

Abstract
Formic acid (HCOOH) is among the most abundant carboxylic acids in the atmosphere, but its budget is poorly understood. We present eddy flux, vertical gradient, and soil chamber measurements from a mixed forest and apply the data to better constrain HCOOH source/sink pathways. While the cumulative above‐canopy flux was downward, HCOOH exchange was bidirectional, with extended periods of net upward and downward flux. Net above‐canopy fluxes were mostly upward during warmer/drier periods. The implied gross canopy HCOOH source corresponds to 3% and 38% of observed isoprene and monoterpene carbon emissions and is 15× underestimated in a state‐of‐science atmospheric model (GEOS‐Chem). Gradient and soil chamber measurements identify the canopy layer as the controlling source of HCOOH or its precursors to the forest environment; below‐canopy sources were minor. A correlation analysis using an ensemble of marker volatile organic compounds suggests that secondary formation, not direct emission, is the major source driving ambient HCOOH.
Keywords: Formic acid, Eddy co‐variance fluxes
Key Points
Bidirectional exchange of HCOOH was observed over a mixed forest, with a canopy‐level source; soil and below‐canopy sources were minor
The gross canopy HCOOH source is 3% and 38% of isoprene and monoterpene C emissions and is underestimated fifteenfold in a current model
Correlation analysis points to secondary formation, not direct emission, as the major HCOOH source in this forest ecosystem
1. Introduction
Organic acids are a significant component of ambient nonmethane organic carbon and contribute to aqueous‐phase acidity and OH reactivity (Galloway et al., 1982; Jacob, 1986). As photochemical products of volatile organic compound (VOC) oxidation, organic acids can also serve as integrated markers of the overall VOC degradation cascade. Formic acid (HCOOH) is among the most abundant carboxylic acids in the atmosphere (Khare et al., 1999; Millet et al., 2015; Paulot et al., 2011); however, its sources are poorly known. Here we present new HCOOH canopy flux, gradient, and soil chamber measurements from a mixed forest in Michigan, United States, and interpret the results in terms of their implications for HCOOH sources and sinks.
Photochemical production is thought to be the dominant source of atmospheric HCOOH globally, contributing 60–80% of its total known budget (Millet et al., 2015; Stavrakou et al., 2012). In particular, the OH‐initiated oxidation of isoprene produces HCOOH via several known pathways, with a total yield that has been estimated at 13% (Millet et al., 2015). HCOOH is also formed from formaldehyde oxide (CH2OO, a stabilized Criegee intermediate derived from terminal alkene ozonolysis), both directly (Nguyen et al., 2016) and via hydroxymethyl hydroperoxide (HMHP; Allen et al., 2018). Ozonolysis precursors of HCOOH thus encompass a wide suite of alkenes including isoprene, many monoterpene isomers, other terpenoids, and simpler alkenes. Reported HCOOH yields from monoterpene photooxidation and ozonolysis range from 1–50% (Larsen et al., 2001; Lee, Goldstein, Kroll, et al., 2006), depending on the species; average yields of 16% (OH) and 8% (ozonolysis) have been assumed in recent large‐scale modeling studies (Millet et al., 2015; Paulot et al., 2011). Additional known secondary HCOOH sources include OH‐driven oxidation of terminal alkynes (Hatakeyama et al., 1986) and photo‐tautomerization of acetaldehyde followed by oxidation of the resulting enol (Millet et al., 2015; Peeters et al., 2015; Shaw et al., 2018).
HCOOH also has several primary sources. Direct emissions from soil (Mielnik et al., 2018; Sanhueza & Andreae, 1991) and plants (Kesselmeier et al., 1998) have been observed, with the latter exhibiting light and temperature dependence. Other identified primary sources include agriculture (Ngwabie et al., 2008) and combustion of biomass (Chaliyakunnel et al., 2016; Goode et al., 2000) and fossil fuels (Kawamura et al., 1985; Talbot et al., 1988). Heterogeneous sources have also been proposed, including in‐cloud formaldehyde oxidation (Jacob, 1986; Lelieveld & Crutzen, 1991), as well as aging of organic aerosol via OH (Vlasenko et al., 2008), O3 (Eliason et al., 2003; Pan et al., 2009), and photolysis (Malecha & Nizkorodov, 2016; Park et al., 2006). Wet and dry deposition are the predominant HCOOH sinks; together with photochemical loss and a minor contribution from dust uptake, these yield an overall atmospheric lifetime of 2–4 days (Chebbi & Carlier, 1996; Paulot et al., 2011; Stavrakou et al., 2012).
Despite the large number of identified primary and secondary HCOOH sources, current chemical transport models typically underestimate observed concentrations by at least twofold (Millet et al., 2015; Paulot et al., 2011; Stavrakou et al., 2012). A number of recent studies have investigated this discrepancy with the aim of characterizing potential missing or underestimated HCOOH sources. For example, Bannan et al. (2014, 2017) measured HCOOH levels over London UK of up to 13 ppb and invoked traffic emissions and biogenic VOC photooxidation as likely sources. In the Colorado Front Range, Mattila et al. (2018) found evidence for photochemical and ground‐level HCOOH sources and speculated that heterogeneous ozonolysis of surface organics might contribute to the latter. Mungall et al. (2018) measured surprisingly high HCOOH concentrations (up to 11 ppb) over the Arctic tundra, which they attributed to unidentified surface‐ or near‐surface‐ sources; elevated concentrations occurred during cold/overcast as well as warm/sunny periods. Nguyen et al. (2015) observed net downward HCOOH fluxes over a deciduous forest in the Southeastern United States; however, observed deposition velocities were only half of predicted values, and the authors invoked a gross daytime canopy source of ~1 nmol/m2/s to account for the difference. Eddy flux measurements by Schobesberger et al. (2016) over a boreal pine/spruce forest revealed a net upward HCOOH flux that was both light and temperature dependent. Neither Nguyen et al. (2015) nor Schobesberger et al. (2016) were able to determine whether the observed canopy source was due to direct HCOOH emissions from vegetation or soil, in‐canopy chemical production, or heterogeneous surface processes.
Here we present measurements of HCOOH and related VOCs by high‐resolution mass spectrometry from the 2016 PROPHET‐AMOS campaign in a mixed deciduous/coniferous forest at the University of Michigan Biological Station. Observations include (i) canopy‐level flux measurements by eddy covariance; (ii) hourly vertical gradients of VOC mixing ratios through the forest canopy; and (iii) diurnal soil fluxes. Together, these measurements provide a more comprehensive picture for understanding the forest‐atmosphere exchange of HCOOH than has been available previously. Based on the flux, gradient, and soil chamber measurements of HCOOH and their correlation patterns with marker VOCs of known origin, we argue that in‐canopy secondary production (rather than direct emission from plants or soils) is the main HCOOH source in this forest ecosystem.
2. Methods
Canopy and vertical gradient measurements were performed on the PROPHET tower during the July 2016 AMOS field campaign at University of Michigan Biological Station, Michigan, United States (Millet et al., 2018). The surrounding forest is dominated by aspen, birch, and red oak (upper canopy) and by white pine, red maple, beech, and red oak (lower canopy; Bryan et al., 2015; Palik & Pregitzer, 1992). Soils at the site are excessively drained spodosols (Seok et al., 2015). Measurements of HCOOH and related VOCs during PROPHET‐AMOS were made with a Proton Transfer Reaction‐Quadrupole Interface Time‐Of‐Flight Mass Spectrometer (PTR‐QiTOF; Ionicon Analytic Gmbh). The hourly sampling procedure included direct ecosystem‐scale VOC flux measurements by eddy covariance, plus vertical profiling to quantify in‐canopy VOC gradients. A detailed description of instrumental performance, calibration, inlet configuration, sampling procedure, flux calculations, and quality control is provided by Millet et al. (2018) and summarized in supporting information S1.
During this study we also measured diurnal fluxes of HCOOH and other VOCs from the forest floor using four dynamic soil chambers. Details on these measurements and on other supporting observations used here are provided in supporting information S1 (Pape et al., 2009).
Finally, we compare the observed HCOOH concentrations and fluxes with predictions from a high‐resolution (0.25° × 0.3125°) GEOS‐Chem model simulation nested over North America, with model details provided in supporting information S1.
3. Results and Discussion
3.1. Observations Reveal Bidirectional HCOOH Exchange
Figure 1 shows the above‐canopy HCOOH mixing ratios, fluxes, and exchange velocities measured throughout the campaign. Mixing ratios ranged from 0.1–5.3 ppb, with an overall mean of 1.3 ppb and a daytime (10:00–17:00 EDT) mean of 1.6 ppb. When aggregated over the entire campaign, the net above‐canopy HCOOH flux was downward (into the canopy), averaging −3.9 × 108 molec/cm2/s (mean exchange velocity: −0.07 cm/s). However, the time series in Figure 1 reveals extended periods of net upward as well as net downward HCOOH exchange, indicating a bidirectional flux with counteracting gross source and sink effects.
Figure 1.
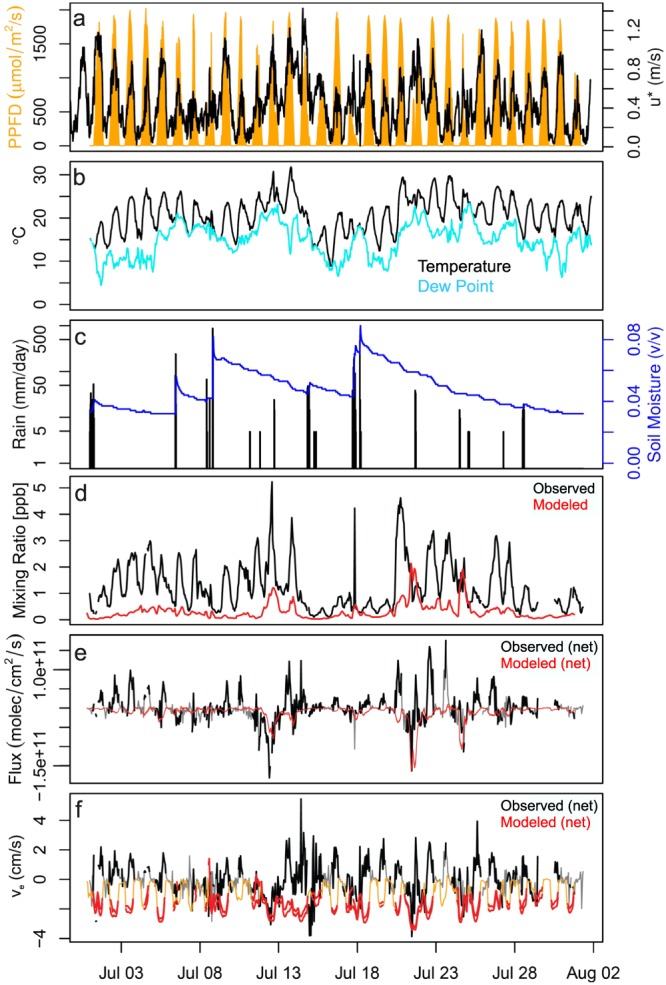
Time series of HCOOH and related measurements during PROPHET‐AMOS. Plotted is the observed (a) photosynthetic photon flux density (PPFD) and friction velocity (u*), (b) temperature and dew point, (c) rainfall and soil moisture, (d) HCOOH mixing ratios, (e) net canopy HCOOH flux, and (f) net canopy HCOOH exchange velocity. Also shown in (d)–(f) are the simulated HCOOH mixing ratios, fluxes, and exchange velocities as predicted by GEOS‐Chem (in red). For (e) and (f), the gray and orange lines show the full observational and model data sets, respectively, while the black and red show the same datasets after filtering for turbulent conditions (u* > 0.3 m/s).
Figure 2 shows the corresponding mean diel profiles of the HCOOH mixing ratios, fluxes, and exchange velocities. On average, the HCOOH mixing ratios peak during afternoon (at ~1.7 ppb) before dropping after sunset to <1 ppb. The diel mean fluxes (which as shown in Figure 1 combine distinct periods with net downward and upward exchange) exhibit net upward exchange during daytime, again peaking during afternoon, with net fluxes into the canopy at night. In‐canopy storage changes (HCOOH burden fluctuations below the sampling inlet; Figure 2b) account for ~15% of the net 24‐hr integrated above‐canopy HCOOH flux and do not significantly change the diel cycle. The weak measured soil fluxes (Figure 2b) argue against an important role for soil HCOOH emissions in this ecosystem.
Figure 2.
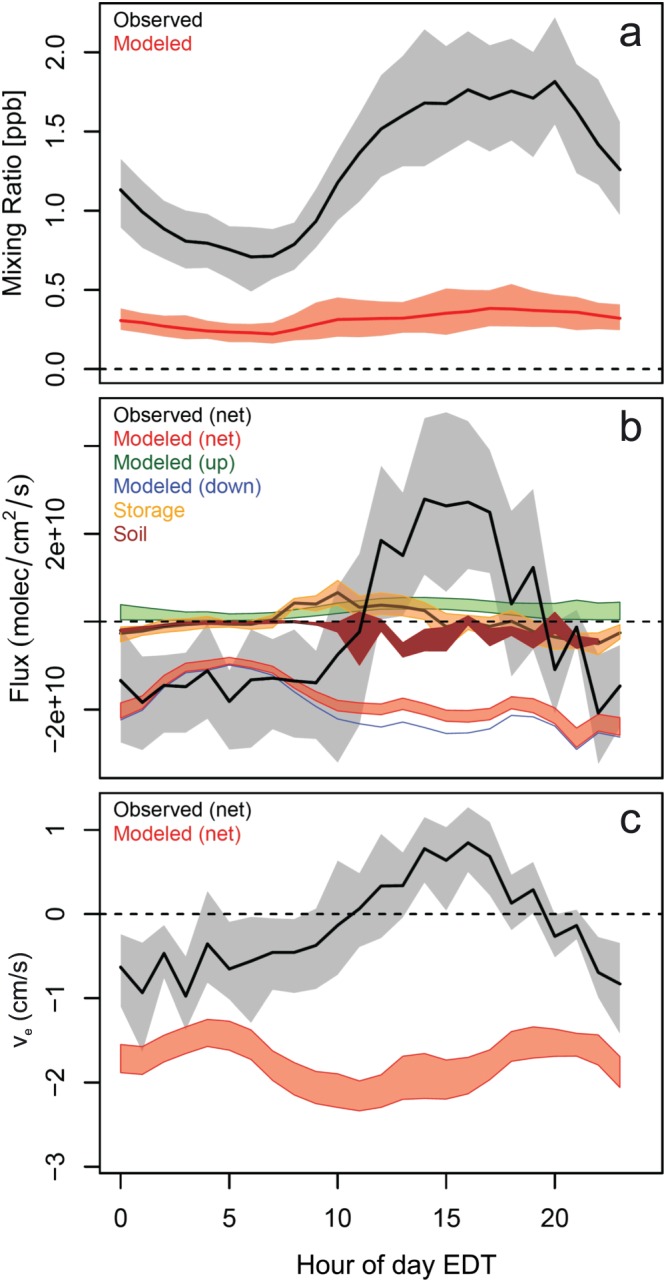
Mean diel profiles of HCOOH (a) mixing ratios, (b) fluxes, and (c) exchange velocities as measured during PROPHET‐AMOS and simulated by GEOS‐Chem. The solid black lines and shaded gray regions show the observed mean and associated 95% confidence interval, with the corresponding model values shown in red. Shaded regions for the model fluxes and exchange velocities span upper and lower limits for in‐canopy production and loss as described in supporting information S1. Along with the measured above‐canopy flux, (b) shows the observed contributions from canopy storage (orange) and soils (brown; the shaded region here spans the range from the three chambers with exposed soil after correcting for wall effects based on the blank chamber). Also shown in (b) are the model‐derived gross upward (green) and downward (blue) fluxes.
Measured net exchange velocities (Figure 2c) follow a similar diel profile as the fluxes. The observed daytime (10:00–17:00 EDT) mean exchange velocity for the campaign of 0.42 ± 0.48 (μ ± σ) cm/s is lower than that reported by Schobesberger et al. (2016) for a boreal forest (0.7 ± 1.7 cm/s) and larger than the net downward exchange observed for a mixed temperate forest in the Southeastern United States (−1.0 ± 0.4 cm/s; Nguyen et al., 2015) and for a tropical forest in Amazonia (−0.2 cm/s; Kuhn et al., 2002).
Also plotted in Figures 1 and 2 are the HCOOH mixing ratios, fluxes (net, gross upward, and gross downward), and exchange velocities predicted by GEOS‐Chem. The observed HCOOH mixing ratios are underpredicted in the model by a mean factor of 3–5, consistent with prior findings (Millet et al., 2015; Paulot et al., 2011; Yuan et al., 2015). Furthermore, while the observations reveal upward as well as downward net HCOOH fluxes over the canopy, the simulated net fluxes are consistently downward (Figures 1 and 2). The net deposition measured at night (Figure 2b) is, on average, well‐captured by the model; however, the model is clearly missing the large gross canopy HCOOH source apparent in the daytime observations.
We see in Figure 2b that the gross canopy HCOOH source estimated from the model output is significantly less than the mean net flux observed during daytime, despite the fact that the latter reflects the sum of gross upward (emission + in‐canopy photochemical production) and downward (deposition + in‐canopy photochemical loss) exchange. We can estimate the gross HCOOH source flux that is implied by the observations by correcting for deposition (in‐canopy chemical loss is negligible for HCOOH due to its ~25‐day photochemical lifetime; Atkinson et al., 2006). To accomplish this, we apply the HCOOH deposition velocities computed by the model to the observed HCOOH number densities and add the resulting loss term to the measured above‐canopy net flux. In this way, we derive from the observations a 24‐hr mean gross HCOOH canopy source of (5.1 ± 0.3) × 1010 molec/cm2/s (mean ± standard error), with corresponding daytime (10:00–17:00 EDT) and nighttime (22:00–05:00 EDT) values of (10.9 ± 0.6) × 1010 and (1.1 ± 0.2) × 1010 molec/cm2/s, respectively. This implied gross canopy HCOOH source corresponds to 2.9% and 38% (on a carbon basis) of the observed 24‐hr mean isoprene and monoterpene fluxes, respectively. It is also 15× larger than the 24‐hr gross HCOOH source (emissions + photochemical production) predicted by GEOS‐Chem (20× and 5× larger for the daytime‐only and nighttime‐only values).
The measured HCOOH mixing ratios and fluxes are the highest (most positive) under warm and low‐relative humidity (RH) conditions. For example, we see in Figure 1 that upward net fluxes tend to occur under large dew point depressions. Figure 3 quantifies this relationship, plotting the measured fluxes as a function of temperature, light, and humidity: On average, the daytime net fluxes become more positive with increasing light and temperature and more negative with increasing RH (or dew point). The daytime temperature dependence for the modeled net HCOOH fluxes is the reverse of that seen in the observations, with fluxes becoming more negative rather than more positive under warmer conditions—a tendency driven by increasing model deposition fluxes at warmer temperatures. Figure 3 also shows that the light dependence for the modeled net HCOOH flux is weaker than observed.
Figure 3.
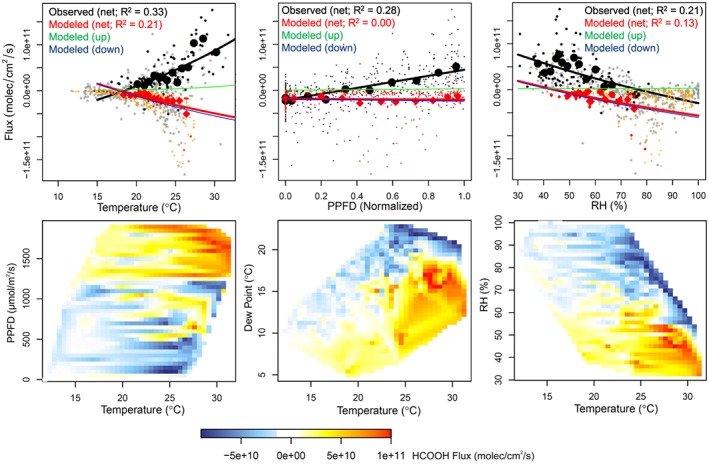
Dependence of canopy HCOOH fluxes on environmental drivers. The top row shows measured (black) and modeled (red) HCOOH fluxes plotted as a function of temperature, light (photosynthetic photon flux density [PPFD]), and relative humidity (RH). For the temperature and RH plots, daytime (PPFD > ~1,500 μmol/m2/s; black and red) and other (gray and orange) values are indicated separately. The large symbols and fit lines show the mean fluxes binned by quantile of the independent variable. The thin green and blue lines show the corresponding dependencies for the modeled gross upward and downward HCOOH fluxes, respectively. The bottom row shows curtain plots of the measured HCOOH fluxes as a function of temperature, light, RH, and dew point.
We see in Figures 3 and S1 that, for the most part, the net canopy HCOOH fluxes are upward for RH ≤ 60% and downward for RH ≥ 70%. It is possible that this behavior partly reflects the adsorption of HCOOH to wet surfaces with subsequent release from thin film water upon drying—as observed for the weak acid HONO at the same site (He et al., 2006). Karl et al. (2004) previously estimated the HCOOH uptake velocity to dew‐wetted surfaces in a tropical forest at 0.06–0.08 cm/s, versus the nighttime deposition velocities seen here approaching ~1 cm/s (Figure 2). However, the extent of aqueous‐phase uptake will depend on several poorly known factors, including leaf surface area, the pH of the liquid, presence of deposited mineral dust, and any HCOOH/formate metabolism by vegetation or surface‐inhabiting microbes.
3.2. In‐Forest HCOOH Gradients Point to a Canopy‐Driven Secondary Source
The bidirectional exchange (and implied in‐forest source) of HCOOH seen in the above‐canopy fluxes is also evident in the measured vertical gradients. Figure 4 shows a diel curtain plot of HCOOH in‐canopy vertical gradients, averaged over the entire PROPHET‐AMOS campaign. The nighttime HCOOH mixing ratios decrease with depth into the canopy, consistent with the net downward exchange measured at that time (Figure 2c). During daytime, an HCOOH maximum appears in the midcanopy (~20 m), implying a direct and/or indirect source from foliage. The gradient measurements do not reveal a surface‐level HCOOH enhancement at any time of day, which supports the soil chamber data in showing that the forest floor is not an appreciable HCOOH source in this environment.
Figure 4.
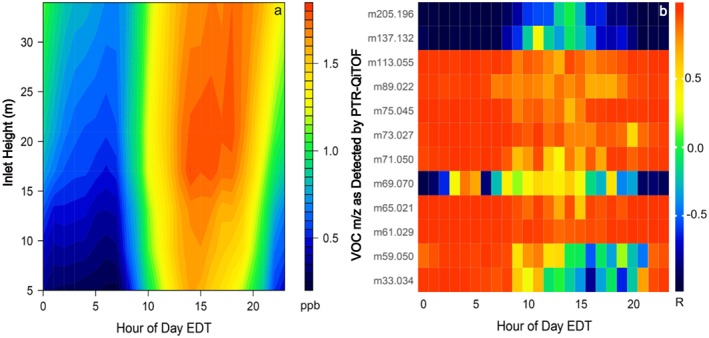
In‐canopy vertical gradients of HCOOH and correlations with tracers of known origin. (a) Curtain plot showing the mean diel cycle of HCOOH vertical gradients for the PROPHET‐AMOS campaign. (b) Mean hourly correlations between the HCOOH vertical gradients and those for selected other VOCs: m/z 33.034 = methanol; m/z 59.050 = acetone; m/z 61.029 = acetic acid/glycolaldehyde; m/z 65.021 = CH5O3 +; m/z 69.070 = isoprene; m/z 71.050 = MVK + MACR; m/z 73.027 = methylglyoxal; m/z 75.045 = hydroxyacetone; m/z 89.022 = C3H5O3 +; m/z 113.055 = C6H9O2 +; m/z 137.132 = monoterpenes; m/z 205.196 = sesquiterpenes.
Along with HCOOH, we also measured hourly vertical gradients for all the VOCs across the mass spectrum detected by PTR‐QiTOF (636 ions). These in‐canopy gradients for other VOCs of known origin can be used as spatial‐temporal fingerprints for understanding the factors driving observed HCOOH fluxes. Figure S2 shows diel mean vertical gradients for selected marker VOCs: isoprene, monoterpenes, methanol, acetone, acetic acid/glycolaldehyde, m/z 65.021 (CH5O3 +), methyl vinyl ketone + methacrolein (MVK + MACR), and m/z 113.055 (C6H9O2 +). For isoprene, we see peak concentrations from midday through afternoon, with a relatively sharp in‐canopy maximum around 20 m (and low concentrations at night), consistent with light‐ and temperature‐driven emissions from the aspen‐dominated overstory. Monoterpenes exhibit a very different pattern, with concentration peaks during morning and evening in the lower canopy, reflecting the influences of temperature‐dependent (and partially light‐independent; Millet et al., 2018; Ortega et al., 2007) emissions, a direct source from the white pine and red maple understory, and atmospheric mixing.
Methanol and acetone are likewise directly emitted by vegetation (Guenther et al., 2012). While both species can also be produced from the oxidation of biogenic precursors, this is thought to be a small source compared to the direct biogenic flux (Fischer et al., 2012; Millet et al., 2008). We should thus expect in‐canopy variability of methanol and acetone during PROPHET‐AMOS to be driven by direct emissions and depositional uptake. Figure S2 shows that very similar gradient patterns are seen for both species. At night, concentrations decrease with depth in the canopy due to deposition, while during daytime, an afternoon peak emerges that appears to originate in both the upper and lower canopies—perhaps because the capacity to emit methanol and acetone is relatively uniform across woody plants (Guenther et al., 2012).
Sources for other species in Figure S2 are believed to be dominated by secondary production, that is, oxidation of precursor VOCs. Acetic acid/glycolaldehyde (produced from monoterpene and isoprene oxidation; Dibble, 2004; Lee, Goldstein, Keywood, et al., 2006; Lee, Goldstein, Kroll, et al., 2006; Paulot et al., 2011), m/z 65.021 (either HMHP or a fragment of a larger oxygenated VOC [OVOC]), m/z 75.045 (hydroxyacetone), and m/z 113.055 (a likely monoterpene oxidation product; Lee, Goldstein, Keywood, et al., 2006; Lee, Goldstein, Kroll, et al., 2006) all exhibit gradient patterns that are very similar to those of HCOOH (Figure 4), with net nighttime uptake and a diffuse midafternoon peak that is centered around midcanopy. The diel cycle for MVK + MACR (first‐generation products of isoprene oxidation) is somewhat distinct, with a peak around 10:00 EST that suggests a larger role for entrainment in this case.
We performed a correlation analysis to quantify the level of agreement between the hourly averaged vertical gradients of HCOOH and those of other species. Results are shown in Figure 4 for the marker VOCs discussed above, plus sesquiterpenes, m/z 73.027 (C3H5O2 +; likely methyl glyoxal), m/z 75.045 (C3H7O2 +; hydroxyacetone/propanoic acid), and m/z 89.022 (C3H5O3 +; possibly pyruvic acid). Figure 4 shows that the monoterpene and sesquiterpene gradients are weakly or negatively correlated with that of HCOOH at all times, indicating dissimilar in‐canopy source/sink distributions. The isoprene gradients exhibit only a weak correlation with HCOOH during daytime (mean R = 0.38 for 10:00–17:00 EDT) that disappears at night. Acetone and methanol both have similar vertical structure to that of HCOOH at night (mean R = 0.94 and 0.99, respectively, for 22:00–05:00 EDT), likely reflecting a common net sink to depositional uptake, but the daytime vertical gradients for these directly emitted VOCs do not match those of HCOOH (mean R < 0.3 for both species; Figure 4).
Overall, of the 636 detected ions, 25 have mean vertical gradients that have R > 0.5 with that of HCOOH during every hour of the day, thus implying similar in‐canopy source‐sink distributions. Of these, 14 can be assigned a molecular formula according to the procedure outlined in Millet et al. (2018), excluding isotopes: C4H5O3 +, C2H5O2 + (acetic acid/glycolaldehyde), C5H7O2 +, C5H7O3 +, C2H3O+ (common OVOC fragment), C3H5O3 +, C6H9O2 + (likely terpene oxidation product), O2N+ (fragment of peroxyacetyl nitrate and other organic nitrates), C3H7O2 + (hydroxyacetone/propanoic acid), C5H5O2 +, C4H7O4 +, CH5O3 + (HMHP or OVOC fragment), C6H11O3 +, and C3H5O2 + (methylglyoxal).
A similar correlation analysis was performed with respect to the HCOOH mean diel cycles at each sampling height, with example results shown in Figure S3. Here, 313 detected ions exhibited R > 0.5 with the HCOOH diel cycle for all heights (75 had minimum R > 0.9), showing that the diel cycles at any given height are a less sensitive diagnostic for resolving VOC source/sink patterns than are the vertical gradients. Finally, since the observed HCOOH flux time series (Figure 1) reveals distinct periods with net upward and net downward exchange, we repeated the above analyses for data subsets reflecting each of those scenarios: 3–5 July + 22–23 July (upward HCOOH flux tendency) and 11–12 July (downward HCOOH flux tendency). Among identified ions, only m/z 61.029 (acetic acid/glycolaldehyde), m/z 65.021 (HMHP or OVOC fragment), and m/z 75.045 (hydroxyacetone/propanoic acid) exhibited high correlation with HCOOH in all cases (i.e., R > 0.5 for vertical gradients at all hours in the diel mean and R > 0.9 for mean diel cycles at all heights). As before, the VOCs with the most similar vertical and diel patterns to HCOOH all correspond to oxidized compounds, most with two or more oxygen atoms.
Together, the above analyses suggest a rapid in‐canopy photochemical (rather than direct emission) HCOOH source: The HCOOH gradients do not match well with those for tracers of direct biogenic emissions (isoprene, monoterpenes, sesquiterpenes, methanol, and acetone) and instead strongly resemble those of oxidized VOCs whose main atmospheric source is thought to be secondary chemical production. Potential mechanisms for such a secondary HCOOH source are discussed in supporting information S2 (Crounse et al., 2012; Finlayson‐Pitts et al., 2003; Jud et al., 2016; Li et al., 2018; Neeb et al., 1997; Paulot et al., 2009; Welz et al., 2014). A separate possibility is that the canopy source of HCOOH arises from direct emissions that are dissimilar from those of any available primary emission tracers (thus causing the poor correlations in Figure 4b). Overall, however, while a mainly primary HCOOH source cannot be definitively ruled out based on the data at hand, a mainly secondary HCOOH source seems the simplest and most likely explanation.
4. Conclusions
We presented new measurements of HCOOH mixing ratios, net above‐canopy fluxes, vertical in‐canopy gradients, and soil fluxes by high‐resolution PTR‐QiTOF over a mixed forest in Michigan, United States, during the PROPHET‐AMOS study. Detected net fluxes were bidirectional, mostly upward for RH < 60% and mostly downward for RH > 70%. A state‐of‐science chemical transport model (GEOS‐Chem CTM) underestimates the implied gross canopy HCOOH source by on average fifteenfold and misdiagnoses the temperature and light dependence of the net exchange (implying a missing T‐ and hν‐sensitive source). Soils are a negligible HCOOH source in this environment. A correlation analysis of the vertical and diel patterns of HCOOH variability based on other VOC tracers points to secondary chemical production, rather than primary emission from plants or soil, as the most likely HCOOH source for this forest ecosystem.
Supporting information
Supporting Information S1
Figure S1
Figure S2
Figure S3
Acknowledgments
This research was supported by NSF (grants AGS‐1428257, AGS‐1148951 and AGS‐1352375). GEOS‐Chem model development and simulations were supported by NASA (grant NNX14AP89G). Computing resources were provided by MSI (www.msi.umn.edu). Soil flux measurements were supported by DOE (grant DE‐SC0014443). We thank Steve Bertman, Phil Stevens, all other PROPHET‐AMOS collaborators, UMBS, the US‐UMB Ameriflux team, and the University of Houston/Rice group for providing meteorological measurements. We acknowledge Timothy Griffis and Julian Deventer for helpful discussions, plus John Poehlman, Jeremy Boshears, and colleagues for help designing and building the chamber system. High‐frequency measurements used here (winds, HCOOH, and select other VOCs) are available online (https://doi.org/10.13020/D6JQ3R). Processed fluxes and other VOC data are available from the authors upon request. GEOS‐Chem model code is available online (www.geos‐chem.org).
Alwe, H. D. , Millet, D. B. , Chen, X. , Raff, J. D. , Payne, Z. C. , & Fledderman, K. (2019). Oxidation of volatile organic compounds as the major source of formic acid in a mixed forest canopy. Geophysical Research Letters, 46, 2940–2948. 10.1029/2018GL081526
References
- Allen, H. M. , Crounse, J. D. , Bates, K. H. , Teng, A. P. , Krawiec‐Thayer, M. P. , Rivera‐Rios, J. C. , Keutsch, F. N. , St. Clair, J. M. , Hanisco, T. F. , Møller, K. H. , Kjaergaard, H. G. , & Wennberg, P. O. (2018). Kinetics and product yields of the OH initiated oxidation of hydroxymethyl hydroperoxide. The Journal of Physical Chemistry A, 122, 6292–6302. 10.1021/acs.jpca.8b04577 [DOI] [PubMed] [Google Scholar]
- Atkinson, R. , Baulch, D. L. , Cox, R. A. , Crowley, J. N. , Hampson, R. F. , Hynes, R. G. , Jenkin, M. E. , Rossi, M. J. , Troe, J. , & IUPAC Subcommittee (2006). Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II—Gas phase reactions of organic species. Atmospheric Chemistry and Physics, 6, 3625–4055. 10.5194/acp-6-3625-2006 [DOI] [Google Scholar]
- Bannan, T. J. , Bacak, A. , Muller, J. B. , Booth, A. M. , Jones, B. , Le Breton, M. , & Percival, C. J. (2014). Importance of direct anthropogenic emissions of formic acid measured by a chemical ionisation mass spectrometer (CIMS) during the Winter ClearfLo Campaign in London, January 2012. Atmospheric Environment, 83, 301–310. 10.1016/j.atmosenv.2013.10.029 [DOI] [Google Scholar]
- Bannan, T. J. , Booth, A. M. , Le Breton, M. , Bacak, A. , Muller, J. B. , Leather, K. E. , Khan, M. A. H. , Lee, J. D. , Dunmore, R. E. , Hopkins, J. R. , Fleming, Z. L. , Sheps, L. , Taatjes, C. A. , Shallcross, D. E. , & Percival, C. J. (2017). Seasonality of formic acid (HCOOH) in London during the ClearfLo campaign. Journal of Geophysical Research: Atmospheres, 122, 12,488–12,498. 10.1002/2017JD027064 [DOI] [Google Scholar]
- Bryan, A. M. , Cheng, S. J. , Ashworth, K. , Guenther, A. B. , Hardiman, B. S. , Bohrer, G. , & Steiner, A. L. (2015). Forest‐atmosphere BVOC exchange in diverse and structurally complex canopies: 1‐D modeling of a mid‐successional forest in northern Michigan. Atmospheric Environment, 120, 217–226. 10.1016/j.atmosenv.2015.08.094 [DOI] [Google Scholar]
- Chaliyakunnel, S. , Millet, D. B. , Wells, K. C. , Cady‐Pereira, K. E. , & Shephard, M. W. (2016). A large underestimate of formic acid from tropical fires: Constraints from space‐borne measurements. Environmental Science & Technology, 50, 5631–5640. 10.1021/acs.est.5b06385 [DOI] [PubMed] [Google Scholar]
- Chebbi, A. , & Carlier, P. (1996). Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review. Atmospheric Environment, 30(24), 4233–4249. 10.1016/1352-2310(96)00102-1 [DOI] [Google Scholar]
- Crounse, J. D. , Knap, H. C. , Ørnsø, K. B. , Jørgensen, S. , Paulot, F. , Kjaergaard, H. G. , & Wennberg, P. O. (2012). Atmospheric fate of methacrolein: 1. Peroxy radical isomerization following addition of OH and O2 . Journal of Physical Chemistry A, 116, 5756–5762. 10.1021/jp211560u [DOI] [PubMed] [Google Scholar]
- Dibble, T. C. (2004). Prompt chemistry of alkenoxy radical products of the double H‐atom transfer of alkoxy radicals from isoprene. Journal of Physical Chemistry A, 108, 2208–2215. 10.1021/jp0312161 [DOI] [Google Scholar]
- Eliason, T. L. , Aloisio, S. , Donaldson, D. J. , Cziczo, D. J. , & Vaida, V. (2003). Processing of unsaturated organic acid films and aerosols by ozone. Atmospheric Environment, 37(16), 2207–2219. 10.1016/S1352-2310(03)00149-3 [DOI] [Google Scholar]
- Finlayson‐Pitts, B. J. , Wingen, L. M. , Sumner, A. L. , Syomin, D. , & Ramazan, K. A. (2003). The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: An integrated mechanism. Physical Chemistry Chemical Physics, 5(2), 223–242. 10.1039/B208564J [DOI] [Google Scholar]
- Fischer, E. V. , Jacob, D. J. , Millet, D. B. , Yantosca, R. M. , & Mao, J. (2012). The role of the ocean in the global atmospheric budget of acetone. Geophysical Research Letters, 39, L01807 10.1029/2011GL050086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway, J. N. , Likens, G. E. , Keene, W. C. , & Miller, J. M. (1982). The composition of precipitation in remote areas of the world. Journal of Geophysical Research, 87(C11), 8771–8786. 10.1029/JC087iC11p08771 [DOI] [Google Scholar]
- Goode, J. G. , Yokelson, R. J. , Ward, D. E. , Susott, R. A. , Babbitt, R. E. , Davies, M. A. , & Hao, W. M. (2000). Measurements of excess O3, CO2, CO, CH4, C2H4, C2H2, HCN, NO, NH3, HCOOH, CH3COOH, HCHO, and CH3OH in 1997 Alaskan biomass burning plumes by airborne Fourier transform infrared spectroscopy (AFTIR). Journal of Geophysical Research, 105(D17), 22,147–22,166. 10.1029/2000JD900287 [DOI] [Google Scholar]
- Guenther, A. B. , Jiang, X. , Heald, C. L. , Sakulyanontvittaya, T. , Duhl, T. , Emmons, L. K. , & Wang, X. (2012). The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): An extended and updated framework for modeling biogenic emissions. Geoscientific Model Development, 5, 1471–1492. 10.5194/gmd-5-1471-2012 [DOI] [Google Scholar]
- Hatakeyama, S. , Washida, N. , & Akimoto, H. (1986). Rate constants and mechanisms for the reaction of OH (OD) radicals with acetylene, propyne, and 2‐butyne in air at 297 +/‐ 2 K. Journal of Physical Chemistry, 90, 173–178. 10.1021/j100273a039 [DOI] [Google Scholar]
- He, E. , Zhou, X. , Hou, J. , Gao, H. , & Bertman, S. B. (2006). Importance of dew in controlling the air‐surface exchange of HONO in rural forested environments. Geophysical Research Letters, 33, L02813 10.1029/2005GL24348 [DOI] [Google Scholar]
- Jacob, D. J. (1986). Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate. Journal of Geophysical Research, 91(D9), 9807–9826. 10.1029/JD091iD09p09807 [DOI] [Google Scholar]
- Jud, W. , Fischer, L. , Canaval, E. , Wohlfahrt, G. , Tissier, A. , & Hansel, A. (2016). Plant surface reactions: An opportunistic defence mechanism impacting atmospheric chemistry. Atmospheric Chemistry and Physics, 16(1), 277–292. 10.5194/acp-16-277-2016 [DOI] [Google Scholar]
- Karl, T. , Potosnak, M. , Guenther, A. , Clark, D. , Walker, J. , Herrick, J. D. , & Geron, C. (2004). Exchange processes of volatile organic compounds above a tropical rain forest: Implications for modeling tropospheric chemistry above dense vegetation. Journal of Geophysical Research, 109, D18306 10.1029/2004JD004738 [DOI] [Google Scholar]
- Kawamura, K. , Ng, L. L. , & Kaplan, I. R. (1985). Determination of organic acids (C1–C10) in the atmosphere, motor exhausts, and engine oils. Environmental Science and Technology, 19(11), 1082–1086. 10.1021/es00141a010 [DOI] [PubMed] [Google Scholar]
- Kesselmeier, J. , Bode, K. , Gerlach, C. , & Jork, E. M. (1998). Exchange of atmospheric formic and acetic acids with trees and crop plants under controlled chamber and purified air conditions. Atmospheric Environment, 32(10), 1765–1775. 10.1016/S1352-2310(97)00465-2 [DOI] [Google Scholar]
- Khare, P. , Kumar, N. , Kumari, K. M. , & Srivastava, S. S. (1999). Atmospheric formic and acetic acids: An overview. Reviews of Geophysics, 37(2), 227–248. 10.1029/1998RG900005 [DOI] [Google Scholar]
- Kuhn, U. , Rottenberger, S. , Biesenthal, T. , Ammann, C. , Wolf, A. , Schebeske, G. , Oliva, S. T. , Tavares, T. M. , & Kesselmeier, J. (2002). Exchange of short‐chain monocarboxylic acids by vegetation at a remote tropical forest site in Amazonia. Journal of Geophysical Research, 107(D20), 8069 10.1029/2000JD000303 [DOI] [Google Scholar]
- Larsen, B. R. , Di Bella, D. , Glasius, M. , Winterhalter, R. , Jensen, N. , & Hjorth, J. (2001). Gas‐phase OH oxidation of monoterpenes: Gaseous and particulate products. Journal of Atmospheric Chemistry, 38(3), 231–276. 10.1023/A:1006487530903 [DOI] [Google Scholar]
- Lee, A. , Goldstein, A. H. , Keywood, M. D. , Gao, S. , Varutbangkul, V. , Bahreini, R. , Ng, N. L. , Flagan, R. C. , & Seinfeld, J. H. (2006). Gas‐phase products and secondary aerosol yields from the ozonolysis of ten different terpenes. Journal of Geophysical Research, 111, D07302 10.1029/2005JD006437 [DOI] [Google Scholar]
- Lee, A. , Goldstein, A. H. , Kroll, J. H. , Ng, N. L. , Varutbangkul, V. , Flagan, R. C. , & Seinfeld, J. H. (2006). Gas‐phase products and secondary aerosol yields from the photooxidation of 16 different terpenes. Journal of Geophysical Research, 111, D17305 10.1029/2006JD007050 [DOI] [Google Scholar]
- Lelieveld, J. , & Crutzen, P. J. (1991). The role of clouds in tropospheric chemistry. Journal of Atmospheric Chemistry, 12, 229–267. 10.1007/BF00048075 [DOI] [Google Scholar]
- Li, G. , Cheng, Y. , Kuhn, U. , Xu, R. , Yang, Y. , Meusel, H. , Ma, N. , Wu, Y. , Li, M. , Williams, J. , Hoffmann, T. , Ammann, M. , Pöschl, U. , Shao, M. , & Su, H. (2018). Physicochemical uptake and release of volatile organic compounds by soil in coated‐wall flow tube experiments with ambient air. Atmospheric Chemistry and Physics Discussions, 1–47. 10.5194/acp-2018-683 [DOI] [Google Scholar]
- Malecha, K. T. , & Nizkorodov, S. A. (2016). Photodegradation of secondary organic aerosol particles as a source of small, oxygenated volatile organic compounds. Environmental Science & Technology, 50, 9990–9997. 10.1021/acs.est.6b02313 [DOI] [PubMed] [Google Scholar]
- Mattila, J. M. , Brophy, P. , Kirkland, J. , Hall, S. , Ullmann, K. , Fischer, E. V. , Brown, S. , McDuffie, E. , Tevlin, A. , & Farmer, D. K. (2018). Tropospheric sources and sinks of gas‐phase acids in the Colorado front range. Atmospheric Chemistry and Physics, 18, 12,315–12,327. 10.5194/acp-18-12315-2018 [DOI] [Google Scholar]
- Mielnik, A. , Link, M. , Mattila, J. , Fulgham, S. R. , & Farmer, D. K. (2018). Emission of formic and acetic acid from two Colorado soils. Environmental Science: Processes & Impacts, 20, 1537–1545. 10.1039/c8em00356d [DOI] [PubMed] [Google Scholar]
- Millet, D. B. , Alwe, H. D. , Chen, X. , Deventer, M. J. , Griffis, T. J. , Holzinger, R. , Bertman, S. B. , Rickly, P. S. , Stevens, P. S. , Léonardis, T. , Locoge, N. , Dusanter, S. , Tyndall, G. S. , Alvarez, S. L. , Erickson, M. H. , & Flynn, J. H. (2018). Bidirectional ecosystem–atmosphere fluxes of volatile organic compounds across the mass spectrum: How many matter? ACS Earth and Space Chemistry, 2, 764–777. 10.1021/acsearthspacechem.8b00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet, D. B. , Baasandorj, M. , Farmer, D. K. , Thornton, J. A. , Baumann, K. , Brophy, P. , Chaliyakunnel, S. , de Gouw, J. A. , Graus, M. , Hu, L. , Koss, A. , Lee, B. H. , Lopez‐Hilfiker, F. D. , Neuman, J. A. , Paulot, F. , Peischl, J. , Pollack, I. B. , Ryerson, T. B. , Warneke, C. , Williams, B. J. , & Xu, J. (2015). A large and ubiquitous source of atmospheric formic acid. Atmospheric Chemistry and Physics, 15, 6283–6304. 10.5194/acp-15-6283-2015 [DOI] [Google Scholar]
- Millet, D. B. , Jacob, D. J. , Custer, T. G. , de Gouw, J. A. , Goldstein, A. H. , Karl, T. , Singh, H. B. , Sive, B. C. , Talbot, R. W. , Warneke, C. , & Williams, J. (2008). New constraints on terrestrial and oceanic sources of atmospheric methanol. Atmospheric Chemistry and Physics, 8, 6887–6905. 10.5194/acp-8-6887-2008 [DOI] [Google Scholar]
- Mungall, E. L. , Abbatt, J. P. D. , Wentzell, J. J. B. , Wentworth, G. R. , Murphy, J. G. , Kunkel, D. , Gute, E. , Tarasick, D. W. , Sharma, S. , Cox, C. J. , Uttal, T. , & Liggio, J. (2018). High gas‐phase mixing ratios of formic and acetic acid in the high Arctic. Atmospheric Chemistry and Physics, 18, 10,237–10,254. 10.5194/acp-18-10237-2018 [DOI] [Google Scholar]
- Neeb, P. , Sauer, F. , Horie, O. , & Moortgat, G. K. (1997). Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour. Atmospheric Environment, 31(10), 1417–1423. 10.1016/S1352-2310(96)00322-6 [DOI] [Google Scholar]
- Nguyen, T. B. , Crounse, J. D. , Teng, A. P. , St Clair, J. M. , Paulot, F. , Wolfe, G. M. , & Wennberg, P. O. (2015). Rapid deposition of oxidized biogenic compounds to a temperate forest. Proceedings of the National Academy of Sciences of the United States of America, 112, E392‐401. 10.1073/pnas.1418702112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, T. B. , Tyndall, G. S. , Crounse, J. D. , Teng, A. P. , Bates, K. H. , Schwantes, R. H. , Coggon, M. M. , Zhang, L. , Feiner, P. , Milller, D. O. , Skog, K. M. , Rivera‐Rios, J. C. , Dorris, M. , Olson, K. F. , Koss, A. , Wild, R. J. , Brown, S. S. , Goldstein, A. H. , de Gouw, J. A. , Brune, W. H. , Keutsch, F. N. , Seinfeld, J. H. , & Wennberg, P. O. (2016). Atmospheric fate of Criegee intermediates in the ozonolysis of isoprene. Physical Chemistry Chemical Physics, 18(15), 10,241–10,254. 10.1039/c6cp00053c [DOI] [PubMed] [Google Scholar]
- Ngwabie, N. M. , Schade, G. W. , Custer, T. G. , Linke, S. , & Hinz, T. (2008). Abundances and flux estimates of volatile organic compounds from a dairy cowshed in Germany. Journal of Environmental Quality, 37(2), 565–573. 10.2134/jeq2006.0417 [DOI] [PubMed] [Google Scholar]
- Ortega, J. , Helmig, D. , Guenther, A. , Harley, P. , Pressley, S. , & Vogel, C. (2007). Flux estimates and OH reaction potential of reactive biogenic volatile organic compounds (BVOCs) from a mixed northern hardwood forest. Atmospheric Environment, 41(26), 5479–5495. 10.1016/j.atmosenv.2006.12.033 [DOI] [Google Scholar]
- Palik, B. J. , & Pregitzer, K. S. (1992). A comparison of pre‐settlement and present‐day forests on two Bigtooth Aspen‐dominated landscapes in northern lower Michigan. The American Midland Naturalist, 127(2), 327–338. 10.2307/2426539 [DOI] [Google Scholar]
- Pan, X. , Underwood, J. S. , Xing, J.‐H. , Mang, S. A. , & Nizkorodov, S. A. (2009). Photodegradation of secondary organic aerosol generated from limonene oxidation by ozone studied with chemical ionization mass spectrometry. Atmospheric Chemistry and Physics, 9(12), 3851–3865. 10.5194/acp-9-3851-2009 [DOI] [Google Scholar]
- Pape, L. , Ammann, C. , Nyfeler‐Brunner, A. , Spirig, C. , Hens, K. , & Meixner, F. X. (2009). An automated dynamic chamber system for surface exchange measurement of non‐reactive and reactive trace gases of grassland ecosystems. Biogeosciences, 6(3), 405–429. 10.5194/bg-6-405-2009 [DOI] [Google Scholar]
- Park, J. , Gomez, A. L. , Walser, M. L. , Lin, A. , & Nizkorodov, S. A. (2006). Ozonolysis and photolysis of alkene‐terminated self‐assembled monolayers on quartz nanoparticles: Implications for photochemical aging of organic aerosol particles. Physical Chemistry Chemical Physics, 8(21), 2506–2512. 10.1039/B602704K [DOI] [PubMed] [Google Scholar]
- Paulot, F. , Crounse, J. D. , Kjaergaard, H. G. , Kroll, J. H. , Seinfeld, J. H. , & Wennberg, P. O. (2009). Isoprene photooxidation: New insights into the production of acids and organic nitrates. Atmospheric Chemistry and Physics, 9(4), 1479–1501. 10.5194/acp-9-1479-2009 [DOI] [Google Scholar]
- Paulot, F. , Wunch, D. , Crounse, J. D. , Toon, G. C. , Millet, D. B. , DeCarlo, P. F. , Vigouroux, C. , Deutscher, N. M. , González Abad, G. , Notholt, J. , Warneke, T. , Hannigan, J. W. , Warneke, C. , de Gouw, J. A. , Dunlea, E. J. , de Mazière, M. , Griffith, D. W. T. , Bernath, P. , Jimenez, J. L. , & Wennberg, P. O. (2011). Importance of secondary sources in the atmospheric budgets of formic and acetic acids. Atmospheric Chemistry and Physics, 11(5), 1989–2013. 10.5194/acp-11-1989-2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters, J. , Nguyen, V. N. , & Müller, J. F. (2015). Atmospheric vinyl alcohol to acetaldehyde tautomerization revisited. The Journal of Physical Chemistry Letters, 6(20), 4005–4011. 10.1021/acs.jpclett.5b01787 [DOI] [PubMed] [Google Scholar]
- Sanhueza, E. , & Andreae, M. O. (1991). Emission of formic and acetic acids from tropical savanna soils. Geophysical Research Letters, 18(9), 1707–1710. 10.1029/91GL01565 [DOI] [Google Scholar]
- Schobesberger, S. , Lopez‐Hilfiker, F. D. , Taipale, D. , Millet, D. B. , D'Ambro, E. L. , Rantala, P. , Mammarella, I. , Zhou, P. , Wolfe, G. M. , Lee, B. H. , Boy, M. , & Thornton, J. A. (2016). High upward fluxes of formic acid from a boreal forest canopy. Geophysical Research Letters, 43, 9342–9351. 10.1002/2016GL069599 [DOI] [Google Scholar]
- Seok, B. , Helmig, D. , Liptzin, D. , Williams, M. W. , & Vogel, C. S. (2015). Snowpack‐atmosphere gas exchanges of carbon dioxide, ozone, and nitrogen oxides at a hardwood forest site in northern Michigan. Elementa Science of the Anthropocene, 3, 000040 10.12952/journal.elementa.000040 [DOI] [Google Scholar]
- Shaw, M. F. , Sztáray, B. , Whalley, L. K. , Heard, D. E. , Millet, D. B. , Jordan, M. J. T. , Osborn, D. L. , & Kable, S. H. (2018). Photo‐tautomerization of acetaldehyde as a photochemical source of formic acid in the troposphere. Nature Communications, 9(1). 10.1038/s41467-018-04824-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrakou, T. , Müller, J.‐F. , Peeters, J. , Razavi, A. , Clarisse, L. , Clerbaux, C. , Coheur, P. F. , Hurtmans, D. , de Mazière, M. , Vigouroux, C. , Deutscher, N. M. , Griffith, D. W. T. , Jones, N. , & Paton‐Walsh, C. (2012). Satellite evidence for a large source of formic acid from boreal and tropical forests. Nature Geoscience, 5(1), 26–30. 10.1038/ngeo1354 [DOI] [Google Scholar]
- Talbot, R. W. , Beecher, K. M. , Harriss, R. C. , & Cofer, W. R. (1988). Atmospheric geochemistry of formic and acetic acids at a mid‐latitude temperate site. Journal of Geophysical Research, 93(D2), 1638–1652. 10.1029/JD093iD02p01638 [DOI] [Google Scholar]
- Vlasenko, A. , George, I. J. , & Abbatt, J. P. D. (2008). Formation of volatile organic compounds in the heterogeneous oxidation of condensed‐phase organic films by gas‐phase OH. The Journal of Physical Chemistry A, 112(7), 1552–1560. 10.1021/jp0772979 [DOI] [PubMed] [Google Scholar]
- Welz, O. , Eskola, A. J. , Sheps, L. , Rotavera, B. , Savee, J. D. , Scheer, A. M. , Osborn, D. L. , Lowe, D. , Murray Booth, A. , Xiao, P. , Anwar H. Khan, M. , Percival, C. J. , Shallcross, D. E. , & Taatjes, C. A. (2014). Rate coefficients of C1 and C2 Criegee intermediate reactions with formic and acetic acid near the collision limit: Direct kinetics measurements and atmospheric implications. Angewandte Chemie International Edition, 53(18), 4547–4550. 10.1002/anie.201400964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, B. , Veres, P. R. , Warneke, C. , Roberts, J. M. , Gilman, J. B. , Koss, A. , Edwards, P. M. , Graus, M. , Kuster, W. C. , Li, S. M. , Wild, R. J. , Brown, S. S. , Dubé, W. P. , Lerner, B. M. , Williams, E. J. , Johnson, J. E. , Quinn, P. K. , Bates, T. S. , Lefer, B. , Hayes, P. L. , Jimenez, J. L. , Weber, R. J. , Zamora, R. , Ervens, B. , Millet, D. B. , Rappenglück, B. , & de Gouw, J. A. (2015). Investigation of secondary formation of formic acid: Urban environment vs. oil and gas producing region. Atmospheric Chemistry and Physics, 15(4), 1975–1993. 10.5194/acp-15-1975-2015 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1
Figure S1
Figure S2
Figure S3