

Abstract
Whole genome sequencing of bacterial isolates has become a daily task in many laboratories, generating incredible amounts of data. However, data acquisition is not an end in itself; the goal is to acquire high‐quality data useful for understanding genetic relationships. Having a method that could rapidly determine which of the many available run metrics are the most important indicators of overall run quality and having a way to monitor these during a given sequencing run would be extremely helpful to this effect. Therefore, we compared various run metrics across 486 MiSeq runs, from five different machines. By performing a statistical analysis using principal components analysis and a K‐means clustering algorithm of the metrics, we were able to validate metric comparisons among instruments, allowing for the development of a predictive algorithm, which permits one to observe whether a given MiSeq run has performed adequately. This algorithm is available in an Excel spreadsheet: that is, MiSeq Instrument & Run (In‐Run) Forecast. Our tool can help verify that the quantity/quality of the generated sequencing data consistently meets or exceeds recommended manufacturer expectations. Patterns of deviation from those expectations can be used to assess potential run problems and plan preventative maintenance, which can save valuable time and funding resources.
Keywords: Forecast, In‐Run, MiSeq, sequencing, tool
1. INTRODUCTION
Current genome sequencing platforms have improved the ease and speed of whole genome sequencing (WGS) for bacterial isolates (Shendure & Ji, 2008), allowing laboratory scientists to complete the sequencing of 24 Salmonella or Listeria genomes in approximately 39 hr on a MiSeq sequencer (Illumina®, San Diego, CA) (Illumina®, 2013). Parallel to improvements in speed, the amount of obtained data is also steadily increasing. The data from a single sequencing run could be sufficient to require days of analysis. However, the time and funding necessary to perform these analyses should be spent wisely. It is important to ensure that the sequences being obtained consistently meet or exceed quality expectations, to reduce the risk of expending effort on inadequate runs. Since it is possible to discern the similarities and differences among individual sequencing instruments, we evaluated which of the many run metrics collected during MiSeq runs could be used to better predict the level of data quality needed for successful downstream analyses. Additionally, in large core laboratories, where sequencers are running large numbers of isolates on a regular basis, it would be beneficial to have tools for routine monitoring of run performance, based on predetermined metrics and how those compare to past trends. Routine monitoring allows abnormal or out‐of‐spec sequencer performance to be flagged in a timely fashion and acted on promptly. Such a tool should offer an interpretation of run metrics as a whole instead of observing them individually. Here, we describe the development of such a tool.
Our laboratory is part of the GenomeTrakr network, a distributed group of laboratories working with the US Food and Drug Administration (FDA) for food pathogen trace‐back and outbreak detection (Allard et al., 2016); we will therefore focus on the Illumina® MiSeq systems and processes we use most often (Allard et al., 2016). Briefly, the Illumina® Nextera XT‐based DNA library preparation process works by using transposons to fragment and tag each sample DNA with a unique combination of two adapter index sequences (i5 and i7) (Caruccio, 2011). Multiple libraries can be pooled together and loaded onto the MiSeq to start a sequencing run. During the cluster generation stage of the run, a single DNA strand is seeded onto the flow cell to serve as a template and is then clonally amplified (Illumina®, 2016a, 2016b). Thus, millions of these clusters will be generated in parallel, each containing approximately one thousand copies of the template DNA. During the sequencing stage of the run, four sequencing reads will be generated for each individual cluster: the forward read (Read 1—R1), the i7 and i5 index read (Index Reads 1 and 2—IR1 and IR2, respectively) and the reverse read (Read 2—R2) (Illumina®, 2016a, 2016b). After the run, all reads sharing the same i7/i5 adapter index combination will represent the total number of reads for that particular sample.
Several important metrics are generated during each sequencing run, including Cluster Density (CD), %≥Q30, Clusters Passing Filter (Clusters PF), Total Number of Reads, Total Number of Reads Passing Filter (Reads PF), Total Yield, Q30 Yield, Phasing, and Prephasing (Illumina®, 2015a, 2015b). Cluster Density indicates the quantity of clusters that are generated per flow cell surface area during the cluster generation stage. Phasing and Prephasing indicate the rate at which singular molecules in a cluster fall behind (“Phasing”) or move ahead (“Prephasing”) of the current cycle during the sequencing stage of a MiSeq run. Together, these two metrics are important in describing the loss of synchrony during sequencing (Kircher, Heyn, & Kelso, 2011). The Phred quality score (“Q Score”) is used to determine the accuracy of sequencing by measuring the base‐calling accuracy during a run (Ewing & Green, 1998). Clusters PF and Reads PF represent the percentage of generated clusters and number of reads, respectively, that pass an internal quality filtering procedure used by Illumina® (Illumina®, 2015a, 2015b).
Although having all these automatically generated run metrics is useful, these may provide a daunting amount of information for researchers who only want to know whether a given run has performed adequately. To address this need, we carried out this comprehensive comparative study, based on 486 MiSeq runs, performed over the course of five years, to explore differences in run metrics across sequencers. Further, we performed statistical analyses of these run metrics to help identify patterns useful for predicting either the quality of a given run or the performance of a specific MiSeq instrument.
2. MATERIALS AND METHODS
2.1. Bacterial strains
Our MiSeq runs comprised 16 different species of bacteria, from ten different genera: Campylobacter (coli and jejuni), Citrobacter braakii, Cronobacter sp., Erwinia amylovora, Escherichia coli, Listeria monocytogenes, Salmonella (bongori and enterica), Shigella (boydii, dysenteriae, flexneri and sonnei), Staphylococcus aureus and Vibrio (cholerae and parahaemolyticus). All bacteria, except for Campylobacter sp., Cronobacter sp. and E. amylovora, were grown in pure cultures overnight in tryptic soy broth (TSB) (BD Biosciences, San Jose, CA, USA) at 37°C under aerobic conditions. Cronobacter sp. and E. amylovora were grown overnight in TSB at 30°C and at 23°C under aerobic conditions, respectively. Campylobacter sp. was first grown on blood tryptic soy agar (TSA with 5% sheep blood medium) plates (BD Biosciences, San Jose, CA, USA) at 42°C under microaerophilic conditions. After roughly 48 hr of growth, cultures of Campylobacter sp. were then resuspended in TSB before DNA extraction.
2.2. Library preparation
All bacterial isolates were extracted using the Gram‐negative and Gram‐positive DNA extraction protocols described in the DNeasy Blood & Tissue Kit User Manual (Qiagen, Germantown, MD, USA). The resulting DNA samples were then constructed into DNA libraries using either the Nextera (2012–2013) or Nextera XT (2013–2017) DNA Library Preparation Kit (Illumina®, San Diego, CA, USA), using an initial DNA input of 0.2 ng/μl, and performed according to the manufacturer's protocol.
2.3. Sequencing
All sequencing described in this study was performed on Illumina® MiSeq desktop sequencers using the 500‐cycle MiSeq Reagent V2 Kits (Illumina®, San Diego, CA, USA).
2.4. Data set
We analysed 486 MiSeq runs, from five MiSeq machines (A, B, C, D and E), operated by at least eight different users, between December 2012 and May 2017. These runs consisted of multiple library pool numbers, resulting in a total DNA library number of 8,518 samples. For each sequencing run, we collected 15 MiSeq run metrics (Table 1) using the on‐board Sequencing Analysis Viewer software (SAV) (Illumina®, San Diego, CA, USA).
Table 1.
Summary of MiSeq run metrics. The following table depicts the run metrics to be compared and analysed in this study. Manufacturer recommended metrics are shown when available as well as a brief description of each run metric
| Run metric | Manufacturer recommended range/value | Brief description |
|---|---|---|
| Q30 yield | N/A | The number of gigabases (Gb) that passed the chastity filter |
| Reads PF | 24–30 million reads | The number of reads that passed the chastity filter |
| Total yield | 7.5–8.5 Gb | The total number of Gb expected to be generated during the sequencing run |
| %≥Q30 (Overall) | ≥75.0% | The percentage of bases having a quality score of 30 or higher |
| %≥Q30 (R1, R2, IR1, IR2) | N/A | The %≥Q30 score broken down into its component parts |
| Phasing (R2), Prephasing (R2) | N/A | The amount of asynchrony during the reverse sequencing read |
| Phasing (R1), Prephasing (R1) | <0.1% | The amount of asynchrony during the forward sequencing read |
| Cluster density (CD) | 1,000–1,200 K/mm2 | The quantity of clusters that are generated per flow cell surface area during the cluster generation stage of a sequencing run |
| Total reads | N/A | The total number of reads generated during a sequencing run |
| Clusters PF | ≥80.0% | The percentage of generated clusters that pass the chastity filter |
2.5. Statistical analysis
First, we compared the data from the 15 metrics on an individual instrument basis, with data expressed as means ± standard error (SE). Statistical differences between categorical variables were analysed using the Student's t test in Microsoft Excel 2010 (Microsoft, Redmond, WA, USA); values equal to or smaller than p < 0.05 were considered to be statistically significant.
Next, we compared the 15 run metrics across all 486 MiSeq runs and used these to create a scree plot and a Pearson correlation matrix. We assessed the magnitude of these correlations following a scale similar to the one described by Evans et al. (Evans, 1996), categorizing the absolute value of each Pearson's correlation coefficient (r) as weak (0.00–0.49), moderate (0.50–0.79) or strong (0.80–1.00). From this matrix, a principal components analysis (PCA) using Origin Pro software (OriginLab Corporation, Northhampton, MA, USA) was performed. PCA can be defined as an orthogonal linear transformation that aims to maintain the same variance of the raw data by reducing the number of variables into a new coordinate system of principal components (PCs) through factor scores (Zhang & Castelló, 2017). PCA allows us to observe what factors are at play, and the extent to which they correlate with each other. To observe if our data set was appropriate for a PCA, we looked at two measures of sampling adequacy. First, we applied Bartlett's sphericity test to the data set. The obtained p value was < 0.0001, which allowed us to reject the null hypothesis, meaning that it is appropriate to expect a correlation to be found. Second, we evaluated the data set using the Kaiser–Meyer–Olkin (KMO) index, resulting in an overall score of 0.698, which provided further confidence supporting the use of our data. A PCA was used to reduce our correlated variables to a smaller set of important independent variables. We used each run metric as a variable (Table 1), and each MiSeq run was treated as an observation.
To explore other possible points of comparison across runs and find similar groups (clusters) in our data, we applied a K‐means clustering algorithm (Tan, Steinbach, & Kumar, 2005). The clustering algorithm begins by randomly initializing K number of clusters, then assigning each MiSeq run into one of these clusters. The centroid of each cluster is updated by calculating a new mean, which, in turn, is used to relocate the position of each cluster centroid. This process is then repeated until all the centroids stop moving, thus allowing the algorithm to converge to a local optimum (Nidheesh, Abdul Nazeer, & Ameer, 2017). We ran this analysis several times using two to five clusters, and with both Euclidean (Kaya, Pehlivanli, Sekizkardes, & Ibrikci, 2017) and Mahalanobis distances (Wang, Hu, Huang, & Xu, 2008).
3. RESULTS AND DISCUSSION
3.1. MiSeq metric comparison by instrument
According to the MiSeq manufacturer specifications, the percentage of clusters passing the chastity filter is typically higher than 80.0% (Illumina®, 2016a, 2016b). In our study, all five MiSeqs exceeded this criterion. Nonetheless, it must be noted that the generated data can be used, even at lower than recommended Clusters PF. In fact, a lower percentage of Clusters PF will impact the total yield of the run and result in overall less output. This is due to the fact that clusters that do not pass this quality check step do not get counted in the final per cent. Among the five MiSeqs examined in terms of Clusters PF, we found that there was only an 8.1% range of difference, with the lowest value observed in MiSeq B (82.1% ± 1.6%) (Table 2). According to the manufacturer's recommendations, the optimal CD range is between 1,000 and 1,200 K/mm2 (Illumina®, 2016a, 2016b). Based on this range, the optimal CD values will be the median value (i.e., 1,100 K/mm2). In our data set, the sequencer most closely approaching this value is MiSeq D (1,048.3 ± 43.5 K/mm2) and the one farthest from the recommended value is MiSeq E (828.5 ± 32.5 K/mm2) (Table 2).
Table 2.
Comparison of 15 MiSeq run metrics according to the 5 MiSeq instruments
| MiSeq instrument (n=) | |||||
|---|---|---|---|---|---|
| A (n = 136) | B (n = 125) | C (n = 67) | D (n = 51) | E (n = 107) | |
| Cluster density | 929.1 ± 31.7 | 886.8 ± 35.8 | 1,032.1 ± 37.1a, b | 1,048.3 ± 43.5 a, b | 828.5 ± 32.5 a, c, d |
| Clusters PF | 86.9% ± 0.8% | 82.1% ± 1.6% a | 90.2% ± 1.2% a, b | 84.4% ± 1.3%c | 86.5% ± 0.8%b, c |
| %≥Q30 (Overall) | 81.6% ± 0.6% | 73.8% ± 1.1% a | 78.9% ± 1.4%b | 76.4% ± 1.7%a | 78.5% ± 0.7%a, b |
| %≥Q30 (R1) | 88.1% ± 0.4% | 82.7% ± 1.0%a | 85.2% ± 1.4%a | 82.0% ± 1.8% a | 85.6% ± 0.6%a, b |
| %≥Q30 (IR1) | 93.8% ± 0.6% | 88.2% ± 1.7% a | 94.7% ± 1.2% b | 93.5% ± 1.2%b | 90.1% ± 1.0%a, c, d |
| %≥Q30 (IR2) | 90.5% ± 0.8% | 82.5% ± 1.8% a | 92.1% ± 1.1% b | 89.1% ± 1.5%b | 82.9% ± 1.2%a, c, d |
| %≥Q30 (R2) | 75.7% ± 0.6% | 64.1% ± 1.4% a | 72.1% ± 1.5%a, b | 70.4% ± 1.7%a, b | 71.4% ± 0.9%a, b |
| Total yield | 7.5 ± 0.2 Gb | 7.0 ± 0.3 Gb | 9.0 ± 0.3 Gb a, b | 8.3 ± 0.3 Gba, b | 6.6 ± 0.2 Gb a, c, d |
| q30 yield | 6.1 ± 0.2 Gb | 5.2 ± 0.2 Gb a | 7.2 ± 0.3 Gb a, b | 6.4 ± 0.3 Gbb | 5.2 ± 0.2 Gb a, c, d |
| Total reads | 1.7 × 107 ± 5.4 × 105 | 1.6 × 107 ± 6.4 × 105 | 2.0 × 107 ± 6.9 × 10 b | 1.9 × 107 ± 7.9 × 10b | 1.5 × 107 ± 5.9 × 10 c, d |
| Reads PF | 1.5 × 107 ± 4.2 × 105 | 1.3 × 107 ± 5.5 × 105 | 1.8 × 107 ± 6.2 × 10 b | 1.6 × 107 ± 6.5 × 10b | 1.3 × 107 ± 4.6 × 10 c, d |
| Prephasing (R1) | 0.055% ± 0.002% | 0.111% ± 0.008%a | 0.113% ± 0.031% | 0.119% ± 0.037% | 0.074% ± 0.004%a, b |
| Prephasing (R2) | 0.116% ± 0.006% | 0.171% ± 0.016%a | 0.169% ± 0.028% | 0.179% ± 0.037% | 0.131% ± 0.007%b |
| Phasing (R1) | 0.074% ± 0.005% | 0.096% ± 0.008% a | 0.092% ± 0.017% | 0.072% ± 0.003% b | 0.087% ± 0.005%d |
| Phasing (R2) | 0.150% ± 0.007% | 0.174% ± 0.012% | 0.172% ± 0.008%a | 0.162% ± 0.007% | 0.172% ± 0.008%a |
“n” is equal to the number of MiSeq runs performed per sequencer. Values represent the means ± SE. Values that are deemed statistically significant are indicated by the corresponding symbol according to the figure legend below. The best and worst values based on manufacturer recommendations for each variable are indicated with underline and bold, respectively
Significant compared to MiSeq A.
Significant compared to MiSeq B.
Significant compared to MiSeq C.
Significant compared to MiSeq D.
The %≥Q30 (Overall), as a measure of base call accuracy, is comprised of its component %≥Q30 scores; that is, the four reads—R1, IR1, IR2 and R2, respectively. According to the manufacturer's specifications, the %≥Q30 (Overall) should be at least >75% (Illumina®, 2015a, 2015b). Four out of our five MiSeqs passed this criterion: The lowest value was from MiSeq B (73.8% ± 1.1%), and the highest value was observed from MiSeq A (81.6% ± 0.6%) (Table 2).
The amount of data generated by a given sequencing run could be evaluated using both the Total Yield and the Total Reads metrics, as these two metrics directly correlate with each other. Among the MiSeqs tested, MiSeq C showed the highest average Total Yield (9.0 ± 0.3 Gb) and Total Reads (2.0 × 107 ± 6.9 × 105 reads), and MiSeq E showed the lowest Total Yield (6.6 ± 0.2 Gb) and Total Reads (1.5 × 107 ± 5.9 × 105 reads), respectively (Table 2). However, Total Yield is not as important as the amount of data that passes the filter. MiSeq C showed the highest Q30 Yield value: 7.2 ± 0.3 Gb (corresponding to 1.8 × 107 ± 6.2 × 105 Reads PF). The lowest values came from MiSeqs B and E: Each gave a Q30 Yield of 5.2 ± 0.2 Gb and Reads PF at 1.3 × 107 reads (with different SEs) (Table 2).
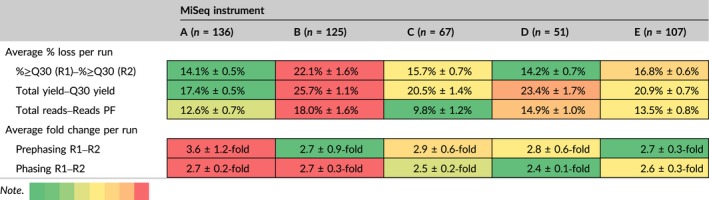
Phasing and Prephasing distortions will increase, as the sequencing read becomes longer (Tan et al., 2005). Prephasing and Phasing can be caused by several factors. Prephasing might be due to a fluorophore‐labelled nucleotide (FLN) that has a defective terminator, which allows two FLNs to adhere to a single molecule, thus promoting the sequencing system to jump ahead during the run. Phasing can occur when the expected terminator cleavage fails to occur during a given cycle and instead happens in the subsequent cycle, causing the sequencing to lag behind the actual genome sequence (Tan et al., 2005). As the instrument continues cycling, the clusters that were initially formed on the flow cell will start to lose their coherence (Ding & He, 2004). Therefore, the Prephasing detected during R1 should be less than what is typically found during R2, which is confirmed by our data as the amount of Prephasing exhibited a 2.7–2.9 fold change (as observed in MiSeqs B, C, D and E) or a 3.6 fold change (as seen in MiSeq A) (Table 3). In all five MiSeqs, the average Phasing change between R1 and R2 exhibited between a 2.4 (MiSeq D) – 2.7 (MiSeq B) fold change (Table 3). Four out of our five instruments (MiSeqs B, C, D and E) demonstrated similar average changes of Prephasing and all five presented similar average changes in Phasing, even though the other run metrics across the five machines were vastly divergent. Therefore, this implies that although cycle synchronicity plays a key role in obtaining a high‐quality run, it cannot be the sole factor to rely on when deciding whether a run has been successful or not.
Table 3.
Heatmap comparison of changes in run metrics between the start and end of a MiSeq sequencing run for each MiSeq instrument. “n” is equal to the number of MiSeq runs performed per sequencer. Values represent the arithmetic means + SE. The color palette in the note indicates the range of the most favorable average % loss/fold changes (green) to least favorable average % loss/fold changes (red), comparatively for the 5 MiSeq instruments [Colour table can be viewed at wileyonlinelibrary.com]
3.2. Pearson's correlation and scree plot
In our analysis, the Pearson correlation matrix allowed us to distinguish two main groups (Table 4). The first group contains both moderate and strong positive correlations among six metrics: Clusters PF, %≥Q30 (Overall) and the %≥Q30 for R1, R2, IR1 and IR2. The second group consisted of five metrics: Total Reads, Reads PF, Total Yield, Q30 Yield and CD. In this second group, all metrics exhibited strong positive correlations with one another, whereas CD exhibited strong positive correlations with Total Reads, Reads PF and Total Yield and only a moderate positive correlation with the Q30 yield. In contrast, the Pearson matrix also showed that Phasing (R1) had weak negative correlations with other run metrics in our analyses. Prephasing (R1) exhibited moderate negative correlations with the %≥Q30 (Overall) and the %≥Q30 (R1) metrics and Phasing (R2) and Prephasing (R2) were found to have a moderate positive correlation with each other (Table 4).
Table 4.
Pearson's correlation matrix for 15 analysed MiSeq run metrics. Strong correlations (values between 0.80–1.00) are indicated in green; moderate correlations (0.50–0.79) are in yellow, and values with no color indicate weak correlations (0.00–0.49)
| % Clusters PF | %≥Q30 (Overall) | %≥Q30 (R1) | %≥Q30 (IR1) | %≥Q30 (IR2) | %≥Q30 (R2) | Phasing (R1) | Prephasing (R1) | Phasing (R2) | Prephasing (R2) | Total reads | Reads PF | Total yield | Q30 yield | Cluster density | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Clusters PF | * | ||||||||||||||
| %≥Q30 (Overall) | 0.54 | * | |||||||||||||
| %≥Q30 (R1) | 0.49 | 0.89 | * | ||||||||||||
| %≥Q30 (IR1) | 0.52 | 0.46 | 0.38 | * | |||||||||||
| %≥Q30 (IR2) | 0.55 | 0.5 | 0.41 | 0.6 | * | ||||||||||
| %≥Q30 (R2) | 0.52 | 0.9 | 0.77 | 0.46 | 0.5 | * | |||||||||
| Phasing (R1) | −0.3 | −0.32 | −0.38 | −0.29 | −0.3 | −0.27 | * | ||||||||
| Prephasing (R1) | −0.14 | −0.58 | −0.67 | −0.16 | −0.15 | −0.48 | 0.26 | * | |||||||
| Phasing (R2) | −0.03 | 0.05 | 0.03 | 0.09 | −0.19 | 0.04 | 0.13 | −0.03 | * | ||||||
| Prephasing (R2) | −0.02 | −0.17 | −0.2 | 0.02 | −0.12 | −0.15 | 0.11 | 0.31 | 0.56 | * | |||||
| Total reads | −0.14 | −0.15 | −0.13 | 0.04 | 0.13 | −0.15 | −0.04 | −0.05 | −0.29 | −0.23 | * | ||||
| Reads PF | 0.16 | 0.02 | 0.02 | 0.23 | 0.32 | 0.02 | −0.16 | −0.1 | −0.29 | −0.23 | 0.94 | * | |||
| Total yield | 0.14 | −0.01 | 0.01 | 0.21 | 0.3 | −0.01 | −0.16 | −0.09 | −0.3 | −0.24 | 0.93 | 0.99 | * | ||
| Q30 yield | 0.23 | 0.2 | 0.19 | 0.3 | 0.4 | 0.2 | −0.22 | −0.18 | −0.28 | −0.34 | 0.86 | 0.96 | 0.96 | * | |
| Cluster density | −0.25 | −0.2 | −0.16 | −0.03 | 0.05 | −0.21 | −0.04 | −0.08 | −0.27 | −0.21 | 0.9 | 0.82 | 0.84 | 0.77 | * |
3.3. Classification of MiSeq runs using PCA and k‐means clustering
The correlations generated from the Pearson matrix can indicate a predictive relationship that can be exploited in reducing the variables (run metrics). Using the scree plot (Figure S1), we see that we can use five PCs to generate the analysis. However, we will use the top 3 PCs, since five PCs cannot be visualized efficiently. The first three PCs (termed PC1, PC2 and PC3) can be used to linearly separate 72.86% of the total variance of the data generated by our 486 MiSeq runs (Table S1). Figure 1 illustrates the PCA loading plot for these three PCs. We can observe that the 15 variables can be sorted into three groups that are highly correlated (Figure 1). The first group contains eight metrics and accounts for over a third (34.43%) of the total variance, the second group contains three metrics and accounts for 28.29% of the variance, and the third group contains of four metrics, which constituted 10.13% of the total variance. In Table S2, we show the coefficient of each metric and its contribution to each principal component.
Figure 1.
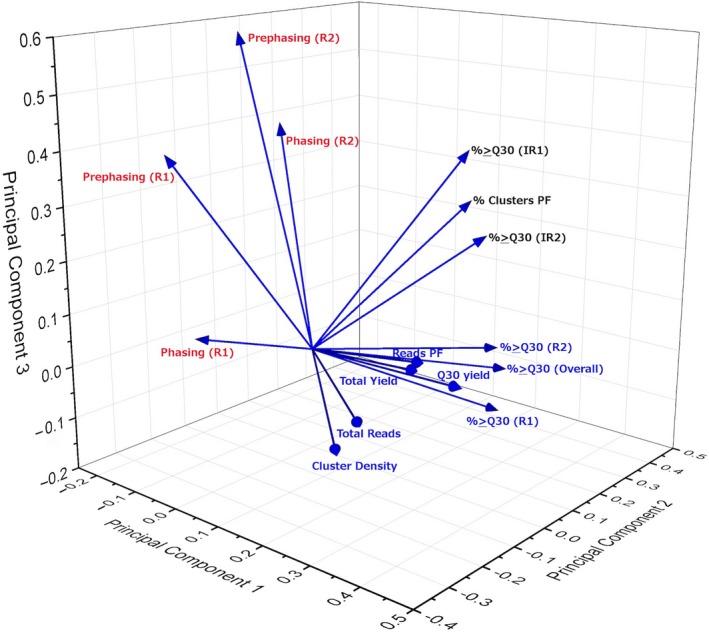
PCA loading plot of the 15 observed MiSeq run metrics across 486 MiSeq runs. Three groups can be distinguished from the plot and are indicated in different colours [Colour figure can be viewed at wileyonlinelibrary.com]
Finally, we generated a three‐dimensional plot using the sets of PC1, PC2 and PC3 from each of our MiSeq runs and colour‐coded each observation (MiSeq run) to represent each of the five MiSeq machines tested: A, B, C, D and E (Figure 2). All our MiSeqs tended to cluster together across the three axes. However, their differences were highly informative. Several MiSeqs had a higher percentage of their runs that gravitated towards the PC1 and PC3 axes, primarily MiSeqs B, C and D. In contrast, the runs from MiSeqs A and E tended to aggregate into really tight groupings (Figure 2). Furthermore, MiSeq B contained several runs that did not fall within the tight run cluster predicted by our PCA. This signals that MiSeq B is not performing adequately compared to the other MiSeqs tested, further confirming the conclusions we could draw from our comparison of instruments (Table 3).
Figure 2.
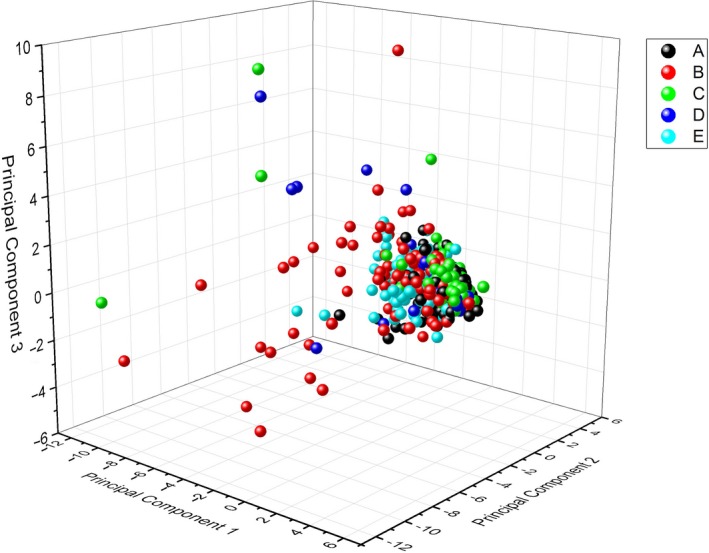
A three‐dimensional PCA plot of each MiSeq instrument. Each colour represents a particular MiSeq desktop sequencer (MiSeq A–E), and each point on the plot represents a single observation (2 × 250 500 cycle V2 MiSeq sequencing run) [Colour figure can be viewed at wileyonlinelibrary.com]
3.4. K‐means cluster analysis
The main drawbacks of using K‐means clustering alone is associated with two well‐established problems such as (a) defining a priori the number of clusters to use and (b) visualizing the obtained clusters in several dimensions. Thus, a typical solution is to preprocess the data using PCA by mapping the data into a new feature space (Laas, Ballester, Cortez, Graesslin, & Daraї, 2017). Afterwards, the k‐means algorithm is applied to the data in the feature space. The final result is able to identify observations that are similar to each other.
After running the K‐cluster analysis, the data points formed three unique clusters (Figure 3a), regardless of MiSeq instrument used. These three unique clusters were colour‐coded green, red and blue (Figure 3a). We used green to label the cluster composed of the adequate runs, and, reassuringly, 93.6% of our MiSeq runs fell into this category (n = 455). The runs in the blue cluster (n = 24) presented very low %≥Q30 values: either %≥Q30 (Overall), %≥Q30 (R1, R2, IR1 and IR2) or a combination of these metrics. The red cluster primarily contained MiSeq runs (n = 7) that exhibited an exorbitant amount of Phasing or Prephasing, well over the acceptable 0.1% threshold.
Figure 3.
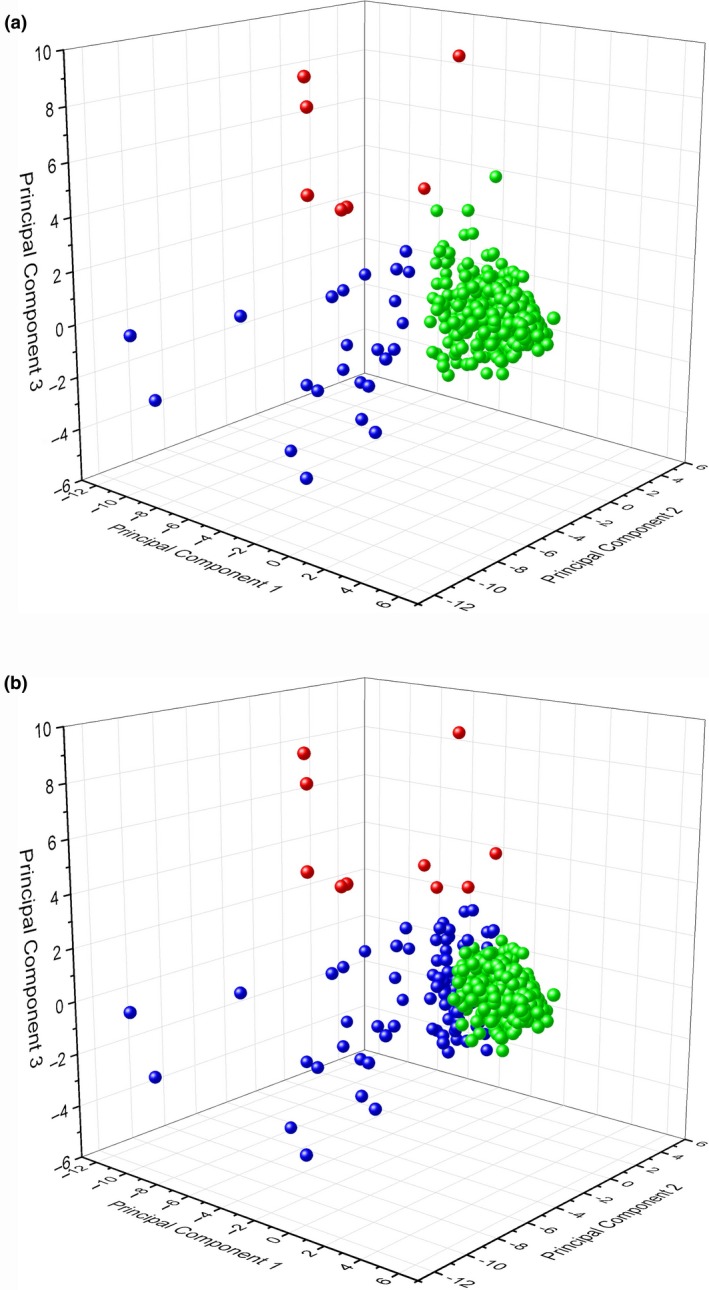
(a) A three‐dimensional k‐means cluster analysis plot of the PCA (Euclidean). The analysis was run several times with a priori number of clusters from 2 to 5. In this plot, higher quality MiSeq runs, which exhibited metrics that met performance criteria, are represented by the green cluster, MiSeq runs that failed primarily due to %≥Q30 issues are represented in blue, and MiSeq runs that failed due to Phasing or Prephasing are depicted in red. (b) A three‐dimensional k‐means cluster analysis plot of the PCA (Mahalanobis). The analysis was run several times with a priori number of clusters from 2 to 5. In this plot, higher quality MiSeq runs, which exhibited metrics that met performance criteria, are represented by the green cluster, MiSeq runs that failed due to %≥Q30 issues and other metrics are represented in blue, and MiSeq runs that failed due to Phasing or Prephasing and other metrics are depicted in red [Colour figure can be viewed at wileyonlinelibrary.com]
These colour‐coded K‐clusters also help us see which metrics gave the most information about performance deficits (Tan et al., 2005). We can observe that most of MiSeq B's problematic runs (Figure 2) were the result of subpar %≥Q30 numbers, as all of the runs falling into this category showed %≥Q30 (Overall) values lower than 70% (below Illumina's recommended values of 75%). Likewise, most of the problematic runs on MiSeq D were due to excessive Phasing and Prephasing. Interestingly, in all MiSeq runs identified as inadequate due to Phasing and Prephasing issues, the Phasing (R1) values were below the 0.1% threshold, whereas one or more of the other three Phasing (R2) and Prephasing (R1 and R2) metrics displayed exceedingly high values, usually at or above 1.0%.
In addition to looking at the runs in relation to the K‐means clustering based on the Euclidean distance (Kaya et al., 2017), we also tested the same data set using the Mahalanobis distance (Wang et al., 2008) and found that it does not fit our data set well as it excludes runs deemed adequate using the Euclidean distance (Figure 3b). K‐means clustering using the Euclidean distance grouped 455 runs out of 486 as adequate (93.6%) while K‐means using the Mahalanobis distance classed 394 runs out of 486 as adequate (81.1%). A MiSeq machine performance check is meant to be used as a first checkpoint, and adequate MiSeq runs will have to eventually pass through downstream Quality Assurance (QA) checks. Therefore, we find the Mahalanobis distance to be too stringent, potentially excluding MiSeq runs that presented viable data for analysis (Figure 3b). Just as well, the Mahalanobis distance also presents a variation to the established clusters and we found that runs were now more difficult to group according to a specific run metric (Figure 3b).
3.5. Development of a quality assessment tool: the “MiSeq Instrument & Run Forecast” (MiSeq In‐Run Forecast)
There is a direct relationship between the Total Yield (in Gb) and the total number of reads passing the filter (Figure S2), also confirmed from the Pearson plot (Table 4). Using the underlying equation of Figure S2:
The Total Yield (in Gb) will be determined by the number of Reads PF multiplied by the appropriate multiplication factor (5.1 × 10−7). As expected, the values predicted with this equation were within 1% of the actual observed numbers produced by SAV in most cases. In such cases, the discrepancy was due to data incorrectly populating into SAV. While this equation correctly classified 97.8% of the adequate runs from our MiSeqs, it only detected 19.4% of the inadequate runs. Thus, this equation alone is not enough to ensure that a run is an adequate one.
Therefore, coupling the Total Yield equation above with the K‐clustering observations mentioned earlier, we developed the “MiSeq In‐Run Forecast” tool, an Excel spreadsheet (Microsoft, Redmond, WA) which can be populated with the SAV run metrics from any MiSeq, provided it used a 500‐cycle MiSeq Reagent V2 kit with prepared Nextera XT libraries. Our spreadsheet calculations encompass both the aforementioned Total Yield equation and an algorithm that uses the values of all fifteen MiSeq run metrics during/after a run to accurately predict whether a given MiSeq run has performed adequately and how it compares to previous runs. The “MiSeq In‐Run Forecast” spreadsheet is available at: https://figshare.com/s/ef7554978305a7089403 ( https://doi.org/10.6084/m9.figshare.5803170). Note that the “MiSeq In‐Run Forecast” tool uses SAV fields of R1, R2, R3 and R4, where R1 and R2 are the forward and reverse sequencing reads (R1 and R4 in SAV), and IR1 and IR2 are the index reads (R2 and R3 in SAV), respectively.
The “MiSeq In‐Run Forecast” tool can start to be populated while the run is sequencing in real time with as few as two to four SAV run metrics. CD and Total Reads are available between the 5th and 12th cycle, while Total Yield and Reads PF are available to users between the 25th and 32nd cycle (approximately 3 to 4 hr into a 39‐hr run). Inserting these run metrics into the tool can provide for a quick preliminary assessment of run quality and performance based off the Total Yield equation. A large change in the “Yield Percent Error” column of the spreadsheet will alert a user whether a run is performing adequately or not in terms of yield. Once the run has finished and all metrics become available in SAV, the “MiSeq In‐Run Forecast” can be fully compiled, to ensure that the whole run is deemed adequate.
Our algorithm is a mathematical representation of the K‐means analysis (Figure 3a) and depicted in Figure 4. The “MiSeq In‐Run Forecast” will designate the run of interest in the chart using the Euclidean distance measurements of each K‐cluster and their centroids, afterwards comparing the smallest distance of the three K‐clusters. If the run maps into the green category, then we can predict that it was an adequate MiSeq run (Figure 3a). If the data from a run trend towards either the red (high Phasing/Prephasing values) or blue cluster (low %≥Q30 values), then we can predict that this run did not perform up to its expected capabilities and was deemed inadequate by the tool (Figure 3a).
Figure 4.
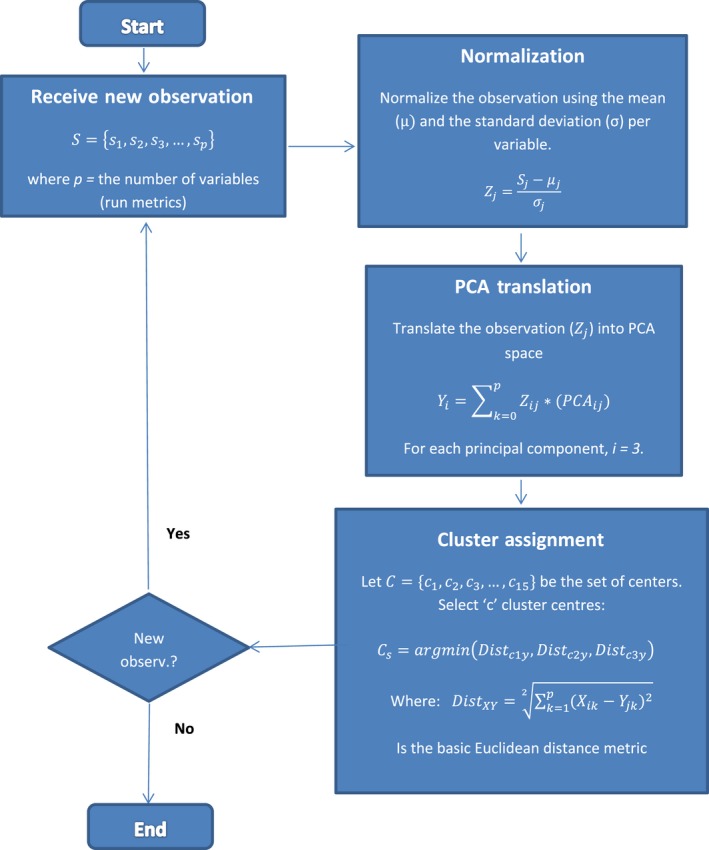
MiSeq In‐Run Forecast Algorithm Flow Chart. A stepwise representation depicting the “MiSeq In‐Run Forecast” operation [Colour figure can be viewed at wileyonlinelibrary.com]
One of the important insights into this work is demonstrating that CD, one of the first run metrics the SAV reports during a sequencing run and often thought to be a primary determining factor for most other run metrics, may not necessarily be so (Illumina®, 2016a, 2016b). As presented above, 455 runs were initially deemed to be adequate runs while 31 runs were considered inadequate (either by %≥Q30 or Phasing/Prephasing). Of the MiSeq runs, we found skewing into the red and blue K‐clusters (n = 31), 93.5% (n = 29) had CD values outside the recommended range (1,000–1,200 K/mm2). Additionally, we observed that an astonishing 81.5% (n = 371) of the adequate runs was outside the recommended CD range (Figure S3). Therefore, this implies that CD cannot be used on its own to predict how a run will perform (See Figure S3). We have seen that other metrics, especially those related to %≥Q30 and Phasing/Prephasing, can critically affect the performance of a MiSeq sequencing run. These metrics are more crucial to consider during a MiSeq run in order to understand if that run is performing up to par.
The algorithm developed herein is reliably able to correctly assess the quality of a run, except in the rare cases where the centroid distances of two different clusters are equidistant. In these circumstances, the run patterns will exhibit traits of both affected clusters. Thus, our “MiSeq In‐Run Forecast” tool will be most effectively used as a quality control measure in the laboratory to assess MiSeq instrument performance. Another advantage of having this tool is that it does not require FASTQ files or any type of post‐data generation processing/transferring. It is a preassembly assessment based solely on the raw sequencing metrics per MiSeq run. Further research is needed to effectively connect these run metrics to actual downstream effects such as sample coverage, assembly quality.
4. CONCLUSION
Using our wealth of MiSeq run data, we have developed an easy to use wet‐lab based QA tool (i.e., the “MiSeq In‐Run Forecast”) that can be run in Excel to provide a rapid instrument‐ and run‐based quality control check. If our algorithm classifies a given MiSeq run as less than adequate, users can then use the tool to assess which factor was the most likely underlying cause (%≥Q30 or Phasing/Prephasing) and thus troubleshoot the instrument. Careful consideration should also be given as to whether the sequence data acquired during such runs should be submitted for downstream assembly and analysis. Our tool can help laboratories achieve a consistent minimum standard quality for data collection and could also potentially save researchers time and money.
Another important use of this QA tool is to support the work of distributed sequencing networks or large core centres. A quality baseline can be established for each laboratory, allowing any deviation from the usual run performance to be rapidly spotted. This tool could detect potential issues with new reagent lots, personnel, protocol changes or indicate that a particular MiSeq instrument is beginning to go out of spec, even before other on‐board QA features detect a problem.
It must be noted that the tool uses the run metrics in order to evaluate the MiSeq sequencing run as a whole, and thus, it cannot currently be used for individual isolate/sample assessment. It is meant to be used as a monitoring tool in order to designate if a MiSeq run performed as expected. Therefore, future projects could use the run metrics from a larger pool of sequencers to observe how machine and run differences could potentially translate into downstream sample differences as well as to zero in on how to effectively troubleshoot the particular run metric(s) at fault.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors. Trade names mentioned in the manuscript do not constitute an endorsement.
AUTHOR CONTRIBUTIONS
G.J.K., E.W.B and M.W.A involved in the study design. G.J.K. partly performed the laboratory work. G.J.K. and L.V.S.Q. performed the data analysis. G.J.K., L.V.S.Q., M.S.L. and S.L. all contributed to interpreting the data and writing the manuscript.
Supporting information
ACKNOWLEDGEMENTS
The authors would like to thank Dwayne Roberson at the US Food and Drug Administration—Center for Food Safety and Applied Nutrition (FDA‐CFSAN) for his insight on bacterial growth, Ruth Timme (FDA‐CFSAN) on her constructive remarks and suggestions, and Daniele Nucera of the Dipartimento di Scienze Agrarie, Forestali e Alimentari (DISAFA) for his knowledge and input on statistical analysis. We thank Lili Fox Vélez, Office of Regulatory Science, for scientific writing support. We also thank Hugh Rand (FDA‐CFSAN) for his statistical expertise and Kelly Hoon (Illumina®, San Diego) for her valuable remarks and recommendations. Additionally, we would also like to greatly thank the members of the sequencing group at the CFSAN for providing sequencing data.
Kastanis GJ, Santana‐Quintero LV, Sanchez‐Leon M, Lomonaco S, Brown EW, Allard MW. In‐depth comparative analysis of Illumina® MiSeq run metrics: Development of a wet‐lab quality assessment tool. Mol Ecol Resour. 2019;19:377–387. 10.1111/1755-0998.12973
Funding information
This work, including the efforts of G.J.K., was funded by the US Food and Drug Administration (FDA), Center for Food Safety and Applied Nutrition (CFSAN). This work was supported by internal research funding from the FDA‐CFSAN. M.S.L. and S.L. received support by appointment to the Research Participation Program at FDA‐CFSAN, administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy and the FDA. The findings and conclusions reported here are those of the authors alone and do not necessarily represent the views of the U.S. Food & Drug Administration.
DATA ACCESSIBILITY
All data presented in this manuscript are available at: https://figshare.com/s/08558c8c51ef01efb318 ( https://doi.org/10.6084/m9.figshare.5950126).
REFERENCES
- Allard, M. W. , Strain, E. , Melka, D. , Bunning, K. , Musser, S. M. , Brown, E. W. , & Timme, R. (2016). Practical value of food pathogen traceability through building a whole‐genome sequencing network and database. Journal of Clinical Microbiology, 54, 1975–1983. 10.1128/JCM.00081-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruccio, N. (2011). Preparation of next‐generation sequencing libraries using nextera technology: simultaneous DNA fragmentation and adaptor tagging by in vitro transposition In: Kwon Y., & Ricke S. (Eds.), High‐throughput next generation sequencing: Methods in molecular biology (pp. 241–255). Totowa, NJ: Humana Press; 10.1007/978-1-61779-089-8_17. [DOI] [PubMed] [Google Scholar]
- Ding, C. , & He, X. (2004). K‐means clustering via principal component analysis. Paper presented at ICML '04: Proceedings of the twenty‐first international conference on Machine learning. Banff, Canada. New York, NY: Association for Computing Machinery; 10.1145/1015330.1015408 [DOI] [Google Scholar]
- Evans, J. D. (1996). Straightforward statistics for the behavioral sciences. Pacific Grove, CA: Brooks/Cole Publishing. [Google Scholar]
- Ewing, B. , & Green, P. (1998). Basecalling of automated sequencer traces using phred. II. Error Probabilities. Genome Research, 8, 186–194. 10.1101/gr.8.3.186. [DOI] [PubMed] [Google Scholar]
- Illumina ® , Inc. (2013). MiSeq® System: The most accurate and easiest‐to‐use benchtop sequencer available [Specification Sheet]. San Diego, CA: Illumina; ®; , Inc; Retrieved from https://support.illumina.com/documents//products/datasheets/datasheet_miseq.pdf. [Google Scholar]
- Illumina ® , Inc. (2015a). Calculating Percent Passing Filter for Patterned and Non‐Patterned Flow Cells: A comparison of methods for calculating percent passing filter on Illumina flow cells [Brochure]. San Diego, CA: Illumina®, Inc; Retrieved from https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/hiseq-x-percent-pf-technical-note-770-2014-043.pdf. [Google Scholar]
- Illumina ® , Inc. (2015b). Illumina MiSeq® System Guide [Product Manual]. San Diego, CA: Illumina; ®; , Inc. [Google Scholar]
- Illumina ® , Inc. (2016a). An Introduction to Next‐Generation Sequencing [Brochure]. San Diego, CA: Illumina; ®; , Inc; Retrieved from https://www.illumina.com/content/dam/illumina-marketing/documents/products/illumina_sequencing_introduction.pdf. [Google Scholar]
- Illumina ® , Inc. (2016b). Optimizing Cluster Density on Illumina Sequencing Systems [Brochure]. San Diego, CA: Illumina; ®; , Inc; Retrieved from https://support.illumina.com/content/dam/illumina-marketing/documents/products/other/miseq-overclustering-primer-770-2014-038.pdf. [Google Scholar]
- Kaya, I. E. , Pehlivanli, A. C. , Sekizkardes, E. G. , & Ibrikci, T. (2017). PCA based clustering for brain tumor segmentation of T1w MRI images. Computer Methods and Programs in Biomedicine, 140, 19–28. 10.1016/j.cmpb.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Kircher, M. , Heyn, P. , & Kelso, J. (2011). Addressing challenges in the production and analysis of Illumina sequencing data. BMC Genomics, 382(12), 1471–2164. 10.1186/1471-2164-12-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laas, E. , Ballester, M. , Cortez, A. , Graesslin, O. , & Daraї, E. (2017). Unsupervised clustering of immunohistochemical markers to define high‐risk endometrial cancer. Pathology Oncology Research. 10.1007/s12253-017-0335-y. [DOI] [PubMed] [Google Scholar]
- Nidheesh, N. , Abdul Nazeer, K. A. , & Ameer, P. M. (2017). An enhanced deterministic K‐Means clustering algorithm for cancer subtype prediction from gene expression data. Computers in Biology and Medicine, 91, 213–221. 10.1016/j.compbiomed.2017.10.014. [DOI] [PubMed] [Google Scholar]
- Shendure, J. , & Ji, H. (2008). Next‐generation DNA sequencing. Nature Biotechnology, 10, 1135–1145. 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Tan, P. , Steinbach, M. , & Kumar, V. (2005). Introduction to data mining, 1st ed Boston, MA: Addison‐Wesley Longman Publishing. [Google Scholar]
- Wang, J. C. , Hu, J. , Huang, X. X. , & Xu, S. C. (2008). Assessment of different genetic distances in constructing cotton core subset by genotypic values. Journal of Zhejiang University. Science. B, 9(5), 356–362. 10.1631/jzus.B0710615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , & Castelló, A. (2017). Principal components analysis in clinical studies. Annals of Translational Medicine, 5(17), 351 10.21037/atm.2017.07.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in this manuscript are available at: https://figshare.com/s/08558c8c51ef01efb318 ( https://doi.org/10.6084/m9.figshare.5950126).