

Abstract
When considering the clinical applications of autologous cell replacement therapy of human induced pluripotent stem cells (iPSC)‐derived cells, there is a clear need to better understand what the immune response will be before we embark on extensive clinical trials to treat or model human disease. We performed a detailed assessment comparing human fibroblast cell lines (termed F1) reprogrammed into human iPSC and subsequently differentiated back to fibroblast cells (termed F2) or other human iPSC‐derived cells including neural stem cells (NSC) made from either retroviral, episomal, or synthetic mRNA cell reprogramming methods. Global proteomic analysis reveals the main differences in signal transduction and immune cell protein expression between F1 and F2 cells, implicating wild type (WT) toll like receptor protein 3 (TLR3). Furthermore, global methylome analysis identified an isoform of the human TLR3 gene that is not epigenetically reset correctly upon differentiation to F2 cells resulting in a hypomethylated transcription start site in the TLR3 isoform promoter and overexpression in most human iPSC‐derived cells not seen in normal human tissue. The human TLR3 isoform in human iPSC‐NSC functions to suppress NF‐KB p65 signaling pathway in response to virus (Poly IC), suggesting suppressed immunity of iPSC‐derived cells to viral infection. The sustained WT TLR3 and TLR3 isoform overexpression is central to understanding the altered immunogenicity of human iPSC‐derived cells calling for screening of human iPSC‐derived cells for TLR3 expression levels before applications. stem cells 2019;37:476–488
Keywords: Cytokine, Human leukocyte antigen, Immune response, Induced pluripotent stem cells, Inflammation, MHC‐I, Neural stem cells, Toll like receptor
Significance Statement.
The cell reprogramming process alters the immune response of most induced pluripotent stem cells (iPSC)‐derived cells highlighting human toll like receptor protein 3 (TLR3) isoform overexpression that functions to suppress innate immune signal transduction and provides an insight for future clinical applications of autologous human iPSC‐derived cell transplantation. The authors propose that screening human iPSC clones and their derived cells for alterations in the genome of patient cells that include expression levels of human TLR3 and its shorter TLR3 isoform identified in this work is essential before embarking on “first in man” clinical trials or human disease modeling using human iPSC.
Introduction
The discovery of methods to reprogram somatic human cells into tissue specific adult stem cells directly or by full reprogramming, to produce induced pluripotent stem cells (iPSC) has presented the possibility of an easy and abundant source of cells for autologous stem cell therapy to treat many human diseases 1, 2, 3. Three major challenges that must be overcome before reprogrammed stem cells can be used for clinical applications are genetic instability, proper differentiation to relevant cell types to regenerate damaged tissues and the immune response of transplanted cells. The interaction of the human immune system with autologous reprogramed human cells remains an unexplored question.
It is well accepted that Toll receptors are a major part of the innate immune system essential for life 4. Likewise, regulation of inflammation cytokines by toll‐like receptor 3 (TLR3 ‐ virus) and toll like receptor 4 (TLR4 ‐ bacteria) is well established 5, 6. Recent publications in animal studies (mice and non‐human primates) indicate that there may be some immune response to autologous reprogramed cells 7, 8, 9, 10, 11, 12, 13, 14. A recent review identified that many aspects of the adaptive and innate immune system mechanisms are yet to be explored in detail for human cells 3, 15. Data from human transplanted autologous reprogramed cells in vivo is limited and only one current clinical trial for iPSC‐RPE cells exists that found edema in the patient retina most likely from the surgical intervention for the transplanted iPSC‐RPE cell sheet 16, 17, 18, 19. As the field grows and moves closer to clinical application the need to understand the extent of the immune response to transplanted human iPSC‐derived cells becomes paramount.
Of the studies conducted to date in mouse models on the immune response to autologous iPSC‐derived cells, the group from Zhao et al. (2011) studied the immunogenicity of both retroviral vector reprogrammed mouse iPSC and episomal vector reprogrammed integration free mouse iPSC. They transplanted the mouse cells under the skin to evaluate the immunogenicity of mouse iPSC‐derived cells in syngeneic mice and found issues with immogenicity 8. Studies that transplanted mouse iPSC‐derived cells into the neural system and kidney found no apparent immunogenicity 7, 9. Further work by Todorova et al. identified one possible mechanism underlying these conflicting conclusions 14. They show that the lack of immunogenicity of mouse iPSC‐derived cells in syngeneic host when transplanted into the kidney is due to the absence of functional antigen presenting cells in mouse kidney, highlighting that the site of iPSC‐derived cell transplantation in vivo is important in experimental design 14. A more recent study was performed in non‐human primates testing iPSC‐derived neurons found no major immune response, although a small immune response was observed 10. In the study by Zhao et al. (2015), iPSC‐derived smooth muscle cells and retinal pigmented epithelial cells were transplanted into humanized mice reconstituted with autologous human immune system, demonstrating differential immunogenicity in vivo 12. Interestingly, IL10 secretion from mouse iPSC‐derived cells has been found to suppress the immune response, and IL10 is known to suppress the activity of activated T cells, suggesting a mechanism for iPSC‐derived cells to by‐pass immune system to survive and engraft 11.
There is a clear need to better understand what the immune response to human autologous reprogramed cells will be before we embark on extensive clinical trials to treat human disease. We have performed a detailed global proteomic and methylome analysis of the possible immune response of matched human patient fibroblast cells (termed F1) reprogramed to iPSC and compared to iPSC‐derived cells (termed F2) for retroviral, episomal, and synthetic mRNA transfection methods to reprogram human fibroblasts to human iPSC. The data shows that in most aspects the immune biology is similar for either viral or non‐viral made iPSC‐derived cells; however, a significantly different higher expression of wild type (WT) human TLR3 in iPSC derived cells. Global methylome analysis in synthetic mRNA made human iPSC revealed a shorter isoform of human TLR3 is over expressed in human reprogrammed cells (the TLR3 isoform is not expressed in mice) not seen in normal human physiological tissues or cells. We confirm in three retroviral made and three episomal made human iPSC from healthy control patient cells that the TLR3 isoform is over expressed in five from six matched pairs of cells (approx. 85% of human iPSC overexpress the human TLR3 isoform). We show that the human TLR3 isoform functions to suppress NF‐KB p65 signaling pathway in response to virus (Poly IC), suggesting suppressed immunity of human iPSC‐derived cells to viral infection. Taken together the data demonstrate that the cell reprogramming process alters specific aspects of the immune response of in vitro generated human iPSC‐derived cells, suggesting a reduction of innate immunogenicity of human iPSC‐derived cells, calling for screening of human iPSC clones for human TLR3 expression levels before use in disease modeling or clinical applications.
Materials and Methods
iPSC Cell Culture
Established human iPSC clones were obtained from collaborators for this project and previously published. Six human skin fibroblast cell lines (termed F1) that have previously been reprogrammed into iPSC, three by retroviral methods 2, 20, 21 and three by non‐viral episomal methods 22 were used. Synthetic mRNA reprogramming to iPSC was performed using Stemgent's microRNA‐enhanced mRNA reprogramming kit (STEMGENT, #00‐0071, UK) protocol using Thermo Fischer defined cell culture media products. Differentiation to various cell lines and the mRNA transfection method is described in full in a separate manuscript. All hiPSC were differentiated between passages 10–15. Feeder‐free “clinical grade” synthetic mRNA iPSC were cultured in defined Flex E8 culture medium (Life Technologies, A2858501, Spain) on Vitronectin (Life Technologies, A14700, Spain) coated plates. Colonies were passaged by gentle dissociation with 0.5 mM EDTA when they were 75%–80% confluence. Cell culture conditions for hiPSC can be found for Episomal and Retroviral made hiPSC in the papers from Refs. 20, 21, 22.
Proteomic Analysis for Global and MHC‐1 Peptide Analysis
Protein Solubilization and Quantification
Protein solubilization was performed independently for each of the eight pelleted cell samples, five with patients fibroblasts (F1) and three with iPSC‐fibroblasts (F2), with 1% (vol/vol) SDS lysis buffer. To eliminate viscosity, the lysates were heated to 60°C for 15 minutes and sonicated at 45% amplitude for 4 seconds. Protein concentration of each sample was estimated using BCA Reagent Compatible kit (Thermo Scientific, Rockford, IL) following the manufacturer's instructions.
Peptides Preparation and Labeling with Isobaric Tags (Tandem Mass Tag 10‐Plex)
For global analysis, 50 g of proteins from each sample were digested and labeled with tandem mass tag (TMT) 10‐plex isotopic label reagent set (Thermo Scientific, Rockford, IL) following the manufacturer's instructions. Nine of the 10 TMT label reagents (tags: 126; 127N; 127C; 128N; 128C; 129N; 129C; 130N; 131) were used. For normalization purposes, an internal control consisting on a mixture of 6.25 g of each of the eight samples was included and labeled with TMT126. The TMT labeled samples including control were then combined at a 1:1:1:1:1:1:1:1:1 ratio. Mixture of samples was cleaned by Pierce C18 Spin columns (Thermo Scientific, Rockford, IL) and stored at −80°C.
Liquid Chromatography–Tandem Mass Spectrometry
For global analysis, labeled peptides were separated by means of nanoliquid chromatography using a nanoLC ULTRA AS2 (Eksigent, Australia) with a flow rate of 400 nl/min, a C18 Pepmap 100 trap column (5 μm, 100 Å, 300 μm i.d. × 5 mm in length), and an Acclaim PepMap C18 analytical column (3 μm, 100 Å, 75 μm i.d. × 15 cm in length).
The following linear gradient, using solvent B (97% ACN, 0.1%formic acid) and solvent A (3% ACN, 0.1% formic acid), was used: 5%–30% buffer B (140 minutes). MS/MS analysis was performed using an LTQ Orbitrap Velos (ThermoFisher Scientific, Rockford, IL) with a nanoelectrospray ion source with precursor ion selection in the Orbitrap at 30,000 of resolution, selecting the 20 most intense precursor ions in positive ion mode. MS/MS data acquisition was completed using Xcalibur 2.1 (ThermoFisher Scientific, Rockford, IL). For identification of TMT labeled peptides, higher energy collisional dissociation (HCD) with 40% fixed collision energy was the fragmentation method used.
Protein Identification and Quantification
Data was processed using Proteome Discoverer 1.4.1.14 (Thermo Fisher Scientific, Rockford, IL). For database searching, raw mass spectrometry files were submitted to the in‐house Homo sapiens UniProtKB/Swiss‐Prot database (released 2015‐02; 20,165 protein entries) using SEQUEST (Thermo FisherScientific, Rockford, IL). The Percolator search node, a machine‐learning supplement to the SEQUEST search algorithm that increases the sensitivity and specificity of peptide identifications, was also used. Searches were performed using the following settings: 2 maximum miss cleavage for trypsin, TMT‐labeled in N‐Terminal, lysine (+229.163 Da) and methionine oxidation (+15.995 Da) as dynamic modifications, cysteine carbamidomethylation (+57.021 Da) as static modification, 20 ppm precursor mass tolerance, 0.05mmu fragment mass tolerance, 5mmu peak integration tolerance, and most confident centroid peak integration method. The criteria used to accept protein identification included a minimum of two peptides matched per protein, with a false discovery rate of 1%. To avoid the possible ambiguity associated with different isoforms of the same protein during the quantification, all of the proteins were treated as ungrouped.
Quantitative analysis of the TMT experiments was performed simultaneously to protein identification using Proteome Discoverer software. Peptide spectral matches (PSMs) were filtered by PD1.4 software with parameters: Min. Precursor Mass: 300 Da, Max. Precursor Mass: 5,000 Da. Reporter ion quantification of HCDMS2 spectra was enabled, and TMT‐10 plex was set as quantification method. To avoid overlap between the isobaric reporter ions a resolution greater than 60,000 was used from m/z 126.1 to m/z 131.1. The quantification values were extracted from the ration between the reporter ions intensities (in; where n represents the reporter ions ranging from m/z 127.1 to m/z 131.1) and the reporter ion intensity from m/z 126.1, and were corrected according to the isotopic purities of the reporter ions provided by the manufacturer. Quantification values were rejected whenever any quantification channel was missing or whenever variability of the reporter intensity was higher than 50% among the different PSMs of the same protein. For protein quantification purposes, only unique peptides were considered, and protein ratios were normalized to protein median. Normalized values for each protein were exported to Microsoft Excel (v14.0.0), and only those proteins with quantification values for all of the samples (i.e., eight samples and the internal control) were considered for further analysis.
Illumina Infinium Methylation EPIC BeadChip
All DNA samples were assessed for integrity, quantity, and purity by electrophoresis in a 1.3% agarose gel, picogreen quantification, and nanodrop measurements. Bisulfite conversion of 500 ng of genomic DNA was performed using EZ DNA methylation kit (Zymo Research. Irvine) following manufacturer's instructions. Bisulfite (200 ng) converted DNA were used for hybridization on the Illumina Infinium MethylationEPIC BeadChip (Illumina, Inc. San Diego). Briefly, samples were whole genome amplified followed by an enzymatic end‐point fragmentation, precipitation, and resuspension. The resuspended samples were hybridized onto the beadchip for 16 hours at 48°C and washed. A single nucleotide extension with labeled dideoxy‐nucleotides was performed and repeated rounds of staining were applied with a combination of fluorescently labeled antibodies differentiating between biotin and DNP. Fluorescent signal from the microarray was measured with a HiScan scanner (Illumina, Inc. San Diego) using iScan Control Software (V 3.3.29).
A three step‐based normalization procedure was performed using the lumi 23 package available for bioconductor 24, under the R statistical environment, consisting in color bias adjustment, background level adjustment and quantile normalization across arrays, as specified in 23. Methylation level (β‐value) for each of the 866,836 CpG sites were calculated as the ratio of methylated signal divided by the sum of methylated and unmethylated signals plus 100. All beta values with an associated p value greater or equal to 0.01 were removed from the analysis.
Bisulfite Pyrosequencing
To validate the methylome array, we performed bisulfite pyrosequencing following published protocols 25. Briefly, specific set of primers for PCR amplification and sequencing were designed using the PyroMark assay design program, version 2.0 (Qiagen, Netherlands); amplification primers hybridize with CpG‐free sites to ensure a methylation‐independent reaction and one primer (opposite the sequencing primer) is biotinylated to convert the PCR product to single‐stranded DNA templates. We used 1 μl of bisulfite‐treated DNA for each PCR. The EZ DNA methylation kit (Zymo Research, Irvine) was used for bisulfite conversion of all DNA samples used 25. To prepare single‐stranded PCR products, we used the Vacuum Prep Tool (Qiagen, Netherlands), following the manufacturer's instructions. Pyrosequencing reactions and methylation quantification were performed in a PyroMark Q24 System version 2.0.6 (Qiagen, Netherlands).
Sequencing
Human TLR3 isoform was sequenced using 50 ng of DNA following a BigDye protocol published by the University of Pompeu Fabra Genomics Core Facility in Barcelona. Primers used listed in Supporting Information Figures.
Lipopolysaccharide and Poly (I:C) Stimulation
F1 and F2 cells were seeded in 24‐well plate and once 80% confluent they were stimulated with 100 ng/ml lipopolysaccharides (LPS) and 1 μg/ml Polyinosinic:polycytidylic acid, (Poly(IC)), to stimulate an innate immune response. After 24 hours supernatants were collected and analyzed with an ilumina cytokine kit. Cells were also pelleted and protein lysates were extracted and separated into cytoplasmic and nuclear fraction.
Cytoplasmic and Nuclear Protein Fraction Extraction
Cells were resuspended on 80 μl of Buffer A (HEPES 10 mM pH 7.9, KCl 10 mM, MgCl2 1.5 mM, DTT 1 mM) with protease inhibitors (PMSF 0.5 mM, aprotinin 5 μg/ml, leupeptin 2 μg/ml, sodium pyrophosphate decahydrate 2 mM, sodium beta‐glycerophosphate pentahydrate 2 mM, sodium orthovanadate 1 mM). It was kept on ice for 15 minutes, then NP40 0.6% was added and centrifuged 30 seconds 16,000g at 4°C. The supernatants (cytoplasmic fraction) were transferred to a new tube. Cell pellets (nuclear fraction) was resuspended in 50 μl of Buffer B (HEPES 20 mM pH 7.9, NaCl 0.46 M, EDTA 1 mM, EGTA 1 mM, DTT 1 mM) with PMSF.
Illuminex Cytokine Detection Array
Cytokine levels were assessed by Luminex (Millipore, Billerica, MA) following the manufacturer's instructions. Briefly, supernatants were incubated for 2 hours with corresponding anticytokine magnetic beads, and then washed with 1× washing buffer and stained with detection antibodies (provided) for 1 hour. Strepatividin‐PE was then added for 30 more minutes. During all incubation steps, the plate was agitated at 650 rpm. After washing, plate was agitated for 15 minutes at 650 rpm and read in the xMAP Luminex reader (Waltham, MA).
Lentiviral Production
Human WT TLR3 Lentiviral Vector was purchased (pLenti‐GIII‐UbC) (Abcam, LV335740, Cambridge, UK) and was used to overexpress human TLR3 full length in iPSC‐derived neural stem cells (NSC). Viral particles were produced with a 293T packaging cell line. NSC were infected once with viral supernatants. At day 3, infected NSC were selected with puromycin for 7 days and placed in experiments. Overexpression of TLR3 was checked by RT‐PCR, FACS, and Western blot.
Western Blot
Protein extracts of cells collected by centrifugation, washed twice in PBS, lysed in 1× lysis buffer (50 nM Tris–HCl, 70 mM 2‐mercaptoethanol, and 1% SDS) and the concentration of total protein was measured by the Bradford assay. Lysates were then boiled for 5 minutes, and subjected to 12% polyacrylamide SDS gels or 4%–12% SDS resolving gels (Invitogen). After electrophoresis, proteins were transferred to a nitrocellulose membrane using a submerged transfer apparatus (BioRad), filled with 25 mM Tris Base, 200 mM glycine, and 20% methanol. After blocking with 5% non‐fat dried milk in TBS‐T (50 mM Tris–HCl [pH 8.0], 150 mM NaCl, and 0.1% Tween 20) the membrane was incubated with the primary antibodies diluted in TBS‐T and washed extensively. For the list of primary antibodies used see Supporting Information Figure S10. The membrane was washed 3 times with TBS‐T and then incubated with the appropriate horseradish peroxidase linked secondary antibody (Amersham, Spain). The detection was performed with the Western Breeze Immunodetection Kit (Invitrogen, Spain).
Flow Cytometry
Cells were stained with antibodies listed in figures. All analyses were performed with a BD FACSCanto‐II flow cytometer or MoFlo cell sorter (Dako Cytomation, USA) running Summit software. See Supporting Information Figure S9 for list of antibodies used.
Real Time PCR
Total mRNA was isolated using TRIZOL and 1 ug was used to synthesize cDNA using the Invitrogen Cloned AMV First‐Strand cDNA synthesis kit. One to two ul of the reaction was used to quantify gene expression by qPCR for genes listed in the table below.
| Human TLR3 | CCTGGTTTGTTAATTGGATTAACGA | TGAGGTGGAGTGTTGCAAAGG |
| Human TLR4 | CCAGTGAGGATGATGCCAGAAT | GCCATGGCTGGATCAGAGT |
| Human TLR3 short lsoform | AAGACACAACCAGGAACTGCC | GCTTCTCTGACCTTCCAGTCC |
| Human human leukocyte antigen (HLA) A | TCCTTGGAGCTGTGATCGCT | AAGGGCAGGAACAACTCTTG |
| Human HLA B | TCCTAG CAGTTGTGGTCATC | TCAAGCTGTGAGAGACACAT |
| Human HLA C | TCCTGGTTGTCCTAG CTGTC | CAGGCTTTACAAGTGATGAG |
| Human B2M | TGACTTTGTCACAG CCCAAGATA | AATCCAAATGCG G CATCTTC |
| Human GAPDH | GCACCGTCAAGGCTGAGAAC | AGGGATCTCGCTCCTGGAA |
TLR3 isoform RT‐PCR primers were tested in both human foreskin fibroblasts (HFF) cells and NSC with R 2 of 0.78465 and 0.92829, respectively (Supporting Information Fig. S10). All other primer sets are previously published.
Statistical Analyses
Unpaired nonparametric t tests were used to evaluate statistical significance at p ≤ .05. All quantitative and qualitative analyses are representative of at least n = 3 biological repeats for all experiments to give significant statistical power for reliability and validity of data. Only significantly different data in the figures are given a p value. Differential Protein Annotation and Bioinformatics Analyses: Proteins identified in different amounts in each of the two experiments were analyzed using the bioinformatics tool DAVID v6.7 (Database for Annotation, Visualization and Integrated Discovery; http://david.abcc.ncifcrf.gov/) to identify overrepresented biological pathways 26, 27. Similarly, differential proteins were analyzed for protein–protein interaction networks using the online tool STRING v10 28. To determine differentially expressed quantified proteins between the groups (F1 vs. F2) for small peptide analysis, statistical analysis was carried out using SPSS for Windows (version 21.0, Chicago, IL). All variables were checked for Levene's test to check variance homogeneity. Comparison between groups (i.e., F1 vs. F2) was performed using Student's t test. p ≤ .05 was considered to be significant.
Results
Global Proteomic Analysis of Patient (F1) and iPSC‐Derived Fibroblasts (F2)
Six human skin fibroblast cell lines (termed F1) that have previously been reprogrammed into iPSC, three by retroviral methods 2, 20, 21 and three by non‐viral episomal methods 22, then differentiated back to fibroblasts (termed F2) were analyzed for their potential immune response (Fig. 1A).
Figure 1.
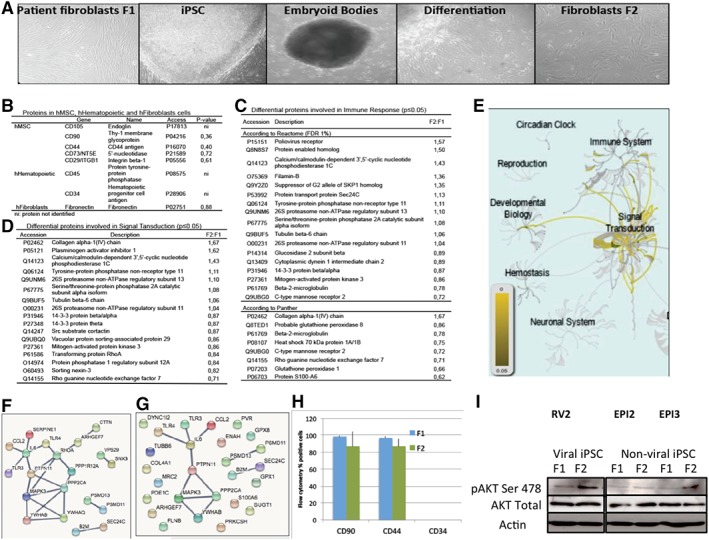
Global proteomic analysis of patient (F1) and human iPSC derived fibroblasts (F2). (A): Panels left to right: Photos representative of the healthy control human fibroblasts termed F1 (human skin fibroblasts), iPSC clone (from previously published studies), differentiation to embryoid bodies and iPSC‐derived fibroblasts, termed F2, at two different passages. A total of six sets of normal healthy control patient cells from previously published studies were used: three RV and three EPI (×100). Five F1 (n = 5) and three F2 (n = 3) cell populations was analyzed for proteomics between passage 3 to 9. (B): Table showing proteins characteristic of human MSC, hematopoietic cells and fibroblasts that have either been identified or not in the F1 and F2 cells. The p value indicates that there is no difference in the amount of these proteins between F1 and F2 indicating that the F1 and F2 cell types are similar. (C): Table showing the differential proteins between F1 and F2 involved in the immune response according to Reactome and Panther. (D): Table showing the differential proteins between F1 and F2 involved in signal transduction according to Reactome. (E): Schematic diagram of the main pathways overrepresented (in yellow) of all proteins detected (Reactome). (F): Interactions among the differential proteins involved in signal transduction and the TLR3, TLR4, IL6, and CCL2 proteins (highest confidence = minimum interaction score of 0.900). (G): Interactions among the differential proteins involved in immune system and the TLR3, TLR4, IL6, and CCL2 proteins (highest confidence = minimum interaction score of 0.900). (H): Validation of proteomic screen in three (n = 3) F1 and three F2 cell populations by flow cytometry for fibroblast markers CD90, CD44 and negative control marker CD34 (MSC marker) in F1 and F2 cells, demonstrating that the two populations of fibroblasts are similar. (I): Validation of proteomic screen in three (n = 3) F1 and three F2 cell populations by Western blot for signal transduction proteins AKT Ser 478, total AKT and loading control Actin. Two from three matched pairs of cells demonstrated increased AKT signaling in F2 cells (EPI‐2 the exception, see Fig. 4B). Abbreviations: EPI, episomal made iPSC; iPSC, induced pluripotent stem cells; MSC, mesenchymal stem cells; RV, retroviral made iPSC.
First, we performed a global proteomic analysis comparing F1 to F2 cells and mapped the protein expression profile. Overall, we detected 1,508 quantifiable proteins of which 86 proteins differed significantly in the expression levels between F1 and F2 cells (Supporting Information Figs. S1, S2). Categorizing proteins based on function, we first demonstrate no significant difference in fibroblast markers CD90, Fibronectin and CD44 between F1 and F2 cells, indicating that they are similar fibroblast cell populations for both viral and non‐viral made iPSC‐derived cells (Fig. 1B). CD90, CD44, and Fibronectin are established markers of fibroblasts as defined by Pilling et al. 29. The main differences in protein expression in F2 cells are signal transduction, cell cycle, and immune system proteins (Fig. 1C–1G; Supporting Information Fig. S1A). Interestingly, the proteomic screen revealed many proteins interacting with the TLR3 and TLR4 pathway (Fig. 1F, 1G).
To verify the global proteomic analysis, we performed flow cytometry analysis of the F1 and F2 populations and confirmed that the two fibroblast cell populations are similar for two fibroblast cell markers CD90, CD44 and negative for stem cell marker CD34 (Fig. 1H). This was the same for retrovirally made iPSC‐derived cells or non‐virally made iPSC‐derived cells and so they are both grouped together (Supporting Information Fig. S3). Given that signal transduction proteins were the most represented differentially expressed proteins between F1 and F2 cells, we also verified that signal transduction pathways for AKT is over expressed in F2 cells compared to F1 cells by Western blot analysis (Fig. 1I). We next assessed the immune response given the differences identified in the proteomic screen.
Characterization of Adaptive and Innate Immune System in Human iPSC
Analysis of HLA type by deep sequencing methods was performed to confirm the HLA type for all cell types used (Fig. 2A). MHCI mRNA gene expression (HLA‐A, B, C, and B2M) for F1, iPSC and F2 cells by RT‐PCR and demonstrates no significant change in expression between F2 cells and F1 cells (Fig. 2B), supported by similar results in a previously published report 30. MHC‐1 protein expression was assessed by flow cytometry in all F2 cell lines and no significant difference in expression levels of MHCI was observed. Expression of MHC‐1 was not significantly different between retrovirally made iPSC‐derived cells (RV1‐3) and non‐virally made iPSC‐derived cells (Epi1‐3) (Fig. 2C).
Figure 2.
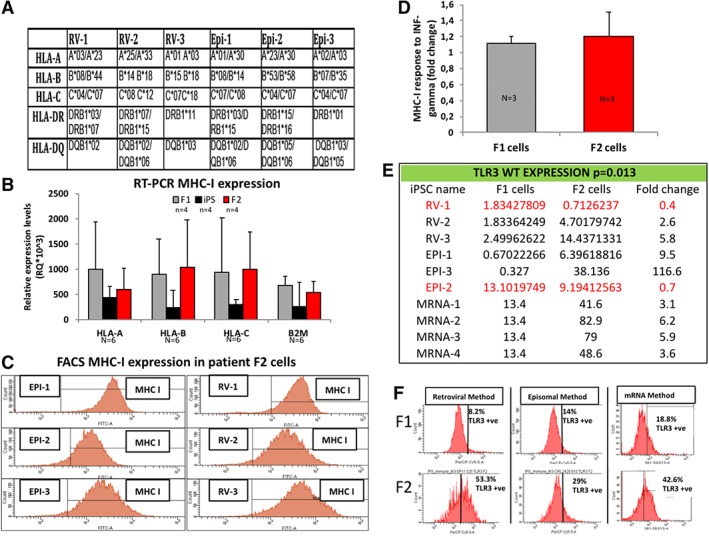
Characterization of adaptive and innate immune system in human iPSC. (A): Genotype of the HLA of the six control healthy patient human fibroblasts (n = 6) previously reprogramed into hiPSC in other published studies. (B): Graph of the RT‐PCR HLA gene expression levels (n = 6) in healthy control patient fibroblasts (F1), subsequent iPSC clones and iPSC‐derived fibroblasts (F2) demonstrating no difference in HLA gene expression between F1 and F2 cells. (C): Flow cytometry histograms of the six patient iPSC derived fibroblasts (F2) (n = 6) for MHC‐I protein expression in three RV and three EPI. (D): Graph representing expression levels of MHC‐I (n = 3) by flow cytometry for F1 cells and F2 cells stimulated with 10 ng/ml INF‐gamma overnight. (E): Table of RT‐PCR analysis of TLR3 gene expression levels in F1 and F2 cells of pairs of cells (n = 10) demonstrating that the wild type (WT) human TLR3 gene is overexpressed between twofold and ninefold more cells in F2 iPSC‐derived cells in eight from 10 matched healthy cells (80%). (F): Representative flow cytometry histograms of F1 and F2 cells stained for TLR3 receptor expression confirming that WT human TLR3 is overexpressed in twofold to fivefold more cells in F2 cells for one RV‐made pair, one EPI‐made pair (for RV and EPI, F1 and F2 cells are fibroblasts) and one synthetic mRNA made iPSC‐made pair (F1 are HFF cells and F2 are neural stem cells) (n = 3). Abbreviations: EPI, episomal made iPSC; HLA, human leukocyte antigen; iPSC, induced pluripotent stem cells; MHC, major histocompatibility complex; RV, retroviral made iPSC; TLR3, toll like receptor protein 3.
To mimic the possible in vivo functional response, we assessed MHCI expression response to inflammation cytokine INF‐gamma (10 ng/ml over 24 hours) in F1 and F2 cells from both viral and non‐viral made iPSC by flow cytometry. Both F1 and F2 cells responded with an upregulation of MHCI expression in response to INF‐gamma and there was no significance difference indicating that the regulation of MHCI expression is not affected in iPSC derived cells (Fig. 2D, Supporting Information Fig. S4). This suggests further that MHCI expression is functionally normal in iPSC‐derived cells.
To further characterize the F1 and F2 cells, we analyzed the expression of small peptides presented by MHC‐1 using mass spectrometry for RV1 clone that revealed differences in the number and type of small peptide expressions between F1 and F2 cells that could implicate functional differences in the adaptive immune response of transplanted iPSC‐derived cells (Supporting Information Fig. S5).
We next analyzed the innate immune response by assessing the expression of 25 cytokines routinely assed by clinical immunologists using the ELISA method in two matched cell lines (F1 vs. F2 cells) for viral (RV) or non‐viral (EPI) made iPSC derived cells, with or without viral ligand stimulation (Poly lC) or bacterial ligand stimulation (LPS) identified the main cytokines involved (Supporting Information Fig. S6A). Further analysis in another focused cytokine array (six cytokines) assessing four matched iPSC lines (F1 vs. F2 cells), we found no significant difference in expression of cytokines between viral and non‐viral made F2 cells when compared to F1 cells. In general, we found high variation between patient cell lines in cytokine levels that suggested altered expression in F2 cells (Supporting Information Fig. S6).
We found proteins associated with the innate immune system with higher WT human TLR3 and WT TLR4 expression found in the proteomic screen in F2 cells (Fig. 1) suggesting a dysfunctional innate cell signaling pathway, prompted us to investigate further human TLR3 expression levels in the healthy patient control matched F1 and F2 cells (Fig. 1, Supporting Information Figs. S1, S2). We observed that four from five patient (80%) control F1 matched F2 cells have increased TLR3 gene expression in F2 cells by RT‐PCR analysis (Fig. 2E, Supporting Information Fig. S7B). We next confirmed by flow cytometry analysis TLR3 receptor protein expression is independent of method used to reprogram cells and overexpressed in F2 cells (Fig. 2F). Taken together the data demonstrate that the WT TLR3 receptor expression is higher in healthy patient control iPSC‐derived cells, suggesting that reprogramed cells have an altered TLR3 pathway.
Human TLR3 Isoform Transcription Start Site Is Hypomethylated in Human iPSC‐Derived Cells and Exclusively Expressed in Humans
To investigate further the increase in TLR3 expression in a wide range of iPSC‐derived cell types, we reprogrammed using non‐viral synthetic mRNA transfection methods HFF (F1) and characterized six iPSC clones differentiated to various cell types (F2) (iPSC characterization data in a separate submitted paper) (Figs. 3, 4, 5). We then performed a methylome array to further characterize and better define the differences between F1 and F2 cells (Fig. 3A). Using the following bioinformatic criteria: (a) more than 20% difference in CpG methylation levels, (b) more than one CpG island affected, and (c) at least two hiPSC clones affected, we analyzed the methylation levels for Toll‐like receptors (Fig. 3B). Interestingly, the methylome array detected that a transcription start site (TSS) site in the CpGs of a shorter genetic isoform of full length TLR3 gene, and not other TLR gene family members (Supporting Information excel methylome data), is hypomethylated in human iPSC and derived NSC (F2), not seen in the starting parent HFF cells (F1) (Fig. 3B). We confirmed the CpG hypomethylation of the TLR3 isoform by bisulfide pyrosequencing (Fig. 3C, Supporting Information Fig. S7). Bioinformatics analysis of the human TLR3 WT and TLR3 Isoform demonstrate that the TSS site is associated with Acetylation and DNAse hypersensitivity sites associated with gene expression and that the TLR3 isoform is uniquely expressed in humans and not in mice or rat (Fig. 3D, 3E).
Figure 3.
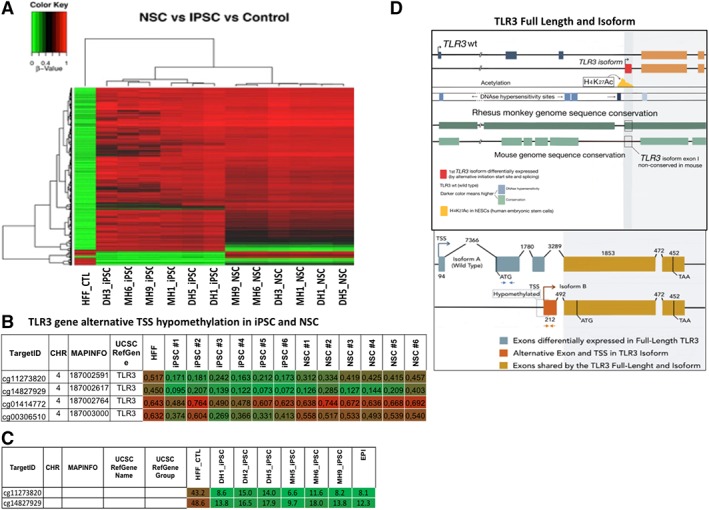
Human TLR3 isoform transcription start site is hypomethylated in human iPSC‐derived cells and exclusively expressed in humans. In addition to human iPSC used in Figures 1 and 2, a HFF cell line (Life Technologies) was reprogrammed by transfection of synthetic mRNA of the reprogramming factors to make six human iPSC lines and differentiated to NSC for further in depth analysis (characterization of mRNA made hiPSC in a separate publication). (A): Methylome array raw data for all toll genes for F1 (HFF), six human iPSC, and six F2 (NSC) cell lines. (B): Methylation profile of HFF (F1), iPSC, and NSC (F2) demonstrating four CpGs that containing a TSS in TLR3 is hypo‐methylated in iPSC and NSC (F2 cells) but not in stating HFF (F1 cells). Methylation regions were considered significant using the following bioinformatic criteria: (a) more than 20% difference in CpG methylation levels between F1 and F2, (b) more than one CpG island affected, and (c) at least two hiPSC or NSC clones affected, (C) validation of hypomethylted TSS by pyrosequencing for two CpGs in human iPSC confirming the CpG TSS region of an isoform TLR3 is hypomethylated. (D): Sketch of bioinformatics of the TLR3 WT and TLR3 isoform demonstrating the TSS site (red) associated with acetylation (yellow) and DNAse hypersensitivity sites (dark blue) that indicate gene expression and homology to other species (green). TLR3 isoform is uniquely expressed in humans and rhesus monkey. Lower panel: Detailed sketch of the exons of the shorter TLR3 isoform discovered from the methylome array, compared to WT TLR3 for comparison. Brown and blue arrows show gene regions showing RT PCR primer sequences. Abbreviations: HFF, human fibroblast; iPSC, induced pluripotent stem cells; NSC, neural stem cells; TLR3, toll like receptor protein 3; TSS, transcription start site; WT, wild type.
Figure 4.
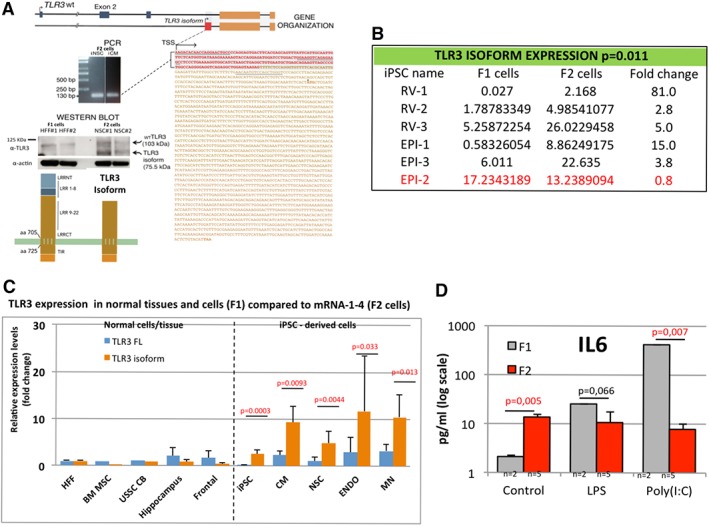
Human TLR3 isoform is overexpressed in most human iPSC derived cells not seen in normal human tissue or cells. (A): Expression of the shorter human isoform TLR3 with unique 5′ UTR in red. Below the PCR of genomic DNA from human iPSC‐derived NSC and CM of the unique 5′ UTR of the TLR3 isoform. Sequencing data of the PCR product for the 5′ UTR of the human TLR3 isoform (in red) with whole sequence found at or XM_017008577.1 transcript variant X1. Underlined regions are RT‐PCR primer sequences used to detect human isoform TLR3. Lower left panel: Western blot of TLR3 expression demonstrating expression of the WT TLR3 at 107 KDa and the putative shorter TLR3 isoform at approximately 80 KDa in F1 cells (HFF) and F2 cells iPSC‐derived NSC with schematic drawing of WT and isoform human TLR3 missing the LRR1‐8 region important in binding viral dsRNA and receptor stability. (B): Table of individual F1 and F2 cell lines for RT‐PCR analysis of gene expression demonstrating that the human TLR3 isoform gene is overexpressed between 3 and 81 fold more in F2 in five from six matched healthy cells (83%). In red, it is highlighted the exception healthy control patient sample (Epi‐2) that also did not have elevated AKT signal transduction protein levels in F2 cells seen in Figure 1. (C): Graph comparing WT TLR3 (FL: full length) and isoform TLR3 gene expression in five normal healthy cells and tissues compared to synthetic mRNA made iPSC‐derived cells (dotted line divides normal from iPSC cells analyzed). Normal cells/tissues include: starting fibroblasts (HFF), normal endogenous BM MSC, cord blood USSC, and normal tissues: normal hippocampus and frontal cortex human brain tissue demonstrating, demonstrating low levels of gene expression of the shorter TLR3 isoform in normal tissues. Dotted line: Compared to the synthetic mRNA made reprogramed human iPSC differentiated into three different iPSC‐derived cells: CM, NSC, ENDO, and MN demonstrating a significant increase in gene expression of the shorter TLR3 isoform in all types of iPSC‐derived cell types tested. (D): Graph of an ELISA assay of the functional response of IL6 in human foreskin fibroblasts (F1, n = 2) and their mRNA made iPSC‐derived neural stem cells (F2, n = 5) to LPS (100 ng/ml over 24 hours) and Poly (I:C) stimulation (1 ug/ml) in triplicate, demonstrating a significantly suppressed IL6 immune response to Poly IC (1 ug/ml over 24 hours) in iPSC‐derived cells (F2 cells). Note: Synthetic mRNA made iPSC and derived NSC, CM, and DE have been characterized and described in another paper currently under consideration for publication. Abbreviations: BM, bone marrow; CB, cord blood; CM, cardiomyocytes; EPI, episomal made iPSC; HFF, human fibroblast; iPSC, induced pluripotent stem cells; LPS, lipopolysaccharide; MN, motor neurons; MSC, mesenchymal stem cells; NSC, neural stem cells; RV, retroviral made iPSC; TLR3, toll like receptor protein 3; TSS, transcription start site; USSC, umbilical stem cells; WT, wild type.
Figure 5.
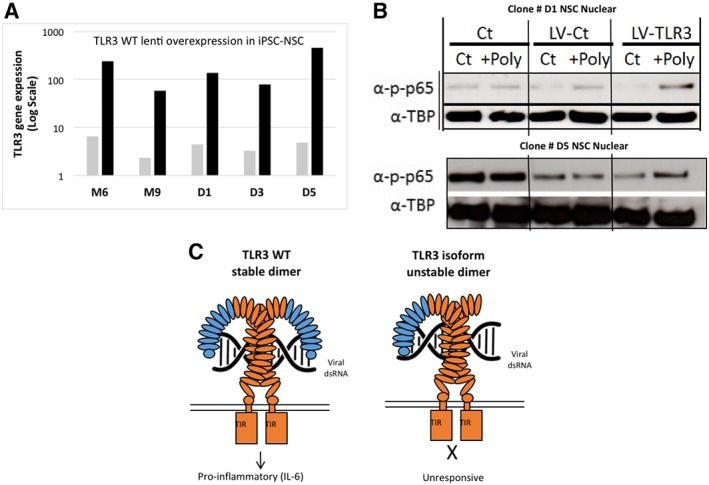
Human TLR3 isoform functions to suppress NF‐KB p65 signaling pathway in response to virus (Poly IC). (A): Graph of RT‐PCR for WT human TLR3 gene lentiviral overexpression in F2 cells (NSC that have high levels of expression of TLR3 isoform, (Black bars)). WT human TLR3 lentivirus was used to overexpress WT TLR3 in five (n = 5) F2 (NSC) clones to functionally outcompete with the TLR3 isoform observed to be overexpressed in F2 cells (B). Western blot analysis of phosphorylated p65 (p‐p65) in two NSC (F2 cells) clones. Gain of function with WT TLR3 in F2 cells demonstrates the immune suppressive function of the short TLR3 isoform on TLR3 signal transduction in response to viral infection (Poly IC). After lentiviral overexpression of full length TLR3, NSC recover responsiveness to stimulation by the viral analog Poly (IC), as seen by the increase of phospho‐p65 in the nuclear fraction. Control NSC were infected with a lentiviral empty vector (ubiquitin promoter only (LV‐Ct) that contained the same ubiquitin promoter as the wild type TLR3‐LV vector). Loading controls are tubulin nuclear protein fraction. Representative pictures of five NSC tested. (C): Schematic proposed model of full length TLR3 and the role of the shorter TLR3 isoform predominately expressed in reprogrammed cells that acts to suppress the immune response. All experiments performed with n = 6 at least twice, unless stated. Abbreviations: iPSC, induced pluripotent stem cells; LV, lenti virus; NSC, neural stem cells; TLR3, toll like receptor protein 3; WT, wild type.
Human TLR3 Isoform Is Overexpressed in Most Human iPSC‐Derived Cells Not Seen in Normal Human Tissue or Cells
We next assessed gene expression levels of human TLR3 isoform in human F1 and F2 cells from the healthy patient control cells (Episomal and Retroviral) described in Figures 1 and 2 and the synthetic mRNA made human iPSC cells described in Figure 3. We next performed PCR of genomic DNA for sequencing in F2 cells from synthetic mRNA made human iPSC‐derived: NSC and cardiomyocytes (CM) to demonstrate the unique 5′ UTR of the human TLR3 isoform in F2 cells. Sequencing data of the PCR product for the 5′ UTR confirms the human TLR3 isoform (in Red) in human F2 cells with whole sequence found at UC011CKZ.2 or XM_017008577.1 transcript variant X1 (Fig. 4A). We next tested protein expression of the human WT TLR3 and shorter TLR3 isoform by Western blot in F1 and F2 cells (from synthetic mRNA made human iPSC‐derived) demonstrating expression of the WT TLR3 at 107 KDa and the putative shorter TLR3 isoform at approximately 80 KDa missing the LRR1‐8 region important in receptor stabilization (Fig. 4A, Supporting Information Fig. S8).
We next performed a comprehensive analysis of human TLR3 gene expression levels in normal endogenous cell lines or normal human neural tissue (frontal cortex or Hippocampus) compared to various human iPSC derived cell types (from synthetic mRNA method) to further confirm overexpression in human iPSC derived cells (F2) and that differentiation or reprograming method does not affect TLR3 expression levels (Fig. 4B). RT‐PCR gene expression analysis revealed that the shorter TLR3 isoform is lower expressed in F1 cells than WT TLR3 and is significantly higher upregulated in all reprogrammed differentiated F2 cell types (CM, p = .0093; Definitive Endoderm – DE, p = .033; NSC, p = .0044 and Motor Neurons – MN, p = .013) (Fig. 4C). Interestingly, in addition to HFF other normal endogenous cells such as bone marrow mesenchymal stem cells (BM MSC), umbilical stem cells, or normal human brain tissue (hippocampus and frontal cortex) have low levels of expression of WT TLR3 and the shorter isoform TLR3, suggesting only reprogramed iPSC‐derived cells have elevated levels of the TLR3 isoform (Fig. 4C).
Therefore, we demonstrate that the overexpression of the shorter TLR3 isoform in iPSC‐derived cells is independent of the methodology used because high levels of the isoform have been found in most human iPSC generated by three different methods (retrovirus, episomal vectors, or mRNA transfections). Moreover, higher expression of the TLR3 isoform is independent of the starting parent cell types because all somatic cell types we tested, including HFF, cord blood cells, patient skin fibroblasts, mesenchymal stem cells had low levels of expression of the human TLR3 isoform. Finally, the differentiation method did not affect levels of the TLR3 isoform expression because we generated many different cell types from iPSC (F2 cells included fibroblasts, NSC, MN, CM, and DE cells) and most had significantly higher levels of expression of the TLR3 isoform (Fig. 4B, 4C).
Given that it has been previously published that a short isoform of mouse TLR3, but different sequence and size to the human TLR3 isoform described in this study can act as a suppressor of the TLR3 innate immune pathway by competing with the full length TLR3 protein in mouse astrocytes 31, 32, 33, we next assessed the functional immune response of inflammation cytokine IL6 in human iPSC‐NSC to Poly (I:C) and LPS. Assessment of IL6 expression levels by ELISA demonstrated that iPSC‐derived cells (F2 cells) have significantly higher levels of expression of IL6 compared to F1 cells and a suppressed innate immune response to viral infection (Poly IC 1ug/ml overnight) in iPSC‐derived cells (Fig. 4D). This suggests that transplanted human iPSC‐NSC would act as a cellular pump producing physiologically higher levels of IL6 cytokine than is normal in circulating blood and tissues and the cells would not respond to viral infection (Fig. 4D).
Human TLR3 Isoform Functions to Suppress NF‐KB p65 Signaling Pathway in Response to Virus (Poly IC)
Next, we performed a gain of function test in human iPSC‐NSC (F2 cells) to demonstrate the possible functional effect of the human TLR3 isoform. We overexpressed WT TLR3 by lentiviral methods in five synthetic mRNA made iPSC‐NSC clones to outcompete the TLR3 isoform and all NSC clones demonstrated high levels of WT human TLR3 expression after infection (Fig. 5A). Analysis of the expression of nuclear NF‐KB p65 protein phosphorylation by Western blot, an established marker of a functional human TLR3 pathway, demonstrated that overexpression of human WT TLR3 in NSC could return sensitivity to Poly (IC) treatment of NSC (Fig. 5B). Note that in control NSC cells and in empty lentiviral control NSC (UB promoter only) the NSC do not respond to Poly IC viral stimulation demonstrated by no change in NF‐KB p65 phosphorylation levels (Fig. 5B). Overexpression of WT TLR3 in NSC and resulting return to sensitivity to Poly IC stimulation supports the model that the shorter TLR3 isoform is suppressing normal TLR3 function in NSC to viral insult (F2 cells) (Fig. 5B). Taken together, we propose the model that in human iPSC‐derived cells that overexpress the shorter isoform of TLR3 competes with full length TLR3 to suppress the innate inflammation signal transduction response in F2 cells to Poly IC (viral infection) (Fig. 5C).
Discussion
There is a clear need to better understand the immune response of autologous reprogramed human cells generated in vitro before we embark on extensive clinical trials using human iPSC to study and treat human disease. With that in mind we posit the question: do patient cells that have been reprogramed into iPSC‐derived cells cause an immune response?
This study is based on a global proteomic and methylome screening we performed using human skin cells (termed F1 cells) compared to their iPSC‐derived cells (F2 cells) made from three different reprogramming methods (retroviral, episomal, and synthetic mRNA transfection). We reveal that WT TLR3 and a isoform of TLR3 are overexpressed in the majority (83%) of F2 cells tested (five from six F1‐F2 match lines tested) and in all types of iPSC‐derived cells (NSC, MN, DE, and CM) warranting screening of iPSC‐derived cell clones and removal in disease modeling studies or clinical applications (Figs. 2D, 3B, 3C).
The data demonstrate that the cell reprogramming process alters specific aspects of the innate immune response resulting in a possible suppression of immunogenicity, supporting similar findings recently described for human iPSC‐derived cells 11, 34. We discover two distinct states in human iPSC‐derived cells related to overexpression WT TLR3 and Isoform TLR3 that could impinge on future clinical applications and disease modeling:
Unstimulated state: increased IL6 associated with increased WT TLR3 expression in F2 cells (iPSC‐derived cells) (Fig. 4C, 4D).
Stimulation with Poly IC: suppressed response to Poly IC (Virus) in F2 cells due to overexpression of an isoform TLR3 that can be rescued by overexpression of WT TLR3 (Fig. 5A–5C).
The higher levels of expression of the human TLR3 isoform is central to understanding the altered immunogenicity of human iPSC‐derived cells. To mediate innate immune response to virus infection and consequently the virus immunogenicity, double‐stranded RNA (dsRNA), a frequent by‐product of virus infection, should be recognized by TLR3. Simultaneously, the PI3K‐Akt (Fig. 1I) and NF‐KB (Fig. 5B) pathways play an essential role in TLR3‐mediated gene induction 35. The loss of inflammation response of cells to bacterial (LPS) or viral (Poly (IC) infection has been shown before in specific mouse neural cell types that overexpress a different mutant mouse TLR3 isoform to the human TLR3 isoform described in this study (not expressed in mice) 31, 32, 33. Interestingly, a recent study found that differentiation of human iPSC results in a loss of immunogenicity and leads to the induction of tolerance, despite expected antigen expression differences between iPSC‐derived versus original somatic cells 11. In further agreement with this study, it has also been demonstrated that neural progenitor cells from human iPSC generated less autogenous immune response, possibly by altered toll like receptor pathway signaling they did not assess 34. This observation among others brings forward the interesting possibility that human iPSC‐derived cells may benefit from a suppressed immunogenic microenvironment favoring initial survival and subsequent engraftment and tissue regeneration.
Research in animal models is well documented and work by Araki et al., working mainly with skin and bone marrow transplant in mice, demonstrates mouse iPSC‐derived CM cause T cell infiltration when transplanted under the skin of syngeneic mice 13. Todorova et al., found that the location of iPSC transplantation can affect the immune outcome because the kidney is immune privileged, highlighting that differences between studies could be explained by experimental procedure and design differences 14. The group by Almeida et al., found that mouse iPSC‐derived endothelial cells are intrinsically immunogenic in syngeneic mice, but are immune tolerated by overexpressing immune suppressive molecule IL‐10; however, their study did not assess TLR3 expression levels 11.
The altered TLR3 innate immune response of human in vitro iPSC‐derived cells raises a number of issues to be considered for future cell transplantation in humans.
This may allow for human iPSC‐derived cells to be used not only as an autologous cell therapy but also in a limited capacity as an allogenic cell therapy because of a suppressed immunogenic microenvironment favoring initial survival and subsequent engraftment and tissue regeneration.
Evasion of the immune system is a key hallmark of cancer and suppression of the immune response of iPSC‐derived cells may impose a long‐term cancer threat not previously considered. This consideration is considerably enhanced by recent work that demonstrates the AKT pathway induces a transcriptional program via C/EBP that promotes immune suppression during inflammation and tumor growth 36. Inhibition of the AKT driven signaling pathway prolongs NFκB activation, which in turn inhibits C/EBPβ activation promoting an immunostimulatory transcriptional program that restores CD8+ T cell activation, cytotoxicity, and synergizes with checkpoint inhibitor therapy to promote tumor regression and extend survival in mouse models of cancer 36.
Transplantation of iPSC‐derived cells that constitutively express physiologically higher levels of inflammatory cytokine IL6 (Fig. 4D) may result in long‐term adverse effects in the body. This may have important implications in future human iPSC clinical trials such as the RIKEN AMD‐RPE clinical trial 17, 18.
The need to screen patient iPSC and derived cells using global gene analysis “omics” for alterations in the genome of patient cells that include expression levels of TLR3 and its shorter TLR3 isoform identified in this work. Indeed, impaired TLR3 function to herpes simple virus (HSV‐1) infection in the CNS has important implications in the pathogenesis of HSV‐1 encephalitis (HSE) in children 37.
Interestingly, activation of innate immunity is required for efficient nuclear reprogramming to iPSC via the TLR3 receptor pathway using either viral or non‐viral methods 38. The authors find that by using gain and loss of function studies, the TLR3 pathway enables efficient induction of pluripotency by viral or non‐viral synthetic mRNA transfection approaches. Stimulation of TLR3 causes rapid and global changes in the expression of epigenetic modifiers to enhance chromatin remodeling and nuclear reprogramming. Furthermore, they conclude that activation of inflammatory pathways are required for efficient nuclear reprogramming in the induction of pluripotency 38. Our finding suggests that the TLR3 pathway, important for achieving cell pluripotency, is not re‐set to the correct expression levels in iPSC‐differentiated cells (F2) and that this then results in altered cytokine secretion leading to possible immune suppression of transplanted iPSC‐derived cells (Figs. 4, 5). We demonstrate that the mechanism of constitutive expression of the shorter TLR3 isoform is most likely at an epigenetic level, resulting in hypomethylation of a TSS producing a shorter TLR3 isoform that competes to suppress normal full length TLR3 signal transduction function (Figs. 3, 4).
In conclusion, the data demonstrate that the cell reprogramming process alters the immune response of most iPSC‐derived cells highlighting human TLR3 isoform overexpression that functions to suppress innate immune signal transduction and provides an insight for future clinical applications of autologous human iPSC‐derived cell transplantation. We propose that screening human iPSC clones and their derived cells for alterations in the genome of patient cells that include expression levels of human TLR3 and its shorter TLR3 isoform identified in this work is essential before embarking on “first in man” clinical trials or human disease modeling using human iPSC.
Ethical Statement
Use of human iPSC and tissue obtained by collaborators from published work funded by approved projects 2012‐StG‐311736‐PD‐HUMMODEL, BFU2016‐80870‐P), Red de Terapia Celular ‐ TerCel RD16/0011/0024 and BFU2011–26596 2, 20, 21, 22. The generation and/or use of human iPSC were approved by the Spanish competent authorities (Commission on Guarantees concerning the Donation and Use of Human Tissues and Cells of the Carlos III National Institute of Health). The human iPSC lines have been (or are in the process of being) deposited at the Spanish National Stem Cell Bank, according to the Spanish legislation.
Author Contributions
J.R., A.B.A.‐P., M.C.‐P., I.A., M.S., A.C., and O.A.B.: collection and/or assembly of data, data analysis and interpretation, manuscript writing; R.D.‐M., S.M., M.E., and A.B.B.: collection and/or assembly of data, data analysis and interpretation; M.J., and J.M.P.: collection and/or assembly of data, data analysis and interpretation, conception and design; E.W. and D.O.: collection and/or assembly of data; D.J.: data analysis and interpretation, manuscript writing; R.O.: collection and/or assembly of data, financial support, data analysis and interpretation, manuscript writing; M.J.E.: conception and design, financial support, provision of study material collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, coordination of national and international teams.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supplemental Figure 1 A. Pathways overrepresented (in yellow) in the 1,508 quantifiable proteins according to Reactome and B. Interactions of 86 proteins at highest confidence (0.900).
Supplemental Figure 2. List of pathways overrepresented in the 86 significantly different proteins between patient fibroblasts (F1) and iPSC‐fibroblasts (F2) for biological process, molecular function and KEGG pathways.
Supplemental Figure 3. Examples of gating for Flow cytometry histograms for fibroblast markers CD90 and CD44 in F1 (patient) and F2 cells (iPSC‐derived) and CD34 stem cell marker for non viral and viral made iPSC demonstrating no differences between methods and both cell populations.
Supplemental Figure 4. Examples of gatings used for flow cytometry histograms for MHC I expression in healthy control patient cells (F1) and iPSC‐derived cells (F2) from clone RV‐1 with or without INF‐gamma stimulation (10 ng/ml for 24 hours) demonstrating no functional difference in MHC1 expression between F1 and F2.
Supplemental Figure 5. List of all small peptides and sequence for MHCI receptor for F1 and F2 cells. Note that the number and type of small peptides presented by MHCI are different between F1 (patient fibroblast cells) and F2 cells (iPSC‐derived fibroblast cells) except for 3 peptides, two RNA polymerases and elongation factor 2 that are the same for both.
Supplemental Figure 6. Screening of cytokine expression. ELISA data from first experiment for two patient cell lines (F1) and two iPSC‐derived fibroblast lines (F2) tested for 25 immune cytokines normally screened in clinical testing of patient samples stimulated with Poly [IC] (1 ug/ml) overnight. Note that from this first experiment that five cytokines were selected (IL6, IL10, IL15, RANTES and MCP1) as the main expressed (in green) and a second ELISA experiment performed (see Figs. 2, 3). B. Four matched cell lines analyzed for cytokines IL6, RANTES, IL15 and MCP‐1 with and without LPS or Poly IC stimulation.
Supplemental Figure 7. A. Primer sequences used for Bisulfide Pyrosequencing to validate CpG hypomethylation on a TSS in TLR3 isoform. B. PCR of WT Human TLR3 gene expression levels in healthy control patient fibroblasts (F1) and iPSC derived fibroblasts (F2)in retroviral (RV) and Episomal (EPI) methods.
Supplemental Figure 8. A. Quantification of Western blot for human WT TLR3 and Isoform TLR3 in human fibroblasts (F1) and iPSC‐derived neural stem cells (F2) B. Gain of function rescue experiment: overexpression of wild type (WT) TLR3 by lentiviral methods in NSC clones to compete with shorter isoform TLR3. Overexpression of TLR3 was checked by Flow cytometry. WT TLR3 overexpression was assessed in NSC after 4–6 days selection with puromycin. Data presented as a table and flow cytometry histogram.
Supplemental Figure 9. List of antibodies used.
Supplemental Figure 10. Graphs of cDNA dilution test for RT‐PCR TLR3 isoform primers and primer sequence list.
Appendix S1: Supplementary Excel methylome data. Methylome array to characterize the differences between F1 and F2 cells. The following bioinformatic criteria: (i) more than 20% difference in CpG methylation levels, (ii) more than one CpG island affected and (iii) at least 2 hiPSC clones affected, we analyzed the methylation levels for Toll‐like receptors. The methylome array detected a transcription start site (TSS) site in the CpGs of a shorter genetic isoform of full length TLR3 gene, and not other TLR gene family members, is hypomethylated in human iPSC and derived NSC (F2), not seen in the starting parent HFF cells (F1) (Fig. 3B).
Acknowledgments
We acknowledge the support of the following people: Angel Raya Centre for Regenerative Medicine Barcelona (CMRB), John Christodoulou of the University of Sydney, Eduard Palou Rivera for HLA analysis, Josep Maria Estanyol of the Technological and Scientific Centre at the University of Barcelona, a member of the ProteoRed network (http://www.proteored.org/) for assistance in the proteomic analysis, Montserrat Pau for technical assistance, Carmen Barrot for RT‐PCR analysis. Isabel Crespo Torres, Responsable Plataforma Citometria IDIBAPS. MJE is supported in part by the Program Ramon y Cajal (RYC‐2010‐06512), FBG project 307900, MINECO project grant BFU2011–26596, BFU2014‐54467‐P, TV3 Marato project grant /FBG309768 and Talent Retention program, University of Barcelona. The proteomic analysis was supported by projects from the Ministerio de Economica y Competitividad P113/00699 and EU‐PF7‐PEOPLE‐2011‐ITN289880 to RO. Dr. Manel Esteller laboratory is supported by, among other institutions, the EU Joint Programme – Neurodegenerative Disease Research (JPND); Cellex Foundation; the Health and Science Departments of the Catalan Government (Generalitat de Catalunya); the E‐Rare (ERA‐Net for research programs on rare diseases) and EuroRETT (a European network on Rett syndrome, funded by the European Commission under its 6th Framework Program since 2006). Dr. Manel Esteller is an ICREA Research Professor. Antonella Consiglio laboratory is supported by the European Research Council‐ERC (2012‐StG‐311736‐ PD‐HUMMODEL), the Spanish Ministry of Economy and Competitiveness‐MINECO (BFU2016‐80870‐P), Instituto de Salud Carlos III‐ISCIII/FEDER (Red de Terapia Celular ‐ TerCel RD16/0011/0024), AGAUR (2017‐SGR‐899). Contact R. Delgado‐Morales and M. Esteller for comments on the methylome array. Contact R. Oliva for comments on the proteomic screen.
References
- 1. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 2. Edel MJ, Menchon C, Menendez S et al. Rem2 GTPase maintains survival of human embryonic stem cells as well as enhancing reprogramming by regulating p53 and cyclin D1. Genes Dev 2010;24:561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palomo AB, Lucas M, Dilley RJ et al. The power and the promise of cell reprogramming: Personalized autologous body organ and cell transplantation. J Clin Med 2014;3:373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoffmann JA, Kafatos FC, Janeway CA et al. Phylogenetic perspectives in innate immunity. Science 1999;284:1313–1318. [DOI] [PubMed] [Google Scholar]
- 5. Romieu‐Mourez R, Francois M, Boivin MN et al. Cytokine modulation of TLR expression and activation in mesenchymal stromal cells leads to a proinflammatory phenotype. J Immunol 2009;182:7963–7973. [DOI] [PubMed] [Google Scholar]
- 6. Bai W, Liu H, Ji Q et al. TLR3 regulates mycobacterial RNA‐induced IL‐10 production through the PI3K/AKT signaling pathway. Cell Signal 2014;26:942–950. [DOI] [PubMed] [Google Scholar]
- 7. Liu P, Chen S, Li X et al. Low immunogenicity of neural progenitor cells differentiated from induced pluripotent stem cells derived from less immunogenic somatic cells. PLoS One 2013;8:e69617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao T, Zhang ZN, Rong Z et al. Immunogenicity of induced pluripotent stem cells. Nature 2011;474:212–215. [DOI] [PubMed] [Google Scholar]
- 9. Guha P, Morgan JW, Mostoslavsky G et al. Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell 2013;12:407–412. [DOI] [PubMed] [Google Scholar]
- 10. Morizane A, Doi D, Kikuchi T et al. Direct comparison of autologous and allogeneic transplantation of iPSC‐derived neural cells in the brain of a nonhuman primate. Stem Cell Reports 2013;1:283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Almeida PE, Meyer EH, Kooreman NG et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self‐tolerance. Nat Commun 2014;5:3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao T, Zhang ZN, Westenskow PD et al. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell 2015;17:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Araki R, Uda M, Hoki Y et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 2013;494:100–104. [DOI] [PubMed] [Google Scholar]
- 14. Todorova D, Kim J, Hamzeinejad S et al. Brief report: Immune microenvironment determines the immunogenicity of induced pluripotent stem cell derivatives. Stem Cells 2016;34:510–515. [DOI] [PubMed] [Google Scholar]
- 15. Hew M, O'Connor K, Edel MJ et al. The possible future roles for iPSC‐derived therapy for autoimmune diseases. J Clin Med 2015;4:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mandai M, Kurimoto Y, Takahashi M. Autologous induced stem‐cell‐derived retinal cells for macular degeneration. N Engl J Med 2017;377:792–793. [DOI] [PubMed] [Google Scholar]
- 17. Mandai M, Watanabe A, Kurimoto Y et al. Autologous induced stem‐cell‐derived retinal cells for macular degeneration. N Engl J Med 2017;376:1038–1046. [DOI] [PubMed] [Google Scholar]
- 18. Kamao H, Mandai M, Okamoto S et al. Characterization of human induced pluripotent stem cell‐derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Reports 2014;2:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alvarez Palomo AB, McLenachan S, Chen FK et al. Prospects for clinical use of reprogrammed cells for autologous treatment of macular degeneration. Fibrogenesis Tissue Repair 2015;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanchez‐Danes A, Consiglio A, Richaud Y et al. Efficient generation of A9 midbrain dopaminergic neurons by lentiviral delivery of LMX1A in human embryonic stem cells and induced pluripotent stem cells. Hum Gene Ther 2012;23:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanchez‐Danes A, Richaud‐Patin Y, Carballo‐Carbajal I et al. Disease‐specific phenotypes in dopamine neurons from human iPS‐based models of genetic and sporadic Parkinson's disease. EMBO Mol Med 2012;4:380–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Briggs JA, Sun J, Shepherd J et al. Integration‐free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells 2013;31:467–478. [DOI] [PubMed] [Google Scholar]
- 23. Du P, Kibbe WA, Lin SM. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008;24:1547–1548. [DOI] [PubMed] [Google Scholar]
- 24. Gentleman RC, Carey VJ, Bates DM et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bibikova M, Le J, Barnes B et al. Genome‐wide DNA methylation profiling using Infinium(R) assay. Epigenomics 2009;1:177–200. [DOI] [PubMed] [Google Scholar]
- 26. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 28. Szklarczyk D, Franceschini A, Wyder S et al. STRING v10: Protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pilling D, Fan T, Huang D et al. Identification of markers that distinguish monocyte‐derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS One 2009;4:e7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Drukker M, Katz G, Urbach A et al. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci USA 2002;99:9864–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farina C, Krumbholz M, Giese T et al. Preferential expression and function of Toll‐like receptor 3 in human astrocytes. J Neuroimmunol 2005;159:12–19. [DOI] [PubMed] [Google Scholar]
- 32. Seo JW, Yang EJ, Kim SH et al. An inhibitory alternative splice isoform of Toll‐like receptor 3 is induced by type I interferons in human astrocyte cell lines. BMB Rep 2015;48:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang E, Shin JS, Kim H et al. Cloning of TLR3 isoform. Yonsei Med J 2004;45:359–361. [DOI] [PubMed] [Google Scholar]
- 34. Huang K, Liu PF, Li X et al. Neural progenitor cells from human induced pluripotent stem cells generated less autogenous immune response. Sci China Life Sci 2014;57:162–170. [DOI] [PubMed] [Google Scholar]
- 35. Sarkar SN, Peters KL, Elco CP et al. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double‐stranded RNA signaling. Nat Struct Mol Biol 2004;11:1060–1067. [DOI] [PubMed] [Google Scholar]
- 36. Kaneda MM, Messer KS, Ralainirina N et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016;539:437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lafaille FG, Pessach IM, Zhang SY et al. Impaired intrinsic immunity to HSV‐1 in human iPSC‐derived TLR3‐deficient CNS cells. Nature 2012;491:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee J, Sayed N, Hunter A et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012;151:547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 A. Pathways overrepresented (in yellow) in the 1,508 quantifiable proteins according to Reactome and B. Interactions of 86 proteins at highest confidence (0.900).
Supplemental Figure 2. List of pathways overrepresented in the 86 significantly different proteins between patient fibroblasts (F1) and iPSC‐fibroblasts (F2) for biological process, molecular function and KEGG pathways.
Supplemental Figure 3. Examples of gating for Flow cytometry histograms for fibroblast markers CD90 and CD44 in F1 (patient) and F2 cells (iPSC‐derived) and CD34 stem cell marker for non viral and viral made iPSC demonstrating no differences between methods and both cell populations.
Supplemental Figure 4. Examples of gatings used for flow cytometry histograms for MHC I expression in healthy control patient cells (F1) and iPSC‐derived cells (F2) from clone RV‐1 with or without INF‐gamma stimulation (10 ng/ml for 24 hours) demonstrating no functional difference in MHC1 expression between F1 and F2.
Supplemental Figure 5. List of all small peptides and sequence for MHCI receptor for F1 and F2 cells. Note that the number and type of small peptides presented by MHCI are different between F1 (patient fibroblast cells) and F2 cells (iPSC‐derived fibroblast cells) except for 3 peptides, two RNA polymerases and elongation factor 2 that are the same for both.
Supplemental Figure 6. Screening of cytokine expression. ELISA data from first experiment for two patient cell lines (F1) and two iPSC‐derived fibroblast lines (F2) tested for 25 immune cytokines normally screened in clinical testing of patient samples stimulated with Poly [IC] (1 ug/ml) overnight. Note that from this first experiment that five cytokines were selected (IL6, IL10, IL15, RANTES and MCP1) as the main expressed (in green) and a second ELISA experiment performed (see Figs. 2, 3). B. Four matched cell lines analyzed for cytokines IL6, RANTES, IL15 and MCP‐1 with and without LPS or Poly IC stimulation.
Supplemental Figure 7. A. Primer sequences used for Bisulfide Pyrosequencing to validate CpG hypomethylation on a TSS in TLR3 isoform. B. PCR of WT Human TLR3 gene expression levels in healthy control patient fibroblasts (F1) and iPSC derived fibroblasts (F2)in retroviral (RV) and Episomal (EPI) methods.
Supplemental Figure 8. A. Quantification of Western blot for human WT TLR3 and Isoform TLR3 in human fibroblasts (F1) and iPSC‐derived neural stem cells (F2) B. Gain of function rescue experiment: overexpression of wild type (WT) TLR3 by lentiviral methods in NSC clones to compete with shorter isoform TLR3. Overexpression of TLR3 was checked by Flow cytometry. WT TLR3 overexpression was assessed in NSC after 4–6 days selection with puromycin. Data presented as a table and flow cytometry histogram.
Supplemental Figure 9. List of antibodies used.
Supplemental Figure 10. Graphs of cDNA dilution test for RT‐PCR TLR3 isoform primers and primer sequence list.
Appendix S1: Supplementary Excel methylome data. Methylome array to characterize the differences between F1 and F2 cells. The following bioinformatic criteria: (i) more than 20% difference in CpG methylation levels, (ii) more than one CpG island affected and (iii) at least 2 hiPSC clones affected, we analyzed the methylation levels for Toll‐like receptors. The methylome array detected a transcription start site (TSS) site in the CpGs of a shorter genetic isoform of full length TLR3 gene, and not other TLR gene family members, is hypomethylated in human iPSC and derived NSC (F2), not seen in the starting parent HFF cells (F1) (Fig. 3B).