

Abstract
Small micromeres of the sea urchin are believed to be primordial germ cells (PGCs), fated to give rise to sperm or eggs in the adult. Sea urchin PGCs are formed at the fifth cleavage, undergo one additional division during blastulation, and migrate to the coelomic pouches of the pluteus larva. The goal of this chapter is to detail classical and modern techniques used to analyze primordial germ cell specification, gene expression programs, and cell behaviors in fixed and live embryos. The transparency of the sea urchin embryo enables both live imaging techniques and in situ RNA hybridization and immunolabeling for a detailed molecular characterization of these cells. Four approaches are presented to highlight small micromeres with fluorescent molecules for analysis by live and fixed cell microscopy: (1) small molecule dye accumulation during cleavage and blastula stages, (2) primordial germ cell targeted RNA expression using the Nanos untranslated regions, (3) fusing genes of interest with a Nanos2 targeting peptide, and (4) EdU and BrdU labeling. Applications of the live labeling techniques are discussed, including sorting by fluorescence-activated cell sorting for transcriptomic analysis, and, methods to image small micromere behavior in whole and dissociated embryos by live confocal microscopy. Finally, summary table of antibody and RNA probes as well as small molecule dyes to label small micromeres at a variety of developmental stages is provided.
1. INTRODUCTION
Primordial germ cells (PGCs) are specialized embryonic cells formed early in development and fated to produce gametes in the adult gonad. PGCs are evolutionarily important as conduits for transmission of the genome across generations (Extavour, 2003), andbiomedicallyrelevanttogermcellcancersandinvitroreproduction. Assuch, their specification (Juliano, Swartz, & Wessel, 2010; Santos & Lehmann, 2004; Wessel et al., 2014) and cell biology (Lehmann, 2012; Raz, 2004; Tarbashevich & Raz, 2010) are of interest to developmental biologists.
Although sea urchin embryos have served as experimental animal models for more than a century, the identity of their primordial germ cells was cryptic until recently. A series of investigations in sea urchins, most of them in the last decade, have established that cells termed small micromeres are the progenitors of the germline in Strongylocentrotus purpuratus. Small micromeres are formed as the result of two asymmetrical cell divisions at the vegetal pole of the early sea urchin embryo. The first asymmetric division occurs at the fourth embryonic cleavage and gives rise to a 16-cell embryo with four micromeres at the vegetal pole (Horstadius, 1937, 1950; Minokawa & Amemiya, 1999). These four micromeres undergo one more cell division to create four large and four small micromeres. The large micromeres become the skeletogenic mesenchyme, while small micromeres contribute to the two coelomic pouches of the pluteus larva (Ettensohn, 2009; Pehrson & Cohen, 1986; Tokuoka, Setoguchi, & Kominami, 2002).
The small micromeres express the genes Vasa, Nanos, and Seawi (Juliano et al., 2006; Voronina et al., 2008), which is characteristic of germ cell progenitors. Small micromere removal results in adult sea urchins lacking germ cells, strongly suggesting that small micromeres are germ line progenitors (Yajima & Wessel, 2011). Given the current convention in the field we refer to the small micromeres as PGCs, although it should be noted that studies to date have not definitively excluded the possibility that these cells could contribute indirectly to the formation of gametes in the adult.
After gastrulation the small micromeres migrate into the coelomic pouches (Campanale et al., 2014) in a characteristic 5 left-3 right pattern that seems to be controlled by a gene regulatory network that produces these asymmetrical distributions (Luo & Su, 2012; Martik & McClay, 2015).
The goal of this chapter is to describe several methods useful for investigating PGC biology in echinoderms, focusing in particular on methods recently used in S. purpuratus and Lytechinus pictus. The methods described here include those for live labeling of the cells with small molecule dyes, fluorescent proteins, or nucleoside analogs at various times during their development. These are followed by protocols to deploy these labeling methods during live imaging experiments, including visualizing and testing hypotheses regarding small micromere migration and incorporation into the coelomic pouches. Finally, a general summary of the current protein and RNA labeling reagents is presented.
2. METHODS FOR LABELING SMALL MICROMERES IN VIVO
Sea urchin small micromeres have a unique cellular, physiological, and gene expression profile that can be used to identify them in living or fixed embryos (Fig. 1). At their formation they undergo rapid changes in plasma membrane organization (Campanale & Hamdoun, 2012) that can be exploited to selectively introduce small molecule dyes, such as calcein, into these cells (Fig. 1). These labeled cells can be tracked in imaging experiments or isolated to purity using flow cytometry (Swartz et al., 2014). The small micromeres will also express, and often selectively retain, exogenous messages making them amenable to labeling (Fig. 1) with fluorescent fusion protein constructs selectively expressed in these cells (Oulhen et al., 2013). Lastly, differences in cell division cycles can be exploited such that incorporation of the nucleoside analogs EdU and BrdU, which can be used to selectively label (Fig. 1) small micromere nuclei for further identification in fixed embryos (Luo & Su, 2012; Pehrson & Cohen, 1986). The subsequent sections provide background and protocols for each of these methods as applied to S. purpuratus. Although these methods are focused on the subclass Euechinoidea that produce small micromere lineages, they may also be applicable to the cidaroid urchins that produce inconsistent numbers of small micromeres (Schroeder, 1981). Some, but not all of these protocols may be useful for Echinoderm classes that do not produce small micromere lineages, including Asteroids (Fresques, Zazueta-Novoa, Reich, & Wessel, 2014).
FIG. 1.
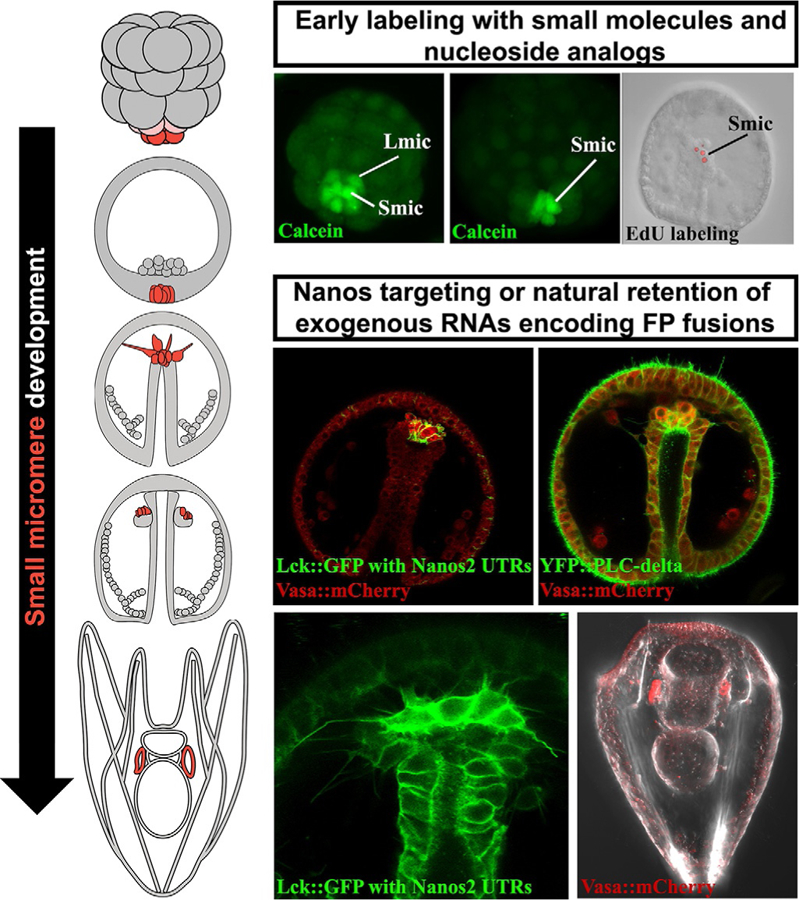
Illustrations of small micromere development from birth during cleavage stages through coelomic pouch residence in pluteus larva. Each stage of small micromere development can be labeled with small molecule dyes such as calcein-AM or nucleoside analogs like EdU. Later stages of small micromere development, including migration behaviors and left/right coelomic pouch distributions, can be observed after injecting RNAs encoding fluorescently tagged germline-specific genes or by engineering messages to contain the Nanos2 UTR retention sequences.
2.1. LABELING PGCs WITH THE SMALL MOLECULE CALCEIN
At their formation, small micromeres undergo a plasma membrane re-organization that includes retrieval and downregulation of efflux transporters. As such, these cells will accumulate fluorescent transporter substrates at a much higher rate than other cells in the embryo (Fig. 2, Campanale & Hamdoun, 2012). This phenomenon has been observed in many species within the class Echinoidea, including species of the Strongylocentrotus, Lytechinus, and Dendraster genera (Fig. 2). Because the transporters downregulated in the PGCs are promiscuous, a number of fluorophores including calcein-AM, Bo-dipyFL verapamil, and vinblastine, and CellTrace RedOrange will accumulate in the small micromeres (Figs. 1 and 2). Described here is the method for quantitative and replicable labeling using calcein-AM up to the mesenchyme blastula stage. Similar procedures will be applicable to labeling with different dyes and/or species, although it is usually necessary to optimize the concentration and incubation time to obtain sufficient contrast between the PGCs and other cells.
FIG. 2.
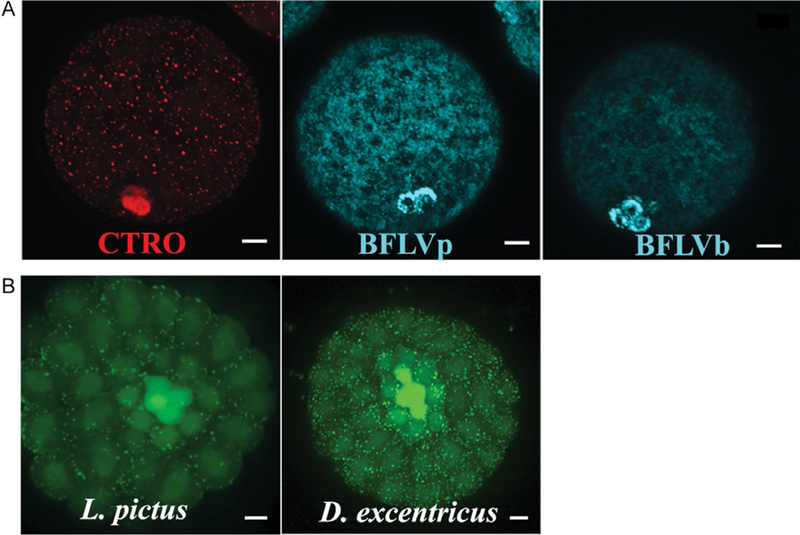
Small molecule dyes are retained in small micromeres of multiple euechinoid species. (A) Calcein-AM, Celltrace RedOrange (CTRO), and Bo-Dipy-FL-Verapamil (BFLVp) and Vinblastine (BFLVb) are retained in small micromeres of blastula stage embryos of (B) Lytechinus pictus and Dendraster excentricus.
Calcein-AM (CAM) has been widely used for measuring transporter activity in cells, and a number of advantages over other transporter substrates used for PGC labeling. Calcein-AM is a neutral, non-fluorescent, plasma membrane permeable substrate of ABCB and ABCC-type transporters present in echinoderm eggs and embryos (Hamdoun, Cherr, Roepke, & Epel, 2004; Roepke, Hamdoun, & Cherr, 2006). Transporters normally exclude CAM, but cells with reduced or inactive transporter activity accumulate it readily. Once CAM enters cells the AM group is cleaved by esterases, producing calcein, which is membrane impermeant and fluorescent (Essodaigui, Broxterman, & Garnier-Suillerot, 1998). In order to achieve consistent and quantitative calcein labeling of small micromeres, the concentration of embryos in exposure solutions should be kept constant at or below 500 embryos/mL.
2.1.1. Calcein labeling protocol
Spawn a female sea urchin according standard procedures in a 100 mL beaker (chapter “Procuring animals and culturing of eggs and embryos” by Adams et al.). Filter eggs through a Nitex mesh to remove any debris. For S. purpuratus eggs, filter with a 120 μm mesh. Ensure that >90% of the eggs are mature and lack germinal vesicles. Wash two times by gravity settling with 30 mL of 0.2 μm filtered seawater (FSW).
Fertilize washed eggs according standard procedures (chapter “Procuring animals and culturing of eggs and embryos” by Adams et al.).
Rehydrate one 50 μg aliquot of CAM (Biotium, catalog# 80011–2, 20 × 50 μg) in 50 μL of fresh molecular grade DMSO (Sigma-Aldrich, D2650–5 × 5 mL). It is important to use good quality DMSO as CAM can undergo hydrolysis to calcein when prepared in DMSO that has absorbed water. We prefer to use fresh CAM with each experiment, but the 1 mM stock solution can be stored as 5 μL subaliquots in microfuge tubes with parafilm sealed lids for 1–2 months at –20°C.
Dilute the 1 mM CAM stock solution to 1 μM in FSW. Vortex briefly.
-
At the desired stage of development begin treatment with CAM. Pipette 1125 μL of gently resuspended embryos into a 1.75 mL microfuge tube containing 375 μL of 1 μM of CAM. By using the stock culture of embryos at 500 embryos/mL, this results in a slight dilution of embryos and keeps the total number of embryos sufficiently low so as to not deplete CAM. Gently rock for 90 min.
Alternately, embryos can be exposed in a glass bottom imaging dish such as a Delta-T (Bioptechs, 04200417B) dish without rocking.
-
Mount embryos between a clay footed coverslip and microscope slide to image on fluorescent microscope according to standard procedures.
Note that CAM does not need to be washed out, it is not fluorescent until trapped intracellularly. Some naturally fluorescent dyes do require washout before imaging.
2.1.2 Modifications and tips for the calcein loading protocol
A 90 min incubation time in CAM alone is usually sufficient to produce fluorescent small micromeres in cleavage stage embryos. The addition of small amounts of ABC transporter inhibitors to the medium can dramatically increase the contrast between small micromeres and other cells in the embryo. This is particularly the case when loading pre-hatched blastula near 16 h post-fertilization. Using ABCB inhibitors cyclosporine A or PSC833 at 1 μM, or the ABCC inhibitor MK571 at 5 μM, alone or in combination, to enhance calcein loading of these cells. The inhibitors and residual CAM can then be washed out of the medium and the calcein will be retained within the PGC for several hours. However, it should be noted that MK571 has higher potential for off target effects and thus PSC833 or cyclosporine is preferred.
-
Intracellular concentrations of calcein in loaded cells can reach 70 μM in inhibitor treated blastulae and thus labeled small micromeres can be imaged in vivo for 6–12 h after labeling. After this time, labeling fades and becomes less useful for imaging. Confocal microscopy usually provides sufficient spatial resolution to resolve the small micromeres. By carefully controlling the time of CAM exposure, the number of embryos loaded with calcein, and the imaging parameters, replicable scans in a specified z-section thickness can be performed quantitatively and reproducibly. For instance, this method has been used to successfully compare the effects of drugs and other plasma membrane perturbations on the accumulation of CAM in the small micromeres versus the rest of the embryo (Campanale & Hamdoun, 2012).
For calculating the absolute intracellular concentration of calcein, a standard curve can be used. In this case, the free fluorescent calcein (Sigma, C-0875) is dissolved in DMSO at a concentration of 1 mM and diluted in FSW to solutions of 125 μM to 30 nM. A standard curve is then created by using eggs or embryos in these solutions to find the imaging plane and take a photo using the same microscope parameters as done with experimental animals. Then quantitative assessments of fluorescent measurements between the rest of the embryo are performed for the calcein containing FSW around the eggs/embryos are performed in ImageJ (chapter “A teaching laboratory on the activation of xenobiotic transporters at fertilization of sea urchins” by Shipp et al.).
Calcein labeling can be combined with other live labeling techniques, including the expression of exogenous mRNAs (Campanale & Hamdoun, 2012; Oulhen & Wessel, 2016) described below, the isolation of PGCs by fluorescence-activated cell sorting (FACS, Swartz et al., 2014), and in combination with additional dyes that label cellular membranes and organelles in live cells. These include red shifted dyes such as the plasma membrane marker FM4–64, Lysotraker, Mitotraker, or with red shifted versions of fluorescent dextran-labeled endosomes.
2.2. METHODS TO EXPRESS EXOGENOUS mRNAs IN SMALL MICROMERES USING NANOS-UTRs
The RNA-binding protein Nanos is essential for development and/or maintenance of the germ line in all animals (Juliano, Swartz, & Wessel, 2010; Juliano & Wessel, 2010). In S. purpuratus, both the Nanos 2 mRNA and protein are highly enriched in the small micromeres (Juliano, Yajima, & Wessel, 2010). Small micromeres have a unique gene expression program that differs from all cells in the embryo, including their siblings, the primary mesenchyme cells (e.g., Alx1 and many skeletogenic genes). RNAs and proteins such as Nanos 2 accumulate selectively in the small micromeres as the result of multiple post-transcriptional controls. That is, Nanos 2 is transcribed broadly in the embryo, but through post-transcriptional and post-translational activities, Nanos 2 mRNA and protein only accumulate in the small micromeres. The basic understanding of how these mechanisms work enabled engineering of molecular tools for efficient and selective expression only in the small micromeres in vivo (Campanale et al., 2014; Oulhen & Wessel, 2013, 2016). For instance, simply injecting RNA encoding S. purpuratus Vasa linked to GFP results in selective fluorescence of the small micromeres (Figs. 3–5).
FIG. 3.
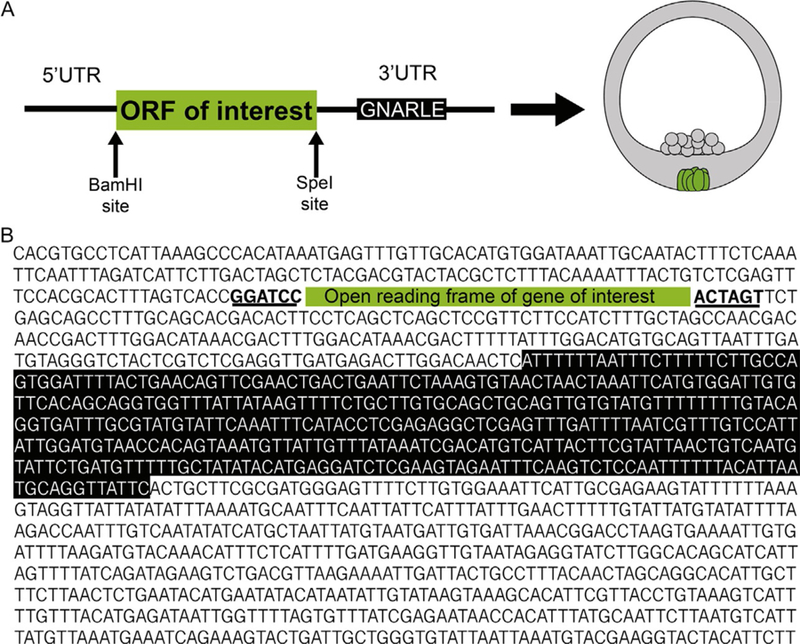
Map (A) and sequence (B) strategy to get mRNAs selectively retained in the small micromeres following injection.
FIG. 5.

Small micromeres expressing engineered or naturally retained RNAs can be used to study cell migration behavior by time lapse imaging. (A) Whole embryos injected with the pH-domain of phospholipase C-delta fused to YFP with tracked small micromere moving to the left coelomic pouch and (B) whole embryos injected with Vasa-mCherry and nanos UTR targeted membrane-YFP before dissociation into single cells showing blebs at 40 h post-fertilization (HPF) and filopodia at 48 HPF.
The PGCs have an inherent ability to retain maternal or xenobiotic mRNAs while somatic cells rapidly turn over the same transcripts. Thus, PGCs selectively maintain high cytoplasmic concentrations of mRNA, either from the egg or from microinjection, well into larval development. Retention of exogenous RNAs is dependent on an element in the Sp nanos2 3′UTR, called GNARLE (Global Nanos Associated RNA Lability Element) (Oulhen et al., 2013). When the GNARLE sequence is present in the 3′UTR of any transcript, the corresponding protein is translated specifically in the small micromeres. The GNARLE sequence and function are conserved between the sea urchins S. purpuratus and Hemicentrotus pulcherimus species that diverged <20 million years ago. The 5′UTR of Sp nanos2 by itself does not provide spatial information for stability to translation, but acts to increase protein synthesis dramatically, compared to the Xenopus β-globin 5′UTR for example. Thus, the 5′UTR of Sp nanos2 linked to a coding region of interest, followed by the GNARLE sequence in the 3′UTR provides high levels of translation selectively in the PGCs (Figs. 3–5).
2.2.1. Cloning strategy
A pGEMTeasy vector (Promega Corporation) containing the Sp nanos2 UTRs flanking a cytoplasmic (Oulhen et al., 2013) or a membrane-targeted GFP (Campanale et al., 2014) have been generated and can be ordered through the plasmid repository Addgene. The original primers used to amplify the Nanos2 UTRs for insertion into the pGEMT vector can be found in Oulhen et al. (2013). This vector was constructed such that the 5′UTR was inserted between the ApaI and BamHI restriction enzymes sites. The GFP is between the BamHI and SpeI sites, and the 3′UTR is between the SpeI and NdeI sites (Fig. 3).
Standard cloning methods can be used to modify this vector to easily insert alternate genes of interest in place of, or in addition to, the GFP. This can be accomplished by digesting these plasmids with BamHI and SpeI to release the GFP fragment from the backbone containing the UTRs and inserting a new gene (and reporter) of interest by several methods of choice including standard sticky end cloning, Infusion cloning, or Gibson cloning.
2.2.2. Producing injection ready mRNA containing the Nanos2UTRs
Plasmids are linearized using Nde1 for 4 h at 37°C. Capped sense RNAs are synthesized using the mMessage mMachine® T7 (ThermoFisher Scientific, Waltham, MA) yielding RNA concentrations of approximately 2 μg/μL (mRNA over-expression chapter “Microinjection of oocytes and embryos with synthetic mRNA encoding molecular probes” by von Dassow et al.). Injection solutions contain 20% glycerol, and 0.66 μg/μL of GFP RNA diluted in nuclease free water. Approximately 2 pL of the injection solution is injected into each fertilized egg. The GFP reporter transcript containing the Sp nanos2 3′UTR is expressed in all the cells at early stages of development but is enriched in the small micromeres at 12 h after fertilization (as observed by in situ hybridization on fixed embryos). The resulting GFP protein becomes specifically enriched in the small micromeres starting at 18 h after fertilization (blastula stage) and can be imaged in live embryos using a confocal microscope. The retention of the GFP reporter transcript containing the Sp nanos2 3′UTR GNARLE only is more difficult to observe in the small micromeres, but the resulting protein specifically accumulates at similar levels as the protein translated from the 3′UTR full-length reporter.
Exogenous mRNAs injected into fertilized sea urchin eggs become slightly enriched in the small micromeres at blastula stage, before becoming selectively retained in the small micromeres during gastrulation, independently of the nanos2 UTRs (Oulhen & Wessel, 2013). The resulting proteins slowly start to accumulate in the small micromeres during gastrulation. Using the nanos2 UTRs is a more efficient system to obtain a stronger and a faster GFP fluorescence selectively in the small micromeres. In contrast, a negative control construct with no GNARLE in its 3′UTR can be used as this does not lead to any reporter enrichment in the small micromeres.
2.3. ENGINEERING SELECTIVE RETENTION OF PROTEIN IN THE PGCs
The Sp nanos2 ORF itself is sufficient to restrict the expression of nanos2 protein in the small micromeres (Oulhen & Wessel, 2016). A GFP reporter will accumulate selectively in the PGCs when designed as follows: Sp nanos2 5′UTR fused to the GFP ORF followed by Sp nanos2 ORF and Sp nanos2 3′UTR ΔGNARLE. The injected transcript remains in the entire embryo because it is missing GNARLE, but by confocal microscopy, the GFP-Nanos2 fused protein is detected only in the small micromeres at blastula stage. This ORF regulation is independent of the sumoylation and ubiquitination pathways. Instead, the Sp nanos2 ORF contains a sequence of 68 amino acids in its N-terminal domain required for enrichment in the small micromeres (Fig. 4).
FIG. 4.
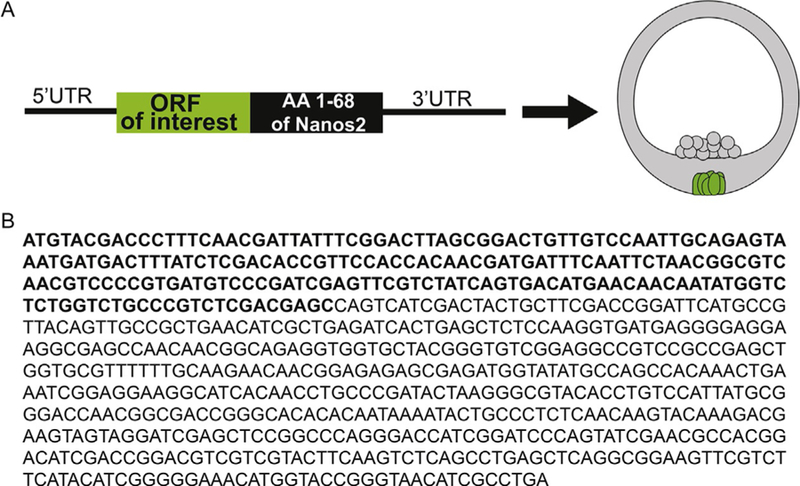
Map (A) and sequence (B) strategy to get proteins selectively translated in the small micromeres following injection of the mRNA.
This 68 amino acid element can be fused to the GFP ORF to label selectively the small micromeres, independently of the 3′UTR (Fig. 4). These constructs are made using Sp nanos2 5′UTR fused to the GFP nanos2 retention element ORF and Sp nanos2 3′UTR ΔGNARLE. The ΔGNARLE 3′UTR was used in this case to study nanos2 ORF regulation independently of its 3′UTR, but if the purpose of the construct is to enrich the GFP fluorescence as much as possible in the small micromeres, using the Sp nanos2 3′UTR full-length or GNARLE could maximize the GFP signal in the small micromeres.
To create a GFP fusion protein, Sp nanos2 ORF was amplified using these primers:
(The nanos2 ORF sequences are in bold and the restriction sites are in italic)
Sp nanos2 N-term F: CGACTAGTATGTACGACCCTTTCAACGATT
Sp nanos2 N-term R: CGACTAGTTCAGCTCGTGAGACGGGCAGA CCAGA
and cloned in frame after the GFP ORF. A stop codon (underlined in the reverse primer) was added to mark the end of the fusion protein. In contrast, the stop at the end of the initial GFP (TAA) was modified into a non-stop codon (TTA) using the QuickChange II Site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA):
GFP stop Fmut: GGACGAGCTGTACAAGTATCTCGAGTTACCAATCAC TAGT.
GFP stop Rmut: ACTAGTGATTGGTAACTCGAGATACTTGTACAGCTC GTCC.
The plasmid was then linearized, capped sense RNAs synthesized and injected as described above. The GFP protein is expressed at low level throughout the embryo until blastula stage (18–20 degrees after fertilization) when the protein starts to be stabilized and highly enriched in only the small micromeres.
2.4. BrdU AND EdU LABELING OF SMALL MICROMERES
During embryogenesis, the four small micromeres divide only once after they are born at the fifth cleavage. As embryogenesis proceeds and most of the cells undergo rapid division, the eight small micromeres remain undivided until the larval stage. This slow cell-cycling property enables small micromeres to be selectively labeled by 5-bromo-2-deoxyuridine (BrdU), a synthetic analog of thymidine (Tanaka & Dan, 1990) or 5-ethynyl-2′-deoxyuridine (EdU, Fig. 1). When fertilized sea urchin eggs are incubated with these compounds during the first embryonic cell cycle, they are incorporated into the newly synthesized DNA of all daughter cells. With development, the signal is diluted in most cells by subsequent cell divisions, but the small micromeres retain the signal for subsequent detection, since they divide infrequently. This method unambiguously labels small micromeres, and thus can be combined with in situ hybridization or immunostaining to definitely identify the small micromeres (Hara & Katow, 2005; Luo & Su, 2012; Voronina et al., 2008).
Using these methods, investigators can choose their favorite Alexa fluor to detect BrdU using antibody labeling and EdU signal using click-it chemistry following fixation without the need for strand-melting the genomic DNA. An example of Click-iT EdU Alexa Fluor 594 image (ThermoFisher Scientific, product # C10339) is shown in a gastrula staged embryo in Fig. 1.
2.4.1. BrdU labeling method
Fertilization is carried out in FSW containing p-aminobenzoic acid (PABA, Sigma A6928; PABA/FSW, 75 mg PABA in 250 mL FSW). After the fertilization envelop is elevated, wash out excessive sperm with fresh PABA/FSW. Incubate the zygotes for 10 min in PABA/FSW to soften the fertilization envelop. For S. purpuratus, filter the zygotes through a 60 μm mesh to remove the fertilization envelop.
Incubate the zygotes in 50 μM 5-bromo-2-deoxyuridine (BrdU; Sigma B9285) in FSW for 1 h.
Wash twice with 500 μM thymidine (Sigma T1895) in FSW and then rinse with FSW.
Culture to a desired stage and fix the embryos with 4% PFA (ref method for fixation in Oulhen, Swartz, Laird, Mascaro, & Wessel, 2017).
After rehydration, embryos are permeabilized with 0.1% Triton X-100 in PBS (PBSTX) for 30 min.
Wash the embryos with 0.1% Tween-20 in PBS (PBST).
Treat the embryos with 1 N HCl in PBST for 30 min, then wash the embryos with PBST.
Incubate the embryos in filtered 3% BSA in PBST (PBST block) for 2 h.
To block endogenous biotin, incubate the embryos in 0.001% avidin (Sigma A9275) in PBST and 0.001% biotin (Sigma B4501) in PBST sequentially for 15 min each. Rinse with PBST between and after the two incubations.
Cells with BrdU labeling are immunolocalized with a biotinylated anti-BrdU antibody (Abcam ab2284) in 1:1000 dilution in PBST block at 4°C overnight. Treat with HiLyte Fluor Streptavidin (AnaSpec 60665 for 488 nm or 60667 for 647 nm) (1:400) for 2 h at room temperature.
2.4.2. EdU labeling method
Labeling EdU using Click-iT technology (https://www.thermofisher.com/us/en/home/life-science/cell-analysis/labeling-chemistry/click-chemistry-labeling-and-detection/click-chemistry-tools-for-biological-assays.html) is an alternative to using BrdU for nascent DNA synthesis and may be easier for some applications. EdU is a nucleoside analog of thymidine and like BrdU is incorporated into DNA during active DNA synthesis. No genomic DNA melting with acid is needed with this approach nor is antibodies needed.
Prepare a 10 mM EdU stock according to the instructions given in the Click-iT kit.
Fertilize the eggs in a 50 mL Falcon tube and remove excess sperm with two washes of sea water.
Add EdU (1:1000 dilution, final concentration of 10 μM) to the 50 mL culture, following the second wash.
Incubate the zygotes with EdU with gentle rocking of frequent inversion.
Following the first cell division, wash the culture six times with filtered sea water to remove the non-incorporated EdU.
Dilute the culture to the desired embryo concentration in filtered seawater and incubate the embryos until the desired stage (shown is gastrula stage (Fig. 1), but the PGCs are readily detectable from mesenchyme blastula until early larval stages).
Concentrate and transfer the embryos in either an Eppendorf tube or a 96-well plate, depending on the desired number of embryos.
-
Fix the embryos by adding PFA to a final concentration of 4%, diluted in filtered sea water.
Note: This protocol works well when the embryos have been fixed with paraformaldehyde (PFA). However, if one needs to fix the embryos with methanol instead of PFA, depending on application with an antibody co-label, it still works, but the EdU staining is not as robust as with PFA fixation.
-
Incubate at room temperature for 15 min. The embryos will settle down without centrifugation during the fixation.
Note: For the following steps, we recommend avoiding centrifugation as it may damage the embryos. It is better to wait for the embryos to settle by gravity between each step.
Discard the supernatant and wash the embryos twice with PBS BSA 3%.
Discard the supernatant and incubate the embryos with PBS/0.5% Triton X-100 at room temperature for 10 min.
Discard the supernatant and wash the embryos three times with PBS/0.05% Tween.
-
Prepare the Click-iT reaction cocktail according to the protocol in the kit.
Note: We suggest using 200 μL of reaction cocktail per sample. This Click-iT reaction cocktail needs to be used within 15 min of preparation. For example, if the investigator has five samples, 1 mL of reaction cocktail is needed as follows:- 860 μL of 1 Click-iT® reaction buffer.
- 40 μL of CuSO4 (component E).
- 2.5 μL Alexa Fluor azide.
- 100 μL of Reaction buffer additive (freshly diluted to one time from the stock).
Incubate the samples in the Click-iT reaction cocktail for 30 min at room temperature, protected from light.
Remove the reaction cocktail and wash the samples three times with PBS/0.05% Tween.
The nuclei can be counterstained using the Hoechst 33342 provided in the kit resulting in a warm purple hue on merged images.
The investigators can either image the samples directly, keep them at 4°C until imaging, or proceed with their specific immunofluorescence protocol. Note that in the early stages of development, the EdU remains abundant in the somatic cells. This somatic signal halves progressively after each cell division.
Modifications of the protocol can be used for multiple other applications. For example, were one to test what cells were cycling during a particular stage or following a particular treatment in comparison to control embryos, one would pulse the EdU reagent by adding to the embryos for 30–60 min at the stage of interest, followed by three washes, immediate fixation, and labeling as given above.
3. METHODS FOR LIVE IMAGING SMALL MICROMERE TRANSLOCATION AND MIGRATION
Using expression of fluorescent markers described above, small micromeres have been found to translocate with the archenteron tip before actively migrating to the coelomic pouches of the early pluteus larva (Campanale et al., 2014; Yajima & Wessel, 2012).
3.1. LIVE IMAGING SMALL MICROMERES IN WHOLE EMBRYOS
Live imaging of small micromeres during gastrulation can be observed and quantitatively measured after injection of fluorescent membrane markers using the Sp-Nanos2 UTRs, fluorescently tagged Sp-Vasa, or mCitrine fused to the pleckstrin homology domain of phospholipase C-delta. Described here are methods for live cell imaging of small micromeres in vivo and in vitro as single cells using confocal microscopy.
3.1.1. Live imaging gastrulae expressing fluorescent proteins
Microinject 200–400 embryos following the protocol above without the addition of glycerol, with 150 ng/μL NTM-mCit and 200 ng/μL Vasa-mCherry.
-
Culture embryos at 15°C in injection dishes and then transfer to culturing dishes (Corning-Falcon brand, 351008) with fresh FSW.
Note: Application of 100 μg/mL of ampicillin to dishes after injection is not necessary but can increase the health and synchrony of the cultures by limiting bacterial growth. Once hatched and transferred to new dishes, ampicillin is no longer necessary.
-
Between 43 and 60 h post-fertilization, transfer 5–10 gastrulae in 10 μL of FSW to a 0.25% protamine sulfate (Calbiochem, 539122) coated Delta-T dish using P20 pipette with a 20 μL tip.
Note: To coat the dishes in protamine sulfate, cover the bottom of the dish in 0.25% solution made in deionized water, allow to sit 1 min, then rinse three times with deionized water. Allow to air dry.
Lightly drag each corner of a 0.25% protamine sulfate coated 15×15 mm2 coverslip in non-toxic silicone clay (available at any craft store) and gently lay over the gastrulae. This small coverslip is made by cutting roughly 30% off of two sides of a 22 × 22 mm2 coverslip with a diamond knife.
Using forceps press down on opposite corners of the coverslip until all of the gastrulae are no longer swimming and snugly stuck between both protamine coated surfaces (Fig. 6).
Quickly pipette 1 mL of fresh FSW directly over the center of the coverslip so as the FSW floods under the coverslip from all sides at once. This will minimize the movement of gastrulae while providing fresh FSW required to keep the embryo healthy for imaging. Wait 1–2 min then check that the embryos are not freely swimming and/or not smashed before applying a chilled stainless-steel Delta-T dish lid (Bioptechs, 04200312) to prevent evaporation during imaging.
The live imaging Delta-T dish with lid is then fitted into a custom built, water chilled, imaging chamber that has been adapted to fit into the universal stage adapters on most confocal microscopes (Figs. 6 and 7). Our stages were built at the Marine Science Development Center, at UC San Diego https://scripps.ucsd.edu/msdc—however similar stages can also be obtained commercially (e.g., Ovo labs). Stages can be cooled adequately using a small Peltier-based chilling system (e.g., Nanotherm Eagle Medical) to the imaging chamber. It is important to use flexible/compliant tubing for delivery of cooled water to the stage, in order to prevent vibrations. The compliant tubing is surrounded by insulation foam to prevent heating of the chilled water before reaching the specimen. Chilled water then circulates through this chamber and returns back to the Nanotherm.
FIG. 6.
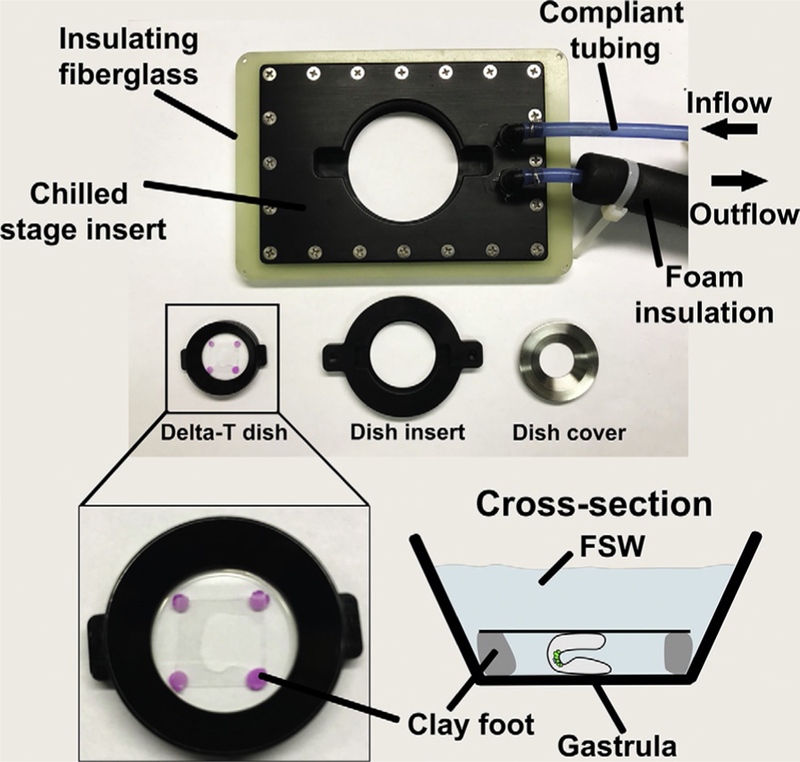
Photo of temperature-controlled stage and imaging dish adapters for long-term live imaging of sea urchin embryos. The stage adapter is designed to fit many microscope stage inserts and is surrounded by insulating fiberglass. The water-cooled insert is fed through insulated compliant tubing that minimizes vibrations during imaging. Insulation is pulled back on inflow to reveal the compliant tubing. Custom made dish inserts serve as adapters between the Delta-T imaging dishes and the stage. The zoomed and illustrated views of the Delta-T dish depicts how gastrulae are mounted between two protamine coverslips and locked into place using clay footed coverslips before being covered by FSW.
FIG. 7.

Photo of the temperature-controlled stage on a Zeiss LSM 700 confocal microscope and labeled to indicate the insulated coolant flow tubes that are fed with recirculating chilled water by a nanotherm unit.
3.2. ADAPTING THE LIVE IMAGING METHOD FOR DISSOCIATED BLASTOMERES
Small micromeres produce dynamic blebs and filipodia during their migration in whole, intact embryos, or in dissociated cells in vitro (Campanale et al., 2014). Culturing small micromeres in vitro offers the advantage of measuring changes in the dynamic properties of migratory cell morphologies often obscured by live imaging deep within an intact embryo. This method relies on embryos injected with and expressing exogenous RNAs in the small micromeres as described in the previous section and cultured to gastrulae.
3.2.1. Protocol for imaging dissociated small micromeres expressing fluorescent RNAs
Roughly 200–500 embryos injected with a fluorescent marker selectively expressed in small micromeres and cultured to the gastrula stage is performed as described above.
-
Embryos are pelleted in 15 mL Falcon tubes using a hand centrifuge at one hand rotation per second (roughly 250 rpms).
Note: A BSA coating is necessary for all plastic and glassware to prevent the cells/gastrulae from sticking to their surface. Coating involves simply rinsing the tube or pipette tip once with a 1% BSA in PBS. BSA solution is discarded before addition of gastrulae or cells.
Gently resuspend gastrulae in 4 mL of hyaline extraction medium (McClay, 2004) and incubate at 15°C for 1 min. Pellet as before and remove HEM.
Gently resuspend gastrulae in calcium-magnesium-free sea water (CMFSW, McClay, 2004) and incubate at 15°C for 1 min. Pellet as before and this time remove all but roughly 100 μL of CMFSW.
Using a P200 fitted with a 200 μL tip, dissociate gastrulae by trituration (10–12 quick passages through the pipette tip near the bottom of the tube).
-
Pipette this solution onto a BSA coated coverslip or Delta-T dish with fresh FSW. Allow the cells to settle for 15 min before starting to image.
Note: Small micromeres are roughly 6–8 μm in diameter and are best imaged by using high NA (typically 1.1) water-immersion 40 × objectives.
3.3. CONSIDERATIONS FOR LIVE IMAGING
Several important practical considerations are required to effectively image live PGCs in whole or dissociated sea urchin embryos/larvae. In general, the culture temperature and photo-toxicity must be controlled. The most commonly used temperatures to culture a variety of species is documented (chapter “Procuring animals and culturing of eggs and embryos” by Adams et al.; Foltz, Adams, & Runft, 2004). Figs. 7 and 8 show a relatively simple system that can be applied across the temperature spectrum necessary to culture the vast majority of species. The actual temperature of the FSW in imaging chambers should be tested empirically and will depend on the ambient temperature of the room housing the microscope. Using this system with 5 ft. tubing in a 22°C room, we have found that if system is set at 12°C, the inside of the Delta-T dish will be maintained between 14 and 15°C.
FIG. 8.
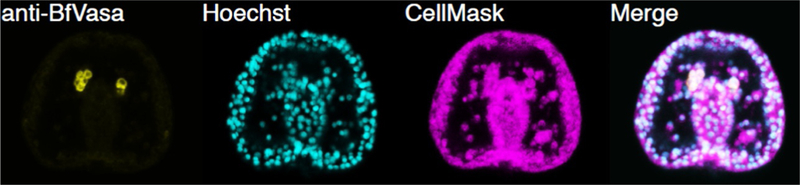
Immunostaining of the sea urchin gastrula with the antiserum made against the amphioxus Vasa (BfVasa). The embryos were fixed in ice-cold methanol for 30 min, and then stored in methanol at —20°C for no longer than 6 months. Embryos were washed in PBST, blocked in 3% BSA for 1.5 h, and incubated in BfVasa antiserum (1:10,000) overnight at 4°C. Embryos were then washed in PBST, incubated in a goat anti-rabbit Alexa Fluor 488-conjugated antibody (Invitrogen, 1:400), washed in PBST, and counterstained with Hoechst (Invitrogen, 1:1000) and CellMask (Invitrogen, 1:2000). This antiserum does not work in embryos fixed in PFA.
In addition to temperature, the imaging modality available for live imaging varies between labs and institutions, from simple stereo/dissection microscopes to sophisticated light sheet fluorescent microscopy (LSFM). Some imaging methods use high intensity lasers, while others use low intensity broad spectrum illumination. This will dictate considerations for the second aspect of live imaging sea urchin embryos/ larvae, light level/exposure. Thus, the imaging modality itself must be empirically tested to limit photo-toxicity and photo-damage, resulting in maximal limits for temporal and spatial resolution.
4. METHODS TO LABEL SMALL MICROMERES IN FIXED EMBRYOS
4.1. CONSIDERATIONS FOR IMMUNOLOCALIZATION AND IN SITU HYBRIDIZATION PROTOCOLS
Despite exhibiting broad transcriptional repression (Swartz et al., 2014), mRNAs of several genes are specifically detected in small micromeres. These transcripts are either products of genes transcribed specifically in small micromeres or maternally deposited mRNAs that are retained in the small micromeres while degraded in other cells. Here, we provide a list of genes that are specifically expressed or the transcripts of which are preferentially enriched in the small micromeres at indicated stages (Tables 1 and 2).
Table 1.
List of Antibodies That Target Small Micromere Specific Proteins
| Antibody Target | Antigen | Species Target | Host Animal | Antibody Type | Working Concentration | Reference |
|---|---|---|---|---|---|---|
| H3K9me3 | AA 1–100 (human) | Strongylocentrotus purpuratus | Rabbit | IgG | 1:500 | Swartz et al. (2014) |
| Vasa | Drosophila melanogaster Vasa | S. purpuratus | Rabbit | IgG | 1:300 | Lasko and Ashburner (1990); used by Luo and Su (2012) |
| Vasa | Branchistoma floridae Vasa | S. purpuratus | Rabbit | Serum fraction | 1:10,000 | Wu et al. (2011) |
| Vasa | D. melanogaster Vasa | S. purpuratus | Mouse | IgM, ascities fluid | 01:30 | Developmental Studies Hybridoma Bank (unpublished) |
Table 2.
Gene Products Detected in Small Micromeres During Embryogenesis
| Gene Name | 60-Cell | MB | LG | PL | Primers Used to Make In Situ Probe (5’ to 3’) | Length (bp) | Reference |
|---|---|---|---|---|---|---|---|
| nanosla,b | F1: T AG AT CATT C AAGACAAGCT CT | 799 | Juliano and Wessel (2010) | ||||
| R3: AGAATGGAGTACTTGCGTAC | |||||||
| F2: GGAAGTACATCGCATTTTACAA | |||||||
| R4: ATACACCCAGCAATCAGTAC | |||||||
| nanos2a | F: AAGGTGATGAGGGGAGGAAG | 748 | Juliano et al. (2006) | ||||
| R: CGCAAATCACCTGTACAAAAA | |||||||
| foxy | F: GT GAT CGCTTT CCTT GCTT C | 1176 | Luo and Su (2012) | ||||
| R: AGAAACCTTTGGTGGTGTCG | |||||||
| nanos3b | F1: GTACACCCGTGTCCGTGAG | 1277 | Juliano and Wessel (2010) | ||||
| R1: TGTCAAAAACTTTGTGCCAGAA | |||||||
| F2: CCAATACAACATTAATCTTCAAG | |||||||
| R2: TACTTCCTACATAGGACGAC | |||||||
| vasa | F: GAT GGT GAT AAT GGGTT CGG | 1218 | Luo and Su (2012) | ||||
| R: AT CAGT CT CT GGAT GTCGGG | |||||||
| seawi | F: GT GT CT CGCCCAAAG AAGAG | 1269 | Luo and Su (2012) | ||||
| R: GCGGACACTCTGACTGATGA | |||||||
| foxc | Left | F: AATGGCGATGCAACCTTATC | 1470 | Luo and Su (2012) | |||
| R: TAGCTGTAATGCGCTGATGG | |||||||
| nodal | Right | Right | Clone upon requestc | 2092 | Luo and Su (2012) | ||
| pitx2 | Right | Right | F: TAGAAGATTTGGTCGCCAGC | 827 | Luo and Su (2012) | ||
| R: GTT GG AGG AAT GGT GCTGTT | |||||||
| not | Right | Right | F: CTCCGCCATCATCCTACAGT | 1180 | Luo and Su (2012) | ||
| R: CGCTCTTCGACCTTCTTCAC |
Orange boxes represent stages at which the transcripts are detected in the small micromeres (MB, mesenchyme blastula; LG, late gastrula; PL, pluteus stage). Side- specific expression in the left or the right groups of small micromeres are also indicated. The primer sequences are based on S. purpuratus genes.
Duc to high sequence similarity, in situ probes of nanosl and nanos2 may cross-hybridize.
Nested PCR is required to amplify nanosl and nanos3.
The nodal clone was originally made by Jongmin Nam. It contains 5’UTR, CDS, and 3’UTR of the S. purpuratus nodal.
The translational regulator nanos1, nanos2, and the forkhead transcription factor, FoxY gene, are among the earliest genes that are known to be expressed exclusively in the small micromeres. Nanos1 and nanos2 transcripts are first detected in the small micromeres at the 60-cell stage, and the exclusive expression pattern continues to the pluteus stage (Juliano, Swartz, & Wessel, 2010). The FoxY gene is activated at a similar stage in the small micromeres by the Delta ligand present in the surrounding large micromere daughter cells (Materna & Davidson, 2012). During gastrulation, FoxY expression expands into mesodermal cells, and the expression in the small micromeres decreases at the late gastrula stage (Materna, Swartz, & Smith, 2013). Nanos3, the third nanos homolog in the S. purpuratus genome, is activated later in the small micromeres at the gastrula stage (Juliano, Yajima, & Wessel, 2010). Vasa and Seawi transcripts are distributed ubiquitously in the embryo initially and then are enriched progressively to the vegetal plate of the mesenchyme blastula stage, to the small micromeres and the adjacent dorsal mesodermal cells at the late gastrula stage, and finally restricted to the small micromeres at the pluteus stage (Fig. 8; Fujii et al., 2009; Juliano et al., 2006; Luo & Su, 2012). Other genes show similar broadly distribution initially and later restricted in the small micromeres include baf250, ctdspl2/SCP2, z62, and mibL (Swartz et al., 2014).
In the larval stage when the small micromeres split into the left and right groups, several genes show side-specific expression in the small micromeres. Expression of the FoxC gene is dynamic in that at the late gastrula stage it is not expressed in the small micromeres but in the adjacent ventral mesodermal cells, and in the larval stage the transcripts can be detected in the small micromeres located in the left coelomic pouch (Luo & Su, 2012). Nodal, pitx2, and not genes, on the other hand, are expressed in the small micromeres of the right coelomic pouch (Luo & Su, 2012).
4.2. TABLE OF REAGENTS
REFERENCES
- Campanale JP, Gokirmak T, Espinoza JA, Oulhen N, Wessel GM, & Hamdoun A (2014). Migration of sea urchin primordial germ cells. Developmental Dynamics, 243(7), 917–927. 10.1002/dvdy.24133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanale JP, & Hamdoun A (2012). Programmed reduction of ABC transporter activity in sea urchin germline progenitors. Development, 139(4), 783–792. 10.1242/dev.076752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essodaigui M, Broxterman HJ, & Garnier-Suillerot A (1998). Kinetic analysis of calcein and calcein-acetoxymethylester efflux mediated by the multidrug resistance protein and P-glycoprotein. Biochemistry, 37(8), 2243–2250. 10.1021/bi9718043. [DOI] [PubMed] [Google Scholar]
- Ettensohn CA (2009). Lessons from a gene regulatory network: Echinoderm skeletogenesis provides insights into evolution, plasticity and morphogenesis. Development, 136(1), 11–21. 10.1242/dev.023564. [DOI] [PubMed] [Google Scholar]
- Extavour CG (2003). Mechanisms of germ cell specification across the metazoans: Epigenesis and preformation. Development, 130(24), 5869–5884. 10.1242/dev.00804. [DOI] [PubMed] [Google Scholar]
- Foltz KR, Adams NL, & Runft LL (2004). Echinoderm eggs and embryos: Procurement and culture. Methods in Cell Biology, 74, 39–74. [DOI] [PubMed] [Google Scholar]
- Fresques T, Zazueta-Novoa V, Reich A, & Wessel GM (2014). Selective accumulation of germ-line associated gene products in early development of the sea star and distinct differences from germ-line development in the sea urchin. Developmental Dynamics, 243, 568–587. 10.1002/dvdy.24038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Sakamoto N, Ochiai H, Fujita K, Okamitsu Y, Sumiyoshi N, et al. (2009). Role of the nanos homolog during sea urchin development. Developmental Dynamics: An Official Publication of the American Association of the Anatomists, 238(10), 2511–2521. 10.1002/dvdy.22074. [DOI] [PubMed] [Google Scholar]
- Hamdoun A, Cherr G, Roepke T, & Epel D (2004). Activation of multidrug efflux transporter activity at fertilization in sea urchin embryos (Strongylocentrotus purpuratus). Developmental Biology, 276(2), 452–462. 10.1016/j.ydbio.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Hara Y, & Katow H (2005). Exclusive expression of hedgehog in small micromere descendants during early embryogenesis in the sea urchin, Hemicentrotus pulcherrimus. Gene Expression Patterns, 5(4), 503–510. 10.1016/j.modgep.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Horstadius S (1937). Investigations as to the localization of the micromere-, the skeleton-, and the entoderm-forming material in the unfertilized egg of arbacia punctulata. Biological Bulletin, 73(2), 295–316. [Google Scholar]
- Horstadius S (1950). The mechanics of sea urchin development. L’ Anne´e Biologique, 26(8), 381–398. [PubMed] [Google Scholar]
- Juliano CE, Swartz S, & Wessel G (2010). A conserved germline multipotency program. Development, 137(24), 4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano CE, Voronina E, Stack C, Aldrich M, Cameron A, & Wessel G (2006). Germ line determinants are not localized early in sea urchin development, but do accumulate in the small micromere lineage. Developmental Biology, 300(1), 406–415. [DOI] [PubMed] [Google Scholar]
- Juliano C, & Wessel G (2010). Developmental biology. Versatile germline genes. Science, 329(5992), 640–641. 10.1126/science.1194037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano CE, Yajima M, & Wessel GM (2010). Nanos functions to maintain the fate of the small micromere lineage in the sea urchin embryo. Developmental Biology, 337(2), 220–232. 10.1016/j.ydbio.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann R (2012). Germline stem cells: Origin and destiny. Cell Stem Cell, 10(6), 729–739. 10.1016/j.stem.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y-J, & Su Y-H (2012). Opposing nodal and BMP signals regulate left-right asymmetry in the sea urchin larva. PLoS Biology, 10(10), e1001402 10.1371/journal.pbio.1001402.s006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martik ML, & McClay DR (2015). Deployment of a retinal determination gene network drives directed cell migration in the sea urchin embryo. eLife, 4, e07343 10.7554/eLife.08827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materna SC, & Davidson EH (2012). A comprehensive analysis of delta signaling in pre-gastrular sea urchin embryos. Developmental Biology, 364(1), 77–87. 10.1016/j.ydbio.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materna SC, Swartz SZ, & Smith J (2013). Notch and nodal control forkhead factor expression in the specification of multipotent progenitors in sea urchin. Development, 140(8), 1796–1806. 10.1242/dev.091157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClay DR (2004). Methods for embryo dissociation and analysis of cell adhesion. Methods in Cell Biology, 74, 311–329. 10.1016/S0091-679X(04)74014-5. [DOI] [PubMed] [Google Scholar]
- Minokawa T, & Amemiya S (1999). Timing of the potential of micromere-descendants in echinoid embryos to induce endoderm differentiation of mesomere-descendants. Development, Growth & Differentiation, 41(5), 535–547. [DOI] [PubMed] [Google Scholar]
- Oulhen N, Swartz SZ, Laird J, Mascaro A, & Wessel GM (2017). Transient translational quiescence in primordial germ cells. Development, 144(7), 1201–1210. 10.1242/dev.144170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, & Wessel GM (2013). Retention of exogenous mRNAs selectively in the germ cells of the sea urchin requires only a 500-cap and a 3–00UTR. Molecular Reproduction and Development, 80(7), 561–569. 10.1002/mrd.22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, & Wessel GM (2016). Differential nanos 2 protein stability results in selective germ cell accumulation in the sea urchin. Developmental Biology, 418(1), 146–156. https://doi.org/10.1016/j.ydbio . https://doi.org/10.1016/j.ydbio2016.07.007. 2016.07.007 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Yoshida T, Yajima M, Song JL, Sakuma T, Sakamoto N, et al. (2013). The 30UTR of nanos2 directs enrichment in the germ cell lineage of the sea urchin. Developmental Biology, 377(1), 275–283. 10.1016/j.ydbio.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson JR, & Cohen LH (1986). The fate of the small micromeres in sea urchin development. Developmental Biology, 113(2), 522–526. [DOI] [PubMed] [Google Scholar]
- Raz E (2004). Guidance of primordial germ cell migration. Current Opinion in Cell Biology, 16(2), 169–173. 10.1016/j.ceb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Roepke TA, Hamdoun AM, & Cherr GN (2006). Increase in multidrug transport activity is associated with oocyte maturation in sea stars. Development, Growth & Differentiation, 48(9), 559–573. 10.1111/j.1440-169x.2006.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AC, & Lehmann R (2004). Germ cell specification and migration in Drosophila and beyond. Current Biology, 14(14), R578–R589. 10.1016/j.cub.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Schroeder T (1981). Development of a “primitive” sea urchin (Eucidaris tribuloides): Irregularities in the hyaline layer, micromeres, and primary mesenchyme. The Biological Bulletin, 161(1), 141–151. [Google Scholar]
- Swartz SZ, Reich AM, Oulhen N, Raz T, Milos PM, Campanale JP, et al. (2014). Deadenylase depletion protects inherited mRNAs in primordial germ cells. Development, 141(16), 3134–3142. 10.1242/dev.110395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, & Dan K (1990). Study of the lineage and cell cycle of small micromeres in embryos of the sea urchin, Hemicentrotus pulcherrimus. Development, Growth & Differentiation, 32(2), 145–156. [DOI] [PubMed] [Google Scholar]
- Tarbashevich K, & Raz E (2010). The nuts and bolts of germ-cell migration. Current Opinion in Cell Biology, 22(6), 715–721. 10.1016/j.ceb.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Tokuoka M, Setoguchi C, & Kominami T (2002). Specification and differentiation processes of secondary mesenchyme-derived cells in embryos of the sea urchin Hemicentrotus pulcherrimus. Development, Growth & Differentiation, 44(3), 239–250. [DOI] [PubMed] [Google Scholar]
- Voronina E, Lopez M, Juliano C, Gustafson E, Song J, Extavour C, et al. (2008). Vasa protein expression is restricted to the small micromeres of the sea urchin, but is inducible in other lineages early in development. Developmental Biology, 314(2), 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel GM, Brayboy L, Fresques T, Gustafson EA, Oulhen N, Ramos I, et al. (2014). The biology of the germ line in echinoderms. Molecular Reproduction and Development, 81, 679–711. 10.1002/mrd.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HR, Chen YT, Su YH, Luo YJ, Holland LZ, & Yu JK (2011). Asymmetric localization of germline markers Vasa and Nanos during early development in the amphioxus Branchiostoma floridae. Developmental Biology, 353(1), 147–159. [DOI] [PubMed] [Google Scholar]
- Yajima M, & Wessel GM (2011). Small micromeres contribute to the germline in the sea urchin. Development, 138(2), 237–243. 10.1242/dev.054940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima M, & Wessel GM (2012). Autonomy in specification of primordial germ cells and their passive translocation in the sea urchin. Development, 139(20), 3786–3794. 10.1242/dev.082230. [DOI] [PMC free article] [PubMed] [Google Scholar]