

Abstract
Despite recent clinical successes for irreversible drugs, potential toxicities mediated by unpredictable modification of off-target cysteines represents a major hurdle for expansion of covalent drug programs. Understanding the proteome-wide binding profile of covalent inhibitors can significantly accelerate their development; however, current mass spectrometry strategies typically do not provide a direct, amino acid level readout of covalent activity for complex, selective inhibitors. Here we report the development of CITe-Id, a novel chemoproteomic approach that employs covalent pharmacologic inhibitors as enrichment reagents in combination with an optimized proteomic platform to directly quantify dose-dependent binding at cysteine-thiols across the proteome. CITe-Id analysis of our irreversible CDK inhibitor THZ1 identified dose-dependent covalent modification of several unexpected kinases, including a previously unannotated cysteine (C840) on the understudied kinase PKN3. These data streamlined our development of JZ128 as a new selective covalent inhibitor of PKN3. Using JZ128 as a probe compound, we identified novel potential PKN3 substrates, thus offering an initial molecular view of PKN3 cellular activity. CITe-Id provides a powerful complement to current chemoproteomic platforms to characterize the selectivity of covalent inhibitors, identify new, pharmacologically addressable cysteine-thiols, and inform structure-based drug design programs.
Graphical Abstract
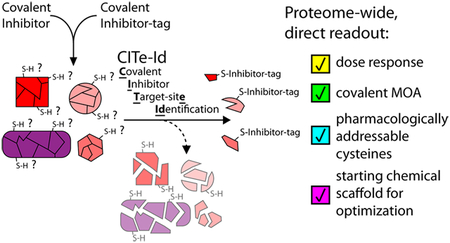
INTRODUCTION
Protein kinases govern many aspects of human physiology, and are associated and/or causatively linked to numerous human diseases. As a result, they are attractive targets for pharmacologic intervention, with most research efforts focused on developing reversible, small molecule kinase inhibitors. More recently, irreversible covalent inhibitors have emerged as compelling alternatives. These compounds permanently disable kinase activity, typically via covalent modification of a nonsequence conserved cysteine residue that lies in or near the ATP-binding pocket. The clinical potential for covalent kinase inhibitors (CKIs) is exemplified by the recent FDA approval of Ibrutinib, which targets BTK,1 and Afatinib, which targets EGFR.2 In fact, there are some 200 human kinases which span major branches of the kinome phylogeny and harbor targetable, active site-proximal cysteines (“cys-kinases”3,4). We recently described a series of CKIs that selectively modify cysteine residues distal to the active site (“remote cysteines”), with THZ15 and THZ5316 as the most advanced examples of this series. These results raise the intriguing possibility that cysteine-directed, selective CKIs may be developed for a much broader range of the human kinome than previously envisioned.4
Despite these promising developments, it remains difficult to predict cysteine reactivity, which represents a bottleneck in the rational design of CKIs.7 More importantly, the potential for idiosyncratic toxicities caused by covalent modification of off-target cysteines drives skepticism for the broad use of irreversible inhibitors. Chemoproteomics, a subset of mass spectrometry (MS) experiments that combines the use of small molecules with the analytical power of proteomics, has been invaluable for interrogation of CKIs and other probe classes. For example, recent chemoproteomic studies have sought to quantify the reactivity of endogenous cysteines across the proteome;8 these data reveal a range of highly reactive cysteine-thiols that represent potential off-target liabilities for CKIs, and highlight the need to include target-site analyses as part of covalent inhibitor development programs.
Tandem Orthogonal Activity-based Protein Profiling (TOP-ABPP, and the quantitative isoTOP-ABPP) is a well-established approach that employs alkyne-derivatized probes to enrich protein targets and identify likely sites of covalent modification.9 An important limitation of this methodology noted by the authors, was the difficulty in obtaining site-level information when using irreversible pharmacologic inhibitors, i.e., chemically complex and target selective compounds.9 Thus, the current standard relies on small, nonselective cysteine probes as surrogates to profile the activity of cysteine-directed selective pharmacologic inhibitors.8,10–17 This type of indirect, nonselective cysteine profiling does not formally confirm covalent ligand-target conjugation and may undersample low-abundance/-stoichiometry targets due to the stochastic nature of LC-MS/MS data acquisition. Recent modifications to the original approach address some of these issues by using affinity-tagged CKIs to identify off-targets and provide a more complete picture of potential toxicity liabilities.18,19 However, as reported this strategy focused on target identification at the protein-level and therefore requires companion biochemical assays to determine the exact site and covalent nature of ligand engagement.
We recently demonstrated that cysteine-directed probes and covalent drugs share common gas-phase dissociation path-ways.20 Pertinent to the limitations noted above, the predictable nature of these fragment ions can be used to improve peptide sequence assignment including the specific site of covalent modification. Here, we build on these results to establish a new chemoproteomic platform that leverages affinity-tagged analogs of pharmacologic CKIs for the biochemical enrichment of targets, along with tunable peptide fractionation and custom spectral processing to identify inhibitor target sites. Our new platform for Covalent Inhibitor Target-site Identification (CITe-Id) enables deeper coverage of cysteines modified by pharmacologic CKIs, while confirming covalent bond formation and providing dose-response data for inhibitor binding at each cysteine-thiol. As a powerful proof-of-concept, we used CITe-Id to identify multiple, unexpected off-targets of our cyclin-dependent kinase (CDK) inhibitor THZ1.5 These new targets included Cys-840 on Protein Kinase N3 (PKN3), an understudied AGC-type kinase linked to metastasis in aggressive prostate tumors.21,22 Residue-level data from CITe-Id facilitated our development of JZ128 as a covalent inhibitor of PKN3. We used JZ128 as a tool compound to identify novel potential PKN3 substrates. Our work exemplifies the utility of CITe-Id to reveal new pharmacologically addressable cysteines and accelerate development of selective, covalent inhibitors.
RESULTS
Developing and Optimizing CITe-Id (Covalent Inhibitor Target-site Identification)
Motivated by the growing clinical impact of CKIs, juxtaposed with key concerns related to potential toxicities as described above, we sought to develop CITe-Id as a robust chemoproteomic strategy that would go beyond protein-level identification of targets, and provide a residue-level direct readout of concentration-dependent covalent binding of a given CKI. We previously described specific gas-phase fragmentation pathways for peptides covalently modified by cysteine-directed, irreversible inhibitors and clinical drugs,20 including our recently described CDK7/12/13 inhibitor, THZ15 (Figure 1A). Building on these insights, we first elaborated THZ1 with a desthiobiotin affinity handle (THZ1-DTB, Figure 1A (1)) and then used Western blot to confirm concentration-dependent labeling and enrichment of CDK7 (Supporting Information, SI, Figure S1A). Beginning with a similar competition-format incubation (Figure S1A), we designed CITe-Id to readout dose-dependent binding of selective, pharmacologic inhibitors based specifically on the analysis of modified peptides (Figure 1B). We performed streptavidin pulldown after tryptic digestion of protein lysate to provide a highly enriched pool of THZ1-DTB labeled peptides for subsequent encoding with iTRAQ reagents, followed by multidimensional chromatography tailored for the hydrophobic nature of inhibitor-modified peptides and also providing wide flexibility for fractionation depth.23–25 In addition, we extended our previous framework for identification of covalent inhibitor-modified peptides to account for the desthiobiotin affinity tag and linker, and confirmed improved sequence scores for ~85% of THZ1-DTB labeled sites20 (Table S1). Finally, we used peptide iTRAQ reporter ion intensities to calculate a competitive dose-response for inhibitor binding at individual cysteine residues.
Figure 1.

CITe-Id directly quantifies covalent probe binding with site-level resolution. (A) Structures of: THZ1 described by Kwiatkowski et al.;5 desthiobiotinylated analog (THZ1-DTB); and THZ531 described by Zhang et al.6 (B) Workflow of CITe-Id using THZ1/THZ1-DTB: (1) Cell lysates were treated with THZ1/THZ1-DTB in a competition format, (2) protease digestion was carried out before streptavidin enrichment to provide a highly enriched pool of CKI-labeled peptides, (3) samples were encoded with iTRAQ stable isotope reagents to enable mass spectrometry quantification of concentration-dependent binding at individual cysteine residues, (4) online, automated RP-SAX-RP chromatography23 was tailored for the increased hydrophobicity of labeled peptides and allowed for ready adjustment of fractionation depth, (5) preprocessing of MS/MS spectra following our previous framework20 improved identification of cysteine residues covalently bound by THZ1- DTB. Competitive binding dose–response curves generated from iTRAQ reporter ions were used to distinguish concentration-dependent/independent binding of CKIs. (C and D) Replicate CITe-Id results for THZ1 and THZ1-DTB in HeLa S3 lysates. (C) THZ1-DTB-labeled cysteine sites rank ordered for competitive dose–response to THZ1. (D) Cysteine residues bound by THZ1-DTB in a concentration-dependent manner as indicated by a dose–response exceeding two standard deviations relative to the mean value of all 527 reproducibly labeled cysteine residues. (E) CITe-Id distinguishes the subset of THZ1 targets for their competitive dose–response to THZ531. CITe-Id analysis conducted as previous but THZ1 treatment was replaced with THZ531 followed by cotreatment with THZ1-DTB. See Supp. Table S2 for full list of THZ1-DTB modified cysteine residues identified by CITe-Id. For D and E, bars are the mean of the ratio of a condition from each biological replicate with standard deviation error bars and individual data points as gray circles.
Our CITe-Id analysis of THZ1/THZ1-DTB identified 527 unique cysteine residues which were covalently modified by THZ1-DTB (Table S2) and detected reproducibly across biological replicates (Figure S1B). Quantitative, competitive dose-response data from CITe-Id (Figure 1C) revealed that THZ1-DTB binding was independent of THZ1 concentration for a majority (>95%) of modified cysteine residues. These sites, which include highly abundant proteins such as glycolytic enzymes and tubulin, as well as many cysteines annotated as “highly reactive”,8 likely represent nonspecific probe binding. Against this null-distribution we identified dose-dependent competitive binding of THZ1 to eight cysteine residues, including the known targets CDKs 7, 12, and 13 (Figure 1C, dashed box). In addition to these positive control data, we identified dose-dependent THZ1 competitive binding on several unexpected kinase targets, PKN3 at C840, protein kinase C theta (PRKCQ) at C661, and glycogen synthase kinase 3B (GSK3B) at C14 (Figure 1D). We next repeated CITe-Id, using THZ531, a THZ1 analog with reported selectivity for CDK12/13 over CDK7,6 as the competing, native inhibitor. These data recapitulated the known CDK selectivity profiles, and further demonstrate that THZ531 has little or no reactivity against the potential new targets of THZ1, PKN3, PRKCQ, or CYP1B1 while gaining some activity against GSK3B (Figure 1E and Table S2). These results confirm the ability of CITe-Id to quantitatively distinguish the target landscape of structurally similar inhibitors.
THZ1 Inhibits PKN3 via Covalent Binding to C840
One of the kinases targeted by THZ1 was PKN3, a kinase that has been functionally linked to metastasis and tumor growth,21,22 and is the target of a liposomal siRNA-based therapeutic currently in clinical trials (NCT0093857426). Despite these data and clinical interest, PKN3 is understudied compared to other disease-associated kinases,27–29 which motivated us to further investigate it as a THZ1 target. A comparison of structural data for CDK7 and PKN3 (Swiss- Model Q6P5Z2) revealed that C840 of PKN3 is positioned close to the kinase active site, similar to the “remote cysteine” C312 of CDK7 targeted by THZ1 (Figure 2A). The targeted cysteines of CDK12 and PRKCQ are similarly positioned (Figure S2A). We therefore sought to validate PKN3 as a covalent target of THZ1 and explore the potential of developing selective tool compounds to study PKN3 biology. To confirm target engagement as well as the covalent nature of PKN3-THZ1 conjugate formation in vivo, we treated HeLa S3 cells with increasing concentrations of THZ1, using the reversible analog (THZ1-R5) as a negative control (Figure 2B). After cell lysis we incubated protein extracts with THZ1-DTB, followed by streptavidin pulldown and detection of bound proteins by Western blot. As expected, THZ1 showed strong competition for CDK7, with similar reactivity profiles observed for CDK12/13 (probing for their obligate cyclin K binding partner as a proxy), as well as two new targets identified by CITe-Id, PKN3 and PRKCQ. The reversible analog THZ1-R competed poorly for binding to all targets.
Figure 2.

THZ1 inhibits PKN3 via specific targeting of C840. (A) Structures of CDK7 and PKN3 facing kinase active sites with cysteine labeling sites of THZ1-DTB highlighted as colored spheres. All other unlabeled cysteine residues are displayed as black spheres. For CDK7, structure for Cys-312 is unavailable, a red dot indicates its approximate location adjacent to the next closest visible residue (Asp-311) (CDK7: PDB 1UA2,31 PKN3: Swiss-Model Q6P5Z2). (B) Live HeLa S3 cell treatments with THZ1 or reversible analog THZ1-R. Lysates treated with THZ1-DTB, followed by streptavidin pulldown and immunoblotting against THZ1 targets. CCNK is used as a proxy for CDK12/13. (C) Summary of PKN3 inhibition by THZ1 and THZ1-R. Middle column is IC50 values for 7-point dose–response kinase assay for PKN3. Right-most column is time-dependent kinase assays for PKN3 with fixed inhibitor concentration (1 μM). THZ1-R did not show time-dependent inhibition. See Figure S2B, C for enzyme inhibition curves. (D) C840 is essential for covalent labeling of PKN3. Dual FLAG-HA tagged PKN3 WT and PKN3 CS constructs were expressed in PC3 PKN3 KO cells. Lysates were treated with THZ1-DTB followed by streptavidin pulldown or HA immunoprecipitation and Western blotting. In = 1% Input, Avi = Streptavidin pulldown, HA = HA pulldown. Blot lanes were rearranged for clarity.
We next sought to confirm that covalent modification of PKN3 by THZ1 inhibited enzyme activity. Using an in vitro kinase assay, we observed that THZ1 inhibited PKN3 in a fixed time-point format with an apparent IC50 of 72 nM, while the reversible analog was more than 50-fold less potent (Figure 2C and Supp. Figure S2B). We also confirmed that inhibition of PKN3 by THZ1 was time dependent, consistent with a covalent mechanism-of-action (Figure 2C and Figure S2C). A similar assay showed THZ1 to have weak potency against PRKCQ (Figure S2D). To demonstrate that C840 is essential for THZ1 binding to PKN3, we expressed tandem FLAG and HA-tagged wild type (WT) or C840S (CS) mutant PKN3 in a clonal PC3 cell line with CRISPR-Cas9-mediated deletion of endogenous PKN3 (PC3 PKN3 KO) (Figure S2E). PC3 is a prostate cancer cell line that has been used previously to investigate PKN3 biology.21,30 Lysates were treated with THZ1-DTB, followed by streptavidin pulldown or immuno-precipitation with HA antibody (Figure 2D). We found that the C840S mutation abrogated THZ1-DTB binding to PKN3. Collectively these data validate PKN3 as a new target of THZ1, and further demonstrate that pharmacologic activity is mediated by the covalent binding to a single cysteine (C840) on the kinase.
Developing an Irreversible Inhibitor of PKN3
THZ1 exhibits good potency against PKN3, but is strongly cytotoxic due to its inhibition of the CDKs, and hence is limited as an inhibitor to study PKN3 biology. Nonetheless, our CITe-Id data suggested that the THZ-scaffold is a reasonable starting point to develop a selective, covalent inhibitor of PKN3. Toward this end we screened a small library of acrylamide-modified THZ-family compounds in our PKN3 kinase assay and compared these data to the activity of each compound against CDK7 (Figure S3A). On the basis of these results we identified JZ128 (2) as a potential selective inhibitor of PKN3 (Figure 3A). We also synthesized a desthiobiotin-tagged analog of JZ128 (JZ128-DTB, 3) for subsequent CITe-Id analysis, as well as a noncovalent analog (JZ128-R, 4), as a control. In vitro kinase assays against PKN3 revealed that JZ128 and the desthiobiotin-modified JZ128-DTB had similar IC50s of ca. 120 nM while the reversible analog was approximately 50-times less potent (Figure 3B and Figure S3B).
Figure 3.

Identifying a selective inhibitor of PKN3. (A) Structures of JZ128 and related compounds. (B) Summary of results for in vitro kinase assays for PKN3. (C) Live PC3 cell treatments with JZ128. After lysis samples were treated with THZ1-DTB or JZ128-DTB, followed by streptavidin pulldown and Western blot. CCNK was used as a proxy for CDK12/13. (D) Live PC3 cell treatments with JZ128-R. After lysis samples were treated with JZ128-DTB, followed by streptavidin pulldown and Western blot. (E) Similar to Figure 2D, dual FLAG-HA tagged PKN3 WT and PKN3 C840S constructs were expressed in PC3 cells lacking endogenous PKN3. Lysates were treated with JZ128-DTB followed by streptavidin pulldown or HA immunoprecipitation and Western blot. In = 1% Input, Avi = Streptavidin pulldown, HA = HA pulldown. Blot lanes were rearranged for clarity. (F) Confluent PC3 PKN3 KO cells expressing PKN3 rescue constructs were serum starved and treated with DMSO, JZ128, or reversible control. A wound was scored across the culture and monitored for 24 h. Shown is the mean ratio of remaining wound area after 24 h. normalized to the mean wound area of the DMSO condition for each cell line with individual replicate values shown as colored circles. See Figure S4 for representative raw images. p-values determined by unpaired 2-sided t test. (n = 6–9 replicates) NS: p-value >0.05, **: p-value = 0.0015, ***: p-value = < 0.0009. Error bars show one standard deviation from the mean.
To confirm cellular target engagement, we treated PC3 cells with increasing concentrations of JZ128. After lysis we incubated protein extracts with JZ128-DTB or THZ1-DTB, followed by streptavidin pulldown and detection of bound proteins by Western blot. As expected, JZ128 bound selectively to PKN3, with no activity against CDK7 or CDK12/13 (probing Cyclin K) (Figure 3C). We performed a similar experiment using the reversible analog (JZ128-R) and other PKN family members which lack an equivalently positioned cysteine to C840 on PNK3 (Figure 3D). JZ128-R did not compete for PKN3, while neither PKN1 nor PKN2 was bound by JZ128-DTB. We also confirmed that JZ128 did not inhibit kinase activity of PKN1 or PNK2 (Supp. Figure S3C, D). To confirm C840 as the only site of JZ128 covalent modification, we again utilized tandem FLAG-HA tagged WT or C840S mutant PKN3 expressed in PC3 PKN3 KO cells. Lysates were treated with JZ128-DTB, after which we performed streptavidin pulldown or immunoprecipitation with HA antibody (Figure 3E). This assay confirmed that C840 is required for JZ128-DTB binding to PKN3.
PKN3 has been functionally linked to cell migration.21,22,30 On the basis of these observations, we asked whether JZ128 could be used to recapitulate a PKN3-dependent phenotype. We first confirmed that JZ128 was not toxic to our set of PKN3 WT and mutant cell lines (Figure S3E). We then performed scratch assays in the presence of vehicle, JZ128, or JZ128-R using the cell lines described in Figure 2D. We observed that JZ128 significantly impaired wound healing in PKN3 WT cells but had no effect in cells that expressed the PKN3 C840S mutant (Figure 3F and Figure S4). Unexpectedly, PKN3 KO cells also exhibited impaired wound healing, indicating the potential for off-target JZ128 activity in the absence of PKN3. Collectively, our in vitro data and cell-based assays confirm PKN3 as a target of JZ128 and that the pharmacologic activity is predominantly mediated by covalent modification of a single cysteine (C840) on PKN3.
Next, we used CITe-Id in PC3 cells to assess the proteome-wide selectivity of JZ128 (Table S2). Quantitative, dose-dependent competitive binding data for 686 reproducible JZ128-DTB-labeled cysteines confirmed C840 of PKN3 as the primary target, with slightly less potency against PIKFYVE and TNK1 (Figure 4A, left and Figure 4B). Twelve other sites showed significant competitive dose-response to JZ128, including the kinases SRC, RIPK2, and RIOK2 (Figure S5A). The JZ128-DTB labeling site for these kinases was positioned adjacent to the kinase active site (Figure S5B). In vitro kinase assays showed JZ128 to have a roughly 200 nM IC50 against SRC and RIPK2 (Figure S5C, D). While our CITe-Id profiling of JZ128 provided information on competitive dose-dependence of covalent bond formation at specific cysteine residues, these data do not necessarily correlate with inhibition of JZ128-labeled enzymes. We used the industry-standard KiNativ platform to interrogate JZ128 with respect to kinase inhibition (ActivX Biosciences, La Jolla, CA).32,33 A Venn diagram in Figure 4A summarizes our complementary data for JZ128 covalent binding (CITe-Id) and kinase inhibition (KiNativ). In total 292 kinases were detected in PC3 cells, with only three showing greater than 50% inhibition at 1 ¼M JZ128 (Figure 4A, right and Table S3). Two of these kinase targets (PIKFYVE and RIPK2) overlapped with CITe-Id data, while a third (PIP4K2C) was identified only by KiNativ. In contrast, kinase targets including PKN3, TNK1, or RIOK2 were detected exclusively by CITe-Id. Further interrogation of these data revealed interesting anomalies. For example, CITe-Id identified SRC as being competitively bound at Cys280, proximal to the kinase active site. Our in vitro kinase assay demonstrated moderate activity of JZ128 for SRC (IC50 = 235 nM), while KiNativ data suggested that JZ128 had little or no SRC-activity in vivo. RIPK2 was also enigmatic. CITe-Id reported competitive binding for JZ128 at multiple cysteines on RIPK2; each of these was outside the active site, but unresolved by available protein structures. Similarly, KiNativ data showed two lysines bound by the ActivX-ATP probe, but with different competition profiles. Our kinase assays for RIPK2 showed nearly equivalent potency for JZ128 and JZ128-R, while biochemical data indicated a substantial difference for RIPK2 engagement for JZ128 compared to its reversible analog (Figure S5F). These observations highlighted the complementary nature of CITe-Id and KiNativ, and also the importance of integrating multiple, orthogonal assays to fully validate the target landscape of small molecule inhibitors.
Figure 4.

Selectivity Profiling of JZ128. (A) CITe-Id profiling compliments KiNativ profiling (Left) Replicate CITe-Id results for JZ128 and JZ128-DTB in PC3 lysates with JZ128-DTB-labeled cysteine sites rank ordered for competitive dose–response to JZ128. Dashed box indicates sites with a competitive dose response threshold two standard deviations relative to the mean value of the null. See Figure S6A for individual treatment condition data and Table S2 for complete CITe-Id results. (Right) KiNativ results rank-ordered by kinase inhibition. Dashed box indicates kinases with >50% inhibition by JZ128 at 1 μM. See Table S3 for complete data set. (Middle) Venn diagram overlaying the kinase results for CITe-Id and KiNativ profiling. *SRC was competively bound by JZ128-DTB (CITe-Id) but not enzymatically inhibited (KiNativ). **JZ128 labeling of PIP4K2C C313 was identified using nontryptic protease digestion of the purified protein. All cysteine labeling sites were determined with CITe-Id except where noted for PIP4K2C. (B) CITe-Id data for kinase cysteines modified by JZ128-DTB with competitive dose responses (>3 std. dev. relative to the mean of the null). Bars represent the mean of the ratio of a condition from each biological replicate with standard deviation error bars and individual data points as gray circles. (C) Live PC3 cells expressing FLAG-HA-PKN WT were treated with JZ128. After lysis samples were treated with JZ128-DTB and FLAG-HA-PKN3 was enriched by tandem affinity purification. Bar plot shows competitive dose–response for the JZ128-DTB modified C840-containing PKN3 target peptide across two biological replicate experiments. (D) Dendrograms of the kinome depicting the covalent targets of JZ128 and THZ1 as identified by CITe-Id. Image generated using TREEspot Software Tool and reprinted with permission from KINOMEscan, a division of DiscoveRx Corporation.
These data also provided clues for potential improvements in our CITe-Id platform. KiNativ detected PIP4K2C as a potential new off-target of JZ128, not reported by CITe-Id. We used cellular target engagement assays to confirm modest competitive binding ofJZ128-DTB for PIP4K2C, in addition to its homologues PIP4K2A/B and PIKFYVE (Figure S5E, F). Closer inspection of PIP4K2C suggested C313 as the most likely JZ128 binding site. However, the tryptic peptide containing C313 comprised 65 amino acids, a size not likely amenable to LC-MS/MS. Importantly, our previous work demonstrated that generation of inhibitor-specific fragments during MS/MS was independent of peptide cleavage specificity.20 Pursuant to this observation we digested purified PIP4K2C-JZ128 conjugates with endoprotease GluC and readily identified the modified C313 peptide (Figure S6). These data suggested that use of a combination of proteolytic enzymes would provide increased target coverage for CITe-Id beyond use of trypsin alone. We also noted a significant difference in competitive dose–response for PKN3 as readout by CITe-Id (Figure 4B) and Western blot target engagement (Figure 3C). To explore this discrepancy, we first used stable incorporation of labeled amino acids (SILAC) in a quantitative proteomic screen to confirm equivalent reactivity ofJZ128 and its DTB-analog against cysteines in protein lysates. As further validation we used the same experimental design to test THZ1/THZ1-DTB (Figure S7 and Table S4). These analyses showed minimal reactivity differences between each pair of analogs for the majority of covalently modified cysteine sites. A second possible explanation was that JZ128 exhibits varying reactivity when incubated in protein lysates (CITe-Id, Figure 4B) versus live cells (WB target engagement, Figure 3C). To test this hypothesis, we treated live FLAG-HA-PKN3 WT PC3 cells with the same concentration range of JZ128 for 3 h, lysed cells and incubated with JZ128-DTB as in CITe-Id, then purified tagged PKN3 using tandem affinity purification,34,35 followed by iTRAQ labeling and LC–MS/MS. Dose–response for competitive JZ128-DTB binding was more robust in this targeted PKN3 analysis (Figure 4C). These data demonstrated the potential improvement afforded by incorporating live cell treatment into our CITe-Id workflow. These platform modifications notwithstanding, our cumulative data suggested that structurally related THZ1 and JZ128 each covalently modify and inhibit a limited set of kinases, with PKN3 as their only shared target (Figure 4D).
Identification of Potential PKN3 Substrates Using JZ128 and THZ1
Mechanistic studies place PKN3 downstream of phosphoinositide 3-kinase (PI3K), and have suggested that biochemical complex formation between RhoC, PKN3, and PDK1 may lead to PDK1-dependent activation of PKN3.22 Previous studies using mouse models and siRNA knock down demonstrated a role for PKN3 in cell migration, invasiveness, and increased metastatic potential of prostate cancer cells,21,30 as well as the dynamics of adherens junctions.36 Interestingly, unlike PKN1 and PKN2 that are ubiquitously expressed, PKN3 is tissue specific.37 Data from various cancer genomics studies suggest that PKN3 is amplified and/or overexpressed in some solid tumors.27–29,37 However, the substrates and downstream effectors of PKN3 are unknown.
We therefore focused on using JZ128 and THZ1 as tool compounds to identify potential PKN3 substrates and get additional insights into PKN3 function. We utilized our set of PC3 cells in a combined quantitative, chemical, and genetic phosphoproteomic analysis (Figure 5A). We incorporated cells expressing the mutant CS PKN3 to control for covalent on-target effects of JZ128, as well as treatment with JZ128-R as a control for potential off-target, noncovalent effects of the inhibitor. We further reasoned that combined use of JZ128 and THZ1 would reduce false-positives, as these probes share PKN3 as their only common target. In total, we reproducibly quantified 14700 phosphorylation sites across 3136 proteins for two biological replicates (Table S5). We used phosphopeptides enriched from untreated PC3 cells to establish reporter ion variance as a function of intensity.38,39 On the basis of this error model we identified 91 discrete phosphorylation sites which exhibited a regulation pattern consistent with a PKN3 substrate under the different conditions tested in our phosphoproteomic analysis (Figure 5B). We next sought to prioritize this set based on similarity with a PKN3 phosphorylation motif. We used recombinant PKN3 with our expanded, randomized peptide library44 to generate a putative PKN3 motif consisting of Leu-X-Arg-X-Pro-*Ser-Phe-Arg-X-X (Figure 5C and Table S6). These data further refine a previously reported PKN3 motif,42,43 placing stronger emphasis on proline at the −1 and arginine at the +2 positions, with reduced dependence on positions −5, −4, −2, + 3, and +4. We scored each of the 91 phosphorylation sites based on sequence-similarity with our PKN3 motif in addition to an estimate of the likelihood of a motif-match for PKN3 compared to a larger set of 191 kinase motifs determined using our peptide library approach (unpublished data). This analysis highlighted 25 phosphorylation sites as putative direct substrates, including a potential site of autophosphorylation (S544) on PKN3 (Figure 5D). Consistent with reported functional roles for PKN336 these putative substrates are enriched for proteins involved in cellular junction related GO terms: cadherin binding (Odds ratio = 23; Padj < 0.001) and cell adhesion molecule binding (odds ratio = 15; Padj = 0.021).40 As a first step in validating these data, we obtained synthetic peptides encompassing the phosphorylation site and surrounding motif and used them as substrates for in vitro PKN3 kinase assays. Our analysis confirmed the original peptide sequence and phosphorylation site assignment (Figure S8A), and furthermore that PKN3 phosphorylated 100% and 50% of motif-serine residues on LAD1 (S375) and EXOC2 (S432), respectively. Pretreatment with either THZ1 or JZ128 partially blocked PKN3-mediated phosphorylation of these sites (Figure 5E). We also tested EGFR (S1064) but failed to detect PKN3-mediated phosphorylation. While we validated the EGFR phosphopeptide assignment identified in our screen (Figure S8B), we noted that the neighboring residues comprised a lower-score sequence for a potential PKN3 motif, compared to LAD1 or EXOC2 (Table S5). Although subject to further validation, these results illustrate a powerful use of our covalent inhibitors to identify promising, candidate PKN3 substrates.
Figure 5.

Phosphoproteomic discovery of potential PKN3 substrates. (A) Illustration of genetic and chemical controls, experimental workflow, and the expected response of a direct PKN3 phosphorylation substrate under the conditions tested. Each experimental treatment was compared to its matched DMSO control (Cntr). Only the JZ128 and THZ1 treatments in the WT PKN3 background were expected to mediate reduced phosphorylation (down arrows). All other treatment conditions were expected to show no change (NC). 91 phospho-sites fit these criteria. (B) Representative volcano plot showing the fold change and log10; p-value of 14,700 reproducibly quantified phosphorylation sites. The p-values were determined by use of a variance model as previously described.38,39 Data for the PKN3 WT JZ128 treatment vs PKN3 WT DMSO treatment is shown. Red dots comprise the 91 phosphorylation sites that met all criteria in A for potential direct substrates of PKN3. See Table S5 for the complete data set and analysis. (C) PKN3 motif derived from analysis of randomized peptide library. See Table S6 for quantified densitometry values. The 91 phosphorylation sites meeting the regulation criteria were further reduced to a set of 25 putative substrates based on a composite motif score: (i) sequence-similarity to the predicted PKN3 motif and (ii) relative match to the predicted PKN3 motif compared to a larger set of 191 ser/thr kinase motifs derived from analysis of the randomized peptide library. (D) Heat map of fold change values for the 25 phosphorylation sites meeting all criteria for potential direct substrates of PKN3. The single red cell (value = −1.148) in the heatmap is outside the range of the legend. (E) PKN3 in vitro phosphorylation of synthetic peptides which exhibited reduced phospho-serine levels in PKN3 WT cells treated with JZ128/THZ1, and closely matched our predicted PKN3 motif. Bar graphs show percent phosphorylation of each site compared to no ATP control conditions across four replicate experiments. Positive control is the same synthetic peptide used for in vitro kinase assays with PKN3. THZ1 and JZ128 treatments were at 1 μM for 30 min prior to starting the assay. (N = 4–10 replicates) NS: p-value >0.05, *: p-value <0.05, **: p-value <0.001, ****: p-value = < 0.0001.
DISCUSSION
Although historically eschewed by drug development programs, irreversible inhibitors are now gaining momentum.41 We and others have used structure-guided synthesis to develop selective CKIs which inhibit kinase activity through covalent modification of nonconserved, active-site cysteine residues.42 We recently extended this paradigm to achieve selective inhibition by targeting “remote” cysteine residues.5,6 Encouragingly these results suggest that the landscape of drug targets amenable to selective covalent inhibition via cysteine reactivity may be significantly larger than previously predicted.3 However, our inability to predict potential off-target reactivity represents a major liability for development of covalent drugs. Therefore, design, development, and characterization of covalent inhibitors must be considered in the context of the entire proteome.8
Activity based protein profiling, exemplified by TOP-ABPP has provided valuable insight for the cellular activity of a variety of small molecules.8,11,13,14,16,17 However, in the specific case of CKIs, these techniques typically rely on the use of promiscuous cysteine probes as surrogate readouts for the reactivity of selective inhibitors.8,9,11,13–15,17 In contrast, our CITe-Id platform repurposes selective CKIs as affinity reagents to directly enrich protein targets. Moreover, incorporation of our new knowledge of gas-phase fragmentation behavior characteristic of covalent probe-peptide adducts20 enables us to focus exclusively on CKI-modified peptides. With CITe-Id we circumvent the difficult task of exhaustively profiling a large set of cysteine residues bound by a biologically irrelevant ABPP probe to identify the modest set of residues which are bound by a selective inhibitor. Importantly, CITe-Id data provide direct evidence of covalent mechanism-of-action while the incorporation of multiplexed isotope labels provides a readout of competitive dose–response at each CKI-modified cysteine-thiol.
In the work described here, we used CITe-Id to profile THZ1 reactivity, and discovered PKN3 as a new selectively bound THZ1 target. PKN3 is a downstream effector of activated phosphoinositide-3-kinase (PI3-K) that mediates metastatic transformation and growth,21 tumor angiogenesis,30 as well as bone resorption.4 A siRNA based therapeutic strategy against PKN3 has been in clinical trials for advanced solid tumors and as a combination treatment for advanced or metastatic pancreatic cancer (ref 26 and NCT01808638), highlighting its relevance as a disease target. However, the biology of PKN3 is understudied, nominating it as a bona fide member of the “dark-kinome”.29 These data, in addition to the lack of commercial assays for this kinase, motivated us to explore PKN3 further. Using THZ1 as a starting scaffold we confirmed JZ128 as a ~100 nM covalent inhibitor for PKN3 with good selectivity over CDK7, 12, and 13. In addition, JZ128 exhibited improved cellular toxicity profiles compared to THZ1, enabling its use in cell-based assays to interrogate PKN3 function. We used CRISPR-engineered prostate cancer-derived PC3 cells to confirm an on-target phenotype for PKN3 WT versus the C840S mutant in a wound healing assay. These results were confounded by reappearance of wound healing defects in matched cells depleted of PKN3. It is difficult to predict the myriad effects of genetic depletion and how they may activate compensatory pathways. It would be interesting for future studies to investigate possible genetic links between PKN3 and the off-targets of JZ128 such as RIPK2 or PIKFYVE, which both play key roles in cellular pathways related to wound healing.44–48 Pending in-depth exploration of PKN3 genetic dependencies, we recommend the PKN3 C840S mutant as the most suitable negative control for pharmacologic studies of JZ128. It also is important to note that installation of the affinity handle may disrupt the kinetics of ligand binding. For the work herein, we attached desthiobiotin to the dimethylaminomethyl (DMAM) moiety of THZ1 and JZ128, respectively. While the DMAM functionality is not formally required for implementation of CITe-Id, we found it a convenient, solvent-exposed anchor point. Ongoing work in our group suggests CITe-Id is compatible with numerous warhead chemistries including unsubstituted acrylamides, haloacetamides, haloketones, and halotetrahydroacridines (unpublished data). For new covalent probe architectures, Western blot target engagement assays can be used to confirm expected competitive binding behavior for new DTB-analogs against known kinase targets. Additionally, when using a new covalent warhead, initial database searches of CITe-Id data should include the possibility for covalent modification on other amino acid side chains.
We used KiNativ to corroborate our CITe-Id results for JZ128; both platforms identified several off-targets, including PIKFYVE, TNK1, RIPK2, RIOK2, and SRC. Consistent with our experience with other kinome profiling services, KiNativ provided no data for PKN3, the primary target of JZ128 as revealed by CITe-Id. KiNativ did uniquely identify PIP4K2C as an off-target. In this case we confirmed that use of an alternative protease enabled identification of the JZ128-modified cysteine residue. In fact, consistent with our previous work,20 it should be straightforward to incorporate multiple enzymes to improve the “proteomic reach” of CITe-Id. In addition, our analysis of enriched FLAG-HA-PKN3 from live cells treated with JZ128, then further processed with our CITe-Id workflow, better replicated our competition-format target engagement Western blot assays. These results provide another promising avenue for further improvement to our CITe-Id platform.
Intrigued that our data apparently represent the first report of a targetable cysteine on PKN3, we performed a metaanalysis based on a compendium of cysteine residues included in eight different TOP-ABPP studies which used broad-reactivity iodoacetamide-alkyne probes8,10–14,16,17 (Figure S9, yellow). In total, these TOP-ABPP data captured approximately 14000 cysteines. Interestingly, 28% of cysteines covalently labeled by THZ1 or JZ128 are not represented in the TOP-ABPP catalog. But strikingly, 12 of the 22 CITe-Id derived cysteine thiols labeled in a concentration-dependent manner were identified exclusively by CITe-Id, including C840 on PKN3 (Figure S9). Taken together, these data highlight the power of CITe-Id to uncover new, pharmacologically addressable regions of the cellular cysteineome.
Our CITe-Id data streamlined development of JZ128 as a new, first-in-class inhibitor of PKN3. In an initial interrogation of PKN3 cellular activity, we used JZ128 in a quantitative phosphoproteomic analysis to identify PKN3 substrates. Numerous chemical and genetic controls along with a refined prediction of a PKN3 kinase motif provided stringent criteria for identification of potential phosphorylation substrates. In vitro phosphorylation assays confirmed PKN3 activity on S375 of LAD1 and to a lesser extent S432 of EXOC2. LAD1 is an attractive candidate for future functional validation given its well-established role in cell junctions.49 In addition to LAD1 and consistent with reports of PKN3,s functional role in normal physiology and tumor metastases,21,36 our set of 25 putative substrates is enriched for proteins involved in adherens junctions, including SCRIB, EGFR, ZC3HAV1, MARCKS, KRT8, PTPN1, and EPHA2. Collectively, our data provide important leads for future mechanistic studies to interrogate the role of PKN3 in cellular junctions as well as the metastatic potential of solid tumors.
It should be noted that binding data from CITe-Id is not directly analogous to inhibition of enzymatic activity, with CITe-Id supplementing but not replacing traditional enzyme-activity assays. Overall, CITe-Id is a complementary methodology that fits well within an irreversible inhibitor development program as it offers information valuable for understanding the precise mechanism of action as well as proteome-wide, covalent off-target effects.
Taken together, CITe-Id represents a conceptually novel strategy for rapid and accurate proteome-wide identification of covalently modified sites targeted by irreversible inhibitors. We expect that CITe-Id profiling will enable rapid irreversible inhibitor screening to inform medicinal chemistry optimization efforts as well as mechanistic studies of these inhibitors and their off-target effects. As illustrated by our PKN3 work, CITe-Id analysis can accelerate discovery of novel selective inhibitors and functional characterization, especially in the context of the understudied kinome.
MATERIALS AND METHODS
Antibodies and Reagents
Antibodies were obtained from the following sources: α-PKN3 (Novus Biologicals, Littleton, CO), α-CDK7 (Cell Signaling Technologies, Danvers, MA), α-CCNK (Bethyl Laboratories, Montgomery, TX), PRKCQ (Thermo Fisher Scientific), α-FLAG (Sigma), α-PKN1 (Invitrogen, Carlsbad, CA), α-PKN2 (Cell Signaling), α-LIMK1 (Cell Signaling), α-PIP4K2A (Cell Signaling), α-PIP4K2B (Cell Signaling), α-PIP4K2C (Sigma), α-PIKFYVE (Millipore), and α-RIPK2 (Novus Biologicals). Sources for supplies and reagents are as follows: Activated Thiol Sepharose 4B beads (GE Healthcare, Piscataway, NJ); Sep-pak tC18 100 mg 96-well plate (tC18; Waters, Milford, MA); SOLA C18 plate (SOLA C18; Thermo Fisher Scientific, Waltham, MA); Ni:NTA magnetic agarose beads (Qiagen, Valencia, CA); Ammonium bicarbonate (AMBIC; Sigma, St. Louis, MO); Triethylammonium bicarbonate buffer (TEAB; Sigma), Ethanol (EtOH; Sigma); Acetonitrile (Macron, Avantor Performance Materials, Center Valley, PA); tris(2-carboxyethyl)phosphine (TCEP; Sigma); S-Methylmethanethiosulfonate (MMTS; Sigma); DL-Dithiothreitol (Sigma); Iodoacetamide (IAA; Sigma); Trifluoroacetic Acid (TFA; Pierce, Rockford, IL); formic acid (FA; Sigma). High-Capacity Streptavidin Resin (streptavidin resin; Pierce). Kinase substrate library (Anaspec). Streptavidin-conjugated membranes (Promega). Synthetic peptides were produced by use of Fmoc chemistry and purified by reversed phase HPLC. THZ1, THZ1-R, and THZ531 were synthesized as described.5,6
Chemical Synthesis
All solvents and reagents were used as obtained. For verification of chemical structures, 1H NMR spectra were recorded with a Varian Inova 500 NMR spectrometer and referenced to dimethyl sulfoxide. Chemical shifts are expressed in ppm. Mass spectra were measured with Waters Micromass ZQ using an ESI source coupled to a Waters 2525 HPLC system operating in reverse mode with a Waters Sunfire C18 5 μm, 4.6 × 50 mm2 column. Purification of compounds was performed with either a Teledyne ISCO CombiFlash Rf system or a Waters Micromass ZQ preparative system. The purity was analyzed on an above-mentioned Waters LC—MS Symmetry (C18 column,4.6 × 50 mm2, 5 μM) using a gradient of 5–95% methanol in water containing 0.035% trifluoroacetic acid (TFA). Detailed synthetic schemes and characterization data are available in Supplementary Chemical Synthesis Details and else-where.5,6
Cell Culture and Cloning
All cell culture was performed using standard techniques. HeLa S3 and HEK 293T cells were cultured in DMEM with 10% FBS, PC3 cells were cultured in RPMI1640 with 10% FBS. Cells were collected using a scraper. The cells were centrifuged at 300g and washed twice with PBS before cell pellets were frozen at −80 °C. Full length PKN3 cDNA with a N-terminal FLAG-HA tag was cloned into the pUC57 vector (GenScript U.S.A., Piscataway, NJ). At the same time, a point mutation was introduced at position 2886 (local sequence: ggccacacagggt) from a → to generate the C840S mutant PKN3. Both PKN3 constructs were excised from pUC57 with EcoRI and XhoI and inserted into a pCDH-CMV backbone.
Target Engagement Pulldown Assays
HeLa S3 or PC3 cells were treated with increasing concentrations of either DMSO, THZ1, THZ1-R, JZ128, or JZ128-R for 4 h. Cells were harvested by scraping and washed twice with PBS before lysis with NP-40 lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol, 0.5% v/v NP-40, protease inhibitors). Protein content was measured by BCA. Samples were treated with 2 μM of either THZ1-DTB or JZ128-DTB for 18 h. at 4 °C. Streptavidin resin was added and incubated for 1 h. at room temperature. The beads were washed three times with NP-40 lysis buffer. Bound protein was eluted by adding SDS-PAGE loading buffer and boiling for 5 min before Western blotting. 1% of sample input was used for loading controls.
Kinase Assays
Kinase assays for PKN3 were performed using the synthetic peptide GGGGPKGPGRRGRRRTSSFAEGG as sub-strate.50 Assay conditions were 26 mM Tris pH 7.5, 0.002% CHAPS, 5.2 mM MgCl2, 2.3 mM DTT, 25 ng PKN3, 20 μM substrate, 68 μM ATP, 1 μCi ATP [γ-32P]. Enzyme was preincubated with compounds for 30 min before adding ATP and reacting for 30 min at room temperature. Reaction was stopped with TCA and spotted in 96 well phosphocellulose plates (Millipore). Wells were washed seven times with 150 mM phosphoric acid, once with 95% isopropanol, and dried before scintillation counting. For the time-course PKN3 kinase assay data in Figure 2C, all compound treatments were at 1 μM and preincubation times were from 0 to 120 min. Kinase assays for PRKCQ, CDK7, LIMK1, PKN1, and PKN2 were performed using SelectScreen (Invitrogen) in a 10-dose titration format.
For in vitro validation of PKN3 substrates, the following changes were made to the above methodology: candidate substrate synthetic peptides were used at 4 μM concentration, ATP was used at 1 mM concentration, and ATP [γ−32P] was omitted. Reactions were performed for 1 h at 37 °C and stopped by adding acetic acid to 1% FC. Ten pmol of each substrate peptide was desalted and mixed with α-cyano-4-hydroxycinnamic acid matrix and analyzed on a 4800 MALDI TOF/TOF (AB SCIEX). Unphosphorylated peptide signal was compared to a control reaction lacking ATP using label free quantitation to indirectly determine the percentage of phosphopeptide conversion. Synthetic peptide sequences: LAD1 S375, RSSPRTISFRMKPKK; EXOC2 S432, ASLKRGSSFQSGRDDTWR; EGFR S1064, NGLQSCPIKEDSFLQRYSSDPT.
PKN3 Rescue Pulldowns
PKN3 constructs were transiently expressed in HEK 293T cells. Cells were harvested and lysed with NP-40 lysis buffer as above. Lysates were initially probed for FLAG signal via Western blotting to determine construct expression in each cell line. Input lysates for subsequent pulldowns and immunoprecipitations was normalized for construct expression with empty vector input equal to highest input used. Lysates were split in four: two were treated with THZ1-DTB or JZ128-DTB for 18 h. at 4 °C, two were treated with DMSO under the same conditions. One probe treated and one DMSO treated sample received streptavidin pulldowns as in target engagement pulldowns above. The remaining two samples were immunoprecipitated with α-HA-agarose resin (Sigma) for 2 h. at room temperature. Resin was washed and bound proteins eluted as for the streptavidin pulldowns. Samples were then analyzed with Western blotting.
Antiproliferation Assays
PC3 cells were plated in 384-well plates at 750 cells/well in 50ul fresh media (RPMI + 15% FBS + 1% Penn/Strep) and treated with 0.1 μL candidate compounds in ten point 4-fold dilution series using the JANUS (PerkinElmer). Cells were incubated with compounds for 72 h in 37 °C 5% CO2. Antiproliferative effects of these compounds were assessed 72 h after compound addition using Cell Titer Glo (Promega cat# G7571) as described in product manual by luminescence measurements using an Envision platereader. All proliferation assays were performed in biological quadruplicate. IC50 values were determined using a nonlinear regression curve fit in GraphPad Prism 7.
Wound Healing Assays
PC3 cells were seeded in 24-well plates (1 × 105 cells/well) and grown to confluency. The media were replaced with serum-free media for 12 h to stop cell division. Wounds were made across the well with a P10 pipet tip and cells washed with PBS before replacing with fresh serum-free media containing the indicated compounds at 300 nM. Images of wound were then captured over 2 days with a Zeiss Axiostar microscope. Wound area was quantified using ImageJ.
PKN3 Phospho Motif Identification
To determine the substrate motif of PKN3, in vitro kinase assays were performed with recombinant PKN3 on the peptide substrate library in the presence of ATP[γ-32P]. These reactions were carried out in Kinase Assay Buffer I (SignalChem) at 30 °C for 90 min. The peptides, which are biotinylated at their C-termini, were blotted onto streptavidin-conjugated membranes and imaged with a Typhoon FLA 7000 phosphorimager. Detailed information on the protocol is provided elsewhere.51 The spot densities from the blot array were quantified and normalized by each row. These values were used to score the amino acid sequence surrounding each identified phosphosite.
Isotopic Reporter Labeling
For CITe-Id experiments, iTRAQ labeling was performed after streptavidin pulldown. Samples were resuspended in 30% 0.5 M TEAB, 70% EtOH. The iTRAQ reagent was added to each corresponding sample. After incubation at room temperature for 1 h, the samples were combined and acidified before vacuum drying. For phosphoproteomics experiments, TMT labeling was performed prior to phosphopeptide enrichment. Samples were resuspended in 50 mM TEAB. ACN was added to each TMT reagent and then mixed with each corresponding sample. After incubation at room temperature for 1 h., the samples were combined and acidified prior to a final desalting step on a SOLA C18 plate.
Phosphoproteome Analysis
Qiagen Ni:NTA magnetic agarose beads stored as a 5% suspension in 30% ethanol were used for phosphopeptide enrichment. The magnetic beads were prepared by rinsing three times with water and then incubating in 100 mM EDTA pH 8 for 30 min. Next, the beads were rinsed three times with water and incubated in 10 mM FeCl3 for 30 min. Prior to incubation with the sample peptides, the bead pellet was rinsed three times with water and once with 80% ACN, 0.1% TFA and finally resuspended with 80% ACN, 0.1% TFA. Reduced, alkylated, desalted, tryptic peptides were TMT-labeled, pooled, and dried prior to phosphopeptide enrichment. Peptides were reconstituted in 80% ACN, 0.1% TFA and incubated with the Fe-activated magnetic agarose NTA bead suspension for 30 min. After incubation, the supernatant was retained and the bead pellet was washed three times with 80% ACN, 0.1% TFA. The bead pellet was eluted with 1.4% ammonia by weight, 3 mM EDTA and water before being vacuum-dried. Phosphopeptides were analyzed using our RP-SAX-RP chromatography platform. Detailed descriptions of the multidimensional chromatography system are provided elsewhere23–25 Peptides were automatically fractionated across first and second dimension high-pH reversed phase and high-pH strong anion exchange chromatographic stages. Peptides were gradient eluted from the third dimension (low-pH RP) into a Q-Exactive HF mass spectrometer (ThermoFisher Scientific) using a 4 h organic gradient (5–30% B in 4 h., A= 0.1% FA, B = acetonitrile with 0.1% FA). The electrospray voltage was set at 3.2 kV. A top-15 data-dependent method was used for precursor selection and MS/MS. A normalized collision energy of 37 eV was used. Data processing of phosphopeptide MS/MS spectra is described below.
Processing of Mass Spectrometry Data
Native .raw data files from the Q-Exactive HF mass spectrometer were processed using our multiplierz Python-based framework52–54 to generate .mgf files for input to Mascot (Matrix Science). For CITe-Id experiments with THZ1-DTB and JZ128-DTB labeled peptides, MS/MS spectra were processed to account for inhibitor-specific fragmentation behavior as described previously (see Table S1 for a list of inhibitor-specific fragment ions).20 Peptide precursor masses were recalibrated on a per-scan basis by correcting all m/z values based on accurate mass recorded for the Si(CH3)O6 peak in each spectrum. All data were searched against a forward–reverse human database assembled from the NCBI Refseq database. For deisotoped HCD spectra, the precursor mass tolerance was set to 10 ppm and the MS/MS fragment ion tolerance was set to 25 mmu. Search parameters included trypsin specificity, with a maximum of two missed cleavages, fixed carbamidomethylation of Cys (+57 Da), variable oxidation on Met (+16 Da with −64 Da neutral loss possible). Depending on the experiment, fixed TMT 6-plex labeling on Lys and peptide N-termini (+229 Da), fixed iTRAQ4-plex labeling on Lys and N-termini (+144 Da), variable phosphorylation on Ser, Thr and Tyr (+80 Da, allowing neutral loss of 98 Da), variable THZ1-DTB labeling of Cys (+993 Da, allowing neutral loss of 993 Da), variable JZ128-DTB labeling of Cys (+987 Da, allowing neutral loss of 987 Da). Reported peptide sequences were filtered based on a 1% FDR.
Phosphopeptide Validation
Synthetic phosphopeptides were TMT labeled and desalted with batch mode C18 cleanup as above. They were then analyzed on a Q Exactive HF mass spectrometer using a 20 min organic gradient as described above. MS/MS spectra of Endogenous and synthetic phosphopeptides were annotated using mzStudio.55
CITe-Id
HeLa S3 or PC3 cell pellets were lysed with NP-40 lysis buffer as above. After lysate clearance centrifugation, protein concentration was determined by BCA. Samples were precleared with streptavidin resin for 1 h. at 4 °C. After centrifugation, the supernatant was split in four and pretreated with DMSO or increasing concentrations of inhibitor (THZ1, THZ531, JZ128) and incubated at room-temperature for 1 h. Two μM of desthiobiotin-tagged analog (THZ1-DTB, THZ1-DTB, JZ128-DTB, respectively) was added to all samples and incubated at 4 °C for 18 h. Urea was added to 8 M final concentration followed by cleanup using Zeba desalting columns. Samples were diluted to 4 M urea with pH 8 lysis buffer and reduced with 10 mM DTT for 30 min at 56 °C. Samples were then alkylated with 20 mM IAA for 30 min at RT. Samples were further diluted to 2 M urea before trypsin digestion at 37 °C overnight. Streptavidin resin was added to the samples and incubated at RT for 1 h. The beads were sequentially washed with lysis buffer three times, PBS three times, and water two times. Peptides were eluted with four sequential incubations of 50% acetonitrile (MeCN), 0.1% trifluoroacetic acid (TFA) for 3 min at RT. Eluted peptides were concentrated by vacuum centrifugation and resuspended in 0.1% TFA before batch mode C18 cleanup as described previously.56 Samples were vacuum-dried before iTRAQ labeling as described above. Peptides were cleaned using the SP3 paramagnetic bead methodology.57 Briefly, dual-Speed Bead Carboxylate Beads (GE Healthcare) were added to peptides reconstituted in 95% MeCN and allowed to bind for 10 min at RT. Beads were washed with MeCN before peptides eluted in 100 mM Ambic, 2% DMSO and vacuum-dried. Samples were reconstituted in 5% DMSO, 100 mM ammonium formate for RP-SAX-RP LC–MS/MS as described above. LC gradients were 5%–60% B for 90 min. Data were processed as above with spectral processing to account for inhibitor-specific fragmentation pathways and searching with variable modifications to cysteine for the respective probe used in each experiment. Normalized reporter ion signal for labeled cysteine residues from multiple PSMs was summed and a ratio was generated for each reporter channel by comparing it to the DMSO-treated control channel. Inhibitor concentrations and ratios were used to generate a trendline for each labeled site with the slope being the competitive dose response for the cysteine site.
Targeted Live Cell PKN3 CITe-Id Using TAP-MS
PC3 cells with CRISPR mediated gene depletion of PKN3 with rescue by FLAG-HA-PKN3 WT expression were treated with JZ128 for 3 h. Cells were lysed and treated with 2 μM JZ128-DTB at 4 °C overnight. Tandem affinity purification of PKN3 was performed as previously described.34,35 Purified proteins were trypsin digested, iTRAQ labeled, and analyzed by LC–MS/MS as described for CITe-Id. Reporter signal for the JZ128-DTB modified C840-containing PKN3 peptide was normalized to total signal for unmodified PKN3 peptides in the experiment.
SILAC Analysis of Compound Reactivity
Lysates were made of SILAC incorporated HeLa S3 cell pellets. Heavy incorporated lysates were treated with a single compound (e.g., THZ1) at 4 °C overnight and light lysates similarly treated with DMSO. The two samples were pooled, trypsin digested, and cysteine peptides enriched using activated thiol resin (GE Healthcare). After LC–MS/MS, each cysteine peptide was quantified based on its reactivity compared to DMSO control. A log-2 fold-change reactivity difference was then determined for cysteine peptides in common between each untagged and tagged compound-pair. Density histograms were plotted for both the entire set of cysteine sites as well as the subset of cysteine sites also detected as inhibitor-modified in the respective CITe-Id experiments for each compound-pair. Density histograms were generated using Prism.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge generous support from NIH grants R21 CA178860 (to J.A.M.), R21CA188881 (to J.A.M.), R35 CA197588 (to L.C.C.), R01 CA179483 (to N.S.G.), and U54 HL127365 (to N.S.G.), T32 GM007306 (to Z.D.), the Chleck Foundation (to Z.D.), and by the Dana-Farber Strategic Research Initiative (to J.A.M.).
Footnotes
DATA AVAILABILITY
All raw mass spectrometry data will be uploaded to MassIVE (ftp://massive.ucsd.edu/MSV000083231). Python scripts for CITe-Id analysis are available for download on GitHub (https://github.com/BlaisProteomics/CITe-Id). All other data are included in this published article and its Supporting Information files.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b07911.
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(PDF)
L.C.C. is a founder and member of the SAB and holds equity in Agios Pharmaceuticals and Petra Pharmaceuticals. L.C.C. is also a member of the BOD of Agios and an observer on the BOD of Petra. Petra provides partial support for his laboratory. Both Agios and Petra are developing drugs for cancer therapies. Jarrod Marto is a member of the scientific advisory board (SAB) of 908 Devices. N.G. is a founder, SAB member and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Her2llc, Deerfield, and Sanofi. N.G., T.Z., and N.K. are inventors on a patent application covering chemical matter in this publication owned by Dana–Farber Cancer Institute.
REFERENCES
- (1).Pan Z; et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. [DOI] [PubMed] [Google Scholar]
- (2).Li D; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu Q; et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhang J; Yang PL; Gray NS Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [DOI] [PubMed] [Google Scholar]
- (5).Kwiatkowski N; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang T; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol 2016, 12, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhao Z; Liu Q; Bliven S; Xie L; Bourne PE Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem 2017, 60, 2879–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Weerapana E; et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Weerapana E; Speers AE; Cravatt BF Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)-a general method for mapping sites of probe modification in proteomes. Nat. Protoc 2007, 2, 1414–1425. [DOI] [PubMed] [Google Scholar]
- (10).Abo M; Weerapana E A Caged Electrophilic Probe for Global Analysis of Cysteine Reactivity in Living Cells. J. Am. Chem. Soc 2015, 137, 7087–7090. [DOI] [PubMed] [Google Scholar]
- (11).Backus KM; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bak DW; Pizzagalli MD; Weerapana E Identifying Functional Cysteine Residues in the Mitochondria. ACS Chem. Biol 2017, 12, 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Deng X; et al. Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe 2013, 13, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Quinti L; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E4676–E4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang C; Weerapana E; Blewett MM; Cravatt BF A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 2014, 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bar-Peled L; et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171, 696–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Blewett MM; et al. Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci. Signaling 2016, 9, rs10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lanning BR; et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol 2014, 10, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Niessen S; et al. Proteome-wide Map of Targets of T790M-EGFR-Directed Covalent Inhibitors. Cell Chem. Biol 2017, 24, 1388–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ficarro SB; Browne CM; Card JD; Alexander WM; Zhang T; Park E; McNally R; Dhe-Paganon S; et al. Leveraging gas-phase fragmentation pathways for improved identification and selective detection of targets modified by covalent probes. Anal. Chem 2016, 88, 12248–12254. [DOI] [PubMed] [Google Scholar]
- (21).Leenders F; et al. PKN3 is required for malignant prostate cell growth downstream of activated PI 3-kinase. EMBO J. 2004, 23, 3303–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Unsal-Kacmaz K; et al. The interaction of PKN3 with RhoC promotes malignant growth. Mol. Oncol 2012, 6, 284–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhou F; et al. Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun 2013, 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ficarro SB; et al. Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol. Cell. Proteomics 2011, 10, No. O111.011064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhou F; Sikorski TW; Ficarro SB; Webber JT; Marto JA Online nanoflow reversed phase-strong anion exchange-reversed phase liquid chromatography-tandem mass spectrometry platform for efficient and in-depth proteome sequence analysis of complex organisms. Anal. Chem 2011, 83, 6996–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schultheis B; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol 2014, 32, 4141–4148. [DOI] [PubMed] [Google Scholar]
- (27).Cerami E; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery 2012, 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gao J; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signaling 2013, 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nguyen DT; et al. Pharos: Collating protein information to shed light on the druggable genome. Nucleic Acids Res. 2017, 45, D995–D1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mukai H; et al. PKN3 is the major regulator of angiogenesis and tumor metastasis in mice. Sci. Rep 2016, 6, 18979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lolli G; Lowe ED; Brown NR; Johnson LN The crystal structure of human CDK7 and its protein recognition properties. Structure 2004, 12, 2067–2079. [DOI] [PubMed] [Google Scholar]
- (32).Patricelli MP; et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol 2011, 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Patricelli MP; et al. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 2007, 46, 350–358. [DOI] [PubMed] [Google Scholar]
- (34).Adelmant G; et al. DNA ends alter the molecular composition and localization of Ku multicomponent complexes. Mol. Cell. Proteomics 2012, 11, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Rozenblatt-Rosen O; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mopert K; Loffler K; Roder N; Kaufmann J; Santel A Depletion of protein kinase N3 (PKN3) impairs actin and adherens junctions dynamics and attenuates endothelial cell activation. Eur. J. Cell Biol 2012, 91, 694–705. [DOI] [PubMed] [Google Scholar]
- (37).Oishi K; Mukai H; Shibata H; Takahashi M; Ona Y Identification and characterization of PKNbeta, a novel isoform of protein kinase PKN: expression and arachidonic acid dependency are different from those of PKNalpha. Biochem. Biophys. Res. Commun 1999, 261, 808–814. [DOI] [PubMed] [Google Scholar]
- (38).Breitwieser FP; et al. General statistical modeling of data from protein relative expression isobaric tags. J. Proteome Res 2011, 10, 2758–2766. [DOI] [PubMed] [Google Scholar]
- (39).Zhang Y; et al. A robust error model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia. Mol. Cell. Proteomics 2010, 9, 780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Berriz GF; Beaver JE; Cenik C; Tasan M; Roth FP Next generation software for functional trend analysis. Bioinformatics 2009, 25, 3043–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bauer RA Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today 2015, 20, 1061–1073. [DOI] [PubMed] [Google Scholar]
- (42).Liu F; et al. Discovery of a Selective Irreversible BMX Inhibitor for Prostate Cancer. ACS Chem. Biol 2013, 8, 1423. [DOI] [PubMed] [Google Scholar]
- (43).Uehara S et al. Protein kinase N3 promotes bone resorption by osteoclasts in response to Wnt5a-Ror2 signaling. Sci. Signaling 2017, 10, eaan0023. [DOI] [PubMed] [Google Scholar]
- (44).Singel SM; et al. Receptor-interacting protein kinase 2 promotes triple-negative breast cancer cell migration and invasion via activation of nuclear factor-kappaB and c-Jun N-terminal kinase pathways. Breast Cancer Res. 2014, 16, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wu S; Kanda T; Nakamoto S; Imazeki F; Yokosuka O Knockdown of receptor-interacting serine/threonine protein kinase-2 (RIPK2) affects EMT-associated gene expression in human hepatoma cells. Anticancer Res. 2012, 32, 3775–3783. [PubMed] [Google Scholar]
- (46).Adams S; Valchanova RS; Munz B RIP2: a novel player in the regulation of keratinocyte proliferation and cutaneous wound repair? Exp. Cell Res 2010, 316, 728–736. [DOI] [PubMed] [Google Scholar]
- (47).Haugsten EM; Oppelt A; Wesche J Phosphatidylinositol 5-phosphate is a second messenger important for cell migration. Commun. Integr. Biol 2013, 6, No. e25446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Oppelt A; et al. PIKfyve, MTMR3 and their product PtdIns5P regulate cancer cell migration and invasion through activation of Rac1. Biochem. J 2014, 461, 383–390. [DOI] [PubMed] [Google Scholar]
- (49).Marinkovich MP; Taylor TB; Keene DR; Burgeson RE; Zone JJ LAD-1, the linear IgA bullous dermatosis autoantigen, is a novel 120-kDa anchoring filament protein synthesized by epidermal cells. J. Invest. Dermatol 1996, 106, 734–738. [DOI] [PubMed] [Google Scholar]
- (50).Falk MD et al. Enzyme kinetics and distinct modulation of the protein kinase N family of kinases by lipid activators and small molecule inhibitors. Biosci. Rep 2014, 34, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Turk BE; Hutti JE; Cantley LC Determining protein kinase substrate specificity by parallel solution-phase assay of large numbers of peptide substrates. Nat. Protoc 2006, 1, 375–379. [DOI] [PubMed] [Google Scholar]
- (52).Parikh JR; et al. multiplierz: an extensible API based desktop environment for proteomics data analysis. BMC Bioinf. 2009, 10, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Alexander WM, Ficarro SB, Adelmant G; Marto JA multiplierz v2.0: A Python-based ecosystem for shared access and analysis of native mass spectrometry data. Proteomics 2017, 17, 1700091. [DOI] [PubMed] [Google Scholar]
- (54).Askenazi M; Parikh JR; Marto JA mzAPI: a new strategy for efficiently sharing mass spectrometry data. Nat. Methods 2009, 6, 240–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ficarro SB, Alexander WM; Marto JA mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data. Proteomes 2017, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Adelmant GO CJ, Ficarro SB, Sikorski TW, Zhang Y, Marto JA in Sample Preparation in Biological Mass Spectrometry; Lazarev AV, Ivanov AR, Eds.; Springer, Ch. 22. [Google Scholar]
- (57).Hughes CS; et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol 2014, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.