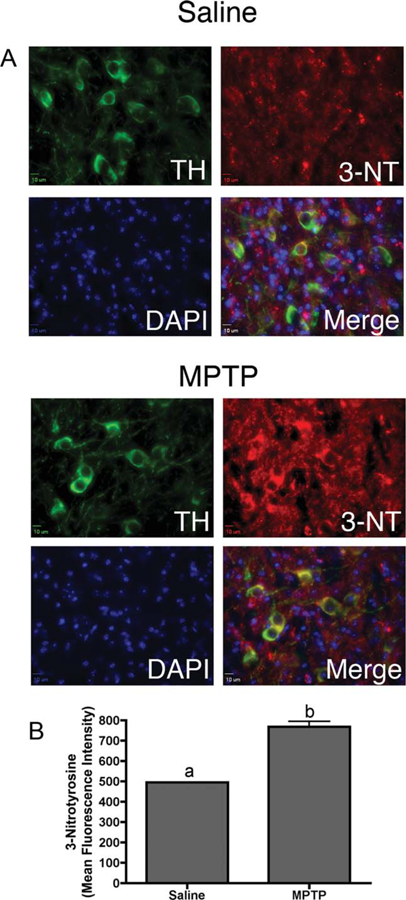
MPTP exposure increased global protein nitration in dopmainergic neurons in the substantia nigra. A: Representative 40× images of tyrosine hydroxylase (TH; green) and 3-nitrotyrosine (3-NT; red) expression in the SNpc of saline control and 30 mg/kg MPTP-treated animals counterstained with DAPI. B: Quantification of 3-NT mean fluorescence intensity (n = 6 individual animals per group). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Astrocyte/Neuron Coculture
Exposure of primary astrocytes to low doses of MPTP + TNF-α + IFNγ resulted in inflammatory activation, as determined by NOS2 gene expression. Adenovirally mediated overexpression of a phosphylation-deficient mutant of IκBα completely suppressed MPTP-and cytokine-dependent induction of Nos2 mRNA (Fig. 6A). Astrocytes grown in permeable cell culture inserts were activated by an 8-hr exposure to MPTP and TNF-α/IFNγ and then incubated for 6 hr with primary striatal neurons, followed by determination of neuronal apoptosis by live-cell fluorescence imaging for caspase activation. Activated astrocytes caused apoptosis in cocultured striatal neurons that was inhibited by overexpression of dominant-negative IκBα (Fig. 6B,C).
Fig. 6.

Suppression of NF-κB signaling in astrocytes prior to inflammatory activation with MPTP protects cocultured striatal neurons. A: Real-time RT-PCR quantification of NOS2 mRNA expression following treatment with 10 µM MPTP, 1 pg/ml TNF-α, and 1 ng/ml IFNγ in the presence of mutant IκBα or empty adenoviral vector (EV). B: Representative 40× images of caspase activity in primary striatal neurons after 6 hr of incubation with stimulated astrocytes expressing mutant IκBα or a control vector. C: Quantitation of caspase activity. Data expressed as mean fluorescence intensity in relative fluorescence units ± SEM (n = 3 independent experiments, with images taken from a minimum of eight microscopic fields per coverslip). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DISCUSSION
A role for glial inflammatory responses in PD and other neurodegenerative disorders is well established (Campbell, 2004; Lucas et al., 2006; Rojo et al., 2008). Both activated astrocytes and reactive microglia have been observed in post-mortem brains of PD patients as well as in parkinsonian animal models (McGeer et al., 1988; Kurkowska-Jastrzebska et al., 1999). Inflammatory activation of glia results in the production of numerous neurotoxic substances, including proinflammatory cytokines and reactive oxygen and nitrogen species (Hirsch and Hunot, 2000). Activation of microglia in response to degenerating neurons has been implicated as a key component in the progressive nature of neurodegenerative disorders, including PD (Liberatore et al., 1999). Damaged dopaminergic neurons can release substances such as α-synuclein and neuromelanin, both of which are able to activate microglia (Block and Hong, 2007). Recent evidence suggests that astrocyte activation may precede microglial activation and ensuing inflammatory injury (Aoki et al., 2009). This contrasts with earlier studies indicating that microglial activation is an early event in acute MPTP intoxication that subsides to control levels prior to peak activation of astrocytes (Liberatore et al., 1999). However, both our results and these previous reports highlight the importance of reactive astrogliosis in MPTP-induced neurotoxic injury. It is clear that exaggerated inflammatory responses of glia contribute to the progressive degeneration encountered in PD, but whether neuroinflammation is merely a consequence of neuronal degeneration or an active participant in the pathogenesis of the disease remains a key question.
One of the most widely used toxicant-induced parkinsonian models is the MPTP mouse model, in which repeated systemic administration of MPTP produces dopaminergic neuron loss with accompanied depletion of striatal dopamine. Typical treatment paradigms with MPTP use an acute dosing strategy, in which relatively high doses of MPTP are employed (total of 80–100 mg/kg in four doses spread over 8 hr). With this dosing regimen, loss of dopaminergic neurons occurs by 4 days after dosing, with up to 90% loss of striatal dopamine after 7 days (Jackson-Lewis and Przedborski, 2007). In the present study, a range of low to moderate doses of MPTP was used in order to identify early changes in glial activation that might occur prior to induction of overt pathology and loss of dopaminergic neurons.
Measurements of neurobehavioral activity using open-field activity monitors did not reveal a significant effect on locomotion at either of the two lower doses (15, 30 mg/kg) of MPTP employed, although the data trended toward a dose-dependent effect. The lack of significant reduction in locomotion at the two lower doses is consistent with previously published reports (Tillerson et al., 2002). In contrast, exposure to the highest dose (60 mg/kg) of MPTP caused a significant reduction in the total number of movements, indicating onset of hypokinesia within 24 hr of dosing. In addition to general locomotor activity, gait analysis revealed a significant reduction in hind-limb stride length by day 7 posttreatment at the highest dose of MPTP, consistent with previous studies reporting this locomotor parameter as a sensitive indicator of striatal dopamine concentrations (Tillerson et al., 2002) and with clinical reports of neurological dysfunction in PD (Fernagut et al., 2002). The inability of the two lowest doses to affect stride length is consistent with the absence of neurochemical changes but contradicts the observed reduction in stride length by Tillerson et al. (2002) in both the 15 mg/kg and the 30 mg/kg dose groups. However, this may be due to the differences in age of the animals employed; their animals were significantly older than those used in the present study. Other studies have also reported age-dependent susceptibility of mice to the neurotoxic effects of MPTP (Mitsumoto et al., 1998). In contrast to stride length, the acute depression of overall locomotor acitivity appeared to be transient, because a recovery in locomotor activity was observed by 7 days after dosing. The reversible nature of MPTP-induced hypokinesia, even in the presence of a significant reduction in striatal dopamine levels, has been shown in various MPTP studies (Sedelis et al, 2000; Hofele et al., 2001). A potential explanation for this outcome could be the involvement of additional neurotransmitter systems, including serotonin, which may compensate for the depletion of dopamine (Sedelis et al., 2001). This hypothesis is supported by the blockage of MPTP-induced hyperactivity by selective ablation of serotonergic neurons with 5,7-dihydroxytryptamine (Chia et al., 1999).
In accordance with the observed neurobehavioral changes, exposure to the highest dose of MPTP resulted in a generalized loss of TH in the SNpc, with noticeable reduction in both the dendritic processess of the ventral tier of the compacta neurons, which project into the reticulata, and the cell bodies of the ventral, medial, and dorsal tiers of neurons. Similarly to the results in SNpc, TH expression in the striatum was also significantly reduced with exposure to the highest dose of MPTP, indicating loss of dopaminergic nerve terminals, which is consistent with the observed depletion of striatal dopamine. Although loss of TH in the SNpc is apparent, stereological counts of dopaminergic neurons showed no significant reduction in the total number of cells. Evidence in the literature suggests that loss of TH expression, especially in dendritic processes, may precede actual loss of the cells, which indicates that the 60 mg/kg dose of MPTP may just precede the initial phases of the pathological progression toward neurodegeneration (Jackson-Lewis et al., 1995; Bywood and Johnson, 2000). Both the neurobehavioral and neuropathological data demonstrate that the dose ranges used in this study encompass the subpathological phase of MPTP-induced dysfunction at which glial inflammatory responses are detectable prior to loss of dopaminergic neurons.
Inflammatory activation of astrocytes has been consistently observed in chronic neurodegenerative disorders and may represent an important event in disease pathogenesis. Initially, this response is believed to be neuro-protective because of the increased production of neuro-trophic factors and antioxidants, which have been shown to increase the survival of dopaminergic neurons in culture as well as in vivo PD models (Mena and García yébenes, 2008). Although astrocyte activation may start out as a protective response, prolonged or exaggerated activation may in fact lead to potentiation of neuronal injury (Mena and García y Yébenes, 2008; McGeer and McGeer, 2008). This most likely is due to overproduction of inflammatory mediators such as TNF-α and IL-1β as well as superoxide and nitric oxide. The production of these and other proinflammatory mediators is significantly regulated by NF-κB (Wang et al., 2002). Using a unique NF-κB-EGFP reporter mouse, we have demonstrated that treatment with 30 and 60 mg/kg MPTP caused an increase in NF-κB activation in astrocytes that correlated with increased expression of the neuroinflammatory gene NOS2. These doses of MPTP also caused a corresponding increase in global protein nitration in dopaminergic neurons, as determined by 3-NT staining. The increase in 3-NT residues was also evident in surrounding glial cells, similar to earlier studies from our laboratory that reported increases in MPTP-induced protein nitration in primary cultured astrocytes (Carbone et al., 2008). Increased nitration of critical proteins may result in loss of neuronal function that could contribute to neuroinflammatory injury. This is supported by studies reporting inactivation of TH by peroxynitrite in MPTP-treated mice (Ara et al., 1998) and detectable nitrated α-synuclein in protein aggregates in post-mortem PD brain tissue (Giasson et al., 2000).
In vitro studies were carried out to determine whether NF-κB signaling is required for inflammatory activation of astrocytes and whether MPTP-activated astrocytes could promote neuronal degeneration. Stimulation of astrocytes with low doses of MPTP and inflammatory cytokines resulted in NF-κB-dependent induction of NOS2 that was suppressed by overexpression of dominant-negative IκBα, a ‘‘super-repressor’’ of NF-κB (Jobin et al., 1998). Coculturing striatal neurons with MPTP/cytokine-stimulated astrocytes resulted in activation of apoptotic cell death, as measured by activation of caspases, an end point shown to correlate closely with additional indicators of apoptosis, including TUNEL staining and nuclear condensation (Liu et al., 2005; Tjalkens et al., 2008; Carbone et al., 2009). Suppressing NF-κB activity in astrocytes with mutant IκBα significantly reduced cell death in cocultured neurons. These results demonstrate that low doses of MPTP and inflammatory cytokines induce an activated phenotype in astrocytes that injures adjacent neurons and that abrogation of NF-κB signaling suppresses astrocyte-dependent neuronal injury.
Collectively, these data demonstrate that increased NF-κB activity is detectable in astrocytes prior to significant neurobehavioral changes or significant loss of dopaminergic neurons in mice treated with low doses of MPTP. This suggests that NF-κB-dependent expression of NOS2 in astrocytes and subsequent neuronal protein nitration represents an early phase in the progression of MPTP-induced neuropathology that occurs prior to overt loss of dopaminergic neurons. This transgenic reporter model may therefore be useful for detecting other exogenous or endogenous activators of NF-κB in glial cells with the potential to cause neuroinflammatory injury. Additionally, these studies suggest that NF-κB activation in glial cells represents an important signaling event early in the development of neuropathology that may be predictive of subsequent neuronal injury in more advanced stages of injury or disease.
ACKNOWLEDGMENTS
The authors thank Dr. Christian Jobin at the University of North Carolina at Chapel Hill for generously providing the NF-κB reporter mouse.
Contract grant sponsors: AstraZeneca Pharmaceuticals and The Michael J. Fox Foundation for Parkinson’s Research.
REFERENCES
- Allen JW, Mutkus LA, Aschner M. 2000. Current protocols in toxicology New York: John Wiley & Sons; 12.4.1–12.4.15. [Google Scholar]
- Amende I, Kale A, McCue S, Glazier S, Morgan JP, Hampton TG. 2005. Gait dynamics in mouse models of Parkinson’s disease and Huntington’s disease. J Neuroengineer Rehab 2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki E, Yano R, Yokoyama H, Kato H, Araki T. 2009. Role of nuclear transcription factor kappa B (NF-kappaB) for MPTP(1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine)-induced apoptosis in nigral neurons of mice. Exp Mol Pathol 86:57–64. [DOI] [PubMed] [Google Scholar]
- Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H. 1998. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc Natl Acad Sci U S A 95:7659–7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Hong JS. 2007. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem Soc Trans 35:1127–1132. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR, Bethea JR. 2009. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol 182:2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywood PT, Johnson SM. 2000. Loss of tyrosine hydroxylase immunore-activity in dendrites is a sensitive index of kainic acid-induced damage in rat substantia nigra neurons in vivo. Neurosci Lett 280:5–8. [DOI] [PubMed] [Google Scholar]
- Camandola S, Mattson MP. 2007. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin Ther Targets 11:123–132. [DOI] [PubMed] [Google Scholar]
- Campbell A. 2004. Inflammation, neurodegenerative diseases, and environmental exposures. Ann N Y Acad Sci 1035:117–132. [DOI] [PubMed] [Google Scholar]
- Carbone DL, Moreno JA, Tjalkens RB. 2008. Nuclear factor kappa-B mediates selective induction of neuronal nitric oxide synthase in astrocytes during low-level inflammatory stimulation with MPTP. Brain Res 1217:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone DL, Popichak KA, Moreno JA, Safe SH, Tjalkens RB. 2009. Suppression of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced nitric-oxide synthase 2 expression in astrocytes by a novel diindolylmethane analog protects striatal neurons against apoptosis. Mol Pharmacol 75:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champney TH, Hanneman WH, Nichols MA. 1992. Gamma-aminobutyric acid, catecholamine and indoleamine determinations from the same brain region by high-performance liquid chromatography with electrochemical detection. J Chromatogr 579:334–339. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernán MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A. 2003. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 60:1059–1064. [DOI] [PubMed] [Google Scholar]
- Chia LG, Ni DR, Cheng FC, Ho YP, Kuo JS. 1999. Intrastriatal injection of 5,7-dihydroxytryptamine decreased 5-HT levels in the striatum and suppressed locomotor activity in C57BL/6 mice. Neurochem Res 24:719–722. [DOI] [PubMed] [Google Scholar]
- Cho I-H, Hong J, Suh EC, Kim JH, Lee H, Lee JE, Lee S, Kim C-H, Kim DW, Jo E-K, Lee KE, Karin M, Lee SJ. 2008. Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death. Brain 131:3019–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart MA, Baeuerle P, Vassalli P. 1990. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 10:1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Labattu B, Tison F. 2002. A simple method to measure stride length as an index of nigrostriatal dysfunction in mice. J Neurosci Methods 113:123–130. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. 2000. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290:985–989. [DOI] [PubMed] [Google Scholar]
- Hald A, Van Beek J, Lotharius J. 2007. Inflammation in Parkinson’s disease: causative or epiphenomenal? Subcell Biochem 42:249–279. [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. 2000. Nitric oxide, glial cells and neuronal degeneration in parkinsonism. Trends Pharmacol Sci 21:163–165. [DOI] [PubMed] [Google Scholar]
- Hofele K, Sedelis M, Auburger GW, Morgan S, Huston JP, Schwarting RK. 2001. Evidence for a dissociation between MPTP toxicity and tyrosinase activity based on congenic mouse strain susceptibility. Exp Neurol 168:116–122. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Przedborski S. 2007. Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc 2:141–151. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. 1995. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurode-generation 4:257–269. [DOI] [PubMed] [Google Scholar]
- Jobin C, Panja A, Hellerbrand C, Iimuro Y, Didonato J, Brenner DA, Sartor RB. 1998. Inhibition of proinflammatory molecule production by adenovirus-mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J Immunol 160:410–418. [PubMed] [Google Scholar]
- Kalinin S, Richardson JC, Feinstein DL. 2009. A PPardelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer’s dsisease. Curr Alzheimer Res (in press). [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. 2000. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol 18:621–663. [DOI] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Wroñska A, Kohutnicka M, Czonkowska A. 1999. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication in mouse. Exp Neurol 156:50–61. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. 1999. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med 5:1403–1409. [DOI] [PubMed] [Google Scholar]
- Liu X, Buffington JA, Tjalkens RB. 2005. NF-kappaB-dependent production of nitric oxide by astrocytes mediates apoptosis in differentiated PC12 neurons following exposure to manganese and cytokines. Brain Res Mol Brain Res 141:39–47. [DOI] [PubMed] [Google Scholar]
- Lucas S-M, Rothwell NJ, Gibson RM. 2006. The role of inflammation in CNS injury and disease. Br J Pharmacol 147(Suppl 1):S232–S240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, Jobin C. 2004. In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol 173:1561–1570. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. 2008. Glial reactions in Parkinson’s disease. Mov Disord 23:474–483. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. 1988. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291. [DOI] [PubMed] [Google Scholar]
- Mena MA, García de Yébenes J. 2008. Glial cells as players in parkinsonism: the ‘‘good,’’ the ‘‘bad,’’ and the ‘‘mysterious’’ glia. Neuroscientist 14:544–560. [DOI] [PubMed] [Google Scholar]
- Mitsumoto Y, Watanabe A, Mori A, Koga N. 1998. Spontaneous regeneration of nigrostriatal dopaminergic neurons in MPTP-treated C57BL/6 mice. Biochem Biophys Res Commun 248:660–663. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. 2000. Cytokines in Parkinson’s disease. J Neural Transm Suppl, p143–151. [PubMed] [Google Scholar]
- Nakano H, Shindo M, Sakon S, Nishinaka S, Mihara M, Yagita H, Okumura K. 1998. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci U S A 95:3537–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND. 2000. The Rel/NF-kappa B family: friend and foe. Trends Biochem Sci 25:434–440. [DOI] [PubMed] [Google Scholar]
- Redfern W, Ewart L, Hammond T, Bialecki R, Kinter L, Lindgren S, Pollard C, Roberts R, Rolf MG, Valentin JP. 2010. Impact and frequency of different toxicities throughout the pharmaceutical drug development process. Toxicologist 14:1081. [Google Scholar]
- Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. 2008. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res 39:1–16. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. 2009. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelis M, Hofele K, Auburger GW, Morgan S, Huston JP, Schwarting RK. 2000. MPTP susceptibility in the mouse: behavioral, neurochemical, and histological analysis of gender and strain differences. Behav Genet 30:171–182. [DOI] [PubMed] [Google Scholar]
- Sedelis M, Schwarting RK, Huston JP. 2001. Behavioral phenotyping of the MPTP mouse model of Parkinson’s disease. Behav Brain Res 125:109–125. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. 2002. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. FASEB J 16:1474–1476. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Zandi PP. 2010. Non-steroidal anti-inflammatory drugs and Alzheimer’s disease: the epidemiological evidence. CNS Neurol Disord Drug Targets 9:132–139. [DOI] [PubMed] [Google Scholar]
- Tillerson JL, Caudle WM, Reverón ME, Miller GW. 2002. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp Neurol 178:80–90. [DOI] [PubMed] [Google Scholar]
- Tjalkens RB, Liu X, Mohl B, Wright T, Moreno JA, Carbone DL, Safe SH. 2008. The peroxisome proliferator-activated receptor-gamma agonist 1,1-bis(3ʹ-indolyl)-1-(p-trifluoromethylphenyl) methane suppresses manganese-induced production of nitric oxide in astrocytes and inhibits apoptosis in cocultured PC12 cells. J Neurosci Res 86:618–629. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, De Wilde G, Notebaert S, Vanden Berghe W, Haegeman G. 2002. Regulation of the transcriptional activity of the nuclear factor-kappaB p65 subunit. Biochem Pharmacol 64:963–970. [DOI] [PubMed] [Google Scholar]
- Wang T, Zhang X, Li JJ. 2002. The role of NF-kappaB in the regulation of cell stress responses. Int Immunopharmacol 2:1509–1520. [DOI] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. 1990. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 296:1–22. [DOI] [PubMed] [Google Scholar]
- Whitton PS. 2007. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol 150:963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilms H, Zecca L, Rosenstiel P, Sievers J, Deuschl G, Lucius R. 2007. Inflammation in Parkinson’s diseases and other neurodegenerative diseases: cause and therapeutic implications. Curr Pharm Des 13:1925–1928. [DOI] [PubMed] [Google Scholar]
- Xie QW, Kashiwabara Y, Nathan C. 1994. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269:4705–4708. [PubMed] [Google Scholar]