

Abstract
Invasive bacterial disease is well described in immunocompromised hosts, including those with malaria infection. One bacterial infection frequently observed in children with Plasmodium falciparum infection is nontyphoidal salmonella (NTS) infection, in which a typically intestinal infection becomes systemic with serious, often fatal, consequences. In this review, we consider the role of malaria‐induced immunoregulatory responses in tipping the balance from tissue homeostasis during malaria infection to risk of invasive NTS. Also, neutrophils are crucial in the clearance of NTS but their ability to mount an oxidative burst and kill intracellular Salmonella is severely compromised during, and for some time after, an acute malaria infection. Here, we summarize the evidence linking malaria and invasive NTS infections; describe the role of neutrophils in clearing NTS infections; review evidence for neutrophil dysfunction in malaria infections; and explore roles of heme oxygenase‐1, IL‐10, and complement in mediating this dysfunction. Finally, given the epidemiological evidence that low density, subclinical malaria infections pose a risk for invasive NTS infections, we consider whether the high prevalence of such infections might underlie the very high incidence of invasive bacterial disease across much of sub‐Saharan Africa.
Keywords: malaria, salmonella, anemia, sepsis, neutrophil, heme oxygenase‐1, IL‐10
Abbreviations
- Hmox1
heme oxygenase‐1 gene
- HO‐1
heme oxygenase‐1
- NETs
neutrophil extracellular traps
- NTS
nontyphoidal Salmonella
- PMN
polymorphonuclear cells
- ROS
reactive oxygen species
- SCV
Salmonella containing vacuoles
1. BACTEREMIA AND MALARIA
Bloodstream bacterial infections remain a global health concern, with high case fatality rates and the potential for long‐term, life‐changing sequelae. Life‐threatening organ dysfunction resulting from systemic bacterial infection, or more commonly sepsis,1 is mediated by a systemic inflammatory response2, 3 wherein septic shock leads to severe tissue damage and death.4, 5 Sepsis is one of the most challenging and most costly conditions to treat in hospital—amassing a bill of $24 billion in the United States for 2013 alone.6
In developed economies, the organisms most frequently isolated from blood include Staphylococcus aureus and Escherichia coli 7 (each accounting for ∼20% of cases). Methicillin‐resistant S. aureus 8 and highly pathogenic E. coli are emerging as major causes of nosocomial infections.9 In contrast, developing nations in Africa see a much greater incidence of community‐acquired bacteremia with Salmonella enterica (often nontyphoidal Salmonella [NTS]) and Streptococcus pneumoniae as the most commonly isolated organisms.10 Laboratory diagnosis for microbiological pathogens in Africa remains poor, with insufficient infrastructure and related funding. Despite challenges in detection, Ao et al. have estimated that NTS causes 3.4 million cases of bacteremia globally each year, of which the majority (1.9 million cases and 380,000 deaths) are in children and young adults in sub‐Saharan Africa.11 In Kenya, 70% of these deaths occur within 2 days of admission to hospital,12 providing a very narrow window for effective intervention. Further, multiple drug‐resistant NTS serotypes have been reported in East and Southern Africa, with sequence type 313 (ST313) seen as a distinct lineage associated with septicemia.13, 14 Increasingly, lack of access to effective and affordable antibiotics may lead to even higher morbidity and mortality in low‐income settings.
NTS thrives in the intestinal environment where, in otherwise healthy hosts, localized gastroenteritis allows NTS to outcompete the microbiota, causing diarrhea and promoting transmission.15 However, the infection can “escape” the gut and invade other tissues, eventually becoming systemic, particularly when the host is immunocompromised. One well‐documented risk factor for invasive NTS is Plasmodium falciparum malaria.16, 17 Plasmodium, the causative agent of malaria, is transmitted to humans through the bite of the female Anopheles mosquito causing a range of clinical manifestations including anemia, metabolic acidosis, and end‐organ failure.18 In The Gambia, the incidence of invasive NTS infection mirrors that of malaria, peaking during the annual rainy season, and in one study, 43% of children with Salmonella bacteremia had concurrent P. falciparum infections.19 In Tanzania, invasive NTS in young children is highly associated with recent malaria infection, with 78% of NTS cases having recently received antimalarial medication and 82% of cases being anemic.20 Intriguingly, recent (past) malaria infection is a higher risk factor for NTS bacteremia than is acute (current) infection.21 Therefore, although children with severe acute malaria have been noted to be at high risk of developing invasive NTS,22 a picture is emerging in which even low‐density or recently cleared malaria infections are a significant contributor to invasive NTS. Finally, evidence that carriage of sickle cell trait (that protects from malarial anemia) reduces the risk of contracting invasive NTS23 and that intensive efforts in the last 15 years to reduce the prevalence and incidence of malaria across Africa have been accompanied by marked falls in the incidence of invasive bacteremia, and especially invasive NTS24, 25 serves to reinforce the clinical observations linking these two diseases and suggests a related underlying pathophysiology.
2. INTESTINAL AND INVASIVE NTS INFECTIONS
Salmonella can infect a broad host range (e.g., pigs, cattle, chickens, and humans) causing varying levels of damage, from enteric fever to severe gastroenteritis to asymptomatic carriage, depending on the particular serovar, typically defined by expression of LPS, flagellar, and capsular Vi antigens.26 With over 2500 known serovars,27 sterile immunity through natural infection or vaccination remains elusive.28, 29 The human‐restricted typhoidal serovars (Salmonella typhi and Salmonella paratyphi) are associated with systemic infection and carriage in the gallbladder30 but, intriguingly, these are not the serovars that are associated with malaria infections. Rather, malaria is associated with invasive disease caused by nontyphoidal serovars that can infect a broad range of different host species and are normally restricted to the intestine.17 Invasion of NTS through the intestinal mucosa can occur via their uptake by dendritic cells extruding dendrites between enterocytes into the intestinal lumen (paracellular uptake), via direct invasion of enterocytes or by passage through M cells of the Peyers Patches31 (Fig. 1), and this is dependent on a degree of inflammation.32, 33 Bacterial invasion triggers the IL‐23/IL‐18 inflammatory axis leading to T cell activation and production of inflammatory cytokines (including IFN‐γ, IL‐17, and IL‐22) and chemokines (CXCL1 and Mip2),34, 35, 36 eventually resulting in edema and infiltration of monocytes and neutrophils into the lamina propria,37 which are hallmarks of NTS pathology.
Figure 1.
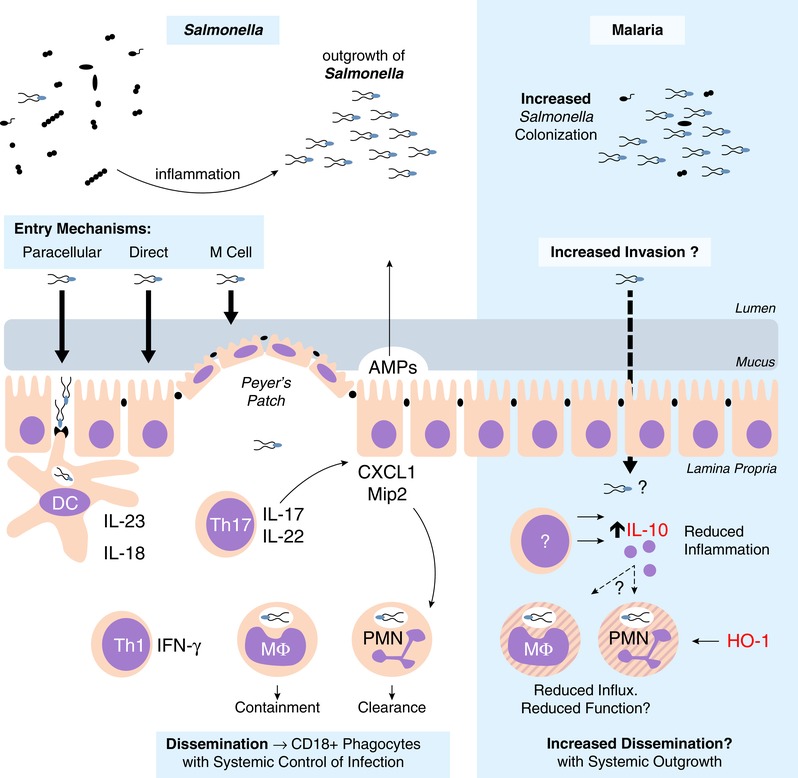
NTS intestinal immune response. NTS is a fecal‐oral pathogen, which thrives in the inflamed intestine. Tissue invasion in the distal intestine can be via direct invasion, uptake by M cells, or through paracellular spaces. Uptake by dendritic cells can initiate inflammation through the IL‐23/IL‐17 axis. Th17 cells promote neutrophil influx via the induction of neutrophil chemokines. NTS is able to persist within the Salmonella‐containing vacuole of macrophages, whereas neutrophils are efficient at NTS clearance. Systemic dissemination to draining lymph nodes is through CD18+ phagocytes. During experimental malaria, NTS colonization resistance is lowered, although it is unclear if there is an increase in tissue invasion. Regardless, inflammation (with reduced PMN influx) is reduced due to increased IL‐10 concentrations. However, the role of IL‐10, and potentially HO‐1, on intestinal neutrophil function and role for increased systemic dissemination are unclear
Although phagocytes, such as neutrophils and macrophages, are efficient in their uptake of NTS, the bacteria can disable the antibacterial machinery of macrophages to create Salmonella containing vacuoles (SCVs) within which they can persist and replicate.38 Proteins encoded within Salmonella pathogenicity island‐2 block lysosomal fusion allowing evasion of ROS‐mediated killing.39 Ultimately, clearance of bacteria from phagocytes is mediated by IFN‐γ, which induces breakdown of the SCV,40 releasing bacteria into the cytosol. Bacterial products now present in the cytosol can induce pyroptosis, a form of cell death involving both canonical and noncanonical inflammasome signaling with caspase‐1 and NLRP3 or caspase‐11 (in mice) and caspase‐4 and caspase‐5 (in humans), which lead to activation of IL‐1 and IL‐18.41, 42, 43 Bacteria released from disintegrating macrophages are cleared by neutrophils, a process that has been termed “phagocyte roulette.”44
Neutrophils, also called polymorphonuclear cells (PMNs), are terminally differentiated leukocytes with distinctive lobulated nuclei and contain antimicrobial cytoplasmic granules. PMNs are the most abundant white blood cell, with 1 × 1011 new cells emerging from the bone marrow daily. They are typically thought to have a very short lifespan in blood (∼7–24 hours45), although infection may delay apoptosis and increase lifespan.46, 47, 48 During infection and inflammation, PMNs are quickly mobilized to sites of injury. In the blood vessel, activated PMNs adhere to endothelium, extravasate and migrate along chemokine gradients to infectious foci.
PMNs are professional phagocytes, which use receptor‐mediated phagocytosis to internalize pathogens and debris into phagolysosomes.49 Intracytoplasmic granules containing cathepsins, elastases, and myeloperoxidases fuse with the phagolysosome to digest internalized pathogens, in a process known as degranulation; leakage of granules or their contents into the extracellular milieu can be a significant cause of tissue damage during infection.49, 50 Release of reactive oxygen species (ROS), produced via an NADPH oxidase‐dependent process, into the phagolysosome is an additional, very important, bactericidal mechanism. PMNs can also kill extracellular pathogens by degranulation, secretion of ROS, or the release of neutrophil extracellular traps (NETs). NETs consist of externalized decondensed chromatin decorated with granular proteins and histones to prevent the dissemination of pathogens.51, 52 Serine proteases and histones provide antimicrobial activity against trapped pathogens. NETs also permit subsequent phagocytosis by proximate phagocytes (Fig. 2).
Figure 2.
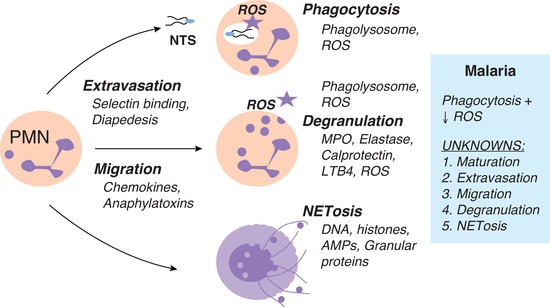
Neutrophil function. PMN extravasation is initiated by inflammatory mediators, which stimulate the upregulation of adhesion molecules, such as selectins, on endothelial cells allowing tethering and rolling of PMN. Chemokine gradients cause PMNs to “crawl” toward the site of infection until an endothelial junction is reached where diapedesis can occur. At the site of infection, neutrophils have numerous functions including phagocytosis, degranulation, and NETosis. Phagocytosis involves the ingestion of a pathogen into a phagolysosome containing granule proteins and ROS, to disable and digest internalized pathogens. Degranulation can also occur extracellularly where granular proteins, such as MPO and elastase, and ROS can be released from PMNs. A novel function of PMNs is NETosis, which is the release of NETs. These decondensed chromatin fibers decorated with histones and granular proteins trap and kill pathogens to prevent further dissemination. It is known that during malaria infection, neutrophils maintain the ability to phagocytose NTS; however, PMNs display impaired ROS production. Mechanisms still to be investigated include: maturation, extravasation, migration, degranulation, and NETosis
Although macrophages can be permissive to NTS growth, inflammatory monocytes and neutrophils are efficient in killing Salmonella through oxidative stress.53 In the intestine, both mucosa‐associated and luminal neutrophils engulf Salmonella.54 However, infiltrating PMNs also promote intestinal inflammation,55 thereby increasing the risk of bacterial invasion, and produce ROS, which can transform carbon sources such as thiosulfate in the intestinal lumen into tetrathionate and the microbial fermentation product 1,2‐propanediol, allowing NTS to outgrow the competing microbiota.32, 33 PMNs also release calprotectin into the intestinal milieu where it sequesters zinc, further restricting the growth of the intestinal microbiota.56 Therefore, although PMNs can limit bacterial growth and prevent overwhelming infection, NTS can exploit neutrophil‐mediated inflammation in the intestine to ensure its survival and eventual transmission.53, 57
3. PATHOPHYSIOLOGY OF NTS–MALARIA COINFECTIONS
Clinically, NTS infections in patients in Africa are not associated with overt diarrheal disease58 suggesting that underlying coinfection may ameliorate the intestinal inflammation typically associated with NTS. Evidence from coinfection models supports this idea (Table 1): intestinal inflammation is markedly reduced in both mice and macaques coinfected with malaria and NTS compared to animals infected with NTS alone and this is associated with reduced neutrophil influx and lower levels of IFN‐γ and IL‐1759 (Fig. 1). In mice, this reduction in intestinal inflammation is mediated by IL‐10,59 a potent anti‐inflammatory cytokine, which is essential for minimizing tissue damage during malaria infections (discussed in detail below). Although reducing intestinal inflammation might be expected to reduce the likelihood of invasion of NTS into the lamina propria and subsequent systemic dissemination, malaria infection has other, less beneficial consequences for the gut. In humans, acute malaria infection, in the apparent absence of gastrointestinal pathogens, is commonly associated with mild to moderate diarrhea, perhaps indicative of dysbiosis and or increased intestinal permeability.60, 61 In mice, malaria infection causes a dysbiosis, changing the composition of the microbiota and providing a foothold for colonization of the intestinal epithelium by NTS and E. coli.62 Increased colonization, taken together with increased gut permeability and reduced availability of neutrophils to control the bacterial infection, may explain why, 48 h after challenge with NTS, bacterial loads in the draining mesenteric lymph nodes are 100‐fold higher in malaria‐coinfected mice than in mice without malaria.59
Table 1.
Animal models of malaria and salmonella coinfection
| Reference | Animal (strain) | Plasmodium spp. | NTS strain (serovar) | Route of NTS challenge | NTS challenge after Plasmodium | Endpoint | NTS‐related outcome |
|---|---|---|---|---|---|---|---|
| Roux et al.77 | Mus (CBA/J) | P. yoelii nigeriensis | IR715 (ATCC 14028) | Intragastric | Day 0 | Day 5 | Increased CFU in Spleen, liver, and Peyer's patch. |
| Cunnington et al.63 | Mus (C57BL/6) | P. yoelii 17XNL | 12023‐GFP | Intraperitoneal | Day 15 | 18 h | Increased CFU in blood, spleen, and liver. |
| Chau et al.124 | Mus (CBA/J) | P. yoelii 17XNL | IR715 (ATCC 14028) | Intragastric | Day 10 | Day 14 | l‐Arginine & l‐Citrulline supplementation during P.y. reduces NTS burden in mesLN |
| Lokken et al.64 | Mus (CBA/J) | P. yoelii nigeriensis | IR715 (ATCC 14028) | Intragastric | Day 10 | Day 12, 14 | Increased CFU in liver (day 12/14), blood (day 14) |
| Mus (CBA/J) | Intraperitoneal | Day 10 | Day 12, 13 | Increased CFU in liver (day 12/13), blood (day 13) | |||
| Mus (C57BL/6J) | Intragastric | Day 10 | Day 12 | No increase in CFU, reduced liver PMN chemokines | |||
| Mus (C57BL/6J) | Intraperitoneal | Day 10 | Day 12 | No increase in CFU | |||
| Mus (C57BL/6J il10–/–) | Intragastric | Day 10 | Day 12 | Reduces CFU in single & co‐infected, restored liver PMN chemokines | |||
| Mus (C57BL/6J il10Rflx:LysMcre) | Intraperitoneal | Day 10 | Day 12 | IL‐10R–/– myeloid cells; loss in increased liver, blood CFU | |||
| Mus (C57BL/6J il10flx:LysMcre) | Intraperitoneal | Day 10 | Day 12 | IL‐10–/– myeloid cells; loss in increased blood CFU | |||
| Mooney et al.59 | Macaca mulatta | P. fragile | IR715 (ATCC 14028) | Ligated ilieal loops | Day 14, 15 | 8 h | Reduced intestinal inflammation to NTS |
| Mus (CBA/J) | P. yoelii nigeriensis | Intragastric | Day 10 | Day 12 | Reduced intestinal inflammation to NTS, increased mesLN CFU | ||
| Mus (C57BL/6J) | Day 10 | Day 12 | Reduced intestinal inflammation & PMN influx | ||||
| Mus (C57BL/6J il10–/–) | Day 10 | Day 12 | Restored intestinal inflammation & PMN influx in IL‐10–/– | ||||
| Mooney et al.125 | Mus (C57BL/6) | P. yoelii 17XNL | BRD509 | Intravenous | Day 14, 28 | Day 17, 31 | Increased CFU in liver |
| Mus (C57BL/6J‐Slc11a1+/+) | Intravenous | Day 14 | Day 17 | No increase in CFU | |||
| Mus (CBA/J) | Intragastric | Day 14 | Day 18 | No increase in CFU | |||
| Mooney et al.62 | Mus (C57BL/6J) | P. yoelii nigeriensis | IR715 (ATCC 14028) | Intragastric | Day 10 | Day 11 | Increased NTS colonization in feces |
| Mus (C57BL/6J) ‐ Germ Free | Day 10 | Day 11 | Increased NTS colonization in feces with fecal donation from P. yoelii‐infected mice into germ‐free mice | ||||
| Lokken et al.79 | Mus (CBA/J) | P. yoelii nigeriensis | IR715 (ATCC 14028) | Intragastric | Day 10 | Day 12, 14 | Increased CFU in liver (day 14 only) |
| Mus (C57BL/6J il10–/–) | Intraperitoneal | Day 10 | Day 12 | Reduced CFU in liver in IL‐10–/– |
Whether increased NTS colonization or lymph node dissemination directly increases the risk of systemic spread is not yet clear but major defects in control of systemic NTS are evident in malaria coinfection: bacterial loads in blood, liver, spleen, and bone marrow after intraperitoneal injection of Salmonella Typhimurium (bypassing any intestinal contribution) were 1000–10,000‐fold higher in Plasmodium yoelii‐infected mice than in malaria‐uninfected mice.63, 64
4. HEMOLYSIS, HEME OXYGENASE‐1, AND NEUTROPHIL FUNCTION DURING MALARIA–NTS COINFECTION
Anemia is a heterogeneous condition defined as a reduction in circulating hemoglobin, the iron containing metalloprotein abundant in RBC, which ferry oxygen through the blood. One important cause of anemia is direct lysis of RBC (hemolysis) due to autoimmune conditions or infection. Some pathogens secrete hemolysins (e.g., Staphylococcus spp., Streptococcus spp., and Clostridium spp.) while other pathogens replicate inside, and ultimately lyse, RBC (e.g., Babesia spp., and Bartonella spp., and Plasmodium spp.). In malaria infection, direct destruction of RBC by parasitization is compounded by eryptosis of large numbers of uninfected RBC by processes that are still incompletely understood;65 however, complement‐mediated opsonization of bystander RBC has been noted.66, 67 Regardless of the mechanism, hemolysis results in the release of hemoglobin, or its breakdown product heme, into the plasma (Fig. 3). Free heme is prooxidant and highly cytotoxic, contributing to endothelial injury and hypertension.68, 69 Extracellular hemoglobin is scavenged by haptoglobin, which then binds to CD163 on the surface of macrophages and is internalized and degraded. Similarly, free heme is sequestered by hemopexin and internalized by binding to CD91. Intracellular heme is then degraded into equimolar amounts of iron, carbon monoxide, and biliverdin through the action of heme oxygenase (HO). Under homeostatic conditions, this process is mediated by the constitutively expressed HO isoform, HO‐2. However, under conditions of hypoxia, oxidative stress, or infection (e.g., presence of LPS), the inducible isoform HO‐1 (encoded by heme oxygenase‐1 gene [hmox1]) is upregulated to prevent heme‐induced pathology and ensure efficient iron recycling.70, 71 Importantly, plasma heme and HO‐1 concentrations are raised during both acute72 and subclinical73 P. falciparum malaria infections in humans, and during acute P. yoelii infection in mice,63 competitive inhibition of HO‐1 enzyme activity restores the ability of P. yoelii‐infected mice to control systemic NTS infections.63 Moreover, hmox1 induction increases NTS growth in murine macrophages,74 indicating that hemolysis (via heme and HO‐1) rather than malaria, per se, may be the true risk factor for invasive NTS disease in malaria patients. In support of this notion, individuals with sickle cell anemia, which gives rise to periodic hemolytic crises, are also highly susceptible to invasive NTS disease75, 76; similarly, induction of acute hemolysis by antibody‐mediated RBC lysis77 or phenylhydrazine treatment63 also renders mice highly susceptible to NTS.
Figure 3.
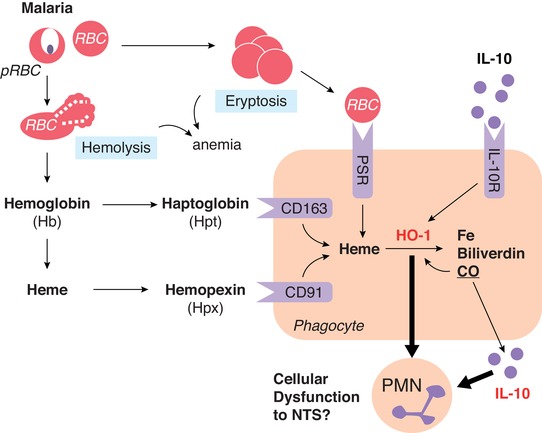
Anemia, HO‐1, and IL‐10. During malaria infection, anemia can occur through direct lysis of parasitized RBCs (hemolysis) or through eryptosis of uninfected RBCs—which are taken up by phagocytes through the phosphatidylserine receptor (PSR). Hemolysis results in release of hemoglobin (Hb), which can be further degraded to free heme. Serum scavenger proteins haptoglobin (Hpt) and hemopexin (Hpx) bind Hb and heme before uptake by phagocytes through CD163 and CD91, respectively. Intracellular heme is then degraded to iron, biliverdin, and carbon monoxide via the enzyme HO‐2 or the inducible isoform, HO‐1. HO‐1 can also be induced by IL‐10 receptor signaling. The impact of IL‐10 and HO‐1 during malarial anemia and hemolysis on phagocyte function remains poorly defined
The first indication that hemolysis has a negative impact on neutrophil function, and thus that neutrophil dysfunction might underlie increased risk of invasive bacterial disease in people with malaria or other hemolytic diseases, came from mouse coinfection studies where circulating neutrophils from malaria‐infected and phenylhydrazine‐treated mice were shown to efficiently phagocytose S. Typhimurium but were unable to kill; viable bacteria persisted and, indeed, replicated inside neutrophils, which were severely deficient in ROS production.63 Of note, in vitro, heme pretreatment reduced phagocytosis of E. coli by human and murine neutrophils78 and high circulating heme during P. falciparum infection can reduce in vitro phagocytosis of Salmonella by neutrophils.72 The observation that treatment with a synthetic heme polymer, hemin, induced similar neutrophil defects and that these defects can be reversed by competitive inhibition of HO‐1, then provided a link among malaria, hemolysis, HO‐1, and neutrophil dysfunction.63 ROS‐defective neutrophils have also been observed in children with acute malaria; importantly, these defects persist for up to 8 weeks after treatment,72 perhaps explaining why children with recent (past) malaria infection remain susceptible to invasive NTS.21
Plasmodium infection also reduces neutrophil mobilization into infected tissues including blood63 and, as discussed above, intestine59 and liver64; infiltration of inflammatory monocytes into the liver is also impaired.79 Although this may be due in part to the anti‐inflammatory effects of IL‐10 (as described below), there are also cell‐intrinsic effects within developing neutrophils. Neutrophil precursors in the bone marrow (i.e., granulocyte macrophage progenitor cells) of malaria‐infected mice express HO‐1 and have unusual surface phenotypes (F4/80 and Gr‐1 expression).63 It has been shown that HO‐1 reduces neutrophil influx into the inflamed lung71 suggesting a causal relationship between HO‐1 and reduced neutrophil migration but much more work is needed to fully characterize neutrophil maturation and function during malaria infection and to determine the extent to which the altered phenotype is mediated by the heme/HO‐1 pathway.
5. THE ROLE OF IL‐10 IN MALARIA NTS COINFECTION
IL‐10 is a potent anti‐inflammatory cytokine and an important regulator of inflammation‐induced pathology,80 it is therefore no surprise that systemic IL‐10 concentrations are elevated in highly inflammatory diseases such as sepsis81, 82 and malaria.83, 84 More surprisingly, however, circulating IL‐10 is also elevated during mild/uncomplicated83, 85 and asymptomatic/subclinical malaria infections73, 86; indeed these cases may represent successful balancing of inflammation‐mediated parasite control and effective regulation of inflammation by IL‐10. In malaria, IL‐10 can come from both innate and adaptive immune cells, including Th1‐derived regulatory T cells that coproduce IFN‐γ and IL‐10,87, 88, 89 and plays an essential role in both adaptive humoral immunity (promoting differentiation of T‐bet+ germinal center B cells)90, 91 and limiting tissue damage.92, 93, 94
The anti‐inflammatory properties of IL‐10 include rendering phagocytes refractory to activation and/or directing macrophage polarization to a regulatory “M2" phenotype.95, 96 The impact of IL‐10 on neutrophil function is well described, reducing recruitment and migration in response to anaphylatoxins and ultimately bacterial clearance.97, 98 Further, neutrophils themselves can be a source of IL‐10, induced by regulatory T cells.99 During malaria NTS coinfection in mice, LysM‐expressing cells are a significant source of IL‐10 and its ablation reduces NTS bacteremia.64 IL‐10 suppresses neutrophil function through the activation of STAT3 and suppressor of cytokine signaling 3,100, 101 leading to the down‐regulation of IFN regulatory factor and NF‐κB family transcription factor.102 In addition, HO‐1 can be directly induced by IL‐10103 (Fig. 3). One hypothesis, therefore, is that malaria‐induced neutrophil dysfunction results from hemolysis‐ and IL‐10‐driven HO‐1 induction. In support of this hypothesis, in a recent study of persistent malaria infections, we have found that heme drives inflammation, that parasite density and inflammation then drives IL‐10 production, and that heme and IL‐10 both then induce HO‐1.73
6. COMPLEMENT DEPLETION DURING MALARIA
Complement proteins play an essential role in orchestrating inflammation and pathogen clearance,104, 105 including during Salmonella infection.106, 107 In brief, the classical pathway is activated by antibody (IgM and IgG) and the alternative pathway is activated by spontaneous hydrolysis of C3.108 C3 becomes membrane bound and cleaves C5, creating soluble C5a and membrane‐bound C5b.109 The complement anaphylatoxin C5a is a potent chemoattractant for neutrophils while also inducing expression of cell adhesion molecules on endothelial cells (such as P‐selectin) that permit neutrophil translocation into tissue.110 C5a also enhances neutrophil resistance to apoptosis,111 allowing the cells more time to perform effector functions. After bacterial uptake, maturation of the neutrophil phagosome requires its fusion with antimicrobial granules and the production of reactive superoxide anions (O2 –) following G oxidase assembly on the phagolysosomal membrane.112 Assembly of NADPH oxidase is primed through the interaction of C5a and its receptor (C5aR and CD88).113, 114 Indeed, the E. coli‐induced oxidative burst in phagocytes is almost entirely dependent on CD88 signaling.115 Opsonization and killing of invasive NTS strains by neutrophils isolated from Malawian children require both antibodies and complement.107
Complement activation on the surface of uninfected RBCs presumably increases the rate of turnover of RBCs during malaria infection,116 possibly due to reduced availability of complement regulatory proteins such as CD55.117, 118, 119 Complement‐mediated red cell lysis not only exacerbates the anemia associated with malaria, but will also increase circulating heme and therefore HO‐1 concentrations and reduce the availability of complement components,120 which may impair ability to control invasive bacterial infection (Fig. 4). Hypothetically, excess C5a in plasma may reduce CD88 on PMNs, and thus reduce the oxidative burst. Further, depletion of complement proteins (with concomitant reduction in expression of endothelial selectin) and reduced generation of anaphylatoxins could reduce neutrophil migration into infected tissues. Further studies are required to determine the significance of complement activation and complement consumption during malaria and risk of invasive NTS.
Figure 4.
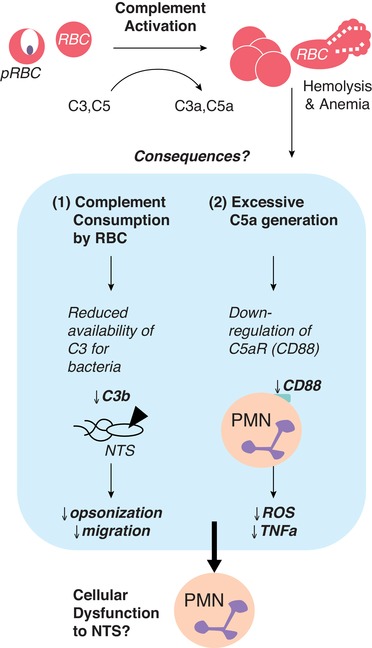
Complement depletion. During malaria infection, systemic complement activation occurs due to deposition on both infected and uninfected RBCs. This results in reduced concentrations of circulating C3 and C5 and generation of anaphylatoxins (C3a and C5a). The consequences that may impact PMN function are 2‐fold; (1) a reduction in C3 available for deposition on extracellular NTS leading to opsonization and migration and/or (2) excessive C5a reducing CD88 (C5aR) on neutrophils leading to reduced ROS and cytokine production. Additional work is needed to clarify the impact of complement activation and subsequent depletion during malaria on risk to NTS bacteremia
7. CONCLUDING REMARKS
We have provided a rational for considering that hemolysis and inflammation, leading to induction of HO‐1 and IL‐10 and activation of complement, during malaria infections might all contribute to the increased susceptibility to bacterial coinfection. It is likely that these pathways synergize to increase risk of invasive NTS: in malaria‐infected mice, both exogenous IL‐10 and anemia were required for increased bacteremia64; HO‐1 is induced both by heme and by IL‐10; and the carbon monoxide generated from heme catabolism can induce both HO‐1 and IL‐10.121, 122 Ultimately, it is important to define how these factors may converge to alter neutrophil biology. Through this, we may begin to understand if targeted treatment, or even prophylactic antimalarial treatment, can improve neutrophil function and so reduce the burden of invasive bacterial disease in malaria endemic populations. Given the importance of neutrophils in clearance of NTS, work is now needed to better describe the impact of malaria on this innate immune cell.
The risk of invasive NTS during malaria in sub‐Saharan Africa is well defined for acute malaria infection. However, the majority of malaria infections in the world are asymptomatic, with chronic, low‐density infections.123 As recent and low‐density malaria infections are a risk factor for NTS bacteremia,21 it is also important to understand if hemolysis, and resulting induction of HO‐1 and IL‐10, seen during these “asymptomatic” infections73 reaches the threshold needed for neutrophil dysfunction. In other words, in addition to defining the pathways leading to neutrophil dysfunction, we also need to identify the point at which the balance tips from these being host protective to increasing the risk to invasive NTS. Importantly, these pathways may contribute to severe bacterial disease even in the absence of malaria infections: in sepsis patients, we have observed that raised concentrations of heme, HO‐1, and IL‐10 are positively correlated with disease severity and mortality.82 Also, while “invasive” NTS is seen in immunocompromised hosts (such as those with malaria infection), it remains unclear if this is due to increased intestinal invasion, increased dissemination from draining lymph nodes, failure to control systemic bacterial replication, or a combination of any of these. Moreover, the extent to which this is primarily a neutrophil defect requires further exploration.
AUTHORSHIP
J.P.M. wrote the first draft of the manuscript. J.P.M., L.G., and E.M.R. contributed to the writing of the manuscript. J.P.M., L.G., and E.M.R. agreed with the manuscript's conclusions. E.M.R. conceived the idea. E.M.R. presented the topic at the Lorne Infection and Immunity 2018 meeting. Review solicited by the Journal of Leukocyte Biology editorial board.
ACKNOWLEDGMENT
J.P.M. and E.M.R. were supported by the United Kingdom Medical Research Council (grant No. MR/P000959/1).
DISCLOSURES
The authors declare no conflicts of interest.
Mooney JP, Galloway LJ, Riley EM. Malaria, anemia, and invasive bacterial disease: A neutrophil problem? J Leukoc Biol. 2019;105:645–655. 10.1002/JLB.3RI1018-400R
REFERENCES
- 1. Shankar‐Hari M, Phillips G, Levy M, et al. Assessment of definition and clinical criteria for septic shock: for the third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA. 2016;315:775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. [DOI] [PubMed] [Google Scholar]
- 3. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. [DOI] [PubMed] [Google Scholar]
- 4. Antonelli M. Sepsis and septic shock: pro‐inflammatory or anti‐inflammatory state. J Chemother. 1999;11:536–540. [DOI] [PubMed] [Google Scholar]
- 5. Tanaka H, Sugimoto H, Yoshioka T, Sugimoto T. Role of granulocyte elastase in tissue injury in patients with septic shock complicated by multiple‐organ failure. Ann Surg. 1991;213:81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Torio CM, Moore BJ. National Inpatient Hospital Costs: the Most Expensive Conditions by Payer, 2013: statistical Brief #204. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs . Rockville, MD: Agency for Healthcare Research and Quality (US); 2016. [PubMed] [Google Scholar]
- 7. Naber CK. Staphylococcus aureus bacteremia: epidemiology, pathophysiology, and management strategies. Clin Infect Dis. 2009;48:S231–S237. [DOI] [PubMed] [Google Scholar]
- 8. Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin‐resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. [DOI] [PubMed] [Google Scholar]
- 9. Pitout JD, Gregson DB, Campbell L, Laupland KB. Molecular characteristics of extended‐spectrum‐beta‐lactamase‐producing Escherichia coli isolates causing bacteremia in the Calgary Health Region from 2000 to 2007: emergence of clone ST131 as a cause of community‐acquired infections. Antimicrob Agents Chemother. 2009;53:2846–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reddy EA, Shaw AV, Crump JA. Community‐acquired bloodstream infections in Africa: a systematic review and meta‐analysis. Lancet Infect Dis. 2010;10:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ao TT, Feasey NA, Gordon MA, Keddy KH, Angulo FJ, Crump JA. Global burden of invasive nontyphoidal salmonella disease. Emerg Infect Dis. 2015;21:941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berkley JA, Lowe BS, Mwangi I, et al. bacteremia among children admitted to a rural hospital in Kenya. N Engl J Med. 2005;352:39–47. [DOI] [PubMed] [Google Scholar]
- 13. Kingsley RA, Msefula CL, Thomson NR, et al. Epidemic multiple drug resistant salmonella Typhimurium causing invasive disease in sub‐Saharan Africa have a distinct genotype. Genome Res. 2009;19:2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. Invasive non‐typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet. 2012;379:2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santos RL, Raffatellu M, Bevins CL, et al. Life in the inflamed intestine, salmonella style. Trends Microbiol. 2009;17:498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Church J, Maitland K. Invasive bacterial co‐infection in African children with Plasmodium falciparum malaria: a systematic review. BMC Med. 2014;12:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takem EN, Roca A, Cunnington A. The association between malaria and non‐typhoid salmonella bacteraemia in children in sub‐Saharan Africa: a literature review. Malar J. 2014;13:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coban C, Lee MSJ, Ishii KJ. Tissue‐specific immunopathology during malaria infection. Nat Rev Immunol. 2018;18:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mabey DC, Brown A, Greenwood BM. Plasmodium falciparum malaria and salmonella infections in Gambian children. J Infect Dis. 1987;155:1319–1321. [DOI] [PubMed] [Google Scholar]
- 20. Mtove G, Amos B, von Seidlein L, et al. Invasive salmonellosis among children admitted to a rural Tanzanian hospital and a comparison with previous studies. PLoS One. 2010;5:e9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Biggs HM, Lester R, Nadjm B, et al. Invasive salmonella infections in areas of high and low malaria transmission intensity in Tanzania. Clin Infect Dis. 2014;58:638–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bronzan RN, Taylor TE, Mwenechanya J, et al. Bacteremia in Malawian children with severe malaria: prevalence, etiology, HIV coinfection, and outcome. J Infect Dis. 2007;195:895–904. [DOI] [PubMed] [Google Scholar]
- 23. Muthumbi E, Morpeth SC, Ooko M, et al. Invasive salmonellosis in Kilifi. Clin Infect Dis. 2015;61:S290–S301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ceesay SJ, Casals‐Pascual C, Erskine J, et al. Changes in malaria indices between 1999 and 2007 in The Gambia: a retrospective analysis. Lancet North Am Ed. 2008;372:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mtove G, Amos B, Nadjm B, et al. Decreasing incidence of severe malaria and community‐acquired bacteraemia among hospitalized children in Muheza, north‐eastern Tanzania, 2006–2010. Malar J. 2011;10:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steve Yan S, Pendrak ML, Abela‐Ridder B, Punderson JW, Fedorko DP, Foley SL. An overview of salmonella typing: public health perspectives. Clin Appl Immunol Rev. 2004;4:189–204. [Google Scholar]
- 27. Brenner FW, Villar RG, Angulo FJ, Tauxe R, Swaminathan B. Salmonella nomenclature. J Clin Microbiol. 2000;38:2465–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dougan G, John V, Palmer S, Mastroeni P. Immunity to salmonellosis. Immunol Rev. 2011;240:196–210. [DOI] [PubMed] [Google Scholar]
- 29. Griffin AJ, McSorley SJ. Development of protective immunity to salmonella, a mucosal pathogen with a systemic agenda. Mucosal Immunol. 2011;4:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez‐Escobedo G, Marshall JM, Gunn JS. Chronic and acute infection of the gall bladder by salmonella Typhi: understanding the carrier state. Nat Rev Microbiol. 2011;9:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Velge P, Wiedemann A, Rosselin M, et al. Multiplicity of salmonella entry mechanisms, a new paradigm for salmonella pathogenesis. Microbiologyopen. 2012;1:243–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winter SE, Thiennimitr P, Winter MG, et al. Gut inflammation provides a respiratory electron acceptor for salmonella. Nature. 2010;467:426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Faber F, Thiennimitr P, Spiga L, et al. Respiration of microbiota‐derived 1,2‐propanediol drives salmonella expansion during colitis. PLoS Pathog. 2017;13:e1006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Godinez I, Raffatellu M, Chu H, et al. Interleukin‐23 orchestrates mucosal responses to salmonella enterica serotype Typhimurium in the intestine. Infect Immun. 2009;77:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Godinez I, Haneda T, Raffatellu M, et al. T cells help to amplify inflammatory responses induced by salmonella enterica serotype Typhimurium in the intestinal mucosa. Infect Immun. 2008;76:2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Srinivasan A, Salazar‐Gonzalez RM, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. Innate immune activation of CD4 T cells in salmonella‐infected mice is dependent on IL‐18. J Immunol. 2007;178:6342–6349. [DOI] [PubMed] [Google Scholar]
- 37. Cheminay C, Chakravortty D, Hensel M. Role of neutrophils in murine salmonellosis. Infect Immun. 2004;72:468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monack DM, Bouley DM, Falkow S. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1 (+) (/) (+) mice and can be reactivated by IFNγ neutralization. J Exp Med. 2004;199:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van der Heijden J, Bosman ES, Reynolds LA, Finlay BB. Direct measurement of oxidative and nitrosative stress dynamics in salmonella inside macrophages. PNAS. 2015;112:560–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shenoy AR, Wellington DA, Kumar P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336:481–485. [DOI] [PubMed] [Google Scholar]
- 41. Sander LE, Davis MJ, Boekschoten MV, et al. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Broz P, Ruby T, Belhocine K, et al. Caspase‐11 increases susceptibility to salmonella infection in the absence of caspase‐1. Nature. 2012;490:288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kayagaki N, Warming S, Lamkanfi M, et al. Non‐canonical inflammasome activation targets caspase‐11. Nature. 2011;479:117. [DOI] [PubMed] [Google Scholar]
- 44. Fenlon LA, Slauch JM. Phagocyte roulette in salmonella killing. Cell Host Microbe. 2014;15:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L. What's your age again? Determination of human neutrophil half‐lives revisited. J Leukoc Biol. 2013;94:595–601. [DOI] [PubMed] [Google Scholar]
- 46. Schwartz JT, Barker JH, Kaufman J, Fayram DC, McCracken JM, Allen LA. Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J Immunol. 2012;188:3351–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sarkar A, Aga E, Bussmeyer U, et al. Infection of neutrophil granulocytes with Leishmania major activates ERK 1/2 and modulates multiple apoptotic pathways to inhibit apoptosis. Med Microbiol Immunol. 2013;202:25–35. [DOI] [PubMed] [Google Scholar]
- 48. Kobayashi SD, Malachowa N, DeLeo FR. Influence of microbes on neutrophil life and death. Front Cell Infect Microbiol. 2017;7:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kruger P, Saffarzadeh M, Weber AN, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015;11:e1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kolaczkowska E, Jenne CN, Surewaard BGJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015;6:6673–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 52. Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell. 2018;44:542–553. [DOI] [PubMed] [Google Scholar]
- 53. Burton NeilA, Schürmann N, Casse O, et al. Disparate impact of oxidative host defenses determines the fate of salmonella during systemic infection in mice. Cell Host Microbe. 2014;15:72–83. [DOI] [PubMed] [Google Scholar]
- 54. Loetscher Y, Wieser A, Lengefeld J, et al. Salmonella transiently reside in luminal neutrophils in the inflamed gut. PLoS One. 2012;7:e34812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spees AM, Kingsbury DD, Wangdi T, Xavier MN, Tsolis RM, Bäumler AJ. Neutrophils are a source of IFN‐γ during acute salmonella colitis. Infect Immun. 2014;82:1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu JanetZ, Jellbauer S, Poe AJ, et al. Zinc sequestration by the neutrophil protein calprotectin enhances salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Behnsen J, Perez‐Lopez A, Nuccio S‐P, Raffatellu M. Exploiting host immunity: the salmonella paradigm. Trends Immunol. 2015;36:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. MacLennan CA, Levine MM. Invasive nontyphoidal salmonella disease in Africa: current status. Expert Rev Anti Infect Ther. 2013;11:443–446. [DOI] [PubMed] [Google Scholar]
- 59. Mooney JP, Butler BP, Lokken KL, et al. The mucosal inflammatory response to non‐typhoidal salmonella in the intestine is blunted by IL‐10 during concurrent malaria parasite infection. Mucosal Immunol. 2014;7:1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sowunmi A, Ogundahunsi OAT, Falade CO, Gbotosho GO, Oduola AMJ. Gastrointestinal manifestations of acute falciparum malaria in children. Acta Trop. 2000;74:73–76. [DOI] [PubMed] [Google Scholar]
- 61. Weinberg W, Wilairatana P, Meddings JB, Ho M, Vannaphan S, Looareesuwan S. Increased gastrointestinal permeability in patients with Plasmodium falciparum malaria. Clin Infect Dis. 1997;24:430–435. [DOI] [PubMed] [Google Scholar]
- 62. Mooney JP, Lokken KL, Byndloss MX, et al. Inflammation‐associated alterations to the intestinal microbiota reduce colonization resistance against non‐typhoidal salmonella during concurrent malaria parasite infection. Sci Rep. 2015;5:14603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cunnington AJ, de Souza JB, Walther M, Riley EM. Malaria impairs resistance to salmonella through heme‐ and heme oxygenase‐dependent dysfunctional granulocyte mobilization. Nat Med. 2012;18:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lokken KL, Mooney JP, Butler BP, et al. Malaria parasite infection compromises control of concurrent systemic non‐typhoidal salmonella infection via IL‐10‐mediated alteration of myeloid cell function. PLoS Pathog. 2014;10:e1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Price RN, Simpson JA, Nosten F, et al. Factors contributing to anemia after uncomplicated falciparum malaria. Am J Trop Med Hyg. 2001;65:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pawluczkowycz AW, Lindorfer MA, Waitumbi JN, Taylor RP. Hematin promotes complement alternative pathway‐mediated deposition of C3 activation fragments on human erythrocytes: potential implications for the pathogenesis of anemia in malaria. J Immunol. 2007;179:5543–5552. [DOI] [PubMed] [Google Scholar]
- 67. Dasari P, Fries A, Heber SD, et al. Malarial anemia: digestive vacuole of Plasmodium falciparum mediates complement deposition on bystander cells to provoke hemophagocytosis. Med Microbiol Immunol. 2014;203:383–393. [DOI] [PubMed] [Google Scholar]
- 68. Balla J, Vercellotti GM, Jeney V, et al. Heme, heme oxygenase and ferritin in vascular endothelial cell injury. Mol Nutr Food Res. 2005;49:1030–1043. [DOI] [PubMed] [Google Scholar]
- 69. Jeney V, Balla J, Yachie A, et al. Pro‐oxidant and cytotoxic effects of circulating heme. Blood. 2002;100:879–887. [DOI] [PubMed] [Google Scholar]
- 70. Ryter SW, Alam J, Choi AMK. Heme oxygenase‐1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. [DOI] [PubMed] [Google Scholar]
- 71. Konrad FM, Knausberg U, Höne R, Ngamsri KC, Reutershan J. Tissue heme oxygenase‐1 exerts anti‐inflammatory effects on LPS‐induced pulmonary inflammation. Mucosal Immunol. 2015;9:98–111. [DOI] [PubMed] [Google Scholar]
- 72. Cunnington AJ, Njie M, Correa S, Takem EN, Riley EM, Walther M. Prolonged neutrophil dysfunction after Plasmodium falciparum malaria is related to hemolysis and heme oxygenase‐1 induction. J Immunol. 2012;189:5336–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mooney JP, Barry A, Gonçalves BP, et al. Haemolysis and haem oxygenase‐1 induction during persistent “asymptomatic” malaria infection in Burkinabé children. Malar J. 2018;17:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mitterstiller A‐M, Haschka D, Dichtl S, et al. Heme oxygenase 1 controls early innate immune response of macrophages to salmonella Typhimurium infection. Cell Microbiol. 2016;18:1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Soothill G, Darboe S, Bah G, Bolarinde L, Cunnington A, Anderson ST. Invasive bacterial infections in Gambians with sickle cell anemia in an era of widespread pneumococcal and hemophilus influenzae type b vaccination. Medicine. 2016;95:e5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Evans C, Orf K, Horvath E, et al. Impairment of neutrophil oxidative burst in children with sickle cell disease is associated with heme oxygenase‐1. Haematologica. 2015;100:1508–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Roux CM, Butler BP, Chau JY, et al. Both hemolytic anemia and malaria parasite‐specific factors increase susceptibility to nontyphoidal salmonella enterica serovar typhimurium infection in mice. Infect Immun. 2010;78:1520–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Martins R, Maier J, Gorki A‐D, et al. Heme drives hemolysis‐induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol. 2016;17:1361–1372. [DOI] [PubMed] [Google Scholar]
- 79. Lokken KL, Stull‐Lane AR, Poels K, Tsolis RM. Malaria parasite‐mediated alteration of macrophage function and increased iron availability predispose to disseminated non‐typhoidal salmonella infection. Infect Immun. 2018. 10.1128/IAI.00301-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Couper KN, Blount DG, Riley EM. IL‐10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. [DOI] [PubMed] [Google Scholar]
- 81. Kasten KR, Muenzer JT, Caldwell CC. Neutrophils are significant producers of IL‐10 during sepsis. Biochem Biophys Res Commun. 2010;393:28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ekregbesi P, Shankar‐Hari M, Bottomley C, Riley EM, Mooney JP. Relationship between anaemia, haemolysis, inflammation and haem oxygenase‐1 at admission with sepsis: a pilot study. Sci Rep. 2018;8:11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Peyron F, Burdin N, Ringwald P, Vuillez JP, Rousset F, Banchereau J. High levels of circulating IL‐10 in human malaria. Clin Exp Immunol. 1994;95:300–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wenisch C, Parschalk B, Narzt E, Looareesuwan S, Graninger W. Elevated serum levels of IL‐10 and IFN‐gamma in patients with acute Plasmodium falciparum malaria. Clin Immunol Immunopathol. 1995;74:115–117. [DOI] [PubMed] [Google Scholar]
- 85. Kurtzhals JAL, Adabayeri V, Goka BQ, et al. Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. Lancet North Am Ed. 1998;351:1768–1772. [DOI] [PubMed] [Google Scholar]
- 86. Guiyedi V, Bécavin C, Herbert F, et al. Asymptomatic Plasmodium falciparum infection in children is associated with increased auto‐antibody production, high IL‐10 plasma levels and antibodies to merozoite surface protein 3. Malar J. 2015;14:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Walther M, Tongren JE, Andrews L, et al. Upregulation of TGF‐beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity. 2005;23:287–296. [DOI] [PubMed] [Google Scholar]
- 88. Freitas d, Rosario AP, Lamb T, et al. IL‐27 promotes IL‐10 production by effector Th1 CD4+ T cells: a critical mechanism for protection from severe immunopathology during malaria infection. J Immunol. 2012;188:1178–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jagannathan P, Eccles‐James I, Bowen K, et al. IFNγ/IL‐10 co‐producing cells dominate the CD4 response to malaria in highly exposed children. PLoS Pathog. 2014;10:e1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Guthmiller JJ, Graham AC, Zander RA, Pope RL, Butler NS. Cutting edge: iL‐10 is essential for the generation of germinal center B Cell Responses and anti‐plasmodium humoral immunity. J Immunol. 2017;198:617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zander RA, Guthmiller JJ, Graham AC, et al. Type I interferons induce T regulatory 1 responses and restrict humoral immunity during experimental malaria. PLoS Pathog. 2016;12:e1005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li C, Sanni LA, Omer F, Riley E, Langhorne J. Pathology of Plasmodium chabaudi chabaudi infection and mortality in interleukin‐10‐deficient mice are ameliorated by anti‐tumor necrosis factor alpha and exacerbated by anti‐transforming growth factor beta antibodies. Infect Immun. 2003;71:4850–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Couper KN, Blount DG, Wilson MS, et al. IL‐10 from CD4+CD25−Foxp3−CD127− adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog. 2008;4:e1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Claser C, De Souza JB, Thorburn SG, et al. Host resistance to plasmodium‐induced acute immune pathology is regulated by interleukin‐10 receptor signaling. Infect Immun. 2017;85:e00941–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL‐10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 96. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. [DOI] [PubMed] [Google Scholar]
- 97. Kulkarni U, Karsten CM, Kohler T, et al. IL‐10 mediates plasmacytosis‐associated immunodeficiency by inhibiting complement‐mediated neutrophil migration. J Allergy Clin Immunol. 2016;137:1487–1497. [DOI] [PubMed] [Google Scholar]
- 98. Bazzoni F, Tamassia N, Rossato M, Cassatella MA. Understanding the molecular mechanisms of the multifaceted IL‐10‐mediated anti‐inflammatory response: lessons from neutrophils. Eur J Immunol. 2010;40:2360–2368. [DOI] [PubMed] [Google Scholar]
- 99. Lewkowicz N, Mycko MP, Przygodzka P, et al. Induction of human IL‐10‐producing neutrophils by LPS‐stimulated Treg cells and IL‐10. Mucosal Immunol. 2015;9:364. [DOI] [PubMed] [Google Scholar]
- 100. Cassatella MA, Gasperini S, Bovolenta C, et al. Interleukin‐10 (IL‐10) selectively enhances CIS3/SOCS3 mRNA expression in human neutrophils: evidence for an IL‐10‐induced pathway that is independent of STAT protein activation. Blood. 1999;94:2880–2889. [PubMed] [Google Scholar]
- 101. Crepaldi L, Gasperini S, Lapinet JA, et al. Up‐regulation of IL‐10R1 expression is required to render human neutrophils fully responsive to IL‐10. J Immunol. 2001;167:2312–2322. [DOI] [PubMed] [Google Scholar]
- 102. Hutchins AP, Takahashi Y, Miranda‐Saavedra D. Genomic analysis of LPS‐stimulated myeloid cells identifies a common pro‐inflammatory response but divergent IL‐10 anti‐inflammatory responses. Sci Rep. 2015;5:9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee TS, Tsai HL, Chau LY. Induction of heme oxygenase‐1 expression in murine macrophages is essential for the anti‐inflammatory effect of low dose 15‐deoxy‐Delta(12,14)‐prostaglandin J(2). J Biol Chem. 2003;278:19325–19330. [DOI] [PubMed] [Google Scholar]
- 104. Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Paolo D, C N, Baldwin LK, et al. IL‐1α and complement cooperate in triggering local neutrophilic inflammation in response to adenovirus and eliminating virus‐containing cells. PLoS Pathog. 2014;10:e1004035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Warren J, Mastroeni P, Dougan G, et al. Increased susceptibility of C1q‐deficient mice to salmonella enterica serovar Typhimurium infection. Infect Immun. 2002;70:551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gondwe EN, Molyneux ME, Goodall M, et al. Importance of antibody and complement for oxidative burst and killing of invasive nontyphoidal salmonella by blood cells in Africans. Proc Natl Acad Sci. 2010;107:3070–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol. 2014;32:433–459. [DOI] [PubMed] [Google Scholar]
- 110. Foreman KE, Vaporciyan AA, Bonish BK, et al. C5a‐induced expression of P‐selectin in endothelial cells. J Clin Invest. 1994;94:1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3‐kinase‐signaling pathway. Kidney Int. 2002;61:456–463. [DOI] [PubMed] [Google Scholar]
- 112. DeLeo FR, Allen LA, Apicella M, Nauseef WM. NADPH oxidase activation and assembly during phagocytosis. J Immunol. 1999;163:6732–6740. [PubMed] [Google Scholar]
- 113. Jiang H, Kuang Y, Wu Y, Smrcka A, Simon MI, Wu D. Pertussis toxin‐sensitive activation of phospholipase C by the C5a and fMet‐Leu‐Phe receptors. J Biol Chem. 1996;271:13430–13434. [DOI] [PubMed] [Google Scholar]
- 114. Hirsch E, Katanaev VL, Garlanda C, et al. Central Role for G protein‐coupled phosphoinositide 3‐kinase γ in inflammation. Science. 2000;287:1049–1053. [DOI] [PubMed] [Google Scholar]
- 115. Mollnes TE, Brekke O‐L, Fung M, et al. Essential role of the C5a receptor in E coli–induced oxidative burst and phagocytosis revealed by a novel lepirudin‐based human whole blood model of inflammation. Blood. 2002;100:1869–1877. [PubMed] [Google Scholar]
- 116. Facer CA, Bray RS, Brown J. Direct Coombs antiglobulin reactions in Gambian children with Plasmodium falciparum malaria. I. Incidence and class specificity. Clin Exp Immunol. 1979;35:119–127. [PMC free article] [PubMed] [Google Scholar]
- 117. Stoute JA, Odindo AO, Owuor BO, Mibei EK, Opollo MO, Waitumbi JN. Loss of red blood cell‐complement regulatory proteins and increased levels of circulating immune complexes are associated with severe malarial anemia. J Infect Dis. 2003;187:522–525. [DOI] [PubMed] [Google Scholar]
- 118. Mahajan RC, Narain K, Mahanta J. Anaemia & expression levels of CD35, CD55 & CD59 on red blood cells in Plasmodium falciparum malaria patients from India. Indian J Med Res. 2011;133:662–664. [PMC free article] [PubMed] [Google Scholar]
- 119. Waitumbi JN, Opollo MO, Muga RO, Misore AO, Stoute JA. Red cell surface changes and erythrophagocytosis in children with severe Plasmodium falciparum anemia. Blood. 2000;95:1481–1486. [PubMed] [Google Scholar]
- 120. Nyakoe NK, Taylor RP, Makumi JN, Waitumbi JN. Complement consumption in children with Plasmodium falciparum malaria. Malar J. 2009;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lin C‐C, Yang C‐C, Hsiao L‐D, Chen S‐Y, Yang C‐M. Heme oxygenase‐1 induction by carbon monoxide releasing molecule‐3 suppresses interleukin‐1β‐mediated neuroinflammation. Front Mol Neurosci. 2017;10:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Otterbein LE, Bach FH, Alam J, et al. Carbon monoxide has anti‐inflammatory effects involving the mitogen‐activated protein kinase pathway. Nat Med. 2000;6:422–428. [DOI] [PubMed] [Google Scholar]
- 123. Lindblade KA, Steinhardt L, Samuels A, Kachur SP, Slutsker L. The silent threat: asymptomatic parasitemia and malaria transmission. Expert Rev Anti Infect Ther. 2013;11:623–639. [DOI] [PubMed] [Google Scholar]
- 124. Chau JY, Tiffany CM, Nimishakavi S, et al. Malaria‐associated L‐arginine deficiency induces mast cell‐associated disruption to intestinal barrier defenses against nontyphoidal salmonella bacteremia. Infect Immun. 2013;81:3515–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mooney JP, Lee S‐J, Lokken KL, et al. Transient loss of protection afforded by a live attenuated non‐typhoidal salmonella vaccine in mice co‐infected with malaria. PLoS Negl Trop Dis. 2015;9:e0004027. [DOI] [PMC free article] [PubMed] [Google Scholar]