

Abstract
Key points
Age‐related arterial dysfunction, characterized by oxidative stress‐ and inflammation‐mediated endothelial dysfunction and arterial stiffening, is the primary risk factor for cardiovascular diseases.
To investigate whether age‐related changes in the gut microbiome may mediate arterial dysfunction, we suppressed gut microbiota in young and old mice with a cocktail of broad‐spectrum, poorly‐absorbed antibiotics in drinking water for 3–4 weeks.
In old mice, antibiotic treatment reversed endothelial dysfunction and arterial stiffening and attenuated vascular oxidative stress and inflammation.
To provide insight into age‐related changes in gut microbiota that may underlie these observations, we show that ageing altered the abundance of microbial taxa associated with gut dysbiosis and increased plasma levels of the adverse gut‐derived metabolite trimethylamine N‐oxide.
The results of the present study provide the first proof‐of‐concept evidence that the gut microbiome is an important mediator of age‐related arterial dysfunction and therefore may be a promising therapeutic target for preserving arterial function with ageing, thereby reducing the risk of cardiovascular diseases.
Abstract
Oxidative stress‐mediated arterial dysfunction (e.g. endothelial dysfunction and large elastic artery stiffening) is the primary mechanism driving age‐related cardiovascular diseases. Accumulating evidence suggests the gut microbiome modulates host physiology because dysregulation (‘gut dysbiosis’) has systemic consequences, including promotion of oxidative stress. The present study aimed to determine whether the gut microbiome modulates arterial function with ageing. We measured arterial function in young and older mice after 3–4 weeks of treatment with broad‐spectrum, poorly‐absorbed antibiotics to suppress the gut microbiome. To identify potential mechanistic links between the gut microbiome and age‐related arterial dysfunction, we sequenced microbiota from young and older mice and measured plasma levels of the adverse gut‐derived metabolite trimethylamine N‐oxide (TMAO). In old mice, antibiotics reversed endothelial dysfunction [area‐under‐the‐curve carotid artery dilatation to acetylcholine in young: 345 ± 16 AU vs. old control (OC): 220 ± 34 AU, P < 0.01; vs. old antibiotic‐treated (OA): 334 ± 15 AU; P < 0.01 vs. OC] and arterial stiffening (aortic pulse wave velocity in young: 3.62 ± 0.15 m s−1 vs. OC: 4.43 ± 0.38 m s−1; vs. OA: 3.52 ± 0.35 m s−1; P = 0.03). These improvements were accompanied by lower oxidative stress and greater antioxidant enzyme expression. Ageing altered the abundance of gut microbial taxa associated with gut dysbiosis. Lastly, plasma TMAO was higher with ageing (young: 2.6 ± 0.4 μmol L−1 vs. OC: 7.2 ± 2.0 μmol L−1; P < 0.0001) and suppressed by antibiotic treatment (OA: 1.2 ± 0.2 μmol L−1; P < 0.0001 vs. OC). The results of the present study provide the first evidence for the gut microbiome being an important mediator of age‐related arterial dysfunction and oxidative stress and suggest that therapeutic strategies targeting gut microbiome health may hold promise for preserving arterial function and reducing cardiovascular risk with ageing in humans.
Keywords: Ageing, arterial stiffness, endothelial function, gut dysbiosis, inflammation
Key points
Age‐related arterial dysfunction, characterized by oxidative stress‐ and inflammation‐mediated endothelial dysfunction and arterial stiffening, is the primary risk factor for cardiovascular diseases.
To investigate whether age‐related changes in the gut microbiome may mediate arterial dysfunction, we suppressed gut microbiota in young and old mice with a cocktail of broad‐spectrum, poorly‐absorbed antibiotics in drinking water for 3–4 weeks.
In old mice, antibiotic treatment reversed endothelial dysfunction and arterial stiffening and attenuated vascular oxidative stress and inflammation.
To provide insight into age‐related changes in gut microbiota that may underlie these observations, we show that ageing altered the abundance of microbial taxa associated with gut dysbiosis and increased plasma levels of the adverse gut‐derived metabolite trimethylamine N‐oxide.
The results of the present study provide the first proof‐of‐concept evidence that the gut microbiome is an important mediator of age‐related arterial dysfunction and therefore may be a promising therapeutic target for preserving arterial function with ageing, thereby reducing the risk of cardiovascular diseases.
Introduction
Advancing age is the primary risk factor for cardiovascular diseases (CVD) (Benjamin et al. 2017). The key pathophysiological events linking ageing to increased CVD risk include the development of vascular endothelial dysfunction and stiffening of the large elastic arteries (Lakatta & Levy, 2003). These adverse changes to arteries with ageing are mediated by increased superoxide‐driven oxidative stress and chronic low‐grade inflammation, which act in a feed‐forward manner to reduce bioavailability of the vasodilatory molecule nitric oxide (NO) and modify structural components of the extracellular matrix (Lakatta, 2003; Fleenor et al. 2010; Seals et al. 2011; Assar et al. 2012). However, the upstream mechanisms driving age‐associated vascular oxidative stress, inflammation and dysfunction are largely unknown.
One possible link between ageing and arterial dysfunction is the gut microbiome, which comprises the collective genomes of the commensal microorganisms that colonize the gut. The gut microbiome is an emerging mediator of host physiology and disease (Clemente et al. 2012), and adverse changes in the gut microbiome, termed ‘gut dysbiosis’, have been causally linked to many diseases, including atherosclerosis (Gregory et al. 2015). Recent studies indicate that high‐fat diet‐induced gut dysbiosis (in young mice) can impair arterial function (Vikram et al. 2016; Battson et al. 2017). Evidence exists for changes in the gut microbiome with primary ageing (i.e. ageing in the absence of overt pathology) (Hopkins et al. 2001; Mariat et al. 2009; Claesson et al. 2011). However, the role of these changes in vascular ageing is presently unknown. Therefore, the primary aim of the present study was to establish proof‐of‐concept evidence that the gut microbiome modulates arterial function with ageing. Accordingly, we treated young and old mice with a cocktail of broad‐spectrum, poorly‐absorbed antibiotics for 3–4 weeks to suppress endogenous microbes. We hypothesized that suppression of the gut microbiome would reverse arterial dysfunction in old mice but would have no effect in young mice. We also examined whether oxidative stress and inflammation, which are key mechanisms of vascular ageing, mediated the effects of the gut microbiome on arterial function.
After determining that the gut microbiome is involved in mediating age‐associated arterial dysfunction, we next aimed to determine what potentially dysbiotic changes occur in the mouse gut microbiome with ageing. Previous studies have observed changes in the gut microbiome with primary ageing consistent with gut dysbiosis; however, the microbial taxa altered most by ageing vary across studies. Investigations in humans are confounded in part by geographical influences (Mueller et al. 2006) and the inclusion of elderly adults who were hospitalized or living in assisted care facilities (Claesson et al. 2012), making it difficult to distinguish the effects of ageing from comorbidities or changing environmental factors. In this regard, studies in rodents offer a better control of environmental factors and isolation of the effects of ageing, although they are limited to date (Langille et al. 2014; Fransen et al. 2017; Scott et al. 2017; Thevaranjan et al. 2017). These investigations reported changes in the abundance of microbial taxa associated with inflammation, suggesting the gut microbiome may be a primary source of systemic pro‐oxidative and pro‐inflammatory signalling with ageing. Therefore, we aimed to confirm that ageing, in a controlled and consistent environment, alters the gut microbiome, and hypothesized that the gut microbiome would differ between young and old mice housed in our facility for the majority of their lifespan (i.e. consistent food, water and environmental factors). Specifically, we expected adverse changes in the abundance of microbial taxa associated with gut dysbiosis.
Lastly, to provide initial insight into a possible signal by which age‐related changes in the gut microbiome may influence oxidative stress, inflammation and arterial function, we measured plasma concentrations of trimethylamine N‐oxide (TMAO) and associated metabolites. TMAO is produced exclusively via microbial conversion of digested precursors (e.g. phosphatidylcholine, betaine and l‐carnitine) into trimethylamine (TMA), which then enters circulation and is converted to TMAO by flavin‐containing monoxygenase (FMO) enzymes in the liver, primarily FMO3 (Bennett et al. 2013). TMAO promotes atherosclerosis (Koeth et al. 2013; Wang et al. 2015) and is associated with an increased CVD risk in humans (Tang et al. 2013). We hypothesized that plasma TMAO would be increased in old mice and suppressed by antibiotic treatment.
Methods
Ethical approval
The Institutional Animal Care and Use Committee at the University of Colorado Boulder reviewed and approved all procedures (protocol #2539). All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and in compliance with the ethical policies and regulations set out in Grundy (2015).
Animals
Young and older male C57BL/6N mice were obtained from Charles River (Wilmington, MA, USA) or the National Institute of Aging colony (maintained by Charles River) at 8–10 weeks or 20–24 months of age, respectively. Male mice of this strain and species demonstrate many clinical aspects of human ageing, including vascular endothelial dysfunction and arterial stiffening (Sindler et al. 2011; Fleenor et al. 2013; LaRocca et al. 2013), whereas female mice do not because their hormone profile is not consistent with female older adult humans. In total, 50 young and 50 older mice were obtained. Data were collected and reported from 35 and 38 mice, respectively; other mice were used for pilot testing, died naturally or were euthanized based on veterinary recommendation prior to completion of the intervention.
Mice were single‐housed in a conventional facility under a 12:12 hour light/dark photocycle with access to standard rodent chow and normal drinking water (or water supplemented with broad‐spectrum antibiotics) available ad libitum from at least 4 weeks prior to baseline testing (antibiotic experiments) or from 8–10 weeks of age (gut microbiome sequencing). For antibiotic experiments, mice were euthanized prior to terminal measures by exsanguination via cardiac puncture during maintained inhaled isoflurane anaesthesia. Mice used for gut microbiome sequencing were not euthanized and were used for other studies.
Antibiotic intervention
To investigate whether the gut microbiome modulates arterial function with ageing, young (5–6 months) and old (26–27 months) C57BL/6N male mice were treated without (control) or with a cocktail of broad‐spectrum, poorly‐absorbed antibiotics in drinking water (1.0 g L−1 ampicillin, 1.0 g L−1 neomycin sulphate, 1.0 g L−1 metronidazole and 0.5 g L−1 vancomycin) to suppress endogenous gut microbes. This antibiotic cocktail has been shown to substantially deplete all detectable commensal bacteria (Rakoff‐Nahoum et al. 2004; Carvalho et al. 2012; Battson et al. 2017). Group abbreviations are: young control (YC) (n = 15); young antibiotic‐treated (YA) (n = 11); old control (OC) (n = 11); and old antibiotic‐treated (OA) (N = 10).
To confirm suppression of the microbiome, fresh faecal samples were collected at baseline and after 3–4 weeks of antibiotics or normal drinking water. All samples were collected immediately upon excretion, were quickly sealed in air‐tight containers (within 20 s after excretion), frozen as soon as possible and stored at –80°C until analysis. DNA was extracted (PureLink Microbiome DNA Purification Kit, A29790; Invitrogen, Carlsbad, CA, USA) and a quantitative PCR was used to determine total microbial DNA counts (reactions optimized for 16s rRNA gene), as described previously (Battson et al. 2017).
To determine which taxa remained after antibiotic treatment, 16S rRNA genes were PCR amplified at the V4 region and subjected to multiplex Illumina sequencing and data processing, as described in Stull et al. (2018). Briefly, paired‐end sequence reads were concatenated and all combined 16S sequences were filtered, trimmed and processed using the default pipeline in DADA2 (R bioconductor package, myPhyloDB, version 1.2.1) (Callahan et al. 2016). Each sequence variant identified in DADA2 was classified to the closest reference sequence contained in the Greengenes reference database, version 13.5.99 (http://greengenes.secondgenome.com) using the usearch_global option (minimum identity of 97%) contained in the open source program VSEARCH (Rognes et al. 2016). Effects of age and treatment on relative abundance of microbial phyla and families were determined by analysis of covariance (ANCOVA) in myPhyloDB (Manter et al. 2016).
Vascular endothelial function
After 3–4 weeks of the intervention, vascular endothelial function was assessed by ex vivo carotid artery endothelium‐dependent dilatation (EDD) in response to increasing doses of ACh, as described previously (Rippe et al. 2010). Briefly, after preconstriction with phenylephrine (2 μmol L−1; Sigma‐Aldrich Corp., St Louis, MO, USA), EDD was assessed by measuring the increase in luminal diameter in response to increasing concentrations of ACh (1 × 10−9 to 1 × 10−4 mol.l−1; Sigma‐Aldrich Corp.), first in the absence and then in the presence of the nitric oxide synthase (NOS) inhibitor NG‐nitro‐l‐arginine methyl ester (l‐NAME; 0.1 mmol.l−1, 30 min pre‐incubation; Sigma‐Aldrich Corp.). NO‐mediated dilatation was calculated as the difference between peak dilatation to ACh alone and in the presence of l‐NAME. Endothelium independent dilatation (EID) was measured as dilatation in response to increasing doses of the exogenous NO donor sodium nitroprusside (SNP) (1 × 10−10 to 1 × 10−4 mol.l−1; Sigma‐Aldrich Corp.). To account for baseline differences in vessel diameter, all dose response data are reported as a percentage of maximal dilatation.
In vivo large elastic artery stiffness and arterial blood pressure
Aortic pulse wave velocity (PWV), the gold standard measure of large elastic artery stiffness, was measured at baseline and after the intervention using Doppler ultrasonography under anaesthesia (2% isoflurane with oxygen adjusted to maintain heart rate between 400–500 beats min–1), as described previously (Fleenor et al. 2012). PWV was calculated as the distance between probes divided by the difference in pre‐ejection times (time between ECG R‐wave and foot of the Doppler signal) of the thoracic and abdominal aortic regions. To examine the potential contribution of changes in arterial blood pressure to any treatment‐related differences in aortic PWV, systolic and diastolic blood pressure were assessed using a CODA non‐invasive tail‐cuff system (Kent Scientific, Torrington, CT, USA), as described previously (Fleenor et al. 2012).
Ex vivo intrinsic mechanical stiffness
Intrinsic mechanical stiffness was assessed in 1–2 mm segments of thoracic aorta via wire myography, as described previously (Fleenor et al. 2012; Gioscia‐Ryan et al. 2018). The thoracic aorta was dissected free of surrounding tissue and sectioned into rings 1 mm in length. After pre‐stretching, the ring diameter was increased to achieve a force of 1 mN force and then incrementally stretched by ∼10% every 3 min until failure. The force corresponding to each stretching interval was recorded and used to calculate stress and strain, and to generate a stress–strain curve:
where D is diameter and d(i) is the initial diameter.
where L is one‐dimensional load, H is wall thickness and D is vessel length.
The elastic modulus of the collagen‐dominant (highest force) region of the stress–strain curve was determined as the slope of a linear equation fit to the final four points of the stress–strain curve (Fleenor et al. 2012; Gioscia‐Ryan et al. 2018). The boundaries of the elastin‐dominant region (low force region where curvature is ∼0) of the stress–strain curve were determined by fitting a seventh‐order polynomial equation to the data (r 2 > 0.99; RStudio, Boston, MA, USA) and then calculating the roots of the equation; the first root was considered the boundary between the very low‐force region and the elastin region, and the second root was considered the boundary between the elastin region and the onset of collagen fibre engagement (Lammers et al. 2008; Gioscia‐Ryan et al. 2018). The elastic modulus of the elastin region was then determined as the slope of a linear equation fit to the stress‐strain data between the two roots.
Aortic superoxide production
Superoxide production was measured in 1 mm segments of thoracic aorta using electron paramagnetic resonance spectroscopy with the superoxide‐specific spin probe 1‐hydroxy‐3methoxycarbonyl‐2,2,5,5‐tetramethylpyrrolidine (0.5 mm) (Enzo Life Sciences Inc., Farmington, NY, USA), as described previously (Rippe et al. 2010).
NOS activity assay
Activity of NOS was determined in aortic lysates using the Ultrasensitive Colorimetric Assay for Nitric Oxide Synthase (Oxford Biomedical Research, Rochester Hills, MI, USA) in accordance with the manufacturer's instructions. Values were normalized to the protein content of each sample and expressed as μmol NO μg–1 aortic protein.
Aortic and liver protein expression
Protein expression was determined in thoracic aorta and liver lysates, loading 20 μg and 2 μg of protein, respectively (larger amounts of liver protein resulted in overexposure of bands), using standard western immunoblotting procedures, as described previously (Rippe et al. 2010; Gioscia‐Ryan et al. 2018). Primary antibodies used for aortic protein were ⍺‐elastin (dilution 1:200; Abcam, Cambridge, MA, USA; catalogue no. ab21607, RRID:AB_446421), collagen type‐1 (dilution 1:1000; EMD Millipore, Burlington, MA, USA; catalogue no. AB765P, RRID:AB_92259), nitrotyrosine (HM.11) (dilution 1:1000; Abcam; catalogue no. ab7048, RRID:AB_305725), extracellular superoxide dismutase (ecSOD; dilution 1:2000; Sigma‐Aldrich Corp.; catalogue no. S4946, RRID:AB_532286), manganese SOD (MnSOD; dilution 1:1000; Enzo Life Sciences; catalogue no. ADI‐SOD‐110, RRID: AB_10616816) and copper/zinc SOD (Cu/ZnSOD; dilution 1:2000; Enzo Life Sciences; catalogue no. ADI‐SOD‐100, RRID: AB_10616253). Primary antibodies used for liver protein were flavin‐containing monoxygenase 3 (EPR6968) (FMO3; dilution 1:5000; Abcam; catalogue no. 126711, RRID: AB_11129340). Relative intensity of bands was normalized to intensity of glyceraldehyde‐3‐phosphate dehydrogenase (14C10) (GAPDH; dilution 1:1000; Cell Signaling Technology, Inc., Beverly, MA, USA; catalogue no. 2118, RRID: AB_561053).
Aortic inflammation
Concentrations of aortic inflammatory cytokines interleukin (IL)‐6, tumor necrosis factor (TNF)‐α and interferon (IFN)‐γ were measured using a commercially‐available multiplex enzyme‐linked immunosorbent assay kit (Ciraplex® Mouse Cytokine Array 1; Aushon BioSystems, Inc., Billerica, MA, USA), using 10–20 μg of thoracic aortic lysate.
Age‐related changes in the gut microbiome
Mice obtained at 8–10 weeks of age were housed in individual cages in the same vivarium room for the remainder of their lifespan. Serial faecal samples were collected from young (n = 8) and older (n = 17) mice at 8 and 10 months of age and 15, 18 and 24 months of age, respectively. Mice from young and older groups were housed at overlapping times and were fed the same batch of standard rodent chow. Thus, environmental factors were controlled as tightly as possible. Microbiome diversity and relative abundance of major microbial phyla remained stable over time within age groups (i.e. from 8–10 months and 15–24 months) and thus all data have been included as separate data points, increasing the samples sizes presented per age group to n = 14 (young) and n = 37 (older).
Bacterial DNA was isolated from faeces using the MO BIO PowerSoil DNA extraction kit (Qiagen, Valencia, CA, USA) and 16S rRNA genes were PCR amplified at the V4 region and subjected to multiplex Illumina sequencing (Caporaso et al. 2011). Raw sequences were deposited in EBI‐ENA (ERP107189), uploaded to Qiita (Study ID: 2248) (González et al. 2018) and processed in Qiita using default parameters via the deblur pipeline. In brief, we demultiplexed and quality control sequenced, as defined by Bokulich et al. 2013. Remaining sequences were denoized via deblur (Amir et al. 2017). Sequences then were inserted in the Greengenes tree using SEPP (Mirarab et al. 2012; Janssen et al. 2018). The resulting files were processed via QIIME2 (Caporaso et al. 2010). Deblurred sequences were assigned a taxonomic classification using the feature‐classifier/classify‐sklearn plugin in QIIME2 using default parameters. For alpha and beta diversity, tables were rarefied at 5000 sequences per sample (Kuczynski et al. 2010). An analysis of composition of microbiomes (ANCOM) was used to determine differential abundance of taxa (Mandal et al. 2015) between young and old mice. This analysis generates a W score, which is the count of the number of subhypotheses that have passed for a given taxon. No rarefaction was performed for this analysis, although samples with less than 5000 sequences were removed.
Plasma TMAO analysis
To investigate a potential link between suppression of the gut microbiome and improved arterial function, levels of TMAO, TMA and l‐carnitine were quantified in heparinized plasma by isocratic ultra‐performance liquid chromatography‐tandem mass spectroscopy using a stable isotope dilution method against internal standards (Wang et al. 2014; Boutagy et al. 2015 a, 2015 b).
Statistical analysis
Statistical analyses were conducted in R, version 3.4.1 (mixed design analyses) (R Foundation for Statistical Computing, Vienna, Austria) and Prism, version 7 (all other analyses) (GraphPad Software, Inc., La Jolla, CA, USA). Data were first assessed for outliers (ROUT method, Q = 1%) and normality (Shapiro–Wilk normality test, P > 0.05) within groups. Differences in PWV, blood pressure (systolic blood pressure, diastolic blood pressure, mean arterial pressure) and carotid artery dose responses were assessed using two‐way mixed design ANOVA with a between factor of group and repeated factor of either time (PWV and blood pressures, pre‐ vs. post‐intervention) or dose (carotid artery EDD and EID). Differences across animal groups in morphological and artery characteristics, water intake, food intake, faecal DNA copies, NO‐mediated dilatation, NOS activity, superoxide production, inflammatory cytokines, and TMAO and related metabolites were assessed using one‐way ANOVA. When significant main effects were detected, pairwise comparisons were made using the Holm–Sidak post hoc test. Differences in western blot protein markers and elastin and collagen elastic moduli were assessed using Student's unpaired t test with the comparisons: YC vs. OC, YC vs. OA and OC vs. OA. Significance was set to α = 0.05. Unless otherwise noted, data are presented as the mean ± SEM.
Results
Animal characteristics
Body mass at death and water and food intake across the intervention are presented in Table 1. Both young and old mice had an initial aversion to the taste of the antibiotic water resulting in a temporary reduction in daily water intake, which has also been reported in previous studies (Reikvam et al. 2011; Battson et al. 2017). However, all mice resumed normal drinking behaviour (i.e. ≥1.5 mL day–1) in 9.1 ± 1.1 days (YA) and 4.7 ± 0.9 days (OA, P < 0.01 vs. YA) and were drinking regularly for at least 3 weeks prior to death and terminal measurements. Food intake was also lower in antibiotic‐treated mice during the first week of the intervention, although this normalized such that there were no differences in food intake across groups by the second week (P = 0.23). In old mice, body weight at death (P = 0.57), daily water intake during the second and last weeks of the intervention (P = 0.56), and daily food intake during the last week of the intervention (P = 0.97) were not different between antibiotic‐treated and control animals. Although young antibiotic‐treated mice drank regularly, average daily intake still tended to be less than control mice (second week: P = 0.12 vs. YC; last week: P = 0.21 vs. YC). As such, YA mice did not fully recover the loss in body weight associated with dehydration that occurred in the first week and body mass at death was significantly lower than YC mice (P = 0.02).
Table 1.
Body mass, water and food intake, artery characteristics and blood pressure
| YC | YA | OC | OA | |
|---|---|---|---|---|
| Body mass (g) | 31.3 ± 1.4 | 25.9 ± 1.0* | 27.1 ± 1.3 | 28.9 ± 1.0 |
| Water intake (mL day−1) | ||||
| First week | 3.8 ± 0.1 | 0.9 ± 0.1* | 4.2 ± 0.5 | 2.1 ± 0.4*† |
| Second week | 3.7 ± 0.2 | 2.6 ± 0.4 | 4.3 ± 0.4 | 3.6 ± 0.5 |
| Last week | 3.7 ± 0.1 | 2.8 ± 0.1 | 4.4 ± 0.5 | 4.0 ± 0.5 |
| Food intake (kcal day−1) | ||||
| First week | 13.0 ± 0.4 | 8.4 ± 0.6* | 12.7 ± 0.7 | 7.7 ± 0.5*† |
| Second week | 12.4 ± 0.4 | 11.0 ± 1.0 | 12.7 ± 0.8 | 11.0 ± 0.9 |
| Last week | 12.3 ± 0.5 | 13.2 ± 0.5 | 11.8 ± 0.7 | 12.0 ± 0.9 |
| Carotid | ||||
| Resting diameter (μm) | 359 ± 7 | 350 ± 10 | 371 ± 11 | 388 ± 11 |
| Maximal diameter (μm) | 401 ± 7 | 419 ± 12 | 435 ± 9* | 443 ± 8* |
| Aorta | ||||
| Diameter (μm) | 674 ± 20 | 619 ± 19 | 662 ± 24 | 708 ± 26 |
| Intima media thickness (μm) | 33 ± 1 | 32 ± 1 | 42 ± 2* | 43 ± 2* |
| Systolic blood pressure (mmHg) | ||||
| Pre | 103 ± 3 | 102 ± 3 | 96 ± 3 | 93 ± 3 |
| Post | 101 ± 2 | 99 ± 2 | 95 ± 3 | 98 ± 3 |
| Diastolic blood pressure (mmHg) | ||||
| Pre | 77 ± 3 | 76 ± 3 | 69 ± 4 | 69 ± 2 |
| Post | 75 ± 2 | 70 ± 3‡ | 66 ± 2* | 66 ± 2* |
Data are the mean ± SEM. * P < 0.05 vs. YC. †P < 0.05 vs. OC. ‡P < 0.05 vs. pre‐intervention within group. Data were averaged across left and right carotid arteries per mouse. Carotid maximal diameter determined as dilatation to sodium nitroprusside and/or calcium‐free media. Non‐invasive in vivo arterial blood pressure measured via tail cuff method. YC, young control; YA, young antibiotic‐treated; OC, old control; OA, old antibiotic‐treated.
Aorta and carotid artery characteristics are presented in Table 1. Relative to young control animals, old mice had greater maximal carotid artery diameter and intima medial aortic wall thickness (see also Fig. 3 G), although there were no effects of antibiotics on these characteristics in either young or old mice.
Figure 3. Antibiotic treatment reverses age‐related arterial stiffening.
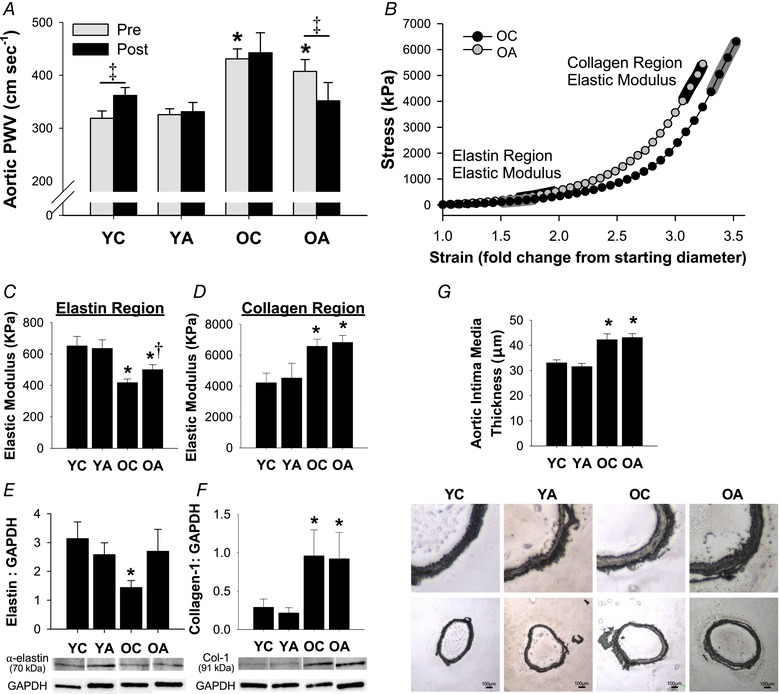
A, aortic pulse wave velocity pre‐ and post‐intervention; n = 9–15 per group. B, representative stress‐strain curve of an aortic ring from OC and OA mice for determination of ex vivo intrinsic mechanical stiffness. C and D, elastic modulus of the elastin (C) and collagen (D) portions of the stress‐strain curve in aortic rings; n = 8–10 per group. E and F, aortic protein expression of elastin (E) and mature‐type collagen‐1 (F) normalized to GAPDH (loading control), with representative western blot images included below; n = 8–10 per group. G, intima media thickness of the aorta, with representative images of whole aortic sections (bottom) and enlargements of the same sections (top) included below the mean data; n = 8–10 per group. Data are the mean ± SEM. * P < 0.05 vs. YC. †P < 0.05 vs. OC. ‡P < 0.05 pre‐ vs. post‐intervention within groups. YC, young control; YA, young antibiotic‐treated; OC, old control; OA, old antibiotic‐treated. [Color figure can be viewed at wileyonlinelibrary.com]
Antibiotic‐mediated suppression of the gut microbiome
Antibiotic treatment was sufficient to suppress the microbiome, as indicated by significantly lower number of faecal DNA copies in both young (P < 0.001 vs. YC) and old (P = 0.04 vs. OC) antibiotic‐treated mice (Fig. 1 A). To confirm the suppressive effects of antibiotics, baseline faeces (i.e. before antibiotic treatment was initiated) were also analysed in a subset of mice, indicating that antibiotic treatment reduced the number of faecal DNA copies within mice (pre‐to‐post treatment: YA, P = 0.001; OA, P = 0.04) (Fig. 1 B).
Figure 1. Antibiotic treatment suppresses the gut microbiome.
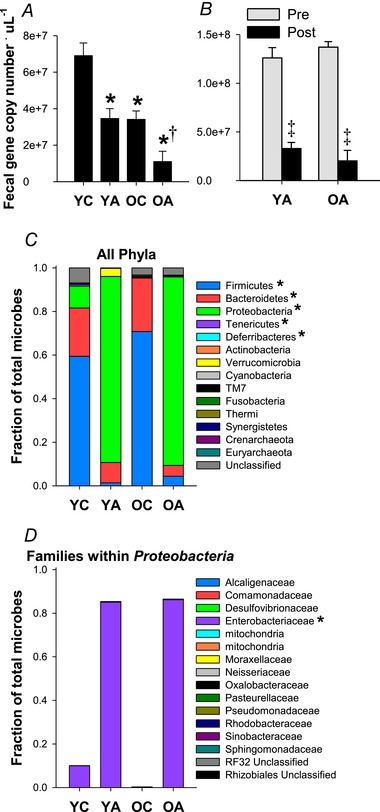
A, number of DNA copies measured by qPCR in faecal samples collected after 3–4 weeks on the intervention. Samples were run in duplicate and these data were confirmed and averaged across two separate qPCR runs. No batch effects were detected (P > 0.05 across batches, within animals, for all groups); n = 10–14 per group. B, number of DNA copies in faecal samples collected pre‐ and 3–4 weeks post‐antibiotic treatment; n = 4–6 per group. Data are the mean ± SEM. * P < 0.05 vs. YC. †P < 0.05 vs. OC. ‡P < 0.05 vs. pre‐intervention within groups. C and D, average relative abundance of all detected microbial phyla (C) and microbial families within the phylum Proteobacteria (D). *Significant effect (P < 0.05) of treatment (control vs. antibiotics) on ANCOVA.
To determine which microbial taxa remained after antibiotic treatment, we performed 16S rRNA sequencing. Relative abundance of most major phyla was significantly lower in antibiotic‐treated mice, including Firmicutes (ANCOVA treatment effect: P < 0.001), Bacteroidetes (P = 0.02), Deferribacteres (P = 0.001) and Tenericutes (P = 0.02); however, relative abundance of the phylum Proteobacteria was significantly higher (P < 0.0001) (Fig. 1 C). This increase was almost entirely attributable to increased relative abundance of unclassified species within the family Enterobacteriaceae (P < 0.0001), which accounted for 85 ± 11% (YA) and 86 ± 33% (OA) of microbes that remained after treatment with antibiotics (Fig. 1 D).
Vascular endothelial function
EDD was significantly impaired in old vs. young control mice (area‐under‐the‐curve EDD: YC, 345 ± 16% vs. OC, 220 ± 34%, P < 0.01) (Fig. 2 A). The age‐related decline in EDD was mediated by reduced NO‐mediated dilatation, as indicated by a lesser reduction in EDD induced by pre‐incubation with the NOS inhibitor l‐NAME (YC vs. OC: P = 0.03) (Fig. 2 B). Antibiotic treatment in old mice restored both EDD (area‐under‐the‐curve EDD, OA: 334 ± 15%, P < 0.01 vs. OC, P = 0.98 vs. YC) and NO‐mediated dilatation (P = 0.87 vs. YC) back to young control levels, whereas no effects of antibiotic treatment were observed in young mice (area‐under‐the‐curve EDD, YA: 340 ± 25%, P = 0.98 vs. YC; NO‐mediated dilatation: P = 0.92 vs. YC). Importantly, these effects were endothelium‐dependent because smooth muscle sensitivity to NO was not different across groups, as assessed by dilatation to increasing doses of the NO donor SNP (main effect of group on SNP dose response: P = 0.52) (Fig. 2 C).
Figure 2. Antibiotic treatment restores vascular endothelial function in old mice via improved NO bioavailability.
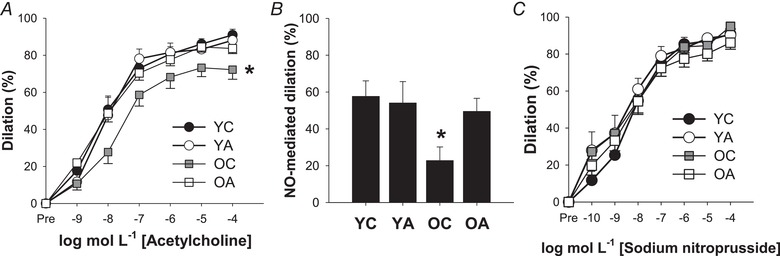
A, dose–response endothelium‐dependent dilatation (EDD) to acetylcholine (ACh). B, peak NO‐mediated dilatation to ACh, assessed as the difference between peak EDD in the absence vs. presence of the NO synthase inhibitor l‐NAME. C, dose–response endothelium independent dilatation to the NO donor SNP. Data are the mean ± SEM. * P < 0.05 vs. YC. YC, young control; YA, young antibiotic‐treated; OC, old control; OA, old antibiotic‐treated.
The total activity of NOS, the enzyme that produces NO, was lower in old vs. young mice (YC: 0.22 ± 0.02 vs. OC: 0.16 ± 0.04 μg NO μg–1 total protein, P = 0.04). However, there were no effects of antibiotic treatment on NOS activity in either young (YA: 0.21 ± 0.02 μg NO μg–1 total protein, P = 0.86 vs. YC) or old mice (OA: 0.13 ± 0.06 μg NO μg–1 total protein, P = 0.33). This finding indicates that improvements in NO bioavailability in old antibiotic‐treated mice were probably mediated by other mechanisms, such as reduced superoxide production and therefore reduced scavenging of NO.
Aortic stiffness
In vivo stiffness: aortic pulse wave velocity
Data are presented in Fig. 3 A. At baseline, old mice had stiffer arteries than young mice, as indicated by higher aortic PWV (P < 0.01 for OC and OA vs. YC). In young mice, we observed a slight but significant increase in aortic PWV across the intervention (P = 0.003), which was prevented in young antibiotic‐treated mice (P = 0.78 vs. pre‐intervention). In old mice, antibiotic treatment reversed age‐related increases in aortic PWV (P = 0.03 vs. pre‐intervention) to levels that were not significantly different from young mice (P = 0.32 vs. YC pre‐intervention). The effects of antibiotics on aortic PWV were accompanied by slight reductions in diastolic blood pressure in young mice, as measured non‐invasively in vivo using the tail cuff method; however, antibiotic treatment did not influence systolic blood pressure in either young nor old animals (Table 1).
Ex vivo intrinsic wall stiffness
To investigate whether reductions in aortic PWV with antibiotic treatment in old mice were associated with structural changes to the arterial wall, we measured the intrinsic mechanical stiffness of the aorta and aortic expression of the major structural proteins, elastin and collagen, via western blotting. Figure 3 B shows a representative stress–strain curve with the collagen and elastin regions of the curve indicated.
The elastic modulus of the elastin region of the stress‐strain curve was lower in old vs. young control mice (P < 0.01). Antibiotic treatment in old mice was associated with a partial improvement back towards young levels (P = 0.047 vs. OC) (Fig. 3 C). Aortic elastin protein expression was lower in old vs. young control mice (P = 0.02) but was restored in old mice treated with antibiotics back to young levels (P = 0.65 vs. YC, P = 0.10 vs. OC) (Fig. 3 E). By contrast, although the elastic modulus of the collagen portion of the stress–strain curve (P = 0.04 vs. YC) and collagen‐1 protein expression (P = 0.02 vs. YC) were or tended to be increased in old control mice, neither were affected by antibiotic treatment (collagen EM: P = 0.71 OC vs. OA, P < 0.01 YC vs. OA; collagen‐1 protein expression: P = 0.76 OC vs. OA) (Fig. 3 D and F). Antibiotic treatment in young mice had no effect on aortic structural properties, nor elastin or collagen protein expression.
Vascular superoxide production and oxidative stress
Aortic superoxide production was greater in old control mice compared to young mice (P = 0.02 vs. YC) and this was normalized in old mice after antibiotic treatment (P = 0.73 vs. YC) (Fig. 4 A). Similarly, aortic expression of nitrotyrosine, a marker of cellular oxidative stress‐mediated protein modification, was higher in old control mice compared to young mice (P < 0.01 vs. YC) but attenuated close to young levels in old antibiotic‐treated mice (Fig. 4 B).
Figure 4. Antibiotic treatment normalizes oxidative stress and inflammation in old mice.
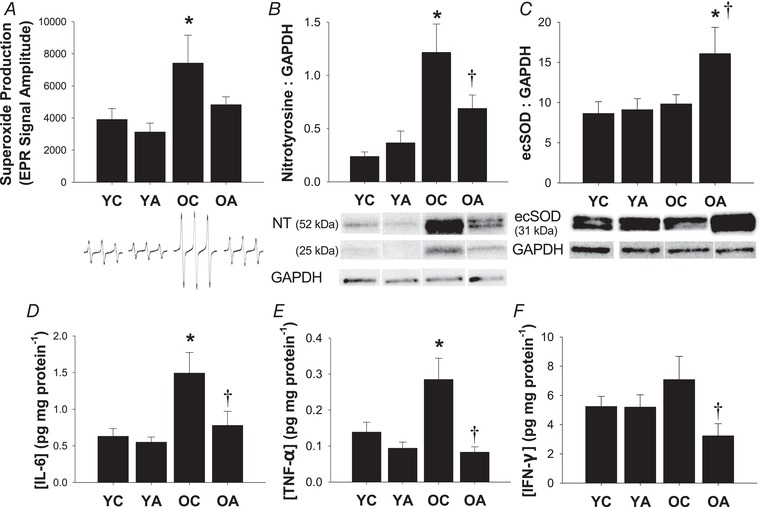
A, whole‐cell superoxide production in aortic segments. Representative electron paramagnetic resonance spectra are shown below. Protein expression of nitrotyrosine (NT) (B) and ecSOD (C). Representative western blot images for NT (data are sum of the two bands), ecSOD and GAPDH (loading control) are shown below. D–F, concentrations of pro‐inflammatory cytokines in aortic lysates: IL‐6 (D), TNF‐α (E) and IFN‐γ (F). Data are the mean ± SEM; n = 7–11 per group. * P < 0.05 vs. YC. †P < 0.05 vs. OC. YC, young control; YA, young antibiotic‐treated; OC, old control; OA, old antibiotic‐treated.
To determine whether reductions in vascular oxidative stress were associated with upregulation of antioxidant enzymes, we measured aortic protein expression of the three isoforms of superoxide dismutase (SOD) via western blotting. Age‐related differences in Cu/ZnSOD (intracellular isoform; YC: 1.0 ± 0.1 vs. OC: 0.7 ± 0.1 AU, P = 0.03) and MnSOD (mitochondrial isoform; YC: 6.5 ± 1.2 vs. OC: 4.0 ± 0.7 AU, P = 0.045), were unaffected by antibiotic treatment (Cu/ZnSOD, YA: 1.1 ± 0.2 AU, P = 0.73 vs. YC, OA: 0.85 ± 0.2 AU, P = 0.40 vs. OC; MnSOD, YA: 6.4 ± 0.9 AU, P = 0.92 vs. YC; OA: 4.5 ± 1.3 AU, P = 0.74 vs. OC). By contrast, despite no age‐related difference in the expression of ecSOD in control mice, ecSOD expression was markedly higher in old antibiotic‐treated mice (P = 0.04 vs. YC, P = 0.06 vs. OC) (Fig. 4 C). Antibiotic treatment in young mice had no effect on aortic markers of oxidative stress, nor aortic antioxidant protein expression.
Vascular inflammation
We measured aortic pro‐inflammatory cytokines because vascular inflammation is a key mechanism underlying arterial ageing that may be influenced by the gut microbiome. Aortic concentrations of IL‐6, TNF‐α and IFN‐γ were or tended to be higher in old vs. young control mice (Fig. 4 D–F). Most importantly, antibiotic treatment reversed aortic inflammation in the old mice, such that aortic concentrations of all pro‐inflammatory cytokines were lower in the old antibiotic‐treated mice compared to the old control animals (P < 0.05) and were not different from the young groups.
Effects of ageing on the gut microbiome
Principal co‐ordinate analysis based on 16S rRNA amplicon unweighted UniFrac distances (Lozupone & Knight, 2005) demonstrated distinct clustering of faecal samples from young and older mice housed in individual cages in the same vivarium room from 8–10 weeks of age (Fig. 5 A). Furthermore, alpha diversity (Faith's phylogenetic diversity) (Faith, 1992), a measure of the number of taxa present within each sample, was increased in older mice (P < 0.0005) (Fig. 5 B).
Figure 5. Ageing alters the mouse gut microbiome.
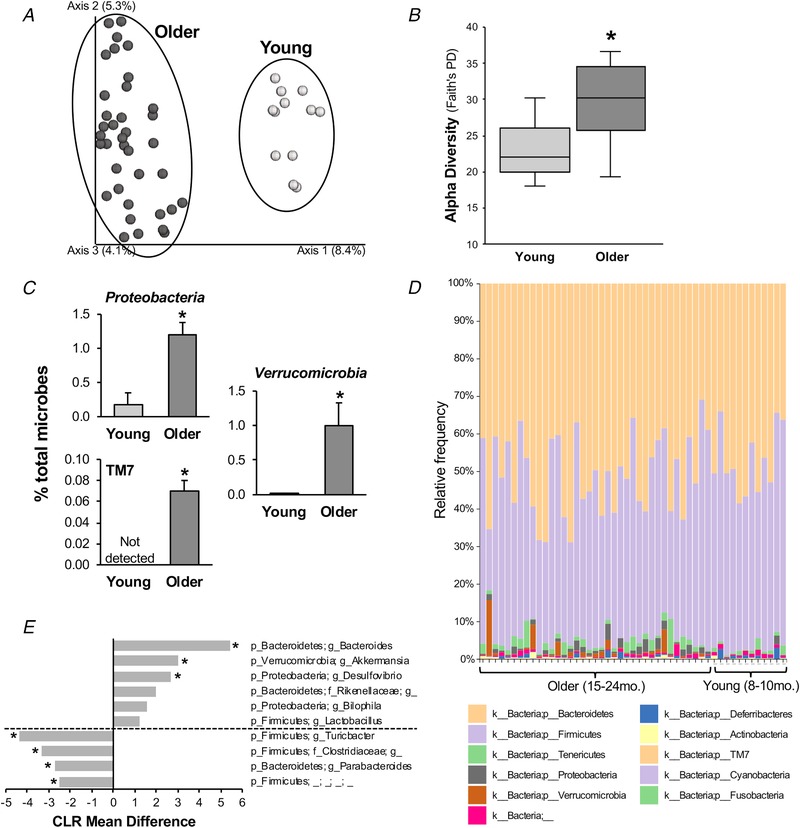
A, principal co‐ordinate analysis plot of 16S rRNA‐based gut microbial profiling with unweighted UniFrac from young (8–10 months) and older (15–24 months) mice fed a standard diet. B, alpha diversity [Faith's phylogenetic diversity (PD)] in young and older mice. Data are box‐and‐whisker plot. * P < 0.05 vs. young. C, average relative abundance (mean ± SE) of bacterial phyla significantly altered by ageing. * P < 0.05 vs. young. D, relative abundance of bacterial phyla across individual samples from older and young mice. E, centred log ratio (CLR) mean differences (F statistic) in differentially abundant bacterial genera between older vs. young mice, as determined by ANCOM analysis. ‘p’, phylum; ‘f’, family; ‘g’, genus. ‘g__’ and the next highest known order are given for operational taxonomic units that do not have a specific genus name. *Statistical significance.
At the phyla level, an ANCOM revealed differential abundance in old vs. young mice in Proteobacteria (W = 9) and the candidate division TM7 (W = 10) (Fig. 5 C), both of which have been associated with gut dysbiosis (Kuehbacher et al. 2008; Ooi et al. 2013), as well as in Verrucomicrobia (W = 9). No significant differences in the abundance of the other phyla analysed were observed (Fig. 5 D and Table 2). Genera that were altered by ageing are summarized in Fig. 5 E and Table 2. Of note, ageing was associated with differential abundance of the pro‐inflammatory and TMA‐producing genus Desulfovibrio (W = 155).
Table 2.
Relative abundance of microbial phyla and genera
| Abundance (%) | ||||
|---|---|---|---|---|
| Young | Older | W score | Centred log ratio mean difference F statistic | |
| Phyla altered by ageing | ||||
| Proteobacteria | 0.02 (0.01, 0.05) | 0.78 (0.38, 1.58) | 9* | 2.569 |
| Verrucomicrobia | 0.01 (0.01, 0.01) | 0.22 (0.13, 0.97) | 9* | 2.377 |
| TM7 | Not detected | 0.08 (0.03, 0.12) | 10* | 0.971 |
| Phyla not significantly altered by ageing | ||||
| Bacteroidetes | 42.3 (32.8, 56.7) | 49.1 (43.1, 57.8) | 3 | –0.663 |
| Firmicutes | 46.5 (41.3, 53.2) | 40.8 (33.1, 54.8) | 3 | –0.884 |
| Tenericutes | 0.75 (0.50, 1.34) | 1.17 (0.75, 2.47) | 3 | –0.423 |
| Deferribacteres | 0.31 (0.11, 0.78) | 0.11 (0.04, 0.38) | 3 | –1.602 |
| Actinobacteria | 0.15 (0.10, 0.20) | 0.25 (0.12, 0.38) | 3 | –0.160 |
| Cyanobacteria | Not detected | 0.01 (0.01, 0.01) | 4 | –0.185 |
| Fusobacteria | Not detected | 0.01 (0.01, 0.01) | 3 | –0.704 |
| Unknown | 0.78 (0.60, 0.96) | 0.41 (0.28, 0.83) | 5 | –1.298 |
| Genera altered by ageing | ||||
| p_Bacteroidetes; g_Bacteroides | 0.01 (0.01, 0.01) | 3.16 (2.38, 4.88) | 164* | 5.413 |
| p_Verrucomicrobia; g_Akkermansia | 0.01 (0.01, 0.01) | 0.22 (0.13, 0.97) | 159* | 3.030 |
| p_Proteobacteria; g_Desulfovibrio | 0.01 (0.01, 0.01) | 0.35 (0.15, 1.16) | 155* | 2.734 |
| p_Firmicutes; g_Turicibacter | 3.99 (0.78, 5.83) | 0.01 (0.01, 0.05) | 163* | –4.352 |
| p_Firmicutues; f_Clostridiaceae; g_ | 3.21 (0.01, 5.86) | 0.01 (0.01, 0.01) | 160* | –3.328 |
| p_Bacteroidetes; g_Parabacteroides | 4.16 (1.81, 8.33) | 0.24 (0.16, 0.57) | 160* | –2.727 |
| p_Firmicutes; _; _; _; _ | 1.80 (0.05, 9.17) | 0.06 (0.03, 0.09) | 153* | –2.508 |
| Genera with a non‐significant trend in difference between young and old mice | ||||
| p_TM7; f_F16; g_ | Not detected | 0.08 (0.03, 0.12) | 137 | 1.623 |
| p_Bacteroidetes; f_Rikenellaceae; g_ | 0.39 (0.01, 3.04) | 2.52 (0.96, 3.53) | 131 | 1.958 |
| p_Proteobacteria; g_Bilophila | Not detected | 0.06 (0.01, 0.14) | 119 | 1.458 |
| p_Firmicutes; g_Lactobacillus | 2.39 (1.22, 4.67) | 8.34 (4.21, 11.40) | 97 | 1.139 |
Median relative abundance (% of total microbes) and analysis W scores determined using ANCOM (old vs. young) of all major microbial phyla and microbial genera with differential abundance between young and old mice. Abundance data are the 50th (25th, 75th) percentiles. The W score indicates the number of statistical subhypotheses that have passed for a given taxon. *Statistical significance. ‘p’, phylum; ‘f’, family; ‘g’, genus. ‘g__’ and the next highest known order are given for operational taxonomic units that do not have a specific genus name.
TMAO and associated metabolites
Plasma TMAO levels were ∼3‐fold higher in old control animals compared to young animals (P < 0.001) (Fig. 6 A), although there was no effect of age on plasma levels of TMA, the precursor to TMAO (P = 0.88) (Table 3). However, hepatic protein expression of FMO3 was significantly higher in old animals (P = 0.03) (Fig. 6 B), indicating a greater capacity to convert TMA into TMAO.
Figure 6. Ageing is associated with increased TMAO.
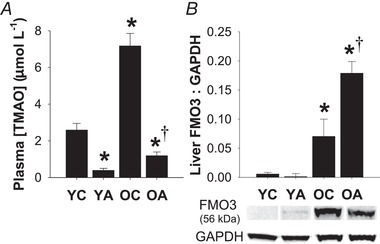
A, plasma concentrations of TMAO were higher in old mice and suppressed by antibiotic treatment. B, both ageing and antibiotic treatment increased protein expression of the TMA‐converting enzyme FMO3 in the liver. Data are the mean ± SEM. * P < 0.05 vs. YC. †P < 0.05 vs. OC. YC, young control; YA, young antibiotic‐treated; OC, old control; OA, old antibiotic‐treated.
Table 3.
TMAO‐related metabolites
| YC | YA | OC | OA | |
|---|---|---|---|---|
| TMA (μmol L−1) | 7.3 ± 1.3 | 0.3 ± 0.1* | 8.3 ± 2.6 | 0.5 ± 0.2* ,† |
| l‐carnitine (μmol L−1) | 20.7 ± 1.0 | 26.8 ± 3.2 | 23.8 ± 1.7 | 20.5 ± 0.7 |
Plasma concentrations of TMA and l‐carnitine, measured by liquid chromatography‐mass spectrometry. Data are the mean ± SEM. * P < 0.05 vs. YC within time point. †P < 0.05 vs. OC. YC, young control; YA, young antibiotic‐treated; OC, old control; OA, old antibiotic‐treated.
As expected, antibiotic treatment suppressed plasma levels of TMA and TMAO in both young and old animals (P < 0.05 vs. YC or OC for both TMA and TMAO). Interestingly, antibiotic treatment increased liver FMO3 protein expression in old (P < 0.001 OA vs. OC) but not young (P = 0.82 YA vs. YC) animals. There were no differences in plasma levels of l‐carnitine (main effect, P = 0.14) (Table 3), a precursor for TMA.
Discussion
Although there is accumulating evidence that the gut microbiome is altered with ageing, the effects of these changes on the vasculature are largely unknown. In the present study, we first established proof‐of‐concept evidence that the gut microbiome is involved in mediating arterial dysfunction with primary ageing because suppression of the gut microbiome (faecal DNA to <30% of baseline) with broad‐spectrum, poorly‐absorbed antibiotics restored arterial function in old mice to levels observed in young animals and also normalized oxidative stress and inflammation. Second, to provide initial insight into changes in the gut microbiome with ageing that could contribute to arterial dysfunction, we sequenced faecal samples from young and old mice housed in our vivarium for the majority of their lifespan and observed an altered abundance in microbial taxa associated with gut dysbiosis.
Suppression of the gut microbiome reverses age‐related arterial dysfunction
To our knowledge, the present study is the first to demonstrate that the gut microbiome is also an important modulator of arterial function with ageing. Our observations are consistent with and extend previous findings showing that experimental manipulation of the gut microbiome, either with antibiotics (Vikram et al. 2016; Battson et al. 2017), transfer of microbiota (Karbach et al. 2016) or targeted dietary interventions (Catry et al. 2018), can mitigate the adverse effects of a high‐fat diet on arterial function in young animals.
The improvements we observed in endothelial function (i.e. EDD) after antibiotic treatment were related to enhanced NO bioavailability. Impaired NO bioavailability is a key underlying cause of endothelial dysfunction with ageing (Seals et al. 2011; Assar et al. 2012). In the present study, impaired endothelial function in the old control mice was associated with an age‐related reduction in NOS activity, which was unaffected by antibiotic treatment. This observation suggests improvements in NO bioavailability were a result of other mechanisms, probably reduced superoxide production and consequent NO scavenging (reduced oxidative stress) vs. direct effects on NO production, as discussed further below.
In the present study, arterial (aortic) stiffness, as assessed by aortic PWV, was increased with ageing and normalized in old mice after antibiotic treatment. Consistent with previous findings from our laboratory in mice (Fleenor et al. 2013; Gioscia‐Ryan et al. 2018), this age‐related aortic stiffening was associated with increased collagen deposition and degradation of elastin fibres. Antibiotic treatment in old mice restored elastin protein expression, such that levels in arteries of old antibiotic‐treated mice were not significantly different from young mice and partially preserved the elastic modulus of the elastin region of the stress‐strain curve, although they had no effects on collagen. It is possible that the effects of antibiotic treatment in reversing aortic stiffening in the old mice may have been the result of a combination of these structural modifications involving elastin, as well as ‘functional’ changes associated with reduced vascular smooth muscle tone because of enhanced NO bioavailability and endothelial function (Zieman et al. 2005).
We and others have shown that oxidative stress is a major mechanism mediating arterial ageing (Seals et al. 2011; Assar et al. 2012). Accordingly, we aimed to determine whether the amelioration of arterial dysfunction with antibiotic treatment in old mice was associated with a reduction in vascular oxidative stress. Antibiotic treatment in old mice reduced aortic oxidative stress as indicated by a decrease in nitrotyrosine, a well‐established marker of oxidant modification of proteins. This reduction in oxidative stress was, in turn, associated with both a decrease in superoxide (reactive oxygen species) production and evidence of greater antioxidant defences. Concerning the latter, we observed a robust increase in aortic protein expression of the antioxidant enzyme ecSOD in old antibiotic‐treated mice. Because we observed no effect of antibiotic treatment on ecSOD protein expression in young mice, the upregulation of ecSOD in old mice with antibiotic treatment may have been secondary to effects on gut‐derived factors/metabolites that are specifically upregulated by ageing.
Another key mechanism of arterial ageing is vascular inflammation (Seals et al. 2011). We observed increased levels of pro‐inflammatory cytokines in old control animals, consistent with previous reports from our laboratory (Rippe et al. 2010; Gioscia‐Ryan et al. 2018), which were reversed by short‐term antibiotic treatment. These results, combined with those showing that oxidative stress was reduced by antibiotic treatment, suggest that age‐related gut dysbiosis impairs arterial function via a combination of superoxide‐driven oxidative stress and inflammation.
Changes in the gut microbiome and related metabolites with ageing
Our second goal was to explore, under controlled environmental conditions, changes in the gut microbiome with primary ageing. We found that ageing was associated with greater alpha diversity (i.e. a measure of the number of species present). To our knowledge, only one other study has reported alpha diversity with ageing in mice, also reporting an increase (Scott et al. 2017), whereas, studies in humans have been inconsistent (Mariat et al. 2009; Claesson et al. 2011). We also observed altered abundance of several microbial taxa, including taxa associated with gut dysbiosis such as the phylum Proteobacteria, its genus Desulfovibrio, and the candidate division TM7 (Kuehbacher et al. 2008; Mukhopadhya et al. 2012). Our observations are consistent with previous reports in mice of a higher abundance of Desulfovibrio (Scott et al. 2017) and TM7 (Fransen et al. 2017; Scott et al. 2017) with ageing. Taken together, our results indicate that old mice demonstrated several bacterial markers of gut dysbiosis and/or inflammation.
To explore potential signalling metabolites that might transduce the effects of changes in bacterial taxa with ageing, we measured circulating levels of TMAO, a gut microbe‐derived factor with known pro‐oxidant/pro‐inflammatory (Seldin et al. 2016) and pro‐atherosclerotic (Wang et al. 2015) properties. TMAO production is dependent on synthesis of its precursor TMA by certain gut microbes, including species within the pro‐inflammatory genus Desulfovibrio (Craciun & Balskus, 2012). In old compared to young mice, we observed higher abundance of the genus Desulfovibrio, as well as greater hepatic protein expression of FMO3, indicating a greater capacity for conversion of TMA into TMAO. Taken together, these differences were associated with an ∼3‐fold age‐related increase in circulating plasma TMAO levels. In both young and old mice, antibiotic treatment suppressed TMA and TMAO levels, consistent with other reports (al‐Waiz et al. 1992; Koeth et al. 2013). Interestingly, liver FMO3 protein expression was greater in antibiotic‐treated mice, indicating that the lower circulating TMAO levels observed after antibiotic treatment were secondary to effects on the gut microbiome and associated suppression of gut microbial TMA synthesis rather than downregulation of FMO3. Overall, these, as well as other recent observations (Li et al. 2017), identify increased circulating concentrations of TMAO associated with changes in the gut microbiome as a possible mechanism contributing to arterial oxidative stress, inflammation and dysfunction with ageing, which should be explored in future investigations.
Limitations
It is important to consider whether the effects that we observed were caused by the direct effects of the antibiotics on the vasculature rather than solely being a result of suppression of the gut microbiome, in that some classes of antibiotic drugs have been shown to influence NO bioavailability and/or have anti‐oxidative effects (Mitsuyama et al. 1998; Al‐Banna et al. 2013). However, to our knowledge, such effects have not been reported with any of the four antibiotics that we used, which were chosen because they are poorly absorbed into circulation (Kunin et al. 1960; Rao et al. 2011). Furthermore, long‐term administration of vancomycin actually impairs endothelial cell function in mice (Dong et al. 2017). Thus, if anything, it is possible that the antibiotic cocktail used may have exerted adverse direct effects on arterial function that countered the beneficial effects of gut microbiome suppression. Furthermore, because we observed no effects of antibiotic treatment in young mice, our antibiotic cocktail probably did not directly influence the vasculature.
YA mice drank significantly less water than other groups, resulting in progressive body weight loss over the course of the intervention. However, the amount of antibiotic‐treated water consumed was sufficient to suppress the microbiome to a similar extent as OA mice. Although the loss in body mass may have caused some of the changes that we observed in YA mice (e.g. lower organ weights and possibly the reduction in blood pressure), endothelial and smooth muscle function in YA mice was not different from YC mice. Importantly, water intake and body weight were not different between OC and OA mice, suggesting that the differences in vascular function and oxidative stress observed between these groups were a result of the suppression of gut microbes.
Our antibiotic cocktail was sufficient to suppress faecal DNA to <30% of baseline. Importantly, all microbial taxa that we reported to be altered by ageing were considerably suppressed by antibiotic treatment. The majority (∼85% on average) of microbes that remained after antibiotic treatment belonged to the family Enterobacteriaceae. These antibiotic‐resistant bacteria are typically considered to be pro‐inflammatory and/or disease causing (Zeng et al. 2017). However, despite the continued presence of these microbes, we observed a reduction in age‐related vascular inflammation. Thus, we do not consider these microbes to have impacted our results.
Lastly, one inconsistency between our data related to the effects of primary ageing on the gut microbiome and previous reports in mice (Thevaranjan et al. 2017) is that we observed an altered abundance of Verrucomicrobia and its genus Akkermansia. Because these taxa are typically reported to be health beneficial and anti‐inflammatory (Derrien et al. 2017), future studies are needed to further explore these differing findings.
Conclusion and perspectives
In the present study, we show for the first time that suppression of the gut microbiome with broad‐spectrum poorly‐absorbed antibiotics in old mice reversed age‐related endothelial dysfunction and aortic stiffening to levels observed in young mice. This amelioration of vascular ageing was associated with restoration of NO bioavailability and normalization of arterial oxidative stress and inflammation. Second, we show that gut microbiome composition is altered by primary ageing in mice, including changes in microbial taxa associated with gut dysbiosis and pro‐inflammatory signalling. Finally, we demonstrate that ageing was associated with an increase in circulating levels of the gut microbe‐dependent metabolite TMAO, which can promote vascular oxidative stress and inflammation.
Taken together, these results suggest that age‐related gut dysbiosis contributes to the development of oxidative stress and inflammation that underlie age‐related arterial dysfunction. As such, our findings support the development of gut microbiome‐targeted agents and interventions aiming to treat/prevent arterial dysfunction with ageing. Because arterial dysfunction is the key antecedent to CVD, as well as a major contributor to other age‐associated disorders, including cognitive impairment/dementia, chronic kidney disease and exercise intolerance, such efforts would have the potential to broadly improve public health and reduce the risk of chronic diseases with human ageing.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All mouse experiments and biochemical analyses were conducted in the laboratory of Dr D. R. Seals at the University of Colorado Boulder. Faecal sample PCR for confirmation of gut microbiome suppression with antibiotic treatment was conducted in the laboratory of Dr T. Weir at Colorado State University. Faecal gut microbiome sequencing was conducted in the laboratory of Dr R. Knight at the University of Colorado Boulder. Plasma TMAO analyses were conducted in the laboratory of Dr A. P. Neilson at Virginia Polytechnic Institute and State University. RAG‐R, MCZ, KPD, RK and DRS conceived the experiments. VEB, RAG‐R, JJR, MCZ, KPD and DRS designed the experiments. VEB, RAG‐R, JJR, MCZ, LMC, AG‐P, YVB, MLB, ATS, ADG, GA, APN and TW collected data. VEB, RAG‐R, JJR, AG‐P, YVB, MLB and TW analysed data. VEB, RAG‐R and DRS interpreted data. VEB, RAG‐R and JJR drafted the manuscript. All authors revised the manuscript critically for intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by R01 HL107120 and R01 HL134887 (D.R.S.); T32 HL007822 and F32 HL140875 (V.E.B.); and F31 AG047784 (R.A.G‐R.).
Acknowledgements
The authors would like to thank Zachary Sapinsley, Nicholas VanDongen, Jacob Frye, Brian Ziemba and Jill Miyamoto‐Ditmon for their assistance with data collection, as well as Jesse Goodrich for assistance with data analysis.
Biographies
Vienna E. Brunt received her PhD in Human Physiology from the University of Oregon in 2016. She is currently a postdoctoral fellow in Dr D. R. Seals' Integrative Physiology of Aging Laboratory at the University of Colorado Boulder. The studies described in the present study represent work carried out as part of an NIH T32 fellowship through the Division of Cardiology at the University of Colorado Denver. Her long‐term research goals are to investigate the efficacy of novel interventions for preserving vascular function with ageing, thereby preventing and/or delaying the progression of cardiovascular diseases.

Rachel A. Gioscia‐Ryan completed her PhD in the Integrative Physiology of Aging Laboratory at the University of Colorado Boulder in 2016 and is currently in medical school at the University of Michigan. She is pursuing a career as a clinician–scientist conducting integrative physiological studies with the aim of improving human health and patient care.
Edited by: Harold Schultz & David Grundy
This is an Editor's Choice article from the 1 May 2019 issue.
Linked articles: This article is highlighted in a Perspectives article by Cryan et al. To read this article, visit https://doi.org/10.1113/JP277784.
References
- Al‐Banna NA, Pavlovic D, Gründling M, Zhou J, Kelly M, Whynot S, Hung O, Johnston B, Issekutz TB, Kern H, Cerny V & Lehmann C (2013). Impact of antibiotics on the microcirculation in local and systemic inflammation. Clin Hemorheol Microcirc 53, 155–169. [DOI] [PubMed] [Google Scholar]
- al‐Waiz M, Mikov M, Mitchell SC & Smith RL (1992). The exogenous origin of trimethylamine in the mouse. Metab Clin Exp 41, 135–136. [DOI] [PubMed] [Google Scholar]
- Amir A, McDonald D, Navas‐Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, González A & Knight R (2017). Deblur rapidly resolves single‐nucleotide community sequence patterns. mSystems 2, e00191–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assar El M, Angulo J, Vallejo S, Peiró C, Sánchez‐Ferrer CF & Rodríguez‐Mañas L (2012). Mechanisms involved in the aging‐induced vascular dysfunction. Front Physiol 3, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battson ML, Lee DM, Jarrell DK, Hou S, Ecton KE, Weir TL & Gentile CL (2017). Suppression of gut dysbiosis reverses western diet‐induced vascular dysfunction. Am J Physiol Endocrinol Metab 49, E96–E34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JHY, Alger HM, Wong SS & Muntner P (2017). Heart disease and stroke statistics – 2017 update: a report from the American Heart Association. Circulation 135, e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL & Lusis AJ (2013). Trimethylamine‐N‐oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab 17, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA & Caporaso JG (2013). Quality‐filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10, 57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutagy NE, Neilson AP, Osterberg KL, Smithson AT, Englund TR, Davy BM, Hulver MW & Davy KP (2015. a). Probiotic supplementation and trimethylamine‐N‐oxide production following a high‐fat diet. Obesity 23, 2357–2363. [DOI] [PubMed] [Google Scholar]
- Boutagy NE, Neilson AP, Osterberg KL, Smithson AT, Englund TR, Davy BM, Hulver MW & Davy KP (2015. b). Short‐term high‐fat diet increases postprandial trimethylamine‐N‐oxide in humans. Nutr Res 35, 858–864. [DOI] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA & Holmes SP (2016). DADA2: high‐resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J & Knight R (2010). QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg‐Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N & Knight R (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108 (Suppl 1), 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho BM, Guadagnini D, Tsukumo DML, Schenka AA, Latuf‐Filho P, Vassallo J, Dias JC, Kubota LT, Carvalheira JBC & Saad MJA (2012). Modulation of gut microbiota by antibiotics improves insulin signalling in high‐fat fed mice. Diabetologia 55, 2823–2834. [DOI] [PubMed] [Google Scholar]
- Catry E, Bindels LB, Tailleux A, Lestavel S, Neyrinck AM, Goossens J‐F, Lobysheva I, Plovier H, Essaghir A, Demoulin J‐B, Bouzin C, Pachikian BD, Cani PD, Staels B, Dessy C & Delzenne NM (2018). Targeting the gut microbiota with inulin‐type fructans: preclinical demonstration of a novel approach in the management of endothelial dysfunction. Gut 67, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Cusack S, O'Sullivan O, Greene‐Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, Stanton C, van Sinderen D, O'Connor M, Harnedy N, O'Connor K, Henry C, O'Mahony D, Fitzgerald AP, Shanahan F, Twomey C, Hill C, Ross RP & O'Toole PW (2011). Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A 108 (Suppl 1), 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, Harris HMB, Coakley M, Lakshminarayanan B, O'Sullivan O, Fitzgerald GF, Deane J, O'Connor M, Harnedy N, O'Connor K, O'Mahony D, van Sinderen D, Wallace M, Brennan L, Stanton C, Marchesi JR, Fitzgerald AP, Shanahan F, Hill C, Ross RP & O'Toole PW (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. [DOI] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW & Knight R (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S & Balskus EP (2012). Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A 109, 21307–21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M, Belzer C & de Vos WM (2017). Akkermansia muciniphila and its role in regulating host functions. Microb Pathog 106, 171–181. [DOI] [PubMed] [Google Scholar]
- Dong X‐H, Peng C, Zhang Y‐Y, Tao Y‐L, Tao X, Zhang C, Chen AF & Xie H‐H (2017). Chronic exposure to subtherapeutic antibiotics aggravates ischemic stroke outcome in mice. EBioMedicine 24, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith DP (1992). Conservation evaluation and phylogenetic diversity. Biological Conservation 61, 1–10. [Google Scholar]
- Fleenor BS, Marshall KD, Durrant JR, Lesniewski LA & Seals DR (2010). Arterial stiffening with ageing is associated with transforming growth factor‐β1‐related changes in adventitial collagen: reversal by aerobic exercise. J Physiol 588, 3971–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor BS, Sindler AL, Eng JS, Nair DP, Dodson RB & Seals DR (2012). Sodium nitrite de‐stiffening of large elastic arteries with aging: role of normalization of advanced glycation end‐products. Exp Gerontol 47, 588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor BS, Sindler AL, Marvi NK, Howell KL, Zigler ML, Yoshizawa M & Seals DR (2013). Curcumin ameliorates arterial dysfunction and oxidative stress with aging. Exp Gerontol 48, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen F, van Beek AA, Borghuis T, Aidy SE, Hugenholtz F, van der Gaast de Jongh C, Savelkoul HFJ, De Jonge MI, Boekschoten MV, Smidt H, Faas MM & de Vos P (2017). Aged gut microbiota contributes to systemical inflammaging after transfer to germ‐free mice. Front Immunol 8, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioscia‐Ryan RA, Battson ML, Cuevas LM, Eng JS, Murphy MP & Seals DR (2018). Mitochondria‐targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. J Appl Physiol 124, 1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González A, Navas‐Molina JA, Kosciolek T, McDonald D, Vázquez‐Baeza Y, Ackermann G, DeReus J, Janssen S, Swafford AD, Orchanian SB, Sander JG, Shorenstein J, Holste H, Petrus S, Robbins‐Pianka A, Brislawn CJ, Wang M, Rideout JR, Bolven E, Dillon M, Caporaso JG, Dorrestein PC & Knight R. (2018). Qiita: rapid, web‐enabled microbiome meta‐analysis. Nat Methods 15, 796–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, Lusis AJ & Hazen SL (2015). Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem 290, 5647–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins MJ, Sharp R & Macfarlane GT (2001). Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut 48, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen S, McDonald D, González A, Navas‐Molina JA, Jiang L, Xu ZZ, Winker K, Kado DM, Orwoll E, Manary M, Mirarab S, Knight R & Chia N (2018). Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 3, e00021–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbach SH, Schönfelder T, Brandão I, Wilms E, Hörmann N, Jäckel S, Schüler R, Finger S, Knorr M, Lagrange J, Brandt M, Waisman A, Kossmann S, Schäfer K, Münzel T, Reinhardt C & Wenzel P (2016). Gut microbiota promote angiotensin II‐induced arterial hypertension and vascular dysfunction. J Am Heart Assoc 5, e003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WHW, Bushman FD, Lusis AJ & Hazen SL (2013). Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19, 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J, Liu Z, Lozupone C, McDonald D, Fierer N & Knight R (2010). Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat Methods 7, 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehbacher T, Rehman A, Lepage P, Hellmig S, Folsch UR, Schreiber S & Ott SJ (2008). Intestinal TM7 bacterial phylogenies in active inflammatory bowel disease. J Med Microbiol 57, 1569–1576. [DOI] [PubMed] [Google Scholar]
- Kunin CM, Chalmers TC, Leevy CM, Sebastyen SC, Lieber CS & Finland M (1960). Absorption of orally administered neomycin and kanamycin with special reference to patients with severe hepatic and renal disease. N Engl J Med 262, 380–385. [DOI] [PubMed] [Google Scholar]
- Lakatta EG (2003). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular and molecular clues to heart and arterial aging. Circulation 107, 490–497. [DOI] [PubMed] [Google Scholar]
- Lakatta EG & Levy D (2003). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a ‘set up’ for vascular disease. Circulation 107, 139–146. [DOI] [PubMed] [Google Scholar]
- Lammers SR, Kao PH, Qi HJ, Hunter K, Lanning C, Albietz J, Hofmeister S, Mecham R, Stenmark KR & Shandas R (2008). Changes in the structure‐function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol 295, H1451–H1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Meehan CJ, Koenig JE, Dhanani AS, Rose RA, Howlett SE & Beiko RG (2014). Microbial shifts in the aging mouse gut. Microbiome 2, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca TJ, Gioscia‐Ryan RA, Hearon CM & Seals DR (2013). The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev 134, 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen Y, Gua C & Li X (2017). Elevated circulating trimethylamine N‐oxide levels contribute to endothelial dysfunction in aged rats through vascular inflammation and oxidative stress. Front Physiol 8, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C & Knight R (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R & Peddada SD (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 26, 27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manter DK, Korsa M, Tebbe C & Delgado JA (2016). myPhyloDB: a local web server for the storage and analysis of metagenomic data. Database (Oxford) 2016, baw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariat D, Firmesse O, Levenez F, Guimarăes V, Sokol H, Doré J, Corthier G & Furet J‐P (2009). The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol 9, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, Nguyen N & Warnow T (2012). SEPP: SATé‐enabled phylogenetic placement. Pac Symp Biocomput, 2012, 247–258. [DOI] [PubMed] [Google Scholar]
- Mitsuyama T, Hidaka K, Furuno T & Hara N (1998). Release of nitric oxide and expression of constitutive nitric oxide synthase of human endothelial cells: enhancement by a 14‐membered ring macrolide. Mol Cell Biochem 181, 157–161. [DOI] [PubMed] [Google Scholar]
- Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, Cresci A, Silvi S, Orpianesi C, Verdenelli MC, Clavel T, Koebnick C, Zunft H‐JF, Doré J & Blaut M (2006). Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross‐sectional study. Appl Environ Microbiol 72, 1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhya I, Hansen R, El‐Omar EM & Hold GL (2012). IBD – what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol 9, 219–230. [DOI] [PubMed] [Google Scholar]
- Ooi JH, Li Y, Rogers CJ & Cantorna MT (2013). Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate‐induced colitis. J Nutr 143, 1679–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff‐Nahoum S, Paglino J, Eslami‐Varzaneh F, Edberg S & Medzhitov R (2004). Recognition of commensal microflora by toll‐like receptors is required for intestinal homeostasis. Cell 118, 229–241. [DOI] [PubMed] [Google Scholar]
- Rao S, Kupfer Y, Pagala M, Chapnick E & Tessler S (2011). Systemic absorption of oral vancomycin in patients with Clostridium difficile infection. Scand J Infect Dis 43, 386–388. [DOI] [PubMed] [Google Scholar]
- Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, McCoy KD, Macpherson AJ, Meza‐Zepeda LA & Johansen F‐E (2011). Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. Plos ONE 6, e17996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippe C, Lesniewski L, Connell M, LaRocca T, Donato A & Seals D (2010). Short‐term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell 9, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes T, Flouri T, Nichols B, Quince C & Mahé F (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4(17), e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott KA., Ida M, Peterson VL, Prenderville JA, Moloney GM, Izumo T, Murphy K, Murphy A, Ross RP, Stanton C, Dinan TG Cryan JF (2017). Revisiting Metchnikoff: age‐related alterations in microbiota‐gut‐brain axis in the mouse. Brain Behav Immun 65, 20–32. [DOI] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL & Donato AJ (2011). Aging and vascular endothelial function in humans. Clin Sci 120, 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, Lusis AJ & Shih DM (2016). Trimethylamine N‐oxide promotes vascular inflammation through signaling of mitogen‐activated protein kinase and nuclear factor‐κB. J Am Heart Assoc 5, e002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ & Seals DR (2011). Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell 10, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull VJ, Finer E, Bergmans RS, Febvre HP, Longhurst C, Manter DK, Patz JA and Weir TL (2018). Impact of edible cricket consumption on gut microbiota in healthy adults, a double‐blind, randomized crossover trial. Sci Rep 8, 10762, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y & Hazen SL (2013). Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, Schertzer JD, Larche MJ, Davidson DJ, Verdu EF, Surette MG & Bowdish DME (2017). Age‐associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikram A, Kim Y‐R, Kumar S, Li Q, Kassan M, Jacobs JS & Irani K (2016). Vascular microRNA‐204 is remotely governed by the microbiome and impairs endothelium‐dependent vasorelaxation by downregulating Sirtuin1. Nat Commun 7, 12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Levison BS, Hazen JE, Donahue L, Li X‐M & Hazen SL (2014). Measurement of trimethylamine‐N‐oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal Biochem 455, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian‐Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ & Hazen SL (2015). Non‐lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163, 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng MY, Inohara N & Nuñez G (2017). Mechanisms of inflammation‐driven bacterial dysbiosis in the gut. Mucosal Immunol 10, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieman SJ, Melenovsky V & Kass DA (2005). Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 25, 932–943. [DOI] [PubMed] [Google Scholar]