

Abstract
Hyoscyamine 6β-hydroxylase (H6H) is an αKG-dependent nonheme iron oxidase that catalyzes the oxidation of hyoscyamine to scopolamine via two separate reactions: hydroxylation followed by oxidative cyclization. Both of these reactions are expected to involve H atom abstraction from each of two adjacent carbon centers (C6 vs C7) in the substrate. During hydroxylation, there is a roughly 85:1 preference for H atom abstraction from C6 versus C7; however, this inverts to a 1:16 preference during cyclization. Furthermore, 18O incorporation experiments in the presence of deuterated substrate are consistent with the catalytic iron(IV)-oxo complex being able to support the coordination of an additional ligand during hydroxylation. These observations suggest that subtle differences in the substrate binding configuration can have significant consequences for the catalytic cycle of H6H.
Graphical Abstract
INTRODUCTION
Nonheme iron oxidases represent an important class of enzymes that catalyze a wide range of oxidative transformations.1–4 The prototypical catalytic cycle of these enzymes involves the use of molecular oxygen to oxidize α-ketoglutarate (αKG) coordinated to an active site iron(II) center (4) to generate succinate, CO2, and an activated iron(IV)-oxo complex (see 5 and 8 in Figure 1).1,5 The iron(IV)-oxo complex then participates in the oxidation of the nominal substrate with the possible involvement of organic radicals confined to the active site. Despite the reactivity of iron(IV)-oxo complexes and radical intermediates, nonheme iron oxidases are able to exert considerable control over the course of the reaction such that they proceed in a regio- and stereoselective manner. Therefore, the mechanistic study of these enzymes can offer significant insights into how enzymes constrain the reactions they catalyze.
Figure 1.

Mechanistic outline of the hydroxylation and cyclization reactions catalyzed by H6H.
Hyoscyamine 6β-hydroxylase (H6H) is one such enzyme of particular interest. H6H is an αKG-dependent nonheme iron oxidase from plants that catalyze two separate and sequential oxidations during biosynthesis of the antimuscarinic natural product scopolamine (3).6–13 The first of these reactions is the C6-hydroxylation of hyoscyamine (1) to generate 6β-hydroxyhyoscyamine (1 → 2, see Figure 1). This is followed by a second reaction where H6H catalyzes the dehydrogenation of 2 to yield the epoxide scopolamine (2 → 3). These two reactions are believed to involve H atom abstraction from the exo-C6 and exo-C7 positions of the corresponding substrates 1 and 2, respectively, by an iron(IV)-oxo intermediary species (5 and 8) as shown in Figure 1. However, H6H can also accept the regioisomer 10 (see Figure 2) as a substrate for the cyclization reaction again yielding scopolamine.14 These observations immediately raise questions regarding how the regiochemistry of the two reactions is controlled within the H6H active site.
Figure 2.

Additional H6H substrate and product analogs studied in the present work.
Analysis of the H6H-catalyzed hydroxylation reaction by LC–MS demonstrates that over 98% of the product is 2, consistent with previous reports.7 However, as described in the Supporting Information, a minor side product can also be identified and was confirmed to be 10. Therefore, while H atom abstraction from C7 is possible during hydroxylation, nearly all of the reaction flux proceeds via H atom abstraction from C6. It follows that from a biological standpoint 2 rather than 10 is the relevant biosynthetic substrate for cyclization to 3 and thus exo-C7–H rather than exo-C6–H is almost always the only option for H atom abstraction during cyclization. However, the regioselectivity of H6H-catalyzed hydroxylation suggests that the H6H iron(IV)-oxo intermediate bound with 2 (i.e., 8) would actually undergo H atom abstraction and subsequent cyclization more slowly than the corresponding complex with 10. In other words, the preferred site of H atom abstraction may in fact be C6 rather than C7 during cyclization as well. In contrast, preferential H atom abstraction from C7 during cyclization would indicate an inversion in the regioselectivity of H6H-catalyzed radical initiation via an unknown mechanism, the details of which underlie precisely how the iron(IV)-complex interacts with the substrate thereby directing the course of the reaction.
RESULTS AND DISCUSSION
In order to properly address the question of regioselectivity during the H6H-catalyzed reactions, it is necessary to disentangle the kinetics of H atom abstraction from all other elementary reactions (e.g., substrate binding) in the two catalytic cycles of H6H. In the case of hydroxylation, H atom abstraction is product-determining, and thus at pseudosteady-state, the 10/2 product ratio is a constant under fixed concentrations of αKG and O2 and given by
| (1) |
In this expression, and represent the net rate constants15 associated with the net decomposition of the iron(IV)-oxo complex bound to 1 via hydroxylation at C7 versus C6, respectively (i.e., 5 → 10 and 5 → 2, see Figure 3). In other words, the net rate constants are indicative of all steps from H atom abstraction up to and including the first “irreversible” downstream elementary reaction. Therefore, 7/6k′hyd reports on the relative partitioning between the two hydroxylation pathways (see Figure 3).
Figure 3.

Partitioning model for the H6H-catalyzed hydroxylation of hyoscyamine (1) across the product determining step of H atom abstraction. The elementary reactions involving H atom abstraction from the exo-C6 vs exo-C7 sites are governed in the “forward” direction by the pseudo-first order rate constants vs , respectively. The steady-state decomposition of intermediate 5 to generate 2 vs 10 is governed by the net rate costants and . As discussed in the text, experimental results suggest that the 5 → 6 and 5 → 14 elementary reactions may be treated as irreversible.
The partitioning ratio 7/6k′hyd was measured by stirring 16 μM H6H in a well-ventilated vial under air (0.21 atm O2) with 100 μM 1 in the presence of 10 mM αKG, 400 μM FeSO4, 4 mM sodium ascorbate, and 50 mM sodium 4-(2-hydroxyethyl)-1-piperazineethanesulfonate (HEPES buffer) at pH 6.9 and room temperature. Under these conditions, the concentrations of the cosubstrates αKG and O2 remain essentially unchanged as the substrate 1 is consumed. Aliquots were taken at three time points within 20 min of initiating the reaction and subjected to LC–MS analysis, whereupon the product peaks in the extracted ion chromatograms (EICs) were integrated to provide the respective signal intensities. Samples were diluted to ensure that both the 2 (high intensity) and 10 (low intensity) signals were simultaneously within the linear dynamic range of the instrument; however, an intercept correction was necessary to account for minimum detection cutoffs. This correction resulted in an approximately 15% increase in the observed ratios versus the uncorrected values. Both 1 and 2 were determined to have the same ionization efficiency, and scopolamine production was found to be negligible. The experiment was replicated five times, yielding an average product ratio of 0.0117 ± 0.0003 (±standard error of the mean, n = 5). Experimental details including examination of controls are provided in the Supporting Information.
A similar experiment was also performed with the regio- and stereospecifically deuterated substrate 11, which was synthesized as described in the Supporting Information. In this case, the observed 13/2 ratio was found to be 0.50 ± 0.04 (n = 5) under the aforementioned reaction conditions. By analogy to eq 1, this product ratio is indicative of the net rate constant ratio given by16
| (2) |
In eq 2, is the net rate constant for steady-state decomposition of the iron(IV)-oxo complex via D atom abstraction from C6 of 11, and is the corresponding kinetic isotope effect. Consequently, this KIE can be readily computed from the ratio
| (3) |
yielding an observed value of 43 ± 3.
While the large primary deuterium KIE on exceeds the semiclassical limit,17 it is consistent with previous reports of H atom abstraction catalyzed by other nonheme iron enzymes.18–21 Moreover, it supports the hypothesis that the elementary reaction involving H atom abstraction (5 → 6) is the most rate-limiting (i.e., has the greatest sensitivity index) with respect to decomposition of the iron(IV)-oxo species bound with substrate to hydroxylated product and may thus be reasonably modeled as “irreversible” in the steady-state (i.e., it has a negligible reverse commitment along this sequence).22–26 Assuming the same holds for H atom abstraction from C7 and decomposition proceeds first via H atom abstraction (see Figure 3), we can make the approximations
| (4a) |
| (4b) |
where and are the respective microscopic rate constants governing H atom abstraction from C6 (5 → 6) and C7 (5 → 14).26 These conclusions are consistent with computational results on the mechanisms of nonheme iron oxidases in general.27 Therefore, 7/6k′hyd reports on not only the relative partitioning between the two hydroxylation pathways but also the regioselectivity of H atom abstraction itself.
In contrast to hydroxylation, the regioselectivity of H atom abstraction during cyclization cannot be inferred from the partitioning across a product-determining step. However, if H atom abstraction is effectively irreversible (see above) and the only elementary reactions to be significantly perturbed by deuteration or OH placement in the substrate are substrate binding and H atom abstraction then the ratio of V-max for 1 versus that for H6H substrate Y = 1, 12, 2, or 10 (see Figure 2) can be approximated as
| (5) |
Here, k1/kY is the ratio of the microscopic rate constants governing H atom abstraction from 1 versus Y. The weighting term α1 lies between 0 and 1 and depends only on the values of the rate constants in the catalytic cycle for 1. The result (eq 5) is trivial when Y = 1 and follows immediately from an analysis by Northrop when Y = 12,22,28 which is generalized to the case where Y = 2, 10 in the Supporting Information. Note that contributions to V1/VY from substrate binding are eliminated because the substrate concentrations are saturating. Therefore, once the weighting term α1 is known, eq 5 provides a means to calculating the regioselectivity of H atom abstraction during cyclization from measurements of V-max.
Compounds 2 and 12 were prepared as described in the Supporting Information, and initial rates were measured for saturating 1, 12, 2, and 10 at fixed 10 mM αKG and 0.21 atm O2. In these experiments, reactions were initiated by the addition of 1.6 μM H6H to solutions containing 100 μM substrate, 10 mM αKG, 400 μM FeSO4, 4 mM sodium ascorbate, and 50 mM HEPES at pH 6.9 and room temperature in a well-ventilated vial under air. Reaction aliquots were taken at three time points (<20% conversion), quenched by addition to MeCN and analyzed by LC–MS to obtain the signal intensities of the product and residual substrate. Since the ionization efficiency of 1 is equal to that of 2 (see Supporting Information), the fraction of reaction in the aliquot could be calculated directly from peak integrations of the corresponding EICs. In contrast, the ionization efficiency of scopolamine (3) was found to be ca. 50% that of either hydroxylated substrate and was thus corrected accordingly prior to determining the fraction of reaction. From the fraction of reaction, the residual substrate concentration could be calculated and linearly regressed versus quench time to obtain the initial rate. The experiment was replicated three times for each substrate. Finally, decreasing or increasing the initial substrate concentration by 50% in all cases induced no significant change to the measured initial rates consistent with substrate saturation and no substrate inhibition at 100 μM for all four substrates under the experimental conditions.
The V-max parameters measured for each of the four substrates are listed in Table 1. We can solve for the weighting term α1 from eq 5 by using the previous result that to obtain
Table 1.
Measurements of V-Max Per Unit Concentration Total Enzyme (e0) for 1, 12, 2, and 10 in the Presence of 10 mM αKG and 0.21 atm O2 at Room Temperature and pH 6.9a
| substrate | V/e0 (min−1) | n |
|---|---|---|
| 1 | 3.90 ± 0.04 | 3 |
| 12 | 0.10 ± 0.01 | 3 |
| 2 | 0.32 ± 0.04 | 3 |
| 10 | 0.020 ± 0.003 | 3 |
Values are reported ± one standard error of the estimate.
As this does not differ significantly from unity, it implies that the primary deuterium isotope effect on H atom abstraction is almost fully expressed in the V-max parameter. This conclusion is consistent with the observation based on the values in Table 1 that
Furthermore, since α1 is measurably greater than 0, eq 5 can also be used to get the ratio
| (6) |
Substituting in the observed parameters, we thus obtain a value of 16 ± 3 for 7/6kcyc. Therefore, the preference for H atom abstraction from C7 versus C6 increases roughly 1400-fold in the H6H-catalyzed cyclization reaction compared to hydroxylation. However, whereas hydroxylation proceeds with a roughly 85:1 selectivity for H atom abstraction from C6 versus C7, the magnitude of the selectivity for C7 versus C6 during cylization is reduced to ca. 16:1. Thus, while there is indeed an inversion in the site-selectivity between hydroxylation and cylization, the effect is strongest during hydroxylation when no substrate hydroxyl group is present.
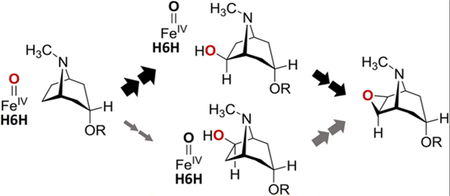
One possible hypothesis to help explain this result is that the hydroxyl groups of 2 and 10 coordinate the ferryl iron during the cyclization reaction as shown in Figure 4. It has been established that the activated iron(IV)-oxo complex of a number of nonheme iron oxidases is pentacoordinate, though computations suggest that octahedral complexes are also feasible.29–32 Consequently, the coordination environment of the iron(IV)-oxo complexes during H6H-catalyzed hydroxylation versus cylization need not necessarily be the same. Thus, the complexes poised for H atom abstraction during cyclization (i.e., 8 and 18) may resemble the putative product complexes expected to follow hydroxyl rebound during hydroxylation (i.e., 7 and 15). Coordination of the substrate hydroxyl group would also permit inner-sphere electron transfer from the substrate to the iron center, thereby facilitating the oxidative cyclizations of 2 and 10. However, given that the ability of iron(IV)-oxo complexes to catalyze H atom abstraction appears to be highly sensitive to orientation,33 direct coordination of a substrate to the ferryl iron may also serve to reorient the reactive oxo-ligand to favor H atom abstraction from the adjacent carbon (8b → 9 and 18b → 9 in Figure 4). In comparison with the unligated case during hydroxylation, the result may thus be a change in preference for abstraction of the C7–H atom but not without the observed reduction in overall regioselectivity.
Figure 4.

Mechanistic hypothesis for the reduction and inversion of the regioselectivity of H6H-catalyzed cyclization with respect to hydroxylation. This mechanism involves direct coordination of the substrate to the iron(IV)-oxo complex to facilitate dehydrogenation of the hydroxylated substrates (A) 2 and (B) 10.
As an initial test of this hypothesis, the ability of the iron(IV)-oxo complex to support an additional aquo or hydroxide ligand during the H6H-catalyzed hydroxylation reaction was investigated. When 1 was incubated with H6H and 18O2 in natural abundance water, the majority (87%) of the hydroxylated product was found to contain 18O consistent with a previous report.9 Likewise, in the converse experiment with natural abundance O2 and 18O-water, only 10% of the product contained 18O. However, when the same experiments were conducted with the deuterated substrate 12, 57% of the product contained 18O with 18O2 and natural abundance water becoming 40% in the presence of natural abundance O2 and 18O-water (see the Supporting Information). Hence, there appears to be significant exchange of the reactive oxo-ligand in the ferryl complex with solvent during the H6H-catalyzed hydroxylation reaction. This implies that the iron(IV)-oxo complex can indeed support an additional ligand34,35 and is thus consistent with the hypothesis that the substrate directly coordinates the iron(IV)-oxo complex during the H6H-catalyzed cyclization reaction while not doing so during hydroxylation.
CONCLUSIONS
H6H catalysis represents a useful system for studying how the course of enzymatic reactions involving highly reactive intermediates can be controlled. During the H6H-catalyzed hydroxylation of hyoscyamine, nearly all of the reaction flux proceeds via H atom abstraction from the exo-C6 position. However, there is more than a thousand-fold inversion in this regioselectivity when a hydroxyl group is present at the adjacent carbon. One possible mechanism to explain this inversion involves direct coordination of the hydroxylated substrate to the Fe(IV)-oxo complex during cyclization (as depicted in 8b and 18b). Thus, in the absence of coordination, the oxo ligand may be directed so as to favor H atom abstraction and subsequent hydroxylation at C6. In the presence of coordination, however, the reactive oxo ligand may be shifted to abstract the H atom from the adjacent carbon being most optimized for abstraction of the exo-C7 H atom in particular. Nevertheless, other explanations for the inversion in regiochemistry that do not necessarily involve direct coordination of the substrate to the iron-center (e.g., substrate repositioning due to changes in H-bonding, steric interactions, or solvent reorganization within the active site) remain very much open possibilities. Further evaluation of these and other mechanistic hypotheses regarding the two H6H-catalyzed reactions is thus expected to provide additional insights into how nonheme iron enzymes direct the course of the reactions they catalyze.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Takashi Hashimoto at Nara Institute of Science and Technology for generously providing the clone of h6h. This work was supported by grants from the National Institutes of Health (GM113106 and GM040541) and the Welch Foundation (F-1511).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11585.
Details regarding H6H assays, analytical controls, synthesis of compounds, and kinetic analysis (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Hausinger RP In 2-Oxoglutarate-dependent oxygenases; Schofield CJ, Hausinger RP, Eds.; The Royal Society of Chemistry: Cambridge, U.K., 2015; pp 1–58. [Google Scholar]
- (2).Kovaleva EG; Lipscomb JD Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol. 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Krebs C; Galonic Fujimori D; Walsh CT; Bollinger JM Jr. Non-heme Fe(IV)-oxointermediates. Acc. Chem. Res. 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Costas M; Mehn MP; Jensen MP; Que L Jr. Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- (5).Bollinger JM; Chang W.-c.; Matthews ML; Martinie RJ; Boal AK; Krebs C Mechanisms of 2-oxoglutarate-dependent oxygenases: the hydroxylation paradigm and beyond In 2-Oxoglutarate-Dependent Oxygenases; Metallobiology; Royal Society of Chemistry, 2015; pp 95–122. [Google Scholar]
- (6).Fodor G; Romeike A; Janzso G; Koczor I Epoxidation experiments in vivo with dehydrohyoscyamine and related compounds. Tetrahedron Lett. 1959, 1, 19–23. [Google Scholar]
- (7).Hashimoto T; Yamada Y Hyoscyamine 6β-hydroxylase, a 2-oxoglutaratedependent dioxygenase, in alkaloid-producing root cultures. Plant Physiol. 1986, 81, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hashimoto T; Yamada Y Purification and characterization of hyoscyamine 6β-hydroxylase from root cultures of Hyoscyamus niger L. Hydroxylase and epoxidase activities in the enzyme preparation. Eur. J. Biochem. 1987, 164, 277–285. [DOI] [PubMed] [Google Scholar]
- (9).Hashimoto T; Kohno J; Yamada Y Epoxidation in vivo of hyoscyamine to scopolamine does not involve a dehydration step. Plant Physiol. 1987, 84, 144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hashimoto T; Kohno J; Yamada Y 6β-Hydroxyhyoscyamine epoxidase from cultured roots of Hyoscyamus niger. Phytochemistry 1989, 28, 1077–1082. [Google Scholar]
- (11).Hashimoto T; Matsuda J; Yamada Y Two-step epoxidation of hyoscyamine to scopolamine is catalyzed by bifunctional hyoscyamine 6β-hydroxylase. FEBS Lett. 1993, 329, 35–39. [DOI] [PubMed] [Google Scholar]
- (12).Li J; van Belkum MJ; Vederas JC Functional characterization of recombinant hyoscyamine 6β-hydroxylase from Atropa belladonna. Bioorg. Med. Chem. 2012, 20, 4356–4363. [DOI] [PubMed] [Google Scholar]
- (13).Matsuda J; Okabe S; Hashimoto T; Yamada Y Molecular cloning of hyoscyamine 6β-hydroxylase, a 2-oxoglutarate-dependent dioxygenase, from cultured roots of Hyoscyamus niger. J. Biol. Chem. 1991, 266, 9460–9464. [PubMed] [Google Scholar]
- (14).Ushimaru R; Ruszczycky MW; Chang W.-c.; Yan F; Liu Y.-n.; Liu H.-w. Substrate conformation correlates with the outcome of hyoscyamine 6β-hydroxylase catalyzed oxidation reactions. J. Am. Chem. Soc. 2018, 140, 7433–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cleland WW Partition analysis and the concept of net rate constants as tools in enzyme kinetics. Biochemistry 1975, 14, 3220–3224. [DOI] [PubMed] [Google Scholar]
- (16).Jones JP; Korzekwa KR; Rettie AE; Trager WF Isotopically sensitive branching and its effect on the observed intramolecular isotope effects in cytochrome P-450 catalyzed reactions: a new method for the estimation of intrinsic isotope effects. J. Am. Chem. Soc. 1986, 108, 7074–7078. [Google Scholar]
- (17).Wiberg KB The deuterium isotope effect. Chem. Rev. 1955, 55, 713–743. [Google Scholar]
- (18).Martinez S; Hausinger RP Catalytic mechanisms of Fe(II)- and 2-oxoglutaratedependent oxygenases. J. Biol. Chem. 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Huang J-L; Tang Y; Yu C-P; Sanyal D; Jia X; Liu X; Guo Y; Chang W.-c. Mechanistic investigation of oxidative decarboxylation catalyzed by two iron(II)- and 2-oxoglutarate-dependent enzymes. Biochemistry 2018, 57, 1838–1841. [DOI] [PubMed] [Google Scholar]
- (20).Chang W.-c.; Guo Y; Wang C; Butch SE; Rosenzweig AC; Boal AK; Krebs C; Bollinger JM Jr. Mechanism of the C5 stereoinversion reaction in the biosynthesis of carbapenem antibiotics. Science 2014, 343, 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Price JC; Barr EW; Glass TE; Krebs C; Bollinger JM Jr. Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:α-ketoglutarate dioxygenase (TauD). J. Am. Chem. Soc. 2003, 125, 13008–13009. [DOI] [PubMed] [Google Scholar]
- (22).Northrop DB Steady-state analysis of kinetic isotope effects in enzymic reactions. Biochemistry 1975, 14, 2644–2651. [DOI] [PubMed] [Google Scholar]
- (23).Cleland WW Use of isotope effects to elucidate enzyme mechanisms. Critical Reviews in Biochemistry 1982, 13, 385–428. [DOI] [PubMed] [Google Scholar]
- (24).Ray WJ Jr. Rate-limiting step: A quantitative definition. Application to steady-state enzymic reactions. Biochemistry 1983, 22, 4625–4637. [DOI] [PubMed] [Google Scholar]
- (25).Ruszczycky MW; Anderson VE Interpretation of V/K isotope effects for enzymatic reactions exhibiting multiple isotopically sensitive steps. J. Theor. Biol. 2006, 243, 328–342. [DOI] [PubMed] [Google Scholar]
- (26).Ruszczycky MW; Liu H.-w. Theory and application of the relationship between steady-state isotope effects on enzyme intermediate concentrations and net rate constants. Methods Enzymol. 2017, 596, 459–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cho K-B; Hirao H; Shaik S; Nam W To rebound or dissociate? This is the mechanistic question in C H hydroxylation by heme and nonheme metal-oxo complexes. Chem. Soc. Rev. 2016, 45, 1197–1210. [DOI] [PubMed] [Google Scholar]
- (28).Northrop DB Minimal kinetic mechanism and general equation for deuterium isotope effects on enzymic reactions: uncertainty in detecting a rate-limiting step. Biochemistry 1981, 20, 4056–4061. [DOI] [PubMed] [Google Scholar]
- (29).Krebs C; Price JC; Baldwin J; Saleh L; Green MT; Bollinger JM Jr. Rapid freeze-quench 57Fe Mossbouer spectroscopy: monitoring changes of an ironcontaining active site during a biochemical reaction. Inorg. Chem. 2005, 44, 742–757. [DOI] [PubMed] [Google Scholar]
- (30).Wong SD; Srnec M; Matthews ML; Liu LV; Kwak Y; Park K; Bell CB III; Alp EE; Zhao J; Yoda Y; Kitao S; Seto M; Krebs C; Bollinger JM Jr.; Solomon EI Elucidation of the Fe(IV)═O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 2013, 499, 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sinnecker S; Svensen N; Barr EW; Ye S; Bollinger JM Jr.; Neese F; Krebs C Spectroscopic and computational evaluation of the structure of the high-spin Fe(IV)- oxo intermediates in taurine: α-ketoglutarate dioxygenase from Escherichia coli and its His99Ala ligand variant. J. Am. Chem. Soc. 2007, 129, 6168–6179. [DOI] [PubMed] [Google Scholar]
- (32).Song X; Lu J; Lai W Mechanistic insights into dioxygen activation, oxygen atom exchange and substrate epoxidation by AsqJ dioxygenase from quantum mechanical/ molecular mechanical calculations. Phys. Chem. Chem. Phys. 2017, 19, 20188–20197. [DOI] [PubMed] [Google Scholar]
- (33).Rohde J-U; In J-H; Lim MH; Brennessel WW; Bukowski MR; Stubna A; Munck E; Nam W; Que L Jr. Crystallographic and spectroscopic characterization of a nonheme Fe(IV)═O complex. Science 2003, 299, 1037–1039. [DOI] [PubMed] [Google Scholar]
- (34).Seo MS; In J-H; Kim SO; Oh NY; Hong J; Kim J; Que L Jr.; Nam W Direct evidence for oxygen-atom exchange between nonheme oxoiron(IV) complexes and isotopically labeled water. Angew. Chem., Int. Ed. 2004, 43, 2417–2420. [DOI] [PubMed] [Google Scholar]
- (35).Puri M; Company A; Sabenya G; Costas M; Que L Jr. Oxygen atom exchange between H2O and non-heme oxoiron(IV) complexes: ligand dependence and mechanism. Inorg. Chem. 2016, 55, 5818–5827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.