

Abstract
Key points
Neurons from the brainstem nucleus of the tractus solitarius (NTS) participate in the counter‐regulatory mechanisms in response to hypoglycaemia.
ATP‐sensitive potassium (KATP) channels are expressed in NTS neurons, and are partially open at rest in normoglycaemic 5 mM glucose.
In normoglycaemic conditions, most NTS neurons depolarize in response to low external glucose (0.5 mM), via a voltage‐dependent mechanism.
Conversely, most NTS neurons incubated in hyperglycaemic 10 mM glucose do not respond to low glucose due to a more positive resting membrane potential caused by the closure of KATP channels following increased intracellular metabolic ATP.
Our findings show that in hyperglycaemic conditions, NTS neurons failed to sense rapid changes in external glucose, which could be related to hypoglycaemia‐associated autonomic failure.
Abstract
The nucleus of the tractus solitarius (NTS) is an integrative centre for autonomic counter‐regulatory responses to hypoglycaemia. KATP channels link the metabolic status of the neuron to its excitability. Here we investigated the influence of KATP channels on the membrane potential of NTS neurons in normo‐ and hyperglycaemic external glucose concentrations, and after switching to a hypoglycaemic concentration, using in vitro electrophysiological recordings in brainstem slices. We found that in normoglycaemic (5 mM) glucose, tolbutamide, a KATP channel antagonist, depolarized the membrane of most neurons, and this effect was observed in more hyperpolarized neurons. All neurons hyperpolarized after pharmacological activation of KATP channels. Most NTS neurons depolarized in the presence of low glucose (0.5 mM), and this effect was only seen in hyperpolarized neurons. The effect of glucose was caused by a cationic current with a reversal potential around −50 mV. In the presence of hyperglycaemic glucose (10 mM), neurons were more depolarized, and fewer neurons responded to KATP blockage. Application of 0.5 mM glucose solution to these neurons depolarized the membrane only in more hyperpolarized neurons. We conclude that NTS neurons present with KATP channels open at rest in normoglycaemic conditions, and their membrane potential is affected by extracellular glucose. Moreover, NTS neurons depolarize the membrane in response to the application of a low glucose solution, but this effect is occluded by membrane depolarization triggered by KATP blockage. Our data suggest a homeostatic regulation of the membrane potential by external glucose, and a possible mechanism related to the hypoglycaemia‐associated autonomic failure.
Keywords: glucose‐sensing neurons, hypoglycemia, ATP‐sensitive potassium channels, nucleus of the tractus solitarius
Key points
Neurons from the brainstem nucleus of the tractus solitarius (NTS) participate in the counter‐regulatory mechanisms in response to hypoglycaemia.
ATP‐sensitive potassium (KATP) channels are expressed in NTS neurons, and are partially open at rest in normoglycaemic 5 mM glucose.
In normoglycaemic conditions, most NTS neurons depolarize in response to low external glucose (0.5 mM), via a voltage‐dependent mechanism.
Conversely, most NTS neurons incubated in hyperglycaemic 10 mM glucose do not respond to low glucose due to a more positive resting membrane potential caused by the closure of KATP channels following increased intracellular metabolic ATP.
Our findings show that in hyperglycaemic conditions, NTS neurons failed to sense rapid changes in external glucose, which could be related to hypoglycaemia‐associated autonomic failure.
Introduction
The brainstem nucleus of the tractus solitarius (NTS) is the primary central site for viscerosensory afferent fibres arising from peripheral neurons, including peripheral chemo‐sensing neurons (Accorsi‐Mendonça et al. 2011; Donovan & Watts, 2014). NTS neurons densely project to the dorsal motor nucleus of the vagus (DMX), where the cell bodies of the preganglionic parasympathetic branch of the vagus are located (Marty et al. 2007; Verberne et al. 2014). Local nutrient and metabolic signals (including glucose and metabolites), as well as direct modulation by receptor agonists/antagonists in NTS neurons, induce physiological responses necessary for the regulation of blood glucose concentration (Ritter et al. 2000; Lam et al. 2010; Zhao et al. 2012). The NTS is associated with the physiological responses to hypoglycaemia, the counter‐regulatory responses, which result in glucagon secretion, increased food intake behaviour, and increased sympathetic tone. For instance, hypoglycaemia induces c‐Fos immunoreactivity in NTS neurons, and portal‐mesenteric deafferentation suppressed this effect (Bohland et al. 2014). Inhibitors of glucose metabolism injected in the NTS increased glucagon secretion and food intake (Ritter et al. 2000; Andrew et al. 2007). Additionally, low glucose increases firing activity in carotid sinus nerve (Gao et al. 2014), which mostly propagates signals into the NTS. Therefore, NTS neurons integrate a variety of synaptic inputs and mediate a plethora of autonomic counter‐regulatory mechanisms in response to food ingestion and signals related to the glycaemic state in order to ensure adequate levels of glucose, including feeding behaviour, gastric motility and hormonal secretion (Marty et al. 2007; Donovan & Watts, 2014; Hermann et al. 2014).
The NTS is an interesting region for detecting changes in extracellular glucose due to the presence of fenestrated capillaries (Gross et al. 1990) and its proximity to the area postrema, a circumventricular organ. Several reports have shown that NTS neurons directly detect fluctuations in glucose levels in the extracellular milieu. For instance, (Mimee & Ferguson 2015) showed that slightly more than half of NTS neurons could depolarize or hyperpolarize their resting membrane potential (RMP) in response to both an increase and a decrease in external glucose in vitro. On the other hand, Balfour et al. (2006) observed that only a minority of the NTS neurons changed membrane potential in response to a zero glucose solution. Lamy et al. (2014) showed a depolarization of the membrane potential in response to 0.5 mM external glucose, only in GLUT‐2/GABAergic neurons in the NTS. Finally, McDougal et al. (2013) showed that a fraction of glia and neurons of NTS increased intracellular calcium in response to low glucose and inhibitors of glycolysis.
The ATP‐sensitive potassium (KATP) channel links metabolic status and electrical excitability (Nichols, 2006), as classically described in pancreatic beta‐cells (Ashcroft & Rorsman, 2013). This channel is a hetero‐octameric complex comprising four pore‐forming Kir6.x subunits and four regulatory sulfonylurea receptor (SURx) subunits, and it conducts an inwardly rectifying potassium current that is inhibited by ATP binding to Kir6.x subunits and stimulated by ADP interaction with nucleotide‐binding sites within SURx subunits (Hibino et al. 2010). In the NTS, the subunits Kir6.2 and SUR1 are expressed in glucose‐sensing neurons (Balfour et al. 2006; Halmos et al. 2015). Glucose‐excited NTS neurons indeed appear to respond to changes in glucose levels using a mechanism similar to pancreatic beta‐cells, in a GLUT/glucokinase/KATP‐dependent system (Thorens, 2012; Ashcroft & Rorsman, 2013), and KATP channel antagonists blunt the responsive of these neurons to increased glucose levels (Balfour et al. 2006; Boychuk et al. 2015).
Here we aimed to investigate how the membrane potential of NTS neurons is affected by decreasing glucose concentrations, and the role of KATP channels. We found that NTS neurons depolarize in response to low external glucose only when their membrane potential is hyperpolarized below −60 mV. Incubation in high glucose external solution depolarizes the neurons by blocking KATP channels, and blunts their response to low glucose. This unexpected finding shows that NTS neurons in the presence of hyperglycaemic glucose concentrations, do not respond to fast reductions in external glucose, with implications for situations of prolonged hyperglycaemia, as in diabetes mellitus.
Methods
Brainstem slices preparation
Animal procedures were performed according to the protocol approved by the Committee on Ethics in Animal Experimentation (CEUA) from the School of Medicine of Ribeirão Preto, University of São Paulo (protocol no. 149/2015). Brainstem slices containing the subpostremal NTS were obtained as previously described (Accorsi‐Mendonça et al. 2011). Male Wistar rats (3 to 11 weeks old) were decapitated following isoflurane anaesthesia, and the brainstem was quickly removed and placed in a dish containing ice‐cold artificial cerebrospinal fluid (aCSF) modified for slicing, containing (in mM): 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 75 sucrose, 25 d‐glucose, 0.2 CaCl2, and 7 MgCl2 (330 mOsm/kg H2O, pH 7.4 when bubbled with a carbogenic mixture of 95% O2 and 5% CO2). The specimen was glued to the sectioning stage, and submerged in ice‐cold slicing aCSF in a vibratome (Vibratome 1000 Plus, Vibratome) chamber. Then, three to four coronal brainstem slices (250 μM) containing the NTS near the level of the area postrema (i.e., ±500 μM rostral and caudal) were sectioned and incubated at 32–33°C for 45 min in aCSF, containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 5 d‐glucose, 2 CaCl2, and 1 MgCl2 (298 mOsm/kg H2O, pH 7.35 when bubbled with a carbogenic mixture of 95% O2 and 5% CO2). In some experiments, the aCSF contained 10 mM glucose. After this period, the slices were stored in the same solution at room temperature until electrophysiological recording. Low glucose (0.5 mM) aCSF, as well as 10 mM glucose aCSF, were made using equimolar amounts of NaCl or sucrose to maintain osmolality equal to the 5 mM glucose aCSF. No differences were observed in both conditions. The use of 5 mM glucose as the basal level, a concentration lower than used previously by our group (see Accorsi‐Mendonça et al. 2011 for instance), and other groups (see Balfour et al. 2006, for instance), is because 5 mM (90 mg/dL) is close to a regular glycaemic situation. We found that the neurons survived well in this extracellular concentration of glucose and were viable for recording.
Electrophysiological recordings
Single brainstem slices were transferred to a chamber mounted on a stage of an upright microscope (BX51WI; Olympus, Japan), and continuously perfused with aCSF at 30–33°C using an inline heating system (TC‐324B; Warner Instruments, Hamden, CT, USA) at a rate of ∼2 mL/min using a gravity‐driven perfusion system. Neurons were then visualized under DIC optics with a 60× immersion objective, and patched with electrodes made with thick‐walled borosilicate glass (BF150‐86‐10; Sutter Instruments, Novato, CA, USA) pulled using a horizontal puller (P‐87; Sutter Instruments). The electrodes were filled with an internal solution, containing (in mM): 128 potassium gluconate, 8 KCl, 10 HEPES, 0.5 EGTA, 4 Mg2ATP, 0.3 Na2GTP, and 10 sodium phosphocreatine (295 mOsm/kg H2O, pH 7.3), resulting in pipette tip resistance between 3 and 7 MΩ.
Whole‐cell patch‐clamp recordings were performed with an EPC‐10 patch‐clamp amplifier (HEKA Elektronik, Lambrecht, Germany) using the PatchMaster acquisition software (HEKA Elektronik), in voltage‐ and current‐clamp mode. Data were acquired at 20 kHz and low‐pass filtered at 5 kHz (Bessel). Cells with series resistance larger than 30 MΩ or showing large variations during whole‐cell recording were discarded. After entering the whole‐cell configuration, neurons were allowed to stabilize their membrane potential for ∼10 min, and during this period depolarizing currents were injected to elicit action potential firing to confirm the neuronal identity of the recorded cell. When tetrodotoxin was used, it was applied only after confirmation that the recorded cell was a neuron. Membrane potential was monitored for 10 min in aCSF with 5 or 10 mM glucose before switching to low glucose (0.5 mM) or drug‐containing solutions for 10 min before returning to 5 or 10 mM glucose aCSF. In some experiments, we perfused the low glucose solution for more prolonged periods (20–30 min). Hyperpolarizing pulses (ranging from −30 to −60 pA; 1000 ms) were applied every 15 s to monitor input resistance. All conditions were recorded for 10 min, and we only recorded one neuron per slice. Based both on previous reports regarding glucose sensing in NTS neurons (Balfour et al. 2006; Lamy et al. 2014; Boychuk et al. 2015) and the response profile to a low glucose challenge observed in the current investigation, we assumed that a neuron was responsive to low glucose and/or drug when they showed a clear membrane potential alteration induced by one of these conditions. We observed that most neurons that presented this behaviour had robust membrane potential changes. Neurons with membrane potential oscillations smaller than ±3 mV did not have a clear distinction from the baseline oscillations. Thus, we considered low glucose and/or drug responsive neurons to be those that presented membrane potential changes bigger than ±3 mV.
For voltage‐clamp experiments, neurons were clamped at −70 mV and the membrane potential varied from −125 mV to −65 mV in seven steps of 500 ms. Series resistance was compensated by 60%.
Drugs
Tetrodotoxin (0.5 μM) was purchased from Alomone Labs (Jerusalem, Israel), and tolbutamide (100 μM) and diazoxide (200 μM) were purchased from Sigma (St Louis, MO, USA). All drugs were diluted at the time of the experiment from 1000× concentrated stock solutions in DMSO or water. DMSO in the final concentration of 0.001% did not affect the membrane potential (not shown).
Data analysis
Electrophysiological data were analysed with custom routines written in IGOR Pro 6.37 (WaveMetrics, Portland, OR, USA). Voltages were corrected for a measured liquid junction potential of 10 mV. Data are shown as means ± SEM. All data values were determined after obtained a plateau response. Resting membrane potential was analysed as the mean of an all points histogram of the recording taken in a segment of the last 3–4 min. Membrane conductances and reversal potentials were calculated by linear regressions of the current‐voltage plots.
Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA), using Student's paired and unpaired two‐tailed t tests, and one‐way ordinary or repeated measures ANOVA with Fisher's LSD test. Correlations were determined using a linear regression. Percentages were compared with Fisher's exact test. The significance level was set at P < 0.05.
Results
NTS neurons incubated in 5 mM glucose express partially closed KATP channels
KATP channels are traditionally associated with coupling the cell energy status and electrical activity, triggering depolarization in a high ATP/ADP ratio, and hyperpolarization in a low ATP/ADP ratio (Nichols, 2006; Hibino et al. 2010). NTS neurons express KATP channels, which are more responsive to metabolic ATP than to ATP provided by the whole‐cell pipette solution, even in intracellular ATP concentrations of 3–4 mM, as used in our recordings (Balfour et al. 2006). In order to know whether KATP channels in the NTS neurons were active in neurons incubated in 5 mM external glucose, a near‐normoglycaemic concentration, we applied their antagonist tolbutamide (100 μM; n = 15, seven animals) and measured the change in RMP. We found that the neurons were viable in aCSF with 5 mM glucose during the whole incubation period of the experiment (up to 5 h).
We verified that in 10 neurons (67%) incubated with aCSF containing 5 mM glucose and tetrodotoxin (TTX), tolbutamide triggered a strong and fast depolarization (16.7 ± 2.7 mV, from a mean of −76.1 ± 2.6 mV to −59.4 ± 1.2 mV; P = 0.0002; Fig. 1 Aa and b), accompanied by robust increased in membrane input resistance (R input;190.5 ± 55.2 MΩ, from a mean of 406.9 ± 79.2 MΩ to 597.4 ± 73.4 MΩ; P = 0.007; Fig. 1 Ac), in accordance with its effect in blocking KATP channels. Additionally, five neurons (33%) did not change their RMP in response to tolbutamide (1.0 ± 0.5 mV, from a mean of −65.1 ± 2.4 mV to −64.1 ± 2.2 mV; P = 0.1; Fig. 1 Ba and Bb) as well as R input (14.8 ± 11.0 MΩ, from a mean of 525.8 ± 121.8 MΩ to 540.6 ± 129.1 MΩ; P = 0.2; Fig. 1 Bc). Interestingly, we verified that neurons unresponsive to tolbutamide were significantly more depolarized than neurons responsive to tolbutamide (−65.2 ± 2.4 mV vs. −76.1 ± 2.5 mV, respectively; P = 0.019; Fig. 1 C). Accordingly, we found a negative correlation between RMP and tolbutamide depolarization (r 2 = 0.79; P < 0.0001; Fig. 1 D). On the other hand, no correlation between the change of R input induced by tolbutamide (ΔR input) and initial R input (r 2 = 0.2; P = 0.19; Fig. 1 E) was observed. Therefore, we conclude that NTS neurons present with active KATP channels, which can control RMP. Because tolbutamide‐insensitive neurons had more depolarized RMP, it is possible that KATP channels are already closed by endogenous ATP in these neurons (see next section).
Figure 1. NTS neurons present active KATP channels.
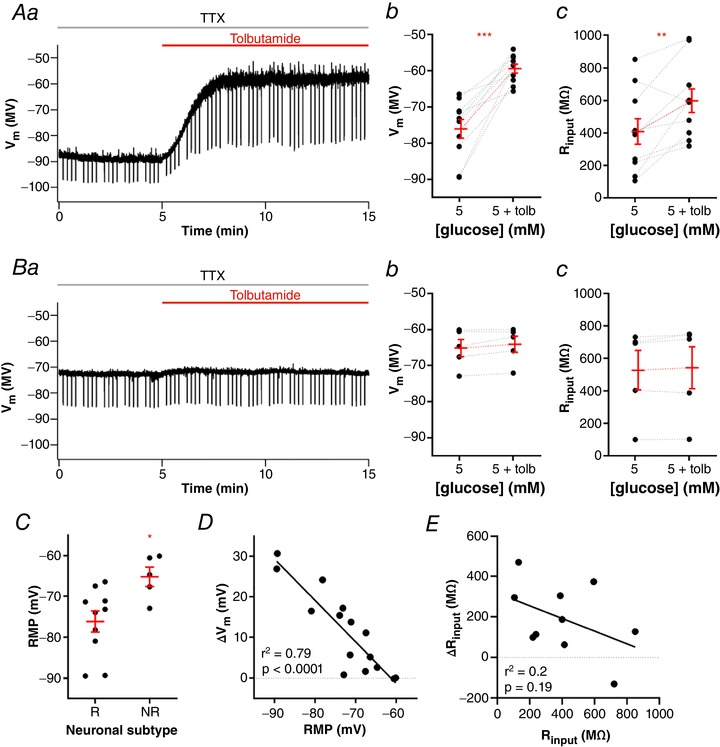
A, representative recording of a neuron responsive to tolbutamide (Aa) and summary of the tolbutamide effect on membrane potential (V m; Ab) and input resistance (R input; Ac) of neurons. B, representative recording of a neuron unresponsive to tolbutamide (Ba) and summary of the tolbutamide effect on V m (Bb) and R input (Bc) of neurons. C, comparison of the resting membrane potential (RMP) between neurons responsive (R) and unresponsive (NR) to tolbutamide. D, linear correlation between the change of V m induced by tolbutamide and the RMP of neurons. E, linear correlation between the change of R input induced by tolbutamide and the basal R input of neurons. Tolb, tolbutamide; TTX, tetrodotoxin. * P < 0.05; ** P < 0.01; *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
The depolarization evoked by tolbutamide is enough to trigger action potential firing in most responsive neurons when recorded in the absence of TTX (from 0.0 to 2.3 ± 0.6 Hz; n = 14; P = 0.0034). Non‐responsive neurons did not change their firing after perfusion of tolbutamide (0.48 ± 0.4 Hz vs. 0.63 ± 0.6 Hz; n = 5; P = 0.29). As in TTX, the non‐responsive neurons were more depolarized than the responsive neurons (responsive neurons: −75.7 ± 2.4 mV; non‐responsive neurons: −66. 3 ± 2 mV; P = 0.02, n = 8 and 5, respectively), and the depolarization was negatively correlated with the membrane potential (r 2 = 0.59; P = 0.0001).
To test whether non‐responsive neurons express KATP channels, we tested the effect of the KATP activator diazoxide (200 μM; n = 7, five animals) on the RMP of NTS neurons. Diazoxide produced a fast and partially reversible hyperpolarization of almost all NTS neurons tested (−8.8 ± 2.1 mV, from a mean of −73.7 ± 1.7 mV to −82.6 ± 3.3 mV; P = 0.005; washout: −79.2 ± 3.1 mV; P = 0.0012; Fig. 2 Aa and Ab). Diazoxide also decreased R input significantly (−230.4 ± 87.7 MΩ, from a mean of 532.3 ± 103.7 MΩ to 301.9 ± 65.3 MΩ; P = 0.04; Fig. 2 Ac), in accordance with the opening of an ion channel, but the reversal of this effect was not significant (400.3 ± 116.5 MΩ; P = 0.13). In contrast to the effect with tolbutamide, we observed an effect of diazoxide in both depolarized and hyperpolarized NTS neurons. In fact, no correlation was observed between the effect of diazoxide on membrane potential and RMP (r 2 = 0.24; P = 0.2; Fig. 2 B). However, we found a significant correlation between the change of R input induced by diazoxide (ΔR input) and initial R input (r 2 = 0.61; P = 0.04; Fig. 2 C), suggesting that the magnitude of the effect of diazoxide is affected by the amount of KATP channels blocked. Thus, because diazoxide can open blocked KATP channels, we conclude that NTS neurons have KATP channels blocked at rest and that modulation of KATP channels can affect RMP of NTS neurons bidirectionally.
Figure 2. KATP channels control the resting membrane potential of NTS neurons.
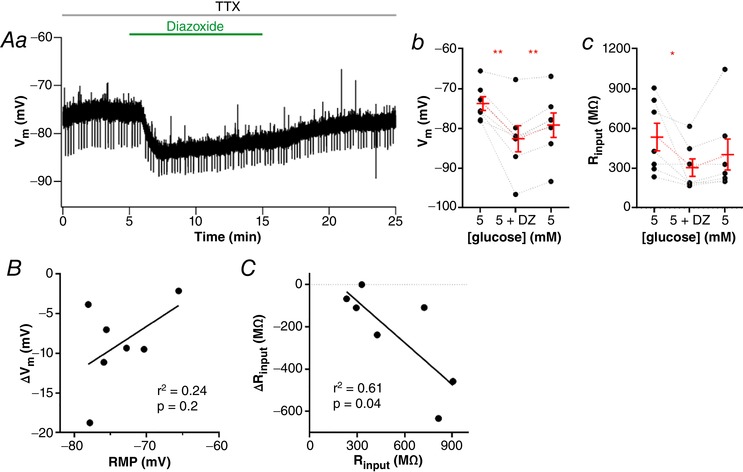
A, diazoxide induces a fast and partially reversible hyperpolarization of neurons, as shown by a representative recording (Aa). The graphs show the summary of the diazoxide effect on membrane potential (V m; Ab) and input resistance (R input; Ac) of neurons. B, linear correlation between the change of V m induced by diazoxide and the resting membrane potential (RMP) of neurons. C, linear correlation between the change of R input induced by diazoxide and the basal R input of neurons. DZ, diazoxide; TTX, tetrodotoxin. * P < 0.05; ** P < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
Most NTS neurons in 5 mM external glucose depolarize in response to low glucose aCSF
NTS neurons are involved in the counter‐regulatory response to hypoglycaemia (Lamy et al. 2014). We showed that partially blocked KATP channels are present in NTS neurons, and thus they can be subjected to modulation by the metabolic ATP/ADP ratio. Other groups showed that a fraction of NTS neurons changed membrane potential in response to changes in extracellular glucose. The effect of diazoxide suggests the existence of KATP channels blocked in our basal conditions. We then tested if perfusion of a low external glucose solution could produce hyperpolarization of NTS neurons by activation of KATP channels, caused by a reduction of the ATP/ADP levels. We then perfused the NTS neurons incubated in 5 mM glucose with a solution containing low glucose (0.5 mM), a concentration that can be achieved in the CSF during periods of hypoglycaemia (Seaquist et al. 2001) and recorded the membrane potential. Surprisingly we found that perfusion of NTS neurons (n = 37, 23 animals) with a low glucose solution depolarized most of NTS neurons (30 neurons; 81%). In these neurons, the RMP was depolarized after perfusion of low glucose aCSF by 9.3 ± 1.0 mV, from −74.3 ± 1.6 mV to −65.0 ± 1.7 mV (P < 0.0001; Fig. 3 Aa and Ab). This effect took on average 383 ± 18 s to reach its peak, being reversible in most neurons (70%), and the RMP after returning to 5 mM glucose was −71.3 ± 1.9 mV (P < 0.0001).
Figure 3. Most NTS neurons depolarize in response to a low glucose challenge.
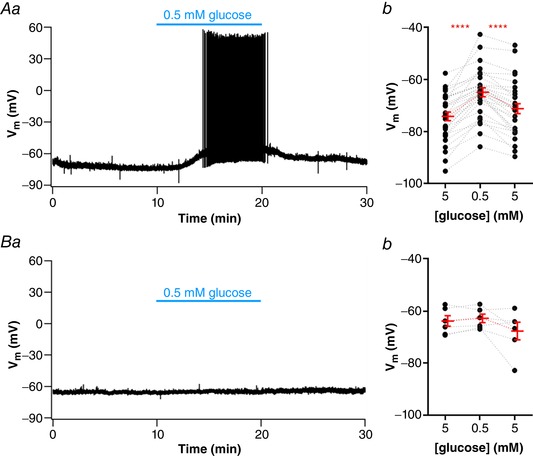
A, subset of neurons depolarized by low glucose, as shown by representative recording of a responsive neuron showing a reversible depolarization and increased firing activity induced by low glucose (Aa) and summary of the low glucose effect on the membrane potential (V m) of neurons (Ab). B, subset of neurons unresponsive to low glucose, as shown by a representative recording (Ba) and summary of the low glucose effect on the V m of neurons (Bb). **** P < 0.0001. [Color figure can be viewed at wileyonlinelibrary.com]
Depolarization induced by low glucose triggered action potential (AP) firing in 16 of 28 silent neurons (57%; 0.6 ± 0.2 Hz; P = 0.007), and an increase in AP frequency in the only two spontaneously active neurons (from a mean of 3.3 ± 2.7 Hz to 8.4 ± 2.0 Hz in low glucose). Surprisingly, only a single cell (3%) was hyperpolarized during perfusion with low glucose solution, from −60 mV to −68 mV, and this effect was partially reversed (41%) on the reinstatement of 5 mM external glucose. Finally, six neurons (16%), were considered unresponsive to low glucose with a mean RMP in 5 mM glucose of −64.1 ± 2.0 mV, and of −63.1 ± 1.6 mV after 0.5 mM glucose, a difference of 1.0 ± 0.7 mV; P = 0.2; Fig. 3 Ba and Bb).
To test the dependence on activation of voltage‐gated sodium channels for low glucose sensing, we performed the low glucose challenge in the presence of TTX (0.5 μM) in a second set of NTS neurons (n = 23 cells, 11 animals). Again, we identified three types of responses. Fourteen cells (61%) were depolarized after low glucose by 8.8 ± 1.0 mV, from a mean of −79.1 ± 1.6 mV to −70.3 ± 1.3 mV (P < 0.0001; Fig. 4 Aa and Ab), and took, on average, 348 ± 18 s to peak. This effect was reversible in most neurons (71%), and the RMP after returning to 5 mM glucose was −77.0 ± 1.8 mV (P < 0.0001). A single cell (4%) was hyperpolarized by −5.5 mV, from −72.1 mV to −77.6 mV in low glucose, and this effect was reversed after returning to 5 mM glucose. Lastly, eight cells (35%) were unresponsive to low glucose solution (1.1 ± 0.5 mV; from a RMP of −66.9 ± 1.6 mV to −65.8 ± 1.7 mV; P = 0.07; Fig. 4 Ba and Bb). Both depolarizing effect and latency to peak response were not different from those observed in experiments conducted with no TTX (P = 0.7, and P = 0.2, respectively), and as in normal aCSF, most neurons depolarized in low glucose solution (84% in normal aCSF, and 65% in TTX). We conclude that the depolarization triggered by low glucose is not dependent on the activation of voltage‐gated sodium channels and action potential firing. Most of the experiments shown from now on were performed in the presence of TTX.
Figure 4. Low glucose sensing of NTS neurons is independent of action potential‐dependent neurotransmission.
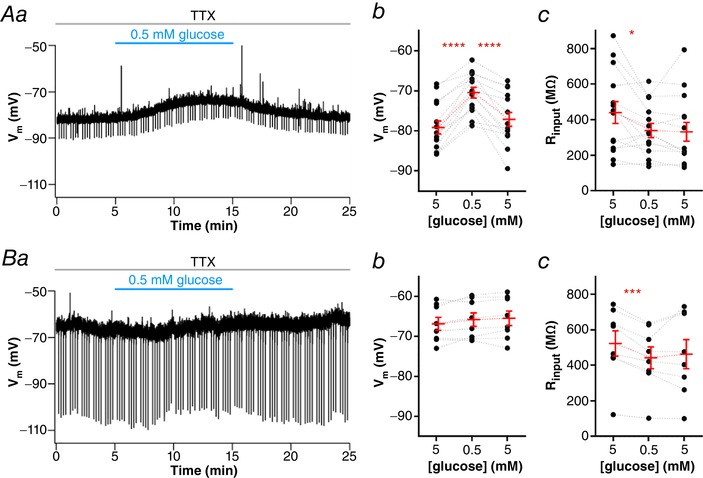
A, subset of neurons depolarized by low glucose in the presence of tetrodotoxin (TTX), as shown by representative recording of a responsive neuron showing a reversible depolarization and decreased input resistance (R input) induced by low glucose (Aa). The graphs show the summary of the low glucose effect on membrane potential (V m; Ab) and R input; Ac) of neurons. B, subset of neurons unresponsive to low glucose in the presence of TTX, as shown by a representative recording (Ba). The graphs show the summary of the low glucose effect on (V m; Bb) and R input; Bc) of neurons. * P < 0.05; *** P < 0.001; **** P < 0.0001. [Color figure can be viewed at wileyonlinelibrary.com]
Lamy et al. (2014) found that the membrane depolarization induced by low glucose in GABAergic NTS neurons was accompanied by an increase in the membrane input resistance (R input) caused by inhibition of a potassium leak conductance. Contrary to the observation of Lamy et al. (2014), we verified that NTS neurons depolarized by low glucose showed a significant decrease in R input (−101.1 ± 37.8 MΩ, from a mean of 440.3 ± 61.6 MΩ to 339.2 ± 40.0 MΩ; P = 0.02; Fig. 4 Ac). In contrast to the effects observed with the membrane potential, these effects did not reverse on reinstatement of 5 mM glucose (P = 0.7). Additionally, neurons unresponsive to low glucose also showed a decrease in R input (−80.3 ± 13.2 MΩ, from a mean of 522.9 ± 71.1 MΩ to 442.6 ± 61.6 MΩ; P = 0.0005; Fig. 4 Bc). As in responsive neurons, R input did not reverse after returning the neurons to 5 mM glucose aCSF in non‐responsive cells (P = 0.5). Interestingly, the single neuron hyperpolarized by low glucose in TTX showed a robust decrease in R input (−629.2 MΩ, from 900.5 MΩ to 271.3 MΩ), suggesting the opening of KATP channels.
Interestingly, we observed that the depolarization caused by low glucose was strongly correlated with neuronal RMP (r 2 = 0.53; P < 0.0001; Fig. 5 A); the more negative the RMP, the greater the membrane potential change in response to low glucose. We then compared the RMP between responsive and non‐responsive neurons, and found that the RMP of non‐responsive cells was significantly more depolarized than responsive cells (−66.8 ± 1.6 mV vs. −78.6 ± 1.6 mV, respectively; P = 0.0001; Fig. 5 C). On the other hand, we found a weak inverse correlation between the low glucose effects on ΔV m and ΔR inpu t (r 2 = 0.18, P = 0.04; Fig. 5 B). We conclude that the depolarization induced by low external glucose uses a voltage‐dependent mechanism, which might involve the opening of a depolarizing membrane conductance.
Figure 5. Low glucose‐induced depolarization of NTS neurons is a voltage‐dependent mechanism.
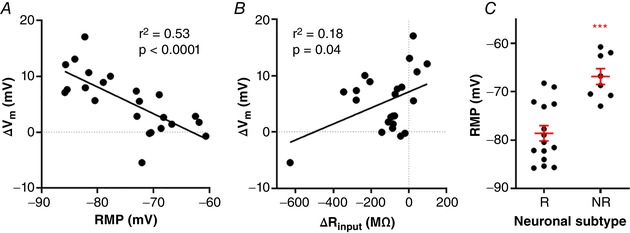
The graphs show the linear correlation between the change of membrane potential (ΔV m) induced by low glucose and resting membrane potential (RMP; A) and input resistance response (ΔR input; B) of neurons. C, comparison of the RMP between neurons responsive (R) and unresponsive (NR) to low glucose. *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
In order to discover whether the decrease in glucose is being sensed by the recorded neuron itself, or is signalled by neighbouring glia as previously suggested (McDougal et al. 2013), we added 3 mM of glucose to the recording pipette and measured the response of the neuron to low glucose (n = 6, four animals). In this condition, perfusion of low glucose external solution did not change the RMP of NTS neurons (−1.0 ± 0.64 mV; from a mean of −76.3 ± 3.6 mV to −75.3 ± 3.2 mV; P = 0.2; Fig. 6 A), showing that the drop in external glucose is being sensed by the recorded neuron. Interestingly, these neurons presented a small but significant drop in R input, from 353 ± 59 MΩ to 323 ± 64 MΩ (P = 0.049). The RMP of the neurons with 3 mM glucose was not significantly different from that of NTS neurons in 5 mM glucose (P = 0.6; Fig. 6 B), and neither was the R input (P = 0.18), showing that the presence of glucose in the pipette did not depolarize the neuron. In fact, when we compared the RMP of the non‐responsive neurons, which were more depolarized, with the RMP in 3 mM internal glucose, we found a significant difference (−66.8 ± 1.6 mV vs. −76.3 ± 3.6 mV, respectively; P = 0.02; Fig. 6 C), confirming that the internal glucose is not depolarizing the NTS neurons.
Figure 6. The presence of glucose in the pipette solution prevents the detection of low external glucose by NTS neurons.
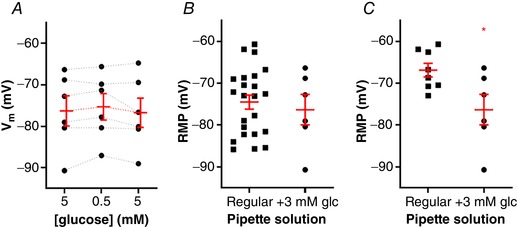
A, summary of the low glucose effect on the membrane potential (V m) of neurons recorded with 3 mM glucose pipette solution. B, comparison of the resting membrane potential (RMP) between neurons recorded with the regular pipette solution and the 3 mM glucose pipette solution. C, comparison of the RMP between unresponsive neurons recorded with the regular pipette solution and the 3 mM glucose pipette solution. Glc, glucose. * P < 0.05. [Color figure can be viewed at wileyonlinelibrary.com]
The age of the animals affects RMP and the response to low glucose
Although most of our recordings were performed on 3‐week‐old animals, we used animals at 6 and 11 weeks old as well. We then compared whether the age of animals could influence n the sensitivity to low glucose. We found that the response to low glucose in responsive neurons was similar in slices from 3‐, 6‐ and 11‐week‐old rats (3 weeks: 8.4 ± 0.7 mV; 6 weeks: 11.4 ± 2.1 mV; 11 weeks: 8.5 ± 1.8 mV; P = 0.22; n = 27, 11 and 6, respectively), but we found a bigger proportion of non‐responsive neurons in slices from 11‐week‐old rats than from 3‐week‐old rats (42% vs. 20%; P = 0.0012) and 6‐week‐old rats (15%; P < 0.001). Accordingly, the RMP of the neurons from 11‐week‐old rats was more depolarized than from 3‐ and 6‐week‐old rats (3 weeks: −74.8 ± 1.4 mV; 6 weeks: −74.6 ± 2.7 mV; 11 weeks: −66.9 ± 2.1 mV; 3 weeks vs. 6 weeks, P = 0.8; 3 weeks vs. 11 weeks, P = 0.007; 6 weeks vs. 11 weeks, P = 0.03; n = 35, 13 and 12 for 3 weeks, 6 weeks and 11 weeks, respectively).
Because the effect of tolbutamide was also voltage dependent, we also compared the number of tolbutamide‐sensitive neurons in slices from 3‐ and 11‐week‐old rats. Similarly, we found more tolbutamide‐non‐responsive neurons in 11‐week‐old animals than in 3‐week‐old animals (45% of non‐responsive neurons in 11‐week‐old animals vs. 13% from 3‐week‐old animals; P < 0.0001, n = 20 and 15, respectively). The RMP of tolbutamide‐non‐responsive neurons from 11‐week‐old animals was more depolarized than that of responsive neurons in both age groups (P = 0.035), but the depolarization induced by tolbutamide was not significantly different in both groups (3 weeks: 14.6 ± 2.1 mV; 6 weeks: 16.5 ± 2.2 mV; P = 0.5). We conclude that NTS neurons from 11‐week‐old animals are more depolarized than from 3‐ or 6‐week‐old animals, and for this reason, they express more low glucose and tolbutamide unresponsive neurons.
Perfusion with low glucose solution produces an inward current
We then investigated which ionic conductance is responsible for the depolarization induced by low glucose. Lamy et al. (2014) showed that in GABAergic neurons from mice NTS, low glucose solution inhibits a potassium leak conductance, leading to depolarization of the neuron. On the other hand, Balfour & Trapp (2007) reported in NTS neurons from rats, an inward current activated by low glucose in half of the neurons, and an inhibition of a potassium conductance or a parallel inward current in the other half of recorded neurons, suggestive of an inhibition of the current of the Na+/K+‐ATPase. We performed current‐voltage relationships voltage‐clamp recordings in NTS neurons (n = 8, five animals), and observed a similar effect to that observed by Balfour & Trapp (2007), with low glucose aCSF perfusion increasing membrane conductance (measured as the slope of the curve) from 3.1 ± 0.1 nS to 4.8 ± 0.1 nS, and this increase was significantly different (P < 0.0001). Low glucose produces a small inward current (−57.6 ± 26 pA at −85 mV) with a reversal around –51 mV (Fig. 7 A and B). Interestingly, the reversal potential is around the membrane potential where the effect of low glucose is not observed. Thus, we conclude that in NTS neurons from rats, the main mechanism of depolarization is the development of an inward current with a reversal potential more positive than the RMP. This is in accordance with the decrease in the R input observed after low glucose external solution in NTS neurons and with the voltage dependency of the depolarization, which is near the reversal potential of this current.
Figure 7. Low glucose‐induced depolarization of NTS neurons is triggered by an inward current.
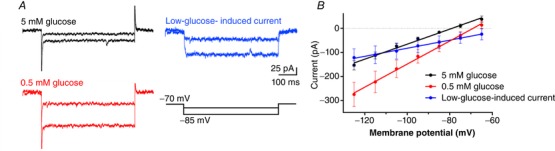
A, representative time course recordings of currents in response to voltage steps obtained in basal (5 mM glucose; black) and low glucose (0.5 mM; red) conditions, and the respective subtraction of currents (blue). B, linear regression of current‐voltage relationship of neurons recorded in the conditions depicted in A. [Color figure can be viewed at wileyonlinelibrary.com]
Depolarization by blocking KATP channels inhibits the effect of low glucose aCSF
We showed that NTS neurons have KATP channels opened at rest and that these channels could strongly affect RMP if inhibited, as demonstrated by the depolarization triggered by their antagonist tolbutamide. Additionally, we found that perfusion with low glucose aCSF depolarized the RMP of these neurons, but this effect decreased with RMP depolarization, and the non‐responsive neurons were more depolarize than the responsive neurons. Thus, two opposite signals, low glucose and block of KATP channels, produce a similar effect, the membrane depolarization. Because the depolarization induced by low external glucose is more prominent in more hyperpolarized neurons, in a situation where KATP channels are blocked, as in a high ATP/ADP ratio, we hypothesized that the neuron could not sense the drop in external glucose.
In order to test this hypothesis, we first tested if a drop in the external glucose could further depolarize a neuron depolarized by tolbutamide. For this experiment, we first tested the responsiveness of the neuron to low glucose, then washed the neuron, and then applied tolbutamide. We found that all neurons responsive to low glucose were responsive to tolbutamide (Fig. 8 Aa and Ab). Low glucose reversibly depolarized the RMP by 9.1 ± 2.2 mV (P = 0.01; n = 5), and application of tolbutamide was able to depolarize the RMP by 18.5 ± 3.0 mV (P = 0.004), showing that neurons responsive to low glucose have KATP channels opened at rest. However, in these neurons, application of low glucose aCSF in the presence of tolbutamide induced only a small, but significant, membrane depolarization (2.0 ± 0.3 mV; P = 0.002), which was smaller than the depolarizing response before tolbutamide application (P = 0.03; Fig. 8 Ac). This result suggests that depolarization induced by blocking KATP channels blunts the neuron's response to low external glucose.
Figure 8. The occlusion of KATP channels suppresses the low glucose‐induced depolarization of NTS neurons.
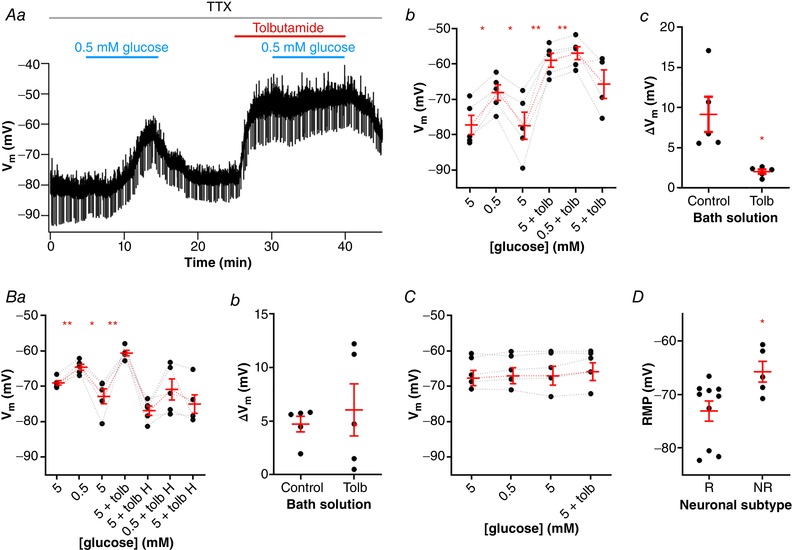
A, application of tolbutamide blunts the depolarizing effect induced by a low glucose challenge, as shown by a representative recording (Aa). Neurons responsive to low glucose are also depolarized by tolbutamide (Ab), but this effect occludes the low glucose sensing (Ac). B, hyperpolarization of neurons responsive to low glucose (Ba) prevents the effect of tolbutamide in blunting the low glucose sensing (Bb). C, neurons unresponsive to low glucose also do not respond to tolbutamide application. D, comparison of the resting membrane potential (RMP) between neurons responsive (R) and unresponsive (NR) to both low glucose and tolbutamide. H, hyperpolarization; Tolb, tolbutamide; TTX, tetrodotoxin; V m, membrane potential. * P < 0.05; ** P < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
To test if this effect was due only to the membrane depolarization induced by tolbutamide, we repeated this protocol, but hyperpolarized the neurons by injecting DC current after the application of tolbutamide, and tested the effect of low glucose aCSF (n = 5; Fig. 8 Ba). Again, we found that the membrane potential is depolarized by both low glucose (4.4 ± 0.7 mV; P = 0.003) and tolbutamide (12.2 ± 2.8 mV; P = 0.001). Next, the injection of −30 to −50 pA of DC current hyperpolarized the membrane potential (from a mean of −60.5 ± 0.8 mV to −76.9 ± 1.3 mV), and in this condition, low glucose aCSF was able to depolarize the membrane by more than 2 mV in three out of five NTS neurons (average for all neurons: 6.0 ± 2.4 mV; P = 0.06). However, this depolarization was similar to that observed in the absence of tolbutamide (P = 0.6; Fig. 8 Bb), showing that tolbutamide is not able to occlude the effect of low glucose if the membrane is hyperpolarized to values similar to the RMP in most NTS neurons.
Interestingly, all neurons unresponsive to low glucose aCSF (0.6 ± 0.5 mV; P = 0.3; n = 5) were also not responsive to tolbutamide (1.0 ± 0.5 mV; P = 0.1; Fig. 8 C). These neurons also had more depolarized RMPs in comparison to responsive neurons (−65.7 ± 1.9 mV vs. −73.1 ± 1.9 mV, respectively; P = 0.03; Fig. 8 D).
Incubation with 10 mM glucose aCSF increases RMP and reduces the number of neurons responsive to low glucose and tolbutamide
Because most NTS neurons express open KATP channels at rest, which can depolarize the membrane when blocked, we asked whether NTS neurons incubated with a higher glucose concentration could lead to more ATP production by the glycolytic/oxidative metabolism, blockage of the KATP channels, and thus membrane depolarization. For this experiment, we incubated the slices in an aCSF solution containing twice the concentration of glucose (10 mM). We found that NTS neurons incubated in 10 mM glucose aCSF (n = 17, eight animals) had more depolarized RMPs than neurons incubated in 5 mM glucose aCSF (−69.0 ± 1.5 mV vs. −74.5 ± 1.7, respectively; P = 0.02; Fig. 9 A. This suggests that NTS neurons incubated in 10 mM glucose produce more ATP, which block KATP channels and depolarize the membrane. Because the response to low glucose was blunted by membrane depolarization, we hypothesized that NTS neurons incubated in 10 mM glucose would be less responsive to a low glucose solution than neurons incubated in 5 mM glucose. In order to test this, we sequentially applied a low glucose solution, followed, after returning to 10 mM glucose, by application of tolbutamide. We found that only three out of 16 cells (19%) were reversible depolarized by perfusion of 0.5 mM glucose (11.2 ± 0.8 mV, from a mean of −74.0 ± 3.7 mV to −62.8 ± 3.3; P = 0.0008; Fig. 9 B and C). This depolarization was similar to what found in neurons incubated in 5 mM external glucose (P = 0.2). Interestingly, the time course of the depolarization was slower, taking on average 435 ± 39 ms to peak in contrast to 349 ± 19 ms in 5 mM glucose aCSF (P = 0.048). After returning to 10 mM glucose, the RMP returned to a value similar to the original RMP (−72.7 ± 5.3 mV; P = 0.9). Additionally, we sequentially applied tolbutamide which triggered a strong depolarization of 18.4 ± 1.6 mV (P = 0.007; Fig. 9 B), a value similar to that produced by tolbutamide in 5 mM glucose (P = 0.5). On the other hand, 13 neurons (81%) were considered unresponsive to low glucose (1.4 ± 0.5 mV, from a mean of −67.5 ± 1.4 mV to −66.2 ± 1.4 mV; Fig. 9 C and E;), and had more depolarized RMPs compared to the responsive cells (responsive neurons: −77.7 ± 0.5 mV; unresponsive neurons: −66.05 ± 2.2 mV; P = 0.01; Fig. 9 D). Moreover, tolbutamide was applied in four of the unresponsive cells and did not induce significant membrane potential change (0.8 ± 0.3 mV; P = 0.08; Fig. 9 E). As in neurons incubated with 5 mM glucose, we found a positive correlation between the effect of low glucose and RMP (r 2 = 0.44, P = 0.005; Fig. 9 F). The proportions of non‐responsive cells were significantly bigger in 10 mM glucose than in 5 mM glucose (81% vs. 39%; P < 0.0001; Fig. 9 C).
Figure 9. Incubation in high glucose (10 mM) decreases the number of NTS neurons sensitive to low glucose.
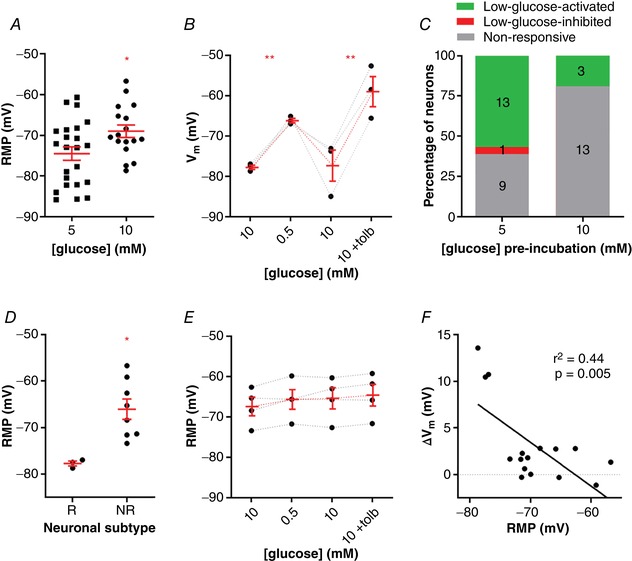
A, comparison of the resting membrane potential (RMP) between neurons incubated in 5 mM and 10 mM glucose. B, neurons incubated in high glucose that are responsive to low glucose are depolarized by tolbutamide application. C, comparison of the distribution of neurons pre‐incubated in different glucose solutions according to their response to low glucose. D, comparison of the RMP of neurons incubated in high glucose that were responsive (R) or unresponsive (NR) to a low glucose challenge. E, neurons incubated in high glucose that are unresponsive to low glucose also do not respond to tolbutamide application. F, linear correlation between the change of membrane potential (V m) and the RMP of neurons incubated in high glucose. Tolb, tolbutamide. * P < 0.05; ** P < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
The lack of effect of both low glucose and tolbutamide on the RMP of NTS neurons suggests that metabolic ATP is blocking KATP in these neurons when incubated in 10 mM glucose, leading to the membrane depolarization and inhibition of the response to low glucose. To test this, we hyperpolarized the depolarized neurons (from −71.3 ± 3.5 mV to 85.1 ± 2.9 mV; n = 5) and applied sequentially low glucose and tolbutamide (Fig. 10 A). Surprisingly, in hyperpolarized neurons, the response to low glucose was present but reduced when compared to regular responsive neurons (3.2 ± 1.1 mV vs. 11.2 ± 0.8 mV, respectively; P = 0.0004; Fig. 10 B), suggesting a diminished response to low glucose in neurons incubated in 10 mM glucose. On the other hand, these neurons continued to be non‐responsive to tolbutamide (−2.4 ± 1.1 mV, from −83.0 ± 1.2 mV to −80.6 ±1.5 mV; P = 0.1; Fig. 10 A). Although some individual neurons presented a smaller response, they were much smaller than in 5 mM glucose (P = 0.03; Fig. 10 C). These results are in accordance with the hypothesis that metabolic ATP is blocking the KATP channels.
Figure 10. Hyperpolarization of NTS neurons incubated in high glucose (10 mM) does not rescue the response to a low glucose challenge and tolbutamide application.
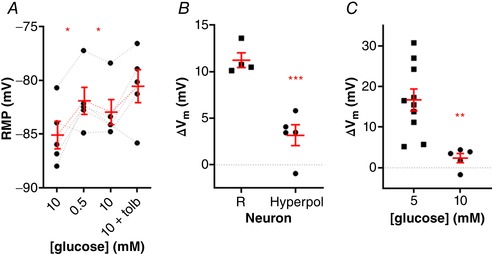
A, neurons incubated in high glucose that had more positive resting membrane potentials (RMP) were hyperpolarized and challenged to a low glucose solution and tolbutamide application, respectively. B, comparison of the membrane potential change (ΔV m) induced by low glucose between high glucose‐incubated responsive neurons (R) and the hyperpolarized neurons (Hyperpol) shown in A. C, comparison of the ΔV m induced by tolbutamide between neurons pre‐incubated in 5 mM and 10 mM glucose shown in A. Tolb, tolbutamide. * P < 0.05; ** P < 0.01; *** P < 0.001 [Color figure can be viewed at wileyonlinelibrary.com]
We conclude that incubating NTS neurons in a high glucose aCSF decreases the number of low glucose‐responsive neurons. Although this effect was not caused by the more depolarized membrane potential, these neurons when hyperpolarized responded more weakly to low glucose. On the other hand, the more depolarized RMPs suggest that the higher extracellular glucose can increase metabolic ATP, which blocks KATP channels and depolarizes neuronal membrane.
The depolarization induced by low glucose is not sustained for prolonged periods
Because of the substantial effect of KATP channels on the RMP modulation of NTS neurons, and the evidence above suggesting that the metabolic ATP can control RMP by modulating KATP channels, we asked if in a situation of prolonged exposure to a low external glucose solution, where metabolic ATP can be reduced, the membrane depolarization could be sustained.
For this purpose, we recorded the membrane potential after perfusion of 0.5 mM glucose for longer than the 10‐min period we used in the previous recordings (n = 4). We observed in three neurons that the depolarization caused by perfusion with 0.5 mM glucose starts to reverse after around 1033 ± 101 s. In two of these neurons, we applied tolbutamide, which depolarized the RMP to values similar to values at the beginning of low glucose aCSF perfusion (Fig. 11 Aa and b). Interestingly, in one non‐responsive neuron, the RMP hyperpolarized after 1300 s of exposure to 0.5 mM glucose, and the addition of tolbutamide reversed the hyperpolarization (Fig. 11 B), suggesting that this hyperpolarization was at least partially caused by the opening of KATP channels. To investigate this further, we applied tolbutamide during the low glucose‐induced depolarization (n = 3). In these three neurons, tolbutamide blocked the hyperpolarization caused by prolonged low glucose exposure (Fig. 11 Ca and b). We conclude that the effect of low external glucose in depolarizing the membrane of NTS neurons is short lived and is probably reversed by depletion of intracellular ATP and opening of KATP channels.
Figure 11. Low glucose‐induced depolarization of NTS neurons is short living and reversed by the opening of KATP channels.
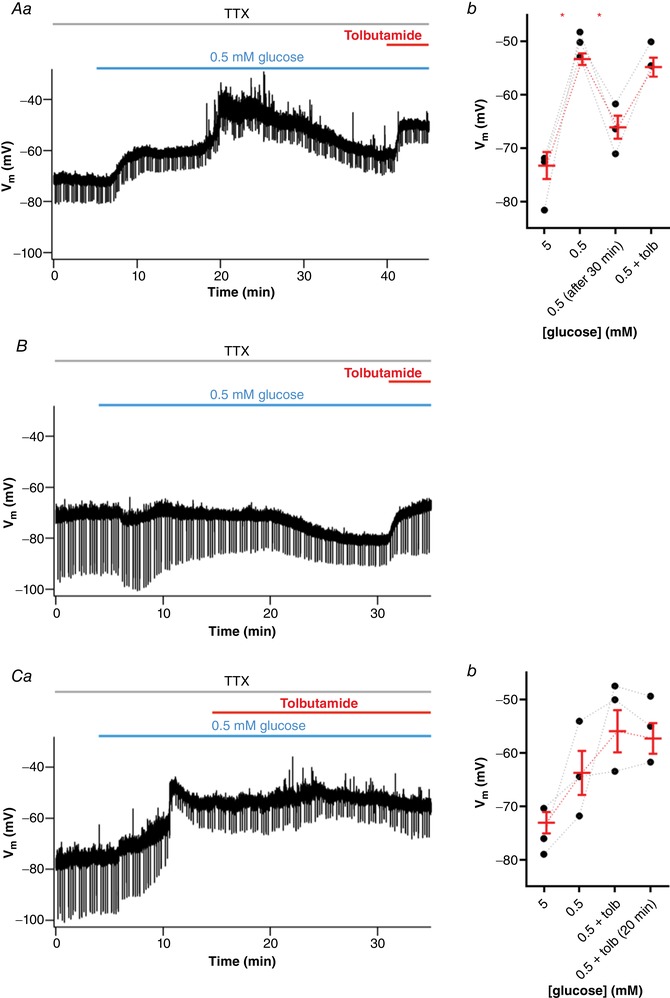
A, subset of neurons initially responsive to low glucose is hyperpolarized after a long period of exposure, as shown by a representative recording (Aa). Note that tolbutamide reverses the hyperpolarizing effect induced by low glucose. Ab, summary of the effect triggered by low glucose and tolbutamide on the membrane potential (V m) of neurons. B, a neuron non‐responsive to a low glucose challenge is hyperpolarized after more than 20 min low glucose exposure. Note that tolbutamide also reverses the hyperpolarizing effect. C, application of tolbutamide suppresses the hyperpolarization induced by a long period of low glucose perfusion, as shown by a representative recording (Ca). Cb, summary of the effect of low glucose and tolbutamide on V m of neurons. Tolb, tolbutamide; TTX, tetrodotoxin. * P < 0.05. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
Glucose is the primary energy source for brain metabolism and survival (Mergenthaler et al. 2013). Due to high levels of energy expenditure for neuronal activity and low content of brain glycogen, the human brain consumes up to 20% of the glucose‐derived energy under physiological conditions (Magistretti & Allaman, 2015). Brain hypoglycaemia, a condition of limited energy availability, can cause neuronal death and may lead to cognitive impairments and loss of consciousness (Cryer, 2007). Therefore, several peripheral and central components act on energy homeostasis regulation to maintain adequate levels of circulating glucose (Marty et al. 2007; Verberne et al. 2014).
Recent studies have demonstrated that glucose‐sensing neurons located in the brainstem NTS can sense glucose levels in the extracellular milieu, using mechanisms which could involve KATP channels (Balfour et al. 2006; Lamy et al. 2014; Boychuk et al. 2015; Halmos et al. 2015; Roberts et al. 2017). Here we found that NTS neurons express KATP channels, as they all responded to diazoxide. In 5 mM external glucose, most of these channels are open, since they depolarized in response to tolbutamide. Thus, in normoglycaemic conditions, NTS neurons have open KATP channels, which could be modulated by local metabolism. In fact, when we incubated the slices in 10 mM glucose, the neurons were more depolarized and unresponsive (or much less responsive) to tolbutamide. These data show that NTS neurons can modulate their membrane potential using KATP channels.
Our experiments were performed in whole‐cell patch clamp using 4 mM Mg‐ATP in the internal solution. Balfour et al. (2006) showed that NTS neurons are more responsive to metabolic ATP than the ATP provided by the electrode solution. Similar results were described in DMX neurons (Müller et al. 2002), and hypothalamic neurons (Song et al. 2001). Although not yet elucidated, this effect is probably due to the compartimentalization of KATP channels in specific membrane domains (Garg et al. 2009) closer to mitochondria and glycolytic enzymes, which have been shown to be concentrated in regions of high metabolic demand and glucose transport activity (Zecchin et al. 2015, Agrawal et al. 2018). Interestingly, this concentration of ATP is more than the micromolar concentrations needed to block Kir6.2/SUR1 channels (Baukrowitz et al. 1998), which is a subunit composition present in NTS neurons (Karschin et al. 1997; Balfour et al. 2006; Halmos et al. 2015). However, the effect of tolbutamide shows that there are KATP channels open even with 4 mM internal ATP. This can be explained by the sensitivity of KATP channels to the ADP/ATP ratio, because they are activated by Mg‐ADP (Nichols et al. 1996; Shyng et al. 1997) and their affinity to ATP is greatly reduced by phosphatidylinositol 4,5‐bis‐phosphate (PIP2; Baukrowitz et al. 1998; Shyng et al. 2000). Thus, our whole‐cell recordings very likely reflect the physiological responses of NTS neurons to metabolic ATP derived from external glucose.
In the current investigation, we showed that low glucose (0.5 mM) induces a voltage‐dependent depolarization in most NTS neurons of rats. These effects were not observed when the internal electrode solution contained 3 mM glucose, showing that the neuron itself is sufficient to detect and respond to a drop in external glucose concentration, although some influence by glial cells cannot be discarded, as suggested by McDougal et al. (2013). Additionally, we showed that the RMP in NTS neurons incubated in 10 mM glucose aCSF is more depolarized and less sensitive to tolbutamide, even when hyperpolarized, suggesting they are depolarized by blocking of KATP channels by metabolic ATP. Therefore, we expected that the depolarization of the RMP induced by the incubation of slices in high glucose could lead to an increase in the number of neurons unresponsive to a low glucose challenge, since our findings demonstrated a voltage‐dependent depolarization by low glucose (i.e. the more negative the RMP, the bigger the membrane potential response amplitude), which was indeed observed. Neurons unresponsive to low glucose accounted for 35% when incubated in normal aCSF, but ∼70% when incubated in high glucose aCSF. Interestingly, these neurons were able to respond to low glucose when hyperpolarized, but the magnitude of the response was significantly smaller, suggesting that other mechanisms are affecting the responsiveness of these neurons to low external glucose.
We also observed that the fraction of low glucose‐ and tolbutamide‐responsive neurons was decreased in slices from 11‐week‐old animals in comparison to slices from 3‐ or 6‐week‐old animals. These neurons were also more depolarized than responsive neurons, as observed in slices from younger animals. Because the low glucose‐ or tolbutamide‐induced depolarizations in responsive neurons from 11‐ and 3‐week‐old animals were similar, we believe the reason for this difference is the presence of more depolarized neurons in 11‐week‐old animals. We don't know the reason for this difference in RMP, which could be related or not to developmental changes. Importantly, 3‐week‐old rats are in the weaning period with (access to the caloric maternal milk) while 11‐week‐old rats are young adult animals feeding on the standard lab rat chow. The differences in caloric intake, type of meals, and food‐related behaviour could lead to distinct metabolic states, and thus changes in passive electrophysiological properties of NTS neurons. Nevertheless, further investigations are needed to elucidate these questions.
Interestingly, Balfour et al. (2006) reported that ∼80% of NTS/DMX neurons of 3‐ to 4‐week‐old rats were unresponsive to glucose removal. However, these authors performed electrophysiological recordings using a control aCSF with 10 mM glucose. They also showed the expression of the KATP channel subunit SUR1 in cells that did not respond to low glucose, which suggests that in these neurons KATP channels are present but could be saturated by high glucose‐derived ATP levels.
Low extracellular glucose induces KATP channels to open following a decrease in intracellular ATP levels (Hibino et al. 2010), but we observed a hyperpolarization response to low glucose in only two of 60 NTS neurons incubated in 5 mM glucose aCSF. Other groups have reported that sensitivity to glucose is not observed in all NTS neurons expressing KATP channel subunits (Dallaporta et al. 2000; Balfour et al. 2006). Lamy et al. (2014) reported an increase in R input with the closure of leak potassium channels in GLUT2‐expressing GABAergic neurons activated by low glucose in mice. This could account for the response we observed, but again, the low glucose‐sensitive current observed in this report was a linear non‐rectifying current, which is not compatible with the voltage dependency we observed, and we observed mainly a decrease in R input, suggesting the opening of an inward conductance. Additionally, our findings demonstrate that most NTS neurons exhibit a voltage‐dependent depolarization and a decrease in R input in response to a low glucose solution, which is suggestive of the opening of a cationic conductance. We found that low glucose induces an inward cationic current with a reversal around −60 mV, which could explain the voltage dependence of the depolarization induced by low glucose.
Interestingly, Balfour & Trapp (2007) showed the opening of an inwardly rectifying current in some neurons of the NTS in response to glucose removal, what could contribute to the depolarizing response seen in our recordings. The opening of HCN channels, which mediate the inwardly rectifying cationic h current, could explain our results, but the activation of the low glucose‐induced current was very fast, not compatible with the slow activation of the HCN channels. Additionally, we performed experiments using the antagonist of these channels, ZD7288 (10 μM), but the results were inconclusive because this drug produced a progressive depolarization of the membrane, masking any effect of low glucose (not shown). In the ventromedial hypothalamus, low glucose led to the closure of chloride channels and membrane depolarization in some neurons (Routh et al. 2014), but the reversal potential of the low glucose‐induced current observed in the current investigation does not suggest a chloride conductance. Ionic mechanisms other than ion channels can contribute to depolarize neurons under low glucose availability. The electrogenic Na+/K+ pumps are important generators of electrochemical gradients in cells. High glucose levels stimulate the Na+/K+‐ATPase pump and trigger membrane hyperpolarization in glucose‐excited neurons in the lateral hypothalamus (Oomura et al. 1974), and brain hypoglycaemia reduces the activity of the Na+/K+‐ATPase pump (Lees, 1991) and may lead to depolarization of neurons (Balfour & Trapp, 2007). However, this mechanism is not compatible with the voltage dependency and the decrease in the R input we observed. Nevertheless, the reduction of the activity of the Na+/K+‐ATPase pump could constitute an additional component altering the RMP of NTS neurons in response to reduced external glucose concentration.
Since KATP blockage and low glucose both produce membrane depolarization, but low glucose is ineffective at depolarizing neurons when the KATP channels are entirely blocked, we believe that the depolarization induced by low external glucose might represent a form of homeostatic regulation of the RMP in order to avoid an excessive hyperpolarization by unblocking KATP channels during a hypoglycaemia episode, which could be essential for the efficient performance of the vital autonomic functions controlled by the NTS. However, when we left the neurons for more than 10—20 min in low external glucose, we started to observe a hyperpolarization that was prevented by tolbutamide. These results show that the low glucose‐induced depolarization does not last for prolonged periods, and the membrane starts to hyperpolarize in response to the decreased metabolic ATP.
Our results clearly show a mechanism of membrane depolarization driven by a rapid reduction in external glucose that could be potentially relevant for neurometabolic responses to hypoglycaemia. However, when neurons are maintained in a hyperglycaemic solution (similar to high post‐prandial circulating glucose levels and diabetic conditions), the number of NTS neurons responsive to low glucose decreases, and the latency for peak response increases. Because NTS neurons are involved in the counter‐regulatory mechanisms in response to hypoglycaemia, which include a decrease in pancreatic insulin secretion, an increase in pancreatic glucagon secretion, and an increase in adrenomedullary adrenaline (epinephrine) secretion (Cryer, 2005), the detection of a rapid drop in glycaemia by these neurons in prolonged hyperglycaemic situations might be blunted. Sudden drops in glycaemia are common occurrences in patients with type 1 and advanced type II diabetes, caused by insulin interventions to control glycaemia (Mergenthaler et al. 2013). In these patients, a failed counter‐regulatory response to hypoglycaemia is commonly observed, a condition named hypoglycaemia‐associated autonomic failure (Cryer, 2005). Interestingly, glucose‐sensing neurons in the ventromedial hypothalamus decrease their response to low glucose after repetitive injections of insulin (Song & Routh, 2006). Our data suggest that the establishment of more depolarized RMPs of NTS neurons by the closure of KATP channels due to increased intracellular ATP levels could be an additional mechanism contributing to reduced brain sensitivity to a low glucose challenge during hyperglycaemic conditions. More studies are needed to support this interesting hypothesis.
Additional information
Competing interests
None declared
Author contributions
CBM and RML designed the experiments. CBM performed the experiments and analyzed the data. CBM and RML interpreted the data, wrote and revised the manuscript. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed
Funding
Research supported by FAPESP grant (2016/01607‐4). C.B.M. is a CNPq PhD scholarship recipient. R.M.L. is a CNPq research fellow.
Acknowledgments
We thank Mr J. Fernando Aguiar and Dr André L. A. Dagostin for technical support, and Drs Daniela Accorsi‐Mendonça, Luiz C. Navegantes and Luiz G. Branco for their criticisms and suggestions.
Biography
Cahuê Murat holds a BSc in Biological Sciences and an MSc in Molecular Biology, and currently is a fourth‐year PhD candidate in physiology at the University of São Paulo, Brazil. Together with Professor Ricardo Leão, he has been studying the electrophysiological mechanisms induced by low external glucose in the neurons of the brainstem nucleus of the tractus solitarius. In his upcoming projects, Cahuê aims to investigate how astrocytes monitor signals related to energy status, and their role in synaptic transmission and information processing involved in brain control of metabolism.

Edited by: Ian Forsythe & Yasuhiko Minokoshi
This article was first published as a preprint. de Bernardis Murat C, Leao R. (2019). A voltage‐dependent depolarization induced by low external glucose in neurons of the nucleus of the tractus solitarius of rats: interaction with KATP channels regulated by external glucose. bioRxiv. https://doi.org/10.1101/328658.
References
- Agrawal A, Pekkurnaz G & Koslover EF (2018). Spatial control of neuronal metabolism through glucose‐mediated mitochondrial transport regulation. Elife 18, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accorsi‐Mendonça D, Castania JA, Bonagamba LG, Machado BH & Leão RM (2011). Synaptic profile of nucleus tractus solitarius neurons involved with the peripheral chemoreflex pathways. Neuroscience 197, 107–120. [DOI] [PubMed] [Google Scholar]
- Andrew, SF , Dinh, TT & Ritter, S (2007). Localized glucoprivation of hindbrain sites elicits corticosterone and glucagon secretion. Am J Physiol Regul Integr Comp Physiol 292, R1792–R1798. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM & Rorsman P (2013). KATP channels and islet hormone secretion: new insights and controversies. Nat Rev Endocrinol 9, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balfour RH, Hansen AM & Trapp S (2006). Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 570, 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balfour RH & Trapp S (2007). Ionic currents underlying the response of rat dorsal vagal neurones to hypoglycaemia and chemical anoxia. J Physiol 579, 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohland M, Matveyenko AV, Saberi M, Khan AM, Watts AG & Donovan CM (2014). Activation of hindbrain neurons is mediated by portal‐mesenteric vein glucosensors during slow‐onset hypoglycemia. Diabetes 63, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boychuk CR, Gyarmati P, Xu H & Smith BN (2015). Glucose sensing by GABAergic neurons in the mouse nucleus tractus solitarii. J Neurophysiol 114, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP & Fakler B (1998). PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282, 1141–1144. [DOI] [PubMed] [Google Scholar]
- Cryer PE (2005). Mechanisms of hypoglycemia‐associated autonomic failure and its component syndromes in diabetes. Diabetes 54, 3592–3601. [DOI] [PubMed] [Google Scholar]
- Cryer PE (2007). Hypoglycemia, functional brain failure, and brain death. J Clin Invest 117, 868–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallaporta M, Perrin J & Orsini JC (2000). Involvement of adenosine triphosphate‐sensitive K+ channels in glucose‐sensing in the rat solitary tract nucleus. Neurosci Lett 278, 77–80. [DOI] [PubMed] [Google Scholar]
- Donovan CM & Watts AG (2014). Peripheral and central glucose sensing in hypoglycemic detection. Physiology (Bethesda) 29, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Ortega‐Sáenz P, García‐Fernández M, González‐Rodríguez P, Caballero‐Eraso C & López‐Barneo J (2014). Glucose sensing by carotid body glomus cells: potential implications in disease. Front Physiol 5, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Jiao J & Hu K (2009). Regulation of ATP‐sensitive K+ channels by caveolin‐enriched microdomains in cardiac myocytes. Cardiovasc Res. 82, 51–58. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Pang JJ, Shaver SW & Wainman DS (1990). Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 259, R1131–R1138 [DOI] [PubMed] [Google Scholar]
- Halmos KC, Gyarmati P, Xu H, Maimaiti S, Jancsó G, Benedek G & Smith BN (2015). Molecular and functional changes in glucokinase expression in the brainstem dorsal vagal complex in a murine model of type 1 diabetes. Neuroscience 306, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Viard E & Rogers RC (2014). Hindbrain glucoprivation effects on gastric vagal reflex circuits and gastric motility in the rat are suppressed by the astrocyte inhibitor fluorocitrate. J Neurosci 34, 10488–10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I & Kurachi Y (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90, 291–366. [DOI] [PubMed] [Google Scholar]
- Karschin C, Ecke C, Ashcroft FM & Karschin A (1997). Overlapping distribution of KATP channel‐forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett 401, 59–64. [DOI] [PubMed] [Google Scholar]
- Lam CK, Chari M, Su BB, Cheung GW, Kokorovic A, Yang CS, Wang PY, Lai TY & Lam TK (2010). Activation of N‐methyl‐D‐aspartate (NMDA) receptors in the dorsal vagal complex lowers glucose production. J Biol Chem 285, 21913–21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy CM, Sanno H, Labouèbe G, Picard A, Magnan C, Chatton JY & Thorens B (2014). Hypoglycemia‐activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab 19, 527–538. [DOI] [PubMed] [Google Scholar]
- Lees GJ (1991). Inhibition of sodium‐potassium‐ATPase: a potentially ubiquitous mechanism contributing to central nervous system neuropathology. Brain Res Brain Res Rev 16, 283–300. [DOI] [PubMed] [Google Scholar]
- McDougal DH, Hermann GE & Rogers RC (2013). Astrocytes in the nucleus of the solitary tract are activated by low glucose or glucoprivation: evidence for glial involvement in glucose homeostasis. Front Neurosci 7, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ & Allaman I (2015). A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. [DOI] [PubMed] [Google Scholar]
- Marty N, Dallaporta M & Thorens B (2007). Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) 22, 241–251. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA & Meisel A (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimee A & Ferguson AV (2015). Glycemic state regulates melanocortin, but not nesfatin‐1, responsiveness of glucose‐sensing neurons in the nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol 308, R690–R699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Brockhaus J & Ballanyi K (2002). ATP‐independent anoxic activation of ATP‐sensitive K+ channels in dorsal vagal neurons of juvenile mice in situ. Neuroscience 109, 313–328. [DOI] [PubMed] [Google Scholar]
- Nichols CG (2006). KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng S‐L, Nestorowicz A, Glaser B, Clement J IV, Gonzalez G, Aguilar‐Bryan L, Permutt AM & Bryan JP (1996). Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272, 1785–1787. [DOI] [PubMed] [Google Scholar]
- Oomura Y, Ooyama H, Sugimori M, Nakamura T & Yamada Y (1974). Glucose inhibition of the glucose‐sensitive neurone in the rat lateral hypothalamus. Nature 247, 284–286. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT & Zhang Y (2000). Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res 856, 37–47. [DOI] [PubMed] [Google Scholar]
- Roberts BL, Zhu M, Zhao H, Dillon C & Appleyard SM (2017). High glucose increases action potential firing of catecholamine neurons in the nucleus of the solitary tract by increasing spontaneous glutamate inputs. Am J Physiol Regul Integr Comp Physiol 313, R229–R239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh VH, Hao L, Santiago AM, Sheng Z & Zhou C (2014). Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci 8, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaquist ER, Damberg GS, Tkac I & Gruetter R (2001). The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 50, 2203–2209 [DOI] [PubMed] [Google Scholar]
- Shyng S, Ferrigni T & Nichols CG (1997). Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol 110, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Barbieri A, Gumusboga A, Cukras C, Pike L, Davis JN, Stahl PD & Nichols CG (2000). Modulation of nucleotide sensitivity of ATP‐sensitive potassium channels by phosphatidylinositol‐4‐phosphate 5‐kinase. Proc Natl Acad Sci U S A 97, 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Levin BE, McArdle JJ, Bakhos N & Routh VH (2001). Convergence of pre‐ and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 50, 2673–2681. [DOI] [PubMed] [Google Scholar]
- Song Z & Routh VH (2006). Recurrent hypoglycemia reduces the glucose sensitivity of glucose‐inhibited neurons in the ventromedial hypothalamus nucleus. Am J Physiol Regul Integr Comp Physiol 291, R1283–R1287. [DOI] [PubMed] [Google Scholar]
- Thorens B (2012). Sensing of glucose in the brain. Handb Exp Pharmacol, 277–294. [DOI] [PubMed] [Google Scholar]
- Verberne AJ, Sabetghadam A & Korim WS (2014). Neural pathways that control the glucose counterregulatory response. Front Neurosci 8, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchin A, Stapor PC, Goveia J & Carmeliet P (2015). Metabolic pathway compartmentalization: an underappreciated opportunity? Curr Opin Biotechnol 34, 73–81. [DOI] [PubMed] [Google Scholar]
- Zhao S, Kanoski SE, Yan J, Grill HJ & Hayes MR (2012). Hindbrain leptin and glucagon‐like‐peptide‐1 receptor signaling interact to suppress food intake in an additive manner. Int J Obes (Lond) 36, 1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]