

Abstract
Receptor‐type protein tyrosine phosphatase κ (PTPRK) is considered to be a candidate tumor suppressor. PTPRK dephosphorylates CD133, which is a stem cell marker; phosphorylated CD133 accelerates xenograft tumor growth of colon cancer cells through the activation of AKT, but the functional significance of this has remained elusive. In this study, we have demonstrated that knockdown of PTPRK potentiates the pro‐oncogenic CD133–AKT pathway in colon cancer cells. Intriguingly, depletion of PTPRK significantly reduced sensitivity to the anti‐cancer drug oxaliplatin and was accompanied by up‐regulation of phosphorylation of Bad, a downstream target of AKT. Together, our present observations strongly suggest that the CD133–PTPRK axis plays a pivotal role in the regulation of colon cancer progression as well as drug resistance.
Keywords: CD133, colon carcinoma, drug resistance, protein tyrosine phosphatase, PTPRK
Abbreviations
- CSC
cancer stem cell
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- PARP
poly(ADP‐ribose) polymerase
- PTP
protein tyrosine phosphatase
- PTPRK
receptor‐type protein tyrosine phosphatase κ
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
Colon cancer is the third leading cause of cancer‐related death and responsible for over 50 000 per year deaths in the USA 1. Although there has been substantial progress in treatment in the past decade, relapse and metastasis to the other tissues/organs are serious issues to be addressed to cure colon cancer patients 2. Various phase 3 clinical trials to evaluate the potential effects of doublet or triplet chemotherapy plus molecular targeting agents such as bevacizumab showed an improvement in the progression‐free survival period of patients with advanced colon cancer; however, their 5‐year survival rate has not been improved 3. In this regard, understanding the precise molecular mechanism(s) behind the aggressiveness and resistance to therapy of advanced colon cancer is an urgent issue to be adequately addressed for better treatment of colon malignancy.
A growing body of evidence suggests that a tyrosine phosphorylation of cellular proteins is one of the critical molecular events regulating numerous cellular processes such as proliferation and cell death; therefore, its abnormality might disrupt the strict regulation of these processes, thereby triggering the initiation/progression of tumors 4. Receptor‐type protein tyrosine phosphatase κ (PTPRK), which belongs to a subfamily of membrane‐bound protein tyrosine phosphatases (PTPs), is expressed as a single precursor protein 5. Upon the single proteolytic cleavage catalyzed by furin protease 6, the resultant carboxyl‐terminal portion (P‐subunit) is anchored to cell membrane, recognizes the phosphorylated tyrosine residues of its target proteins, and catalyzes their dephosphorylation. The amino‐terminal portion (E‐subunit) acts as the extracellular subunit and includes a meprin/A5/m (MAM) domain, an immunoglobulin‐like domain and four fibronectin III‐like repeats; it associates with the P‐subunit non‐covalently. Like the other PTPs, PTPRK might attenuate the activation of various signaling pathways evoked by tyrosine phosphorylation of cellular proteins through the removal of their phosphate groups. For instance, PTPRK has been shown to dephosphorylate epidermal growth factor receptor (EGFR) and subsequently prohibits epidermal growth factor (EGF)‐mediated cell proliferation of human mammary epithelial cells and keratinocytes 7, 8, 9.
Notably, several lines of evidence indicate that PTPRK acts as a putative tumor suppressor. In support of this notion, it has been described that a loss of 6q22‐23 where the PTPRK locus is located is frequently detectable in patients with certain malignancies such as sporadic endocrine pancreatic tumors and juvenile intestinal carcinoma regardless of hereditary and inflammatory disease‐related factors 10, 11. Agarwal et al. 12 found a loss of function mutation within PTPRK in glioblastoma patients. Stevenson et al. 13 reported the presence of DNA methylation‐induced epigenetic silencing of PTPRK in certain blood malignancies. Consistent with these observations, a transposon‐mediated mutagenic screening revealed that mutation and/or dysregulation of Ptprk as well as Apc, Pten, and Smad4 increases the susceptibility to intestinal lesions including intraepithelial neoplasia, adenoma, and adenocarcinoma 14. In addition, Sun et al. 15, 16 described that knockdown of PTPRK promotes proliferation and migration of human breast and prostate cancer cells.
The cancer stem cell (CSC) hypothesis has become increasingly accepted and might provide a clue to the understanding of the precise molecular basis underlying cancer initiation, progression, metastasis, and recurrence 17, 18, 19. Similar to normal tissue stem cells, CSC‐like cells with a higher tumorigenic potential are resistant to anti‐cancer drugs as well as irradiation 20, 21, and thus reliable molecular marker(s) for identifying CSCs might be a promising molecular target to develop a novel therapeutic strategy for cancers. CD133 (also known as prominin‐1/prominin‐like 1) is a unique pentaspan‐transmembrane glycoprotein initially identified in CD34‐positive hematopoietic stem cells 22, 23. Recently, CD133 has been recognized as one of the molecular markers of stem/progenitor cells in various tissues including kidney, neuron, and pancreas 24, 25, 26, 27. For example, Zhu et al. described that CD133‐positive cells exist at the base of the intestinal crypts and have a capability to differentiate into intestinal epithelial cells and goblet cells 24, 25, 26, 27. In addition to cellular differentiation, CD133‐positive tumor cells have displayed CSC‐like behaviors such as a higher tumorigenic as well as self‐renewal potential and chemo‐resistance, indicating that CD133 is one of the reliable CSC markers 28, 29, 30, 31. In good agreement with these functional properties, the expression level of CD133 has a strong prognostic impact on patients with various malignant tumors 32, 33, 34.
Since Boivin et al. 35 found tyrosine phosphorylation of CD133 by Src family kinases, its biological significance in tumorigenesis has been investigated. Recently, Wei et al. 36 found that the phosphorylation level of CD133 is positively associated with a progression of human brain tumor through the activation of the phosphoinositide 3‐kinase–AKT pathway. According to our previous study, forced expression of a phosphomimetic mutant of CD133 potentiated the aggressive phenotypes of colon cancer cells. In accordance with these results, a lower expression of PTPRK, which dephosphorylates CD133 in vitro, was significantly correlated to a short progression‐free survival of the patients with colon cancer expressing a higher level of CD133 37. In the present study, we sought to examine the functional significance of the PTPRK–CD133 axis in colon cancer progression and found for the first time that PTPRK‐mediated dephosphorylation of CD133 suppresses the progression of colon cancer cells and also improves their anti‐cancer drug sensitivity at least in part through down‐regulation of pro‐oncogenic CD133–AKT pathway.
Materials and methods
Cell culture
Human colon cancer‐derived HT‐29 and SW480 cells, and human embryonic kidney‐derived 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma‐Aldrich, St Louis, MO, USA) supplemented with 10% heat‐inactivated FBS (Invitrogen, Carlsbad, CA, USA) and 50 μg·mL−1 of penicillin/streptomycin (Sigma‐Aldrich) in a humidified atmosphere with 5% CO2 at 37 °C. Their identities were verified by a short tandem repeat assay.
Forced expression and knockdown by lentiviral vectors
Lentivirus‐mediated transduction was performed as described previously 37, 38. In brief, 293T cells were co‐transfected with combinations of lentivirus packaging plasmids (MISSION Lentiviral packaging mix; Sigma‐Aldrich) together with the transducing plasmid carrying cDNA for enhanced green fluorescent protein (eGFP) or CD133, or for the expression of short hairpin RNA (shRNA) against PTPRK (pLKO.1; Sigma‐Aldrich) using FuGENE HD transfection reagent (Promega, Madison, WI, USA) according to the manufacturer's instructions. Following the preparation of the cell‐free culture supernatants that contain virus vectors, the indicated colon cancer cells were cultured with the conditioned medium supplemented with 25% (v/v) of the virus‐containing culture supernatants for 24 h at 37 °C. These shRNA‐transfected cells were selected by puromycin (1 μg·mL−1; Sigma‐Aldrich).
Semi‐quantitative RT‐PCR
Total RNA was extracted from cells using Isogen reagent (Nippon gene, Tokyo, Japan) and 5 µg of total RNA was reverse‐transcribed by Superscript III reverse transcriptase (Invitrogen) according to the manufacturers' protocols. The resultant cDNA was used for PCR. Oligonucleotide primer sets used in this study were as follows: CD133, 5′‐TTCCAGAAGCTCTGAGGCAG‐3′ (forward) and 5′‐AGAAATACCCCACCAGAGGC‐3′ (reverse); PTPRK, 5′‐TTGTTGAGACCAGCACAAGC‐3′ (forward) and 5′‐AAGCCTTGGTAGGTCCGATT‐3′ (reverse); GAPDH, 5′‐ACCACAGTCCATGCCATCAC‐3′ (forward) and 5′‐TCCACCACCCTGTTGCTGTA‐3′ (reverse). GAPDH was used as an internal control. PCR products were separated on 1% agarose gels and visualized by ethidium bromide staining.
Western blot analysis
Cells were lysed in a lysis buffer containing 50 mm Tris/HCl (pH 7.5), 150 mm NaCl, 1% NP‐40, 1 mm EDTA and a protease inhibitors cocktail (Calbiochem, San Diego, CA, USA). Equal amounts of cell lysates were separated by 10% SDS/PAGE under reducing condition and electro‐transferred onto a poly(vinylidene difluoride) membrane (Merck Millipore, Billerica, MA, USA). The membrane was probed with the primary antibodies against CD133 (W6C3B1; Miltenyi Biotec, Bergisch Gladbach, Germany), PTPRK (HPA054822; Sigma‐Aldrich), phospho‐AKT at Ser‐473 (no. 4060; Cell Signaling Technology, Danvers, MA, USA), AKT (no. 9272; Cell Signaling Technology), phospho‐Bad at Ser‐136 (no. 4366; Cell Signaling Technology), Bad (no. 9239; Cell Signaling Technology), cleaved caspase‐3 (no. 9664; Cell Signaling Technology), caspase‐9 (no. 9502; Cell Signaling Technology), poly(ADP‐ribose) polymerase (PARP) (no. 9532; Cell Signaling Technology), eGFP (GTX26673; Gene Tex, Irvine, CA, USA) or with actin (A5060; Sigma‐Aldrich) followed by incubation with the appropriate horseradish peroxidase‐conjugated anti‐mouse IgG (no. 7074; Cell Signaling Technology) or with anti‐rabbit IgG antibody (no. 7076; Cell Signaling Technology). Immuno‐reactive signals were visualized with the Immunostar LD detection system (Wako, Osaka, Japan) and ImageQuant LAS4000 mini Imager (GE Healthcare Bioscience, Pittsburgh, PA, USA) according to the manufacturer's protocols.
Immunoprecipitation and western blot analysis
Cells were treated with pervanadate [0.3% (w/w) H2O2 and 100 μm sodium orthovanadate in PBS] and then lysed in the lysis buffer as described above. Cell lysates were incubated with monoclonal anti‐CD133 antibody (AC133; Miltenyi Biotech) and protein G‐Sepharose beads at 4 °C for 2 h. The immunoprecipitates were analyzed by western blotting with monoclonal antibodies against CD133 (W6C3B1; Miltenyi Biotech), phospho‐tyrosine (no. 9411; Cell Signaling Technology) and PTPRK (HPA054822; Sigma‐Aldrich) as described above.
Soft agar colony formation
Cells (5 × 102) were suspended in 2 mL of top layer containing DMEM/10% FBS/0.33% agar and poured into 30‐mm cell culture dishes with 2 mL of basal layer including DMEM/10% FBS/0.5% agar. Fourteen days after incubation, the number of colonies larger than 100 μm in diameter was counted under the microscope.
Tumor xenograft model
Cells (1 × 106) were subcutaneously inoculated into BALB/c nude mice (CREA Japan, Shizuoka, Japan) as described previously 37. All of the animal experiments were performed in accordance with the guidelines of Chiba Cancer Center Research Institute.
Sphere formation
Cells were seeded into 96‐well round bottom low‐attachment plates (Sumitomo Bakelite Co., Ltd, Tokyo, Japan) at a density of 10 cells per well and cultured for 9 days in sphere‐forming medium composed of DMEM/Ham's F‐12 (Wako) supplemented with 2% MACS Neurobrew‐21 (Miltenyi Biotec), 20 ng·mL−1 of EGF (R&D Systems, Minneapolis, MN, USA) and 20 ng·mL−1 of fibroblast growth factor‐2 (FGF‐2; R&D Systems) in a humidified atmosphere containing 5% CO2 at 37 °C. Images were taken under the phase‐contrast microscope and then measured diameters of spheres using imagej software 39. Sphere volume (V) was calculated by the formula: V = 4/3π × [long diameter (μm)] × [short diameter (μm)]2.
WST assay
Cells were seeded into 96‐well plates at a density of 500 cells per well and allowed to attach overnight. Cells were then incubated at 37 °C in a humidified atmosphere with 5% CO2. Cell viability was examined by using Cell Counting Kit‐8 reagent (Dojindo Molecular Technologies, Rockville, MD, USA) according to the manufacturer's suggestions.
Trypan blue‐dye exclusion assay
Cells were seeded into six‐well plates at a density of 1.5 × 105 cells per well and exposed to oxaliplatin at a final concentration of 20 μm. Forty‐eight hours after treatment, floating and adherent cells were collected and washed in ice‐cold 1× PBS. After brief centrifugation, cells were resuspended in fresh medium, mixed with an equal volume of 0.4% trypan blue solution (Sigma‐Aldrich), and then analyzed by automatic cell counter (TC20; Bio‐Rad, Hercules, CA, USA).
Statistical analysis
Results are presented as mean ± SD. Data were compared using unpaired t‐test, one‐way ANOVA, and repeated‐measures two‐way ANOVA. Analyses were performed using ekuseru‐tokei 2010 software (Social Survey Research Information Co., Ltd, Tokyo, Japan) and a P‐value < 0.05 was considered to be significant.
Results
Knockdown of PTPRK potentiates tumorigenic ability of CD133‐positive colon cancer HT‐29 cells
To ask whether PTPRK could participate in the regulation of CD133‐mediated colon cancer progression, human colon cancer HT‐29 cells, which express endogenous CD133 (Fig. S1A), were infected with lentiviral vectors carrying shRNA against PTPRK (shPTPRK#1 or shPTPRK#2) or with the empty vector (pLKO). As shown in Fig. 1A, our shRNAs successfully reduced PTPRK expression at both mRNA and protein levels. Since shPTPRK#2 down‐regulated PTPRK much more effectively than shPTPRK#1, we have employed shPTPRK#2 for further experiments.
Figure 1.
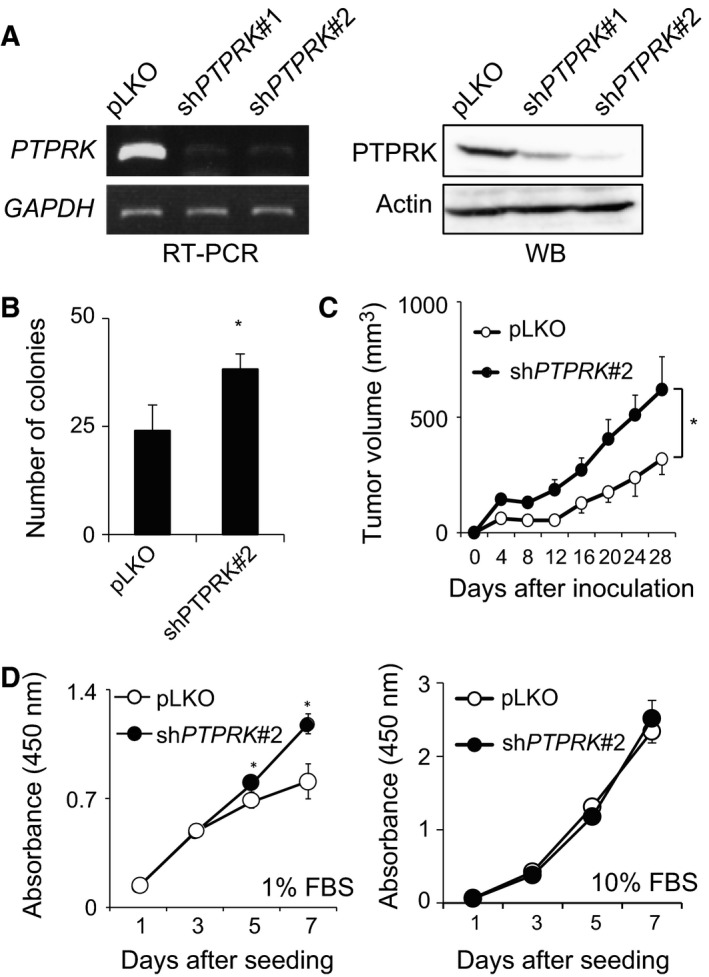
Knockdown of PTPRK potentiates the tumorigenic ability of CD133‐expressing colon cancer cells. (A) Establishment of PTPRK‐knocked down cells. Colon cancer HT‐29 cells were infected with the lentiviral vector harboring PTPRK‐targeting shRNAs (shPTPRK#1 or shPTPRK#2) or with the empty control (pLKO), and finally, puromycin‐resistant cells were obtained. Total RNA and cell lysates were prepared from the indicated cells (HT‐29/pLKO, HT‐29/shPTPRK#1 and HT‐29/shPTPRK#2) and analyzed for PTPRK by semi‐quantitative RT‐PCR (left panels) and western blotting (right panels), respectively. GAPDH or actin was used as an internal or a loading control, respectively. (B) Anchorage‐independent proliferation. pLKO and shPTPRK#2 cells were cultured in soft agar medium (5 × 102 cells/dish). Two weeks after incubation, a number of colonies (larger than 100 μm) were scored. Data show mean ± SD (n = 3) and the asterisk indicates the statistical significance of difference (P < 0.05, t‐test). (C) Xenograft tumor growth. The indicated cells (5 × 106 cells) were subcutaneously inoculated into the flanks of BALB/c nude mice. The volumes of tumor‐derived pLKO (open circles) and shPTPRK#2 (closed circles) cells were measured at the indicated time periods after inoculation. Data show mean ± SD (n = 3) and the asterisk indicates the statistical significance of difference (P < 0.05, t‐test). (D) HT‐29/pLKO (open circles) or HT‐29/shPTPRK#2 (closed circles) were seeded into 96‐well plates (500 cells/well) and cultured with 1% FBS (left panel) or with 10% FBS (right panel) containing culture medium. Their proliferation rates were measured by WST assay at the indicated time points. Data show mean ± SD (n = 5) and asterisk indicates statistical significance of difference (P < 0.05, repeated‐measures two‐way ANOVA).
Firstly, we examined the possible effect of PTPRK depletion on colony formation in soft agar medium. PTPRK‐depleted (HT‐29/shPTPRK#2) or non‐depleted HT‐29 (HT‐29/pLKO) cells were maintained for 2 weeks in soft agar medium and then their colony numbers were scored after incubation. As seen in Fig. 1B, the number of colonies derived from HT‐29/shPTPRK#2 cells was larger than that from HT‐29/pLKO cells. To confirm these results in vivo, HT‐29/pLKO and HT‐29/shPTPRK#2 cells were inoculated into flanks of BALB/c nude mice. At the indicated time points after inoculation, their subcutaneous xenograft tumors were measured. As expected, the volume of tumors arising from HT‐29/shPTPRK#2 cells was larger than that of HT‐29/pLKO cells (Fig. 1C), indicating that PTPRK gene silencing contributes to the tumor progression of colon cancer cells.
Since tumor growth promotion was observed in HT‐29/shPTPRK#2 cells, their proliferation rate was determined by WST cell proliferation assay. As shown in Fig. 1D, the proliferation rate of HT‐29/shPTPRK#2 cells was significantly faster than that of HT‐29/pLKO cells under a low serum concentration (1% of FBS), while knockdown of PTPRK had an undetectable effect on the proliferation rate of HT‐29 cells cultured under the standard serum concentration (10% of FBS).
PTPRK gene silencing stimulates CD133‐mediated colon cancer growth
To address whether PTPRK could attenuate the aggressive growth of colon cancer cells through the inhibition of CD133, we have employed CD133‐negative human colon cancer SW480 cells (Fig. S1B). SW480 cells were infected with the lentiviral vector for eGFP or CD133 to give SW480/eGFP or SW480/CD133 cells, respectively. These cells were subsequently infected with the lentiviral vector carrying shPTPRK#2 or with pLKO. As shown in Fig. 2A, forced expression of eGFP and CD133, and forced depletion of PTPRK were confirmed by western blot analysis. We then determined the proliferation rates of these cells by WST cell proliferation assay at a low serum concentration (1% of FBS). Similar to HT‐29/shPTPRK#2 cells, the proliferation rate of PTPRK‐depleted SW480/CD133 cells was significantly faster than that of non‐depleted SW480/CD133 cells (Fig. 2B, left panel). Additionally, PTPRK depletion had a negligible effect on the proliferation of SW480/eGFP cells under the lower serum condition (Fig. 2B, left panel). By contrast, knockdown of PTPRK had an undetectable effect on the proliferation rate of SW480/CD133 cells cultured with the standard 10% FBS‐containing medium (Fig. 2B, right panel).
Figure 2.
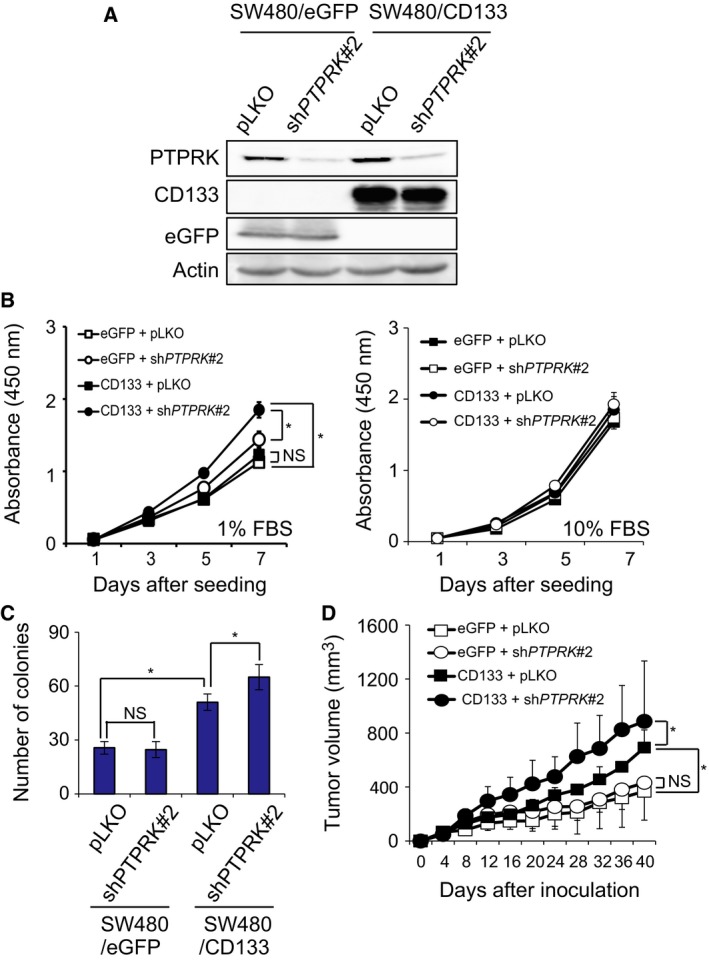
PTPRK gene silencing enhances pro‐oncogenic activity of CD133. (A) Forced expression of CD133 and silencing of PTPRK. CD133‐negative colon cancer SW480 cells were transduced with the lentiviral vector for CD133 or eGFP together with the shPTPRK#2 or pLKO. Cell lysates were prepared from the indicated cells and analyzed for CD133, eGFP and PTPRK by western blotting. Actin was used as a loading control. (B) Aforementioned SW480 derivatives were seeded into 96‐well plates (500 cells per well) and cultured with 1% FBS (left panel) or with 10% FBS (right panel) containing culture medium. Their proliferation rates were examined by WST assay at the indicated time points. Data show mean ± SD (n = 3) and the asterisk indicates the statistical significance of difference (P < 0.05, repeated‐measure two‐way ANOVA). NS, not significant. (C) Anchorage‐independent proliferation. The indicated cells (500 cells per dish) were grown in soft agar medium for 2 weeks, and then the number of colonies (larger than 100 μm) was scored. Data show mean ± SD (n = 3) and the asterisk indicates the statistical significance of difference (P < 0.05, one‐way ANOVA). NS, not significant. (D) Xenograft tumor growth. The indicated cells (5 × 106 cells) were subcutaneously inoculated into the flanks of BALB/c nude mice. Tumor volumes were measured at the indicated time periods after the inoculation. SW480/eGFP plus pLKO (open squares); SW480/eGFP plus shPTPRK #2 (closed squares); SW480/CD133 plus pLKO (open circles); SW480/CD133 plus shPTPRK #2 (closed circles). Data show mean ± SD (n = 3) and the asterisk indicates the statistical significance of difference (P < 0.05, one‐way ANOVA). NS, not significant.
We next determined the possible effect of PTPRK depletion on colony formation in soft agar medium. As shown in Fig. 2C, the number of colonies derived from SW480/CD133 cells was larger than that from SW480/eGFP control cells. Notably, knockdown of PTPRK in SW480/CD133 cells obviously increased number of colonies, whereas SW480/eGFP cells were unaffected by PTPRK depletion. Consistent with these results, SW480/CD133 cells developed larger xenograft tumors than SW480/eGFP cells, and silencing of PTPRK accelerated the growth of SW480/CD133‐derived tumors (Fig. 2D). In contrast to SW480/CD133‐derived tumors, SW480/eGFP‐derived tumors were unaffected by knockdown of PTPRK.
We have further checked the possible effect of PTPRK knockdown on in vitro sphere formation. Sphere culture has been considered to be one of the surrogates of in vitro culture systems, which mimic the complicated microenvironments in tumor tissues in vivo 40. For this purpose, SW480/eGFP and SW480/CD133 cells were grown in the serum‐free sphere‐forming medium. Seven days after incubation, the volume of the spheres was measured. As shown in Fig. 3A,B, SW480/CD133 cells formed larger spheres relative to SW480/eGFP cells. Depletion of PTPRK had an undetectable effect on SW480/eGFP spheres, whereas PTPRK‐knocked down SW480/CD133 cells formed larger spheres as compared to non‐depleted SW480/CD133 cells. Similarly, PTPRK‐depleted HT‐29 spheres were larger than non‐depleted HT‐29 spheres (Fig. S2A,B).
Figure 3.
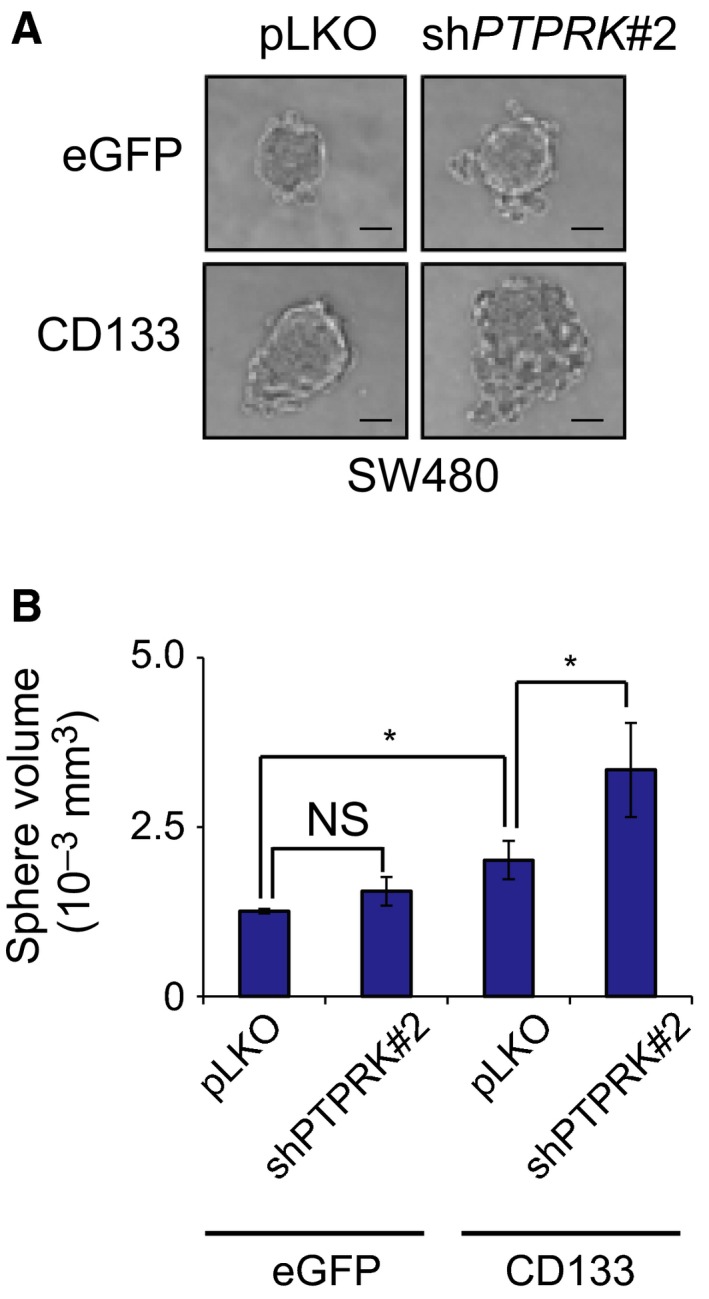
Knockdown of PTPRK enhances CD133‐dependent sphere‐forming ability of SW480 cells. (A,B) Sphere growth. SW480/eGFP plus pLKO, SW480/eGFP plus shPTPRK #2, SW480/CD133 plus pLKO and SW480/CD133 plus shPTPRK #2 (10 cells/well) were grown in serum‐free conditioned medium. Nine days after incubation, the representative images of spheres were taken (A), and their volumes were calculated (B). Bars indicate 50 μm (A). Data show mean ± SD (n = 3) and the asterisk indicates the statistical significance of difference (P < 0.05, one‐way ANOVA). NS, not significant.
Together, these observations suggest that PTPRK suppresses CD133‐mediated colon cancer growth both in vitro and in vivo.
Silencing of PTPRK enhances CD133‐dependent phosphorylation of AKT and its downstream target Bad in SW480 cells
Recently, we have described that tyrosine phosphorylation of CD133 (p‐CD133) contributes to aggressive tumor growth of colon cancer cells through the activation of the pro‐oncogenic AKT pathway 37. These findings prompted us to examine whether PTPRK‐mediated dephosphorylation of CD133 could attenuate the pro‐oncogenic AKT pathway in colon cancer cells. Towards this end, cell lysates prepared from non‐depleted and PTPRK‐depleted SW480/CD133 cells were immunoprecipitated with control IgG or with anti‐CD133 antibody followed by western blotting with anti‐phospho‐tyrosine antibody. As clearly seen in Fig. 4A,B, silencing of PTPRK significantly increased the p‐CD133 level. Consistent with these results, forced expression of PTPRK in SW480/CD133 cells resulted in a remarkable reduction of p‐CD133 level (Fig. 4C,D).
Figure 4.
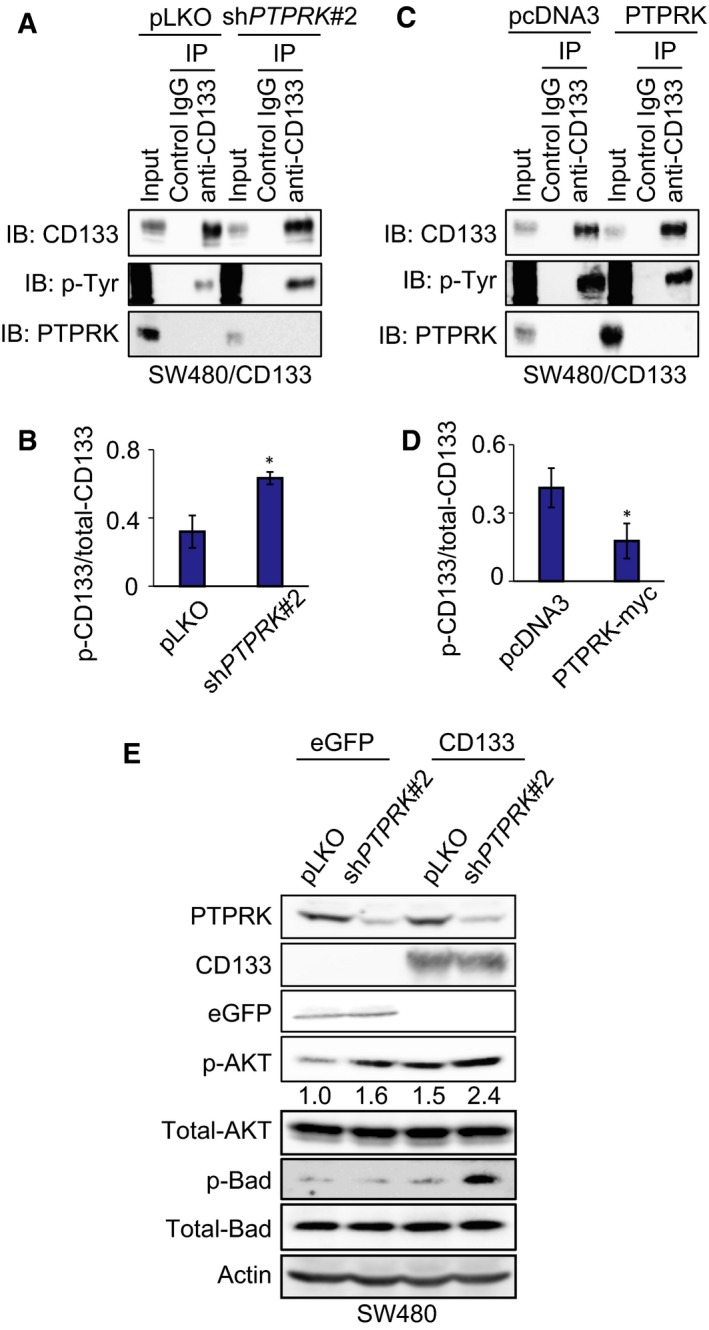
Depletion of PTPRK increases the level of phospho‐CD133 and leads to stimulated phosphorylation of AKT and a downstream target protein, Bad, in colorectal cancer cells. (A–D) Immunoprecipitation analysis. SW480/CD133 cells were infected with the lentiviral vector harboring PTPRK‐targeting shRNAs (shPTPRK#2) or with mock control (pLKO). Cell lysates prepared from these SW480 derivatives were immunoprecipitated with anti‐CD133 antibody or with control IgG. Resultant immunoprecipitates were analyzed by western blotting with the indicated antibodies (A). The ratios of phospho‐CD133 (p‐CD133) to total CD133 from three independent experiments are indicated in (B). Alternatively, SW480/CD133 cells transfected with the expression plasmid for PTPRK or with pcDNA3 empty vector were processed for immunoprecipitation‐immunoblot analysis as mentioned above (C) and the ratios of p‐CD133/CD133 are shown in (D). Asterisks indicate statistical significance of difference (P < 0.05, t‐test). (E) Western blot analysis. Cell lysates (30 μg) prepared from the indicated SW480 derivatives were analyzed by immunoblot with the indicated antibodies. Actin was used as a loading control. Relative band intensities of p‐AKT standardized to those of total‐AKT were also indicated.
We next examined whether PTPRK could modulate AKT phosphorylation (p‐AKT) in CD133‐expressing SW480 cells. For this purpose, cell lysates were prepared from the indicated cells and analyzed for p‐AKT by western blotting. As shown in Fig. 4E, forced expression of CD133 in SW480 cells enhanced p‐ATK level at Ser‐473 as compared to that of SW480/eGFP cells. As expected, CD133‐induced phosphorylation of AKT at Ser‐473 was further stimulated in PTPRK‐depleted SW480/CD133 cells. The expression level of total AKT remained unchanged in response to the exogenous CD133 and PTPRK gene silencing.
To determine whether the pro‐oncogenic AKT signaling pathway could be activated under our experimental conditions, we checked the phosphorylation status of its downstream target Bad at Ser‐136 (p‐Bad). As shown in Fig. 4E, a remarkable up‐regulation of p‐Bad was detectable in PTPRK‐depleted SW480/CD133 cells (Fig. 4E). Forced expression of CD133 alone had an undetectable effect on p‐Bad. Similar results were also obtained in HT‐29/shPTPRK#2 cells (Fig. S3). These observations suggest that PTPRK has the ability to impair the pro‐oncogenic CD133–AKT signaling pathway in colon cancer cells.
Depletion of PTPRK confers resistance of CD133‐expressing SW480 cells to oxaliplatin
Given that p‐Bad inhibits apoptotic cell death induced by the intrinsic mitochondrial pathway 41, we sought to investigate whether PTPRK could be implicated in CD133‐associated anti‐cancer drug resistance of colon cancer cells. For this purpose, non‐depleted and PTPRK‐depleted SW480/CD133 cells were treated with the indicated concentrations of oxaliplatin, which is commonly used in a standard chemotherapy for colon cancer patients. Forty‐eight hours after treatment, cell viability was assessed by WST assay. As shown in Fig. 5A, SW480/CD133 cells showed an enhanced viability as compared to SW480/eGFP cells following oxaliplatin exposure. Moreover, the viability of SW480/CD133 cells was further increased by PTPRK gene silencing in response to oxaliplatin. In support of these results, oxaliplatin‐induced cell death was obviously prohibited by knockdown of PTPRK as examined by trypan blue dye‐exclusion assay (Fig. 5B).
Figure 5.

Silencing of PTPRK reduces oxaliplatin sensitivity of CD133‐expressing colon cancer cells. (A) WST cell survival assay. SW480 derivatives were treated with indicated concentrations of oxaliplatin for 48 h and their viability was measured by WST assay. Data represent mean ± SD (n = 5) and asterisks indicate statistical significance of difference (P < 0.05, one‐way ANOVA). (B) Trypan blue dye‐exclusion assay. The indicated cells were exposed to 20 μm of oxaliplatin (blue bars) or left untreated (open bars). Forty‐eight hours after treatment, floating and attached cells were harvested and processed for trypan blue assay. Data represent mean ± SD (n = 3) and asterisks indicate statistical significance of difference (P < 0.05, one‐way ANOVA). (C) Western blot analysis. The indicated cells were exposed to 20 μm of oxaliplatin or left untreated. Forty‐eight hours after treatment, cell lysates (30 μg) were prepared from floating and attached cells and analyzed by immunoblot with the indicated antibodies. Actin was used as a loading control.
The trypan blue dye‐exclusion assay is an insensitive assay of cell death, particularly for early apoptosis, when compared with annexin V and propidium iodide staining method. To examine whether the oxaliplatin‐induced cell death would be mediated by apoptotic cell death, the indicated cells were treated with or without 20 μm of oxaliplatin, and then cell death‐related cleaved caspase‐9, caspase‐3 and PARP were examined by western blotting. As shown in Fig. 5C, oxaliplatin‐induced proteolytic cleavage of caspase‐9, caspase‐3 and PARP was detectable in both SW480/eGFP and SW480/CD133. Of note, PTPRK depletion in SW480/CD133 cells resulted in a massive decrease in the amounts of cleaved caspase‐9, caspase‐3 and PARP in response to oxaliplatin as compared to those of non‐depleted SW480/CD133 cells exposed to oxaliplatin.
Collectively, these results indicate that PTPRK‐mediated prohibition of the pro‐oncogenic CD133–AKT pathway improves oxaliplatin sensitivity of CD133‐expressing cancer cells.
Discussion
In the present study, we have found for the first time that shRNA‐mediated knockdown of PTPRK significantly augments tumor growth and markedly reduces the anti‐cancer drug oxaliplatin sensitivity of CD133‐expressing colon cancer cells. Our expression studies revealed that PTPRK silencing enhances the phosphorylation of CD133, AKT and its downstream target Bad. Thus, this is suggestive that forced depletion of PTPRK contributes to the aggressive properties of CD133‐positive colon cancer cells through the potentiation of the pro‐oncogenic CD133–AKT pathway (Fig. 6).
Figure 6.
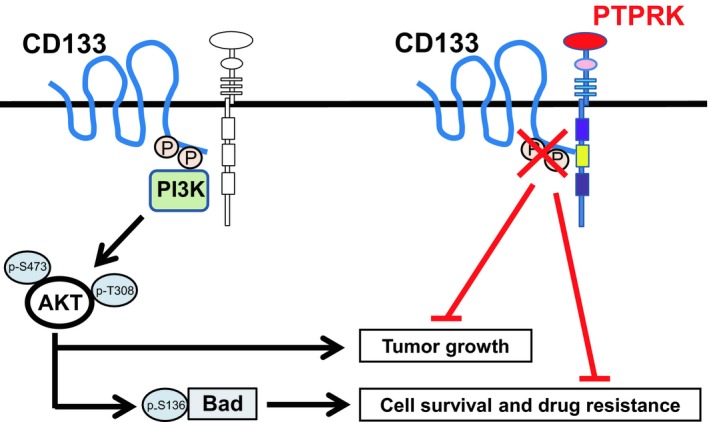
Summary of the present study.
According to our present results, depletion of PTPRK conferred resistance to oxaliplatin in SW480/CD133 cells in association with the marked accumulation of p‐CD133. Our previous study demonstrated that phosphor‐mimetic CD133 mutant protein leads to an increase in phosphorylation of AKT and subsequently activates its enzymatic activity 37. In support of this observation, PTPRK‐silenced SW480/CD133 cells showed the highest level of p‐Bad at Ser‐136, which is phosphorylated by AKT 42. Bad can be also phosphorylated at Ser‐112 mediated by the Ras–mitogen‐activated protein kinase pathway 43, and the phosphorylation of both serine residues inhibits the pro‐apoptotic function of Bad via the interaction with 14‐3‐3 proteins but not with BCL‐XL 44. Our present result, however, demonstrated that neither knockdown of PTPRK nor forced expression of CD133 affects the level of p‐Bad at Ser‐112 in SW480 cells (Fig. S4). Consistent with these observations, Ma et al. 45 described that the activation of the pro‐survival AKT–Bad pathway but not of the mitogen‐activated protein kinase pathway is tightly linked to the drug‐resistant phenotype of the CD133‐positive subset in hepatocellular carcinoma HuH7 cells. We have therefore proposed a model in which a PTPRK–CD133 axis exists and prevents cell death induced by certain anti‐tumor drugs through activation of the AKT–Bad pro‐survival pathway. Furthermore, we have demonstrated that knockdown of PTPRK enhances proliferation of SW480/CD133 cells but not of SW480/eGFP cells under low serum conditions. Edinger et al. 46 described that mammalian target of rapamycin, another downstream target of AKT, plays a pivotal role in cell survival through increased nutrient uptake. These studies collectively suggest that the PTPRK–CD133 axis is also implicated in cell survival and an adaptation of cancer cells to poor microenvironment in tumor tissue in vivo; however, further examination of metabolic pathway activation in PTPRK‐depleted CD133‐positive cancer cells under lower nutrient and/or hypoxic conditions is required.
The present study showed that knockdown of PTPRK accelerated xenografted tumor growth of SW480/CD133 cells but not of CD133‐negative SW480/eGFP cells accompanied by a larger amount of p‐AKT. Bläker et al. 10 described that PTPRK is recurrently mutated in precancerous lesions of certain intestinal carcinoma patients. Zhu et al. 47 demonstrated that CD133‐positive stem/progenitor cells possess the traits of CSCs in response to oncogenic insults. Although natural ligands for CD133, to our knowledge, have not been identified, our present results are suggestive that the abnormality of PTPRK caused by loss‐of‐function mutation and/or deletion is one of the critical triggers for CD133‐positive stem/progenitor cells to transform cancerous cells in vivo. According to our previous study 37, EGF could stimulate phosphorylation of CD133 in colon cancer cells. The pro‐oncogenic EGF/EGFR signaling pathway, which activates AKT phosphorylation 48, is suppressed by PTPRK through the promotion of EGFR dephosphorylation 8, 49. Intriguingly, a clear induction of p‐AKT was observed in PTPRK‐silenced SW480/eGFP cells, but it is insufficient to further augment in vivo tumor growth of CD133‐negative SW480/eGFP cells. Additionally, a sharp increase in p‐AKT was observed in PTPRK‐silenced SW480/CD133 cells. We therefore hypothesized that the PTPRK–CD133 axis is implicated in the pro‐oncogenic EGF/EGFR signaling pathway. Detailed analysis of EGF/EGFR signal status in PTPRK‐silenced colon cancer cells might help to better understand mechanisms of CD133 stimulation by EGF.
Another important finding of our present study was that PTPRK‐depleted SW480/CD133 cells form tumorspheres much more efficiently than non‐depleted SW480/CD133 cells. Unexpectedly, PTPRK has been shown to be highly expressed in breast and prostate cancer tissues as compared to their corresponding non‐tumor ones 15, 16, raising the possibility that the tumor suppressive function(s) but not the expression of PTPRK might be impaired in cancer cell‐derived spheres as well as cancer tissues. Previously, it has been shown that the active site within the phosphatase domain of PTPs is highly susceptible to oxidation caused by reactive oxygen species (ROS), and the oxidized PTPs are no longer able to catalyze dephosphorylation 9, 50. It has been well documented that various types of cancer cells produce a large amount of ROS, which is implicated in the growth promotion of cancers 51. For instance, Le Belle et al. 52 demonstrated that a higher level of ROS stimulates the proliferation and self‐renewal of neuronal stem cells. Qu et al. 53 found that prostate cancer cell spheres generate a large amount of ROS relative to their parental adherent monolayer cultures, which augments their growth through ROS‐mediated activation of the interleukin 6–signal transducer and activator of transcription 3 pathway. Based on our current results, the intracellular ROS level was significantly increased during sphere formation of SW480 cells (Fig. S5), suggesting that ROS inhibits PTPRK and thereby facilitates CD133‐mediated sphere formation. We also speculate that the reactivation of PTPRK might suppress the sphere forming ability of SW480/CD133 cells. Further studies of ROS scavengers, such as N‐acetyl cysteine and melatonin, will be required to adequately address this issue.
In conclusion, our present results strongly suggest that PTPRK‐mediated dephosphorylation of CD133 plays an essential role in the progression and drug resistance of colon cancer at least in part through down‐regulation of the pro‐oncogenic AKT–Bad pathway, and this regulatory mechanism might provide a clue to develop a novel and promising strategy for the better treatment of colon cancer patients.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
OS conceived and supervised the study; MM and OS designed experiments; MM, YM, KU, TOg, MN and HY performed experiments; OS, TOz, HS, NT and YN analyzed and interpreted data; MM, OS and TOz wrote the manuscript.
Supporting information
Fig. S1. Expression levels of PTPRK and CD133 in human colon cancer cells.
Fig. S2. Knockdown of PTPRK accelerates sphere growth of HT‐29 cells.
Fig. S3. Knockdown of PTPRK stimulates phosphorylation of AKT and its target proteins in HT‐29 cells.
Fig. S4. Knockdown of PTPRK and forced‐expression of CD133 has a negligible effect on phosphorylation of Bad at Ser‐112 in SW480 cells.
Fig. S5. A higher production of reactive oxygen species (ROS) in colon cancer‐derived spheres.
Acknowledgements
We thank H. Nagase (Chiba Cancer Center Research Institute) for valuable discussions. This work was supported in part by Japan Society for the Promotion of Science KAKENHI Grant Numbers JP21790397, JP23591978 and JP16K10559.
References
- 1. Siegel RL, Miller KD and Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65, 5–29. [DOI] [PubMed] [Google Scholar]
- 2. Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B and Starling N (2010) Colorectal cancer. Lancet 375, 1030–1047. [DOI] [PubMed] [Google Scholar]
- 3. Cremolini C, Schirripa M, Antoniotti C, Moretto R, Salvatore L, Masi G, Falcone A and Loupakis F (2015) First‐line chemotherapy for mCRC‐a review and evidence‐based algorithm. Nat Rev Clin Oncol 12, 607–619. [DOI] [PubMed] [Google Scholar]
- 4. Ostman A, Hellberg C and Bohmer FD (2006) Protein‐tyrosine phosphatases and cancer. Nat Rev Cancer 6, 307–320. [DOI] [PubMed] [Google Scholar]
- 5. Jiang YP, Wang H, D'Eustachio P, Musacchio JM, Schlessinger J and Sap J (1993) Cloning and characterization of R‐PTP‐kappa, a new member of the receptor protein tyrosine phosphatase family with a proteolytically cleaved cellular adhesion molecule‐like extracellular region. Mol Cell Biol 13, 2942–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anders L, Mertins P, Lammich S, Murgia M, Hartmann D, Saftig P, Haass C and Ullrich A (2006) Furin‐, ADAM 10‐, and gamma‐secretase‐mediated cleavage of a receptor tyrosine phosphatase and regulation of beta‐catenin's transcriptional activity. Mol Cell Biol 26, 3917–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang SE, Wu FY, Shin I, Qu S and Arteaga CL (2005) Transforming growth factor β (TGF‐β)‐Smad target gene protein tyrosine phosphatase receptor type kappa is required for TGF‐β function. Mol Cell Biol 25, 4703–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu Y, Tan LJ, Grachtchouk V, Voorhees JJ and Fisher GJ (2005) Receptor‐type protein‐tyrosine phosphatase‐kappa regulates epidermal growth factor receptor function. J Biol Chem 280, 42694–42700. [DOI] [PubMed] [Google Scholar]
- 9. Xu Y, Shao Y, Voorhees JJ and Fisher GJ (2006) Oxidative inhibition of receptor‐type protein‐tyrosine phosphatase kappa by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. J Biol Chem 281, 27389–27397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bläker H, Mechtersheimer G, Sutter C, Hertkorn C, Kern MA, Rieker RJ, Penzel R, Schirmacher P and Kloor M (2008) Recurrent deletions at 6q in early age of onset non‐HNPCC‐ and non‐FAP‐associated intestinal carcinomas. Evidence for a novel cancer susceptibility locus at 6q14‐q22. Genes Chromosom Cancer 47, 159–164. [DOI] [PubMed] [Google Scholar]
- 11. Barghorn A, Speel EJ, Farspour B, Saremaslani P, Schmid S, Perren A, Roth J, Heitz PU and Komminoth P (2001) Putative tumor suppressor loci at 6q22 and 6q23‐q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. Am J Pathol 158, 1903–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Agarwal S, Al‐Keilani MS, Alqudah MA, Sibenaller ZA, Ryken TC and Assem M (2013) Tumor derived mutations of protein tyrosine phosphatase receptor type k affect its function and alter sensitivity to chemotherapeutics in glioma. PLoS One 8, e62852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stevenson WS, Best OG, Przybylla A, Chen Q, Singh N, Koleth M, Pierce S, Kennedy T, Tong W, Kuang SQ et al (2014) DNA methylation of membrane‐bound tyrosine phosphatase genes in acute lymphoblastic leukaemia. Leukemia 28, 787–793. [DOI] [PubMed] [Google Scholar]
- 14. Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, Gupta M, O'Sullivan MG, Matise I, Dupuy AJ et al (2009) A transposon‐based genetic screen in mice identifies genes altered in colorectal cancer. Science 323, 1747–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun PH, Ye L, Mason MD and Jiang WG (2013) Protein tyrosine phosphatase kappa (PTPRK) is a negative regulator of adhesion and invasion of breast cancer cells, and associates with poor prognosis of breast cancer. J Cancer Res Clin Oncol 139, 1129–1139. [DOI] [PubMed] [Google Scholar]
- 16. Sun PH, Ye L, Mason MD and Jiang WG (2013) Receptor‐like protein tyrosine phosphatase kappa negatively regulates the apoptosis of prostate cancer cells via the JNK pathway. Int J Oncol 43, 1560–1568. [DOI] [PubMed] [Google Scholar]
- 17. Reya T, Morrison SJ, Clarke MF and Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. [DOI] [PubMed] [Google Scholar]
- 18. Wang JC and Dick JE (2005) Cancer stem cells: lessons from leukemia. Trends Cell Biol 15, 494–501. [DOI] [PubMed] [Google Scholar]
- 19. Dick JE (2008) Stem cell concepts renew cancer research. Blood 112, 4793–4807. [DOI] [PubMed] [Google Scholar]
- 20. Dean M, Fojo T and Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5, 275–284. [DOI] [PubMed] [Google Scholar]
- 21. Ozaki T, Nakamura M and Shimozato O (2015) Novel implications of DNA damage response in drug resistance of malignant cancers obtained from the functional interaction between p53 family and RUNX2. Biomolecules 5, 2854–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK and Buck DW (1997) A novel five‐transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 90, 5013–5021. [PubMed] [Google Scholar]
- 23. Yin AH, Miraglia S, Zanjani ED, Almeida‐Porada G, Ogawa M, Leary AG, Olweus J, Kearney J and Buck DW (1997) AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90, 5002–5012. [PubMed] [Google Scholar]
- 24. Lee HJ, You DD, Choi DW, Choi YS, Kim SJ, Won YS and Moon HJ (2011) Significance of CD133 as a cancer stem cell markers focusing on the tumorigenicity of pancreatic cancer cell lines. J Korean Surg Soc 81, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH and Weissman IL (2000) Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA 97, 14720–14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bussolati B, Bruno S, Grange C, Buttiglieri S, Deregibus MC, Cantino D and Camussi G (2005) Isolation of renal progenitor cells from adult human kidney. Am J Pathol 166, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW and Gilbertson RJ (2009) Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 457, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ricci‐Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C and De Maria R (2007) Identification and expansion of human colon‐cancer‐initiating cells. Nature 445, 111–115. [DOI] [PubMed] [Google Scholar]
- 29. Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, Richel DJ, Stassi G and Medema JP (2008) Single‐cell cloning of colon cancer stem cells reveals a multi‐lineage differentiation capacity. Proc Natl Acad Sci USA 105, 13427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G, Medema JP et al (2007) Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin‐4. Cell Stem Cell 1, 389–402. [DOI] [PubMed] [Google Scholar]
- 31. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. [DOI] [PubMed] [Google Scholar]
- 32. Kojima M, Ishii G, Atsumi N, Fujii S, Saito N and Ochiai A (2008) Immunohistochemical detection of CD133 expression in colorectal cancer: a clinicopathological study. Cancer Sci 99, 1578–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Horst D, Scheel SK, Liebmann S, Neumann J, Maatz S, Kirchner T and Jung A (2009) The cancer stem cell marker CD133 has high prognostic impact but unknown functional relevance for the metastasis of human colon cancer. J Pathol 219, 427–434. [DOI] [PubMed] [Google Scholar]
- 34. Artells R, Moreno I, Diaz T, Martinez F, Gel B, Navarro A, Ibeas R, Moreno J and Monzo M (2010) Tumour CD133 mRNA expression and clinical outcome in surgically resected colorectal cancer patients. Eur J Cancer 46, 642–649. [DOI] [PubMed] [Google Scholar]
- 35. Boivin D, Labbe D, Fontaine N, Lamy S, Beaulieu E, Gingras D and Beliveau R (2009) The stem cell marker CD133 (prominin‐1) is phosphorylated on cytoplasmic tyrosine‐828 and tyrosine‐852 by Src and Fyn tyrosine kinases. Biochemistry 48, 3998–4007. [DOI] [PubMed] [Google Scholar]
- 36. Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu N, Xu W, Cui C, Xing Y, Liu Y et al (2013) Activation of PI3K/Akt pathway by CD133‐p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci USA 110, 6829–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shimozato O, Waraya M, Nakashima K, Souda H, Takiguchi N, Yamamoto H, Takenobu H, Uehara H, Ikeda E, Matsushita S et al (2015) Receptor‐type protein tyrosine phosphatase kappa directly dephosphorylates CD133 and regulates downstream AKT activation. Oncogene 34, 1949–1960. [DOI] [PubMed] [Google Scholar]
- 38. Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, Ohira M, Nakagawara A and Kamijo T (2011) CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene 30, 97–105. [DOI] [PubMed] [Google Scholar]
- 39. Schneider CA, Rasband WS and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thoma CR, Zimmermann M, Agarkova I, Kelm JM and Krek W (2014) 3D cell culture systems modeling tumor growth determinants in cancer target discovery. Adv Drug Deliv Rev 69–70, 29–41. [DOI] [PubMed] [Google Scholar]
- 41. Kennedy SG, Kandel ES, Cross TK and Hay N (1999) Akt/protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol 19, 5800–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y and Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell 91, 231–241. [DOI] [PubMed] [Google Scholar]
- 43. Tan Y, Ruan H, Demeter MR and Comb MJ (1999) p90(RSK) blocks bad‐mediated cell death via a protein kinase C‐dependent pathway. J Biol Chem 274, 34859–34867. [DOI] [PubMed] [Google Scholar]
- 44. Zha J, Harada H, Yang E, Jockel J and Korsmeyer SJ (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14‐3‐3 not BCL‐X(L). Cell 87, 619–628. [DOI] [PubMed] [Google Scholar]
- 45. Ma S, Lee TK, Zheng BJ, Chan KW and Guan XY (2008) CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 27, 1749–1758. [DOI] [PubMed] [Google Scholar]
- 46. Edinger AL, Thompson CB and Raff M (2002) Akt maintains cell size and survival by increasing mTOR‐dependent nutrient uptake. Mol Biol Cell 13, 2276–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu L, Finkelstein D, Gao C, Shi L, Wang Y, Lopez‐Terrada D, Wang K, Utley S, Pounds S, Neale G et al (2016) Multi‐organ mapping of cancer risk. Cell 166, 1132–1146.e1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Engelman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE and Cantley LC (2005) ErbB‐3 mediates phosphoinositide 3‐kinase activity in gefitinib‐sensitive non‐small cell lung cancer cell lines. Proc Natl Acad Sci USA 102, 3788–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu Y, Baker D, Quan T, Baldassare JJ, Voorhees JJ and Fisher GJ (2010) Receptor type protein tyrosine phosphatase‐kappa mediates cross‐talk between transforming growth factor‐beta and epidermal growth factor receptor signaling pathways in human keratinocytes. Mol Biol Cell 21, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK and Barford D (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl‐amide intermediate. Nature 423, 769–773. [DOI] [PubMed] [Google Scholar]
- 51. Liou GY and Storz P (2010) Reactive oxygen species in cancer. Free Radical Res 44, 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H and Kornblum HI (2011) Proliferative neural stem cells have high endogenous ROS levels that regulate self‐renewal and neurogenesis in a PI3K/Akt‐dependant manner. Cell Stem Cell 8, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qu Y, Oyan AM, Liu R, Hua Y, Zhang J, Hovland R, Popa M, Liu X, Brokstad KA, Simon R et al (2013) Generation of prostate tumor‐initiating cells is associated with elevation of reactive oxygen species and IL‐6/STAT3 signaling. Can Res 73, 7090–7100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression levels of PTPRK and CD133 in human colon cancer cells.
Fig. S2. Knockdown of PTPRK accelerates sphere growth of HT‐29 cells.
Fig. S3. Knockdown of PTPRK stimulates phosphorylation of AKT and its target proteins in HT‐29 cells.
Fig. S4. Knockdown of PTPRK and forced‐expression of CD133 has a negligible effect on phosphorylation of Bad at Ser‐112 in SW480 cells.
Fig. S5. A higher production of reactive oxygen species (ROS) in colon cancer‐derived spheres.