

Abstract
Gentamicin B (GB), a valuable starting material for the preparation of the semisynthetic aminoglycoside antibiotic isepamicin, is produced in trace amounts by the wild-type Micromonospora echinospora. While the biosynthetic pathway to GB has remained obscure for decades, we have now identified three hidden pathways to GB production via seven hitherto unknown intermediates in M. echinospora. The narrow substrate specificity of a key glycosyltransferase and the C6′-amination enzymes, in combination with the weak and unsynchronized gene expression of the 2′-deamination enzymes, limit GB production in M. echinospora. The crystal structure of the aminotransferase involved in C6′-amination explains its substrate specificity. Some of the new intermediates displayed similar premature termination codon readthrough activity but with reduced toxicity compared to the natural aminoglycoside G418. This work not only led to the discovery of unknown biosynthetic routes to GB, but also demonstrated the potential to mine new aminoglycosides from nature for drug discovery.
Aminoglycosides are one of the oldest classes of antibiotics and have been widely used against mycobacteria, staphylococci, and Gram-negative bacteria1. However, as with other classes of antibiotics, aminoglycoside resistance among microorganisms has emerged and spread rapidly2. To address this challenge, chemically modified aminoglycosides have been developed to overcome deactivation by aminoglycoside-modifying enzymes3. For example, dibekacin (1)4 is the first semisynthetic aminoglycoside derived from kanamycin B (2). Amikacin (3)5 was developed from kanamycin A (4) in the 1970s, and netilmicin (5)6 and isepamicin (6)7 were developed in the 1980s from sisomicin (7) and gentamicin B (GB, 8), respectively. Arbekacin (9)8 was derived from kanamycin B and marketed in 1990 (Fig. 1). Since then, no new aminoglycoside antibiotics have been approved despite the fact that multi-drug resistant (MDR) Gram-negative bacteria are on the rise at an alarming rate9. Importantly, the potential for aminoglycosides in the treatment of viral infection10 and human genetic diseases caused by premature termination codons (PTCs)11 has been demonstrated, emphasizing the need for the development of more new aminoglycosides.
Figure 1. Structures of DOS-containing aminoglycoside antibiotics.
Kanamycin A, kanamycin B, sisomicin, and gentamicin B are natural products, while dibekacin, amikacin, arbekacin, netilmicin, and isepamicin are semisynthetic aminoglycosides.
To generate new aminoglycoside antibiotics or to improve the production of valuable natural congeners for use in the synthesis of semisynthetic aminoglycosides, a detailed knowledge of the biosynthetic pathways and the involved enzymes are required12. A good example is the recent characterization of the entire kanamycin biosynthetic pathway13,14. Interestingly, manipulation of the kanamycin biosynthetic genes has permitted switching between the major products produced in Streptomyces kanamyceticus13,15. As for gentamicin biosynthesis, gentamicin C congeners are the major products of the wild-type Micromonospora echinospora with virtually no production of GB16. Since GB is an important congener used in the preparation of isepamicin, a better understanding of the gentamicin biosynthetic pathways could allow for engineering of bacterial strains to produce GB as the major product. Unfortunately, much less is known about the biosynthesis of gentamicins than kanamycins, even though both share a biosynthetic route from D-Glc-6-phosphate via 2-deoxystreptamine (DOS, 10) to the common pseudodisaccharide intermediate paromamine (PM, 11)1.
Previous studies of gentamicin biosynthesis have shown that the glycosyltransferase GenM1 of M. echinospora catalyzes the transfer of N-acetyl-D-glucosamine (GlcNAc) onto DOS to give 2′-N-acetyl-PM (AcPM, 12), which is converted to PM by the deacetylase GenD17. The second glycosyltransferase GenM2 catalyzes glycosylation of PM with xylose to give the fpseudotrisaccharide gentamicin A2 (GA2, 13)17. Conventionally, the DOS ring is designated as ring ΙΙ, and the sugar attached at C4 and C6 of DOS are labeled as rings Ι and ΙΙΙ, respectively (Fig. 1). Subsequent amination and then N-methylation at C3″ of ring ΙΙΙ of GA2 are catalyzed by the dehydrogenase GenD2, the aminotransferase GenS2, and the N-methyltransferase GenN in sequence to give gentamicin A (GA, 14). This is followed by C4″-methylation by the C-methyltransferase GenD1 to generate gentamicin X2 (GX2, 15)18, and C6′-methylation of GX2 by the C-methyltransferase GenK to give G418 (16)19 (Supplementary Fig. 1). Next, the dehydrogenase GenQ and the aminotransferase GenB1 catalyze the amination at C6′ of GX2 and G418, yielding JI-20A (17) and JI-20Ba (18)/JI-20B (19), respectively20,21. In addition, the substrate-flexible GenB2 can catalyze the epimerization between both JI-20Ba and JI-20B, and gentamicin C2a (20) and C2 (21)20,21. Recently, GenL has been identified to be responsible for 6′-N-methylation of gentamicin C2 and gentamicin C1a (22) to gentamicin C1 (23) and gentamicin C2b (24), respectively22 (Supplementary Fig. 1). Although substantial progress has been made, the biosynthetic pathway to GB remains elusive despite being first discovered in 197223. This is mainly because the general gene knockout approach to identify the biosynthetic genes and to detect the accumulated intermediates cannot be applied in this case due to the low production of GB by the wild-type M. echinospora. Moreover, GB cannot be biotransformed to gentamicin C components by M. echinospora24, indicating that GB is not an intermediate in the gentamicin C series pathway but rather a shunt metabolite of gentamicin biosynthesis.
To better understand gentamicin biosynthesis, we have completed, and report herein, the in vitro reconstitution of GB biosynthesis, revealing seven hitherto-unknown gentamicin biosynthetic intermediates and three enigmatic pathways leading to GB. The limiting factors which contribute to the low production yield of GB were also identified. Among the new gentamicin intermediates, 2′-deamino-2′-hydroxy-GA2 (2′DGA2, 25), 2′-deamino-2′-hydroxy-GA (2′DGA, 26), and 2′-deamino-2′-hydroxy-GX2 (2′DGX2, 27) exhibited reduced in vitro nephrotoxicity but similar PTC readthrough activity ex vivo compared to G418. Overall, our results uncover both the long-sought hidden biosynthetic pathways to GB and a number of bioactive aminoglycosides that had eluded detection in the past. This study demonstrates that the elucidation of minor biosynthetic pathways can be an alternative strategy to mine for new aminoglycosides which are difficult to access from nature.
Results
Discovery of GA2 analogs
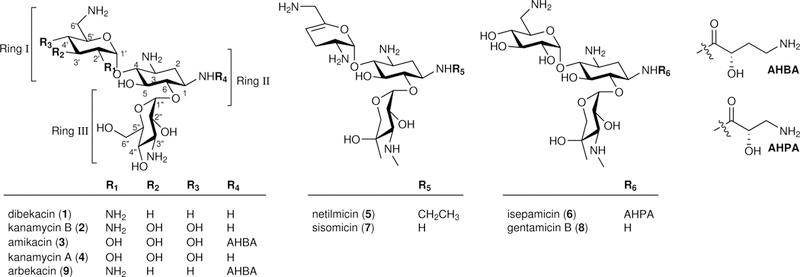
We first reasoned that gentamicin biosynthesis may proceed in a fashion analogous to that of kanamycin. In kanamycin biosynthesis, the substrate-flexible glycosyltransferase KanM1 catalyzes the transfer of either UDP-GlcNAc or UDP-Glc onto DOS to generate PM (via AcPM by the deacetylase KanN1) and 2′-deamino-2′-hydroxy-PM (2′DPM, 28), respectively. These two intermediates, PM and 2′DPM, are further 6′-aminated to neamine (6′-amino-6′-deoxy-PM, NM, 29) and 2′-deamino-2′-hydroxy-NM (2′DNM, 30), respectively, by another flexible C6′-dehydrogenase-aminotransferase KanQ-KanB13. Thus, it is conceivable that GB, which is a 2′-deaminated analog of JI-20A, may be generated in a similar manner from 2′DNM through xylosyltransfer by GenM2 (2′DNM → 6′-amino-2′-deamino-6′-deoxy-2′-hydroxy-GA2 (6′A2′DGA2, 31)), C3″-methylamination by GenD2-GenS2-GenN (6′A2′DGA2 → 6′-amino-2′-deamino-6′-deoxy-2′-hydroxy-GA (6′A2′DGA, 32)), and C4″-methylation by GenD1 (6′A2′DGA → GB), assuming that these enzymes possess reasonable substrate promiscuity typically observed for secondary metabolism enzymes (Fig. 2a). Therefore, we opted to examine the above hypothesis by in vitro reconstitution of the entire biosynthetic pathway of GB from DOS, whose biosynthesis is well known1. Soluble histidine-tagged GenM1 and GenM2 were obtained by expression of the corresponding genes in Escherichia coli and Streptomyces venezuelae, respectively (Supplementary Fig. 2). When the purified GenM1 was incubated overnight with DOS in the presence of UDP-GlcNAc or UDP-Glc, the products predicted as AcPM and 2′DPM were separately produced (Fig. 2a–c). The identities of AcPM and 2′DPM were confirmed by comparing their UPLC-qTOF-HR-MS chromatograms and MS/MS fragmentations with those previously reported for AcPM13 and those of chemically synthesized authentic 2′DPM, respectively (see Supplementary Note for the chemical synthesis and structural characterization of gentamicin intermediates). Under the same incubation conditions, the corresponding conversion yields of DOS to AcPM and 2′DPM were approximately 54% and 13%, respectively, which suggests that UDP-GlcNAc may be a better glycosyl donor compared to UDP-Glc in the GenM1-catalyzed reaction. If correct, this would stand in contrast to results for KanM1 of the kanamycin biosynthetic pathway, where UDP-Glc appears to be the preferred substrate13.
Figure 2. Two glycosylation steps to GA2 and its new analogs.
(a) Biosynthetic pathways to GA2, 6′AGA2, 6′A2′DGA2, and 2′DGA2 catalyzed by the glycosyltransferases GenM1 and GenM2. Each colored functional group represents those formed by the enzymes indicated by the same color. The predicted C6′-amination steps from PM and 2′DPM to NM and 2′DNM, respectively, are depicted by dashed lines. (b,c) Chromatograms of the GenM1-catalyzed production of AcPM (selected for m/z = 366.1871) from DOS (selected for m/z = 163.1077) and UDP-GlcNAc (b) and 2′DPM (selected for m/z = 325.1605) from DOS and UDP-Glc (c). (d–g) Chromatograms of the GenM2-catalyzed production of GA2 (selected for m/z = 456.2188) from PM (selected for m/z = 324.1765) and UDP-Xyl (d), 6′AGA2 (selected for m/z = 455.2348) from NM (selected for m/z = 323.1925) and UDP-Xyl (e), 6′A2′DGA2 (selected for m/z = 456.2188) from 2′DNM (selected for m/z = 324.1765) and UDP-Xyl (f), and 2′DGA2 (selected for m/z = 457.2028) from 2′DPM and UDP-Xyl (g). Lower chromatograms show the reactions without enzymes as controls. The dotted line in the upper chromatograms indicates the remaining substrate. Chromatograms show representative results of n>5 independent reactions.
In the kanamycin pathway, the conversion of PM and 2′DPM to NM and 2′DNM is catalyzed by KanQ–KanB. It is thus likely that the KanQ–KanB counterparts in the gentamicin pathway, GenQ–GenB1, have similar activities despite that GenQ–GenB1 had only been shown to catalyze the C6′-amination of pseudotrisaccharide GX2 in an early experiment20. If GenQ–GenB1 are indeed effective to generate NM/2′DNM from PM/2′DPM, it would be interesting to know whether these four pseudodisaccharides can all be recognized and processed by the glycosyltransferase GenM2. To probe this possibility, we expressed and purified GenM2 and acquired the four pseudodisaccharides: NM from a commercial source, PM by hydrolysis of paromomycin (33), and 2′DPM as well as 2′DNM by chemical synthesis (Supplementary Note). Incubation of GenM2 overnight with PM, NM, 2′DNM, and 2′DPM plus UDP-xylose (UDP-Xyl) led to the formation of new UPLC peaks corresponding to their respective 6-O-xylosylated products GA2, 6′-amino-6′-deoxy-GA2 (6′AGA2, 34), 6′A2′DGA2, and 2′DGA2 at conversion yields of approximately 93%, 13%, 8%, and 9%, respectively (Fig. 2a,d,e,f,g). The structures of GA2, 6′AGA2, 6′A2′DGA2, and 2′DGA2 were identified by UPLC and MS/MS analysis in comparison with the synthesized standards (Supplementary Note). These results showed that GenM2 accepts all four pseudodisaccharides as glycosyl acceptors being apparently most active with PM. It is important to note that of the pseudotrisaccharide products, 6′AGA2, 6′A2′DGA2, and 2′DGA2 have not previously been identified as gentamicin biosynthetic intermediates.
The diverse pathways to analogs of GA and GX2
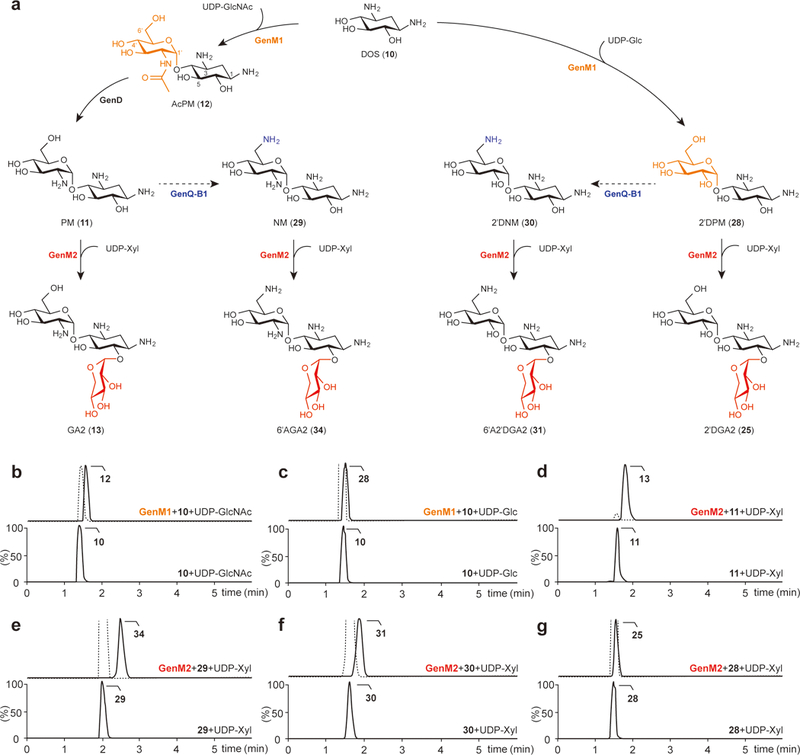
The C3″-methylamination catalyzed by GenD2-GenS2-GenN and subsequent C4″-methylation catalyzed by GenD1 are required for the biosynthesis of GX2 via GA from GA2 (ref. 18). The same set of enzymes could also catalyze the conversion of 6′AGA2, 6′A2′DGA2, and 2′DGA2 to JI-20A, GB, and 2′DGX2, respectively, via the intermediates 6′-amino-6′-deoxy-GA (6′AGA, 35), 6′A2′DGA, and 2′DGA (Fig. 3a). To assess this possibility, the histidine-tagged GenD2, GenS2, GenN, and GenD1 were each expressed in E. coli (Supplementary Fig. 2). When GenD2, GenS2, and GenN were incubated overnight with GA2 in the presence of NAD+, L-glutamine, and S-adenosyl-L-methionine (SAM), the complete conversion of GA2 to a new product was achieved (Fig. 3b). This product was consistent with GA based on UPLC retention time as well as HR-MS/MS analysis in comparison with an authentic standard (Supplementary Note). Because the product of GenD2-GenS2 (3″-amino-3″-deoxy-GA2) could inhibit the activities of GenD2/GenS2 in vitro at a very low concentration18, these three enzymes need to be assayed together to drive the C3″ modification to complete. As expected, 6′AGA2, 6′A2′DGA2, and 2′DGA2 were respectively converted, in equal efficiency as the turnover of GA2 to GA, to a series of new gentamicin derivatives, 6′AGA, 6′A2′DGA, and 2′DGA by GenD2-GenS2-GenN (Fig. 3a,c,d,e). These new compounds were isolated from large-scale enzyme reactions and their structures were confirmed by NMR and UPLC-qTOF-HR-MS spectroscopy (Supplementary Note). Upon treatment with GenD1, GA, 6′AGA, 6′A2′DGA, and 2′DGA all underwent C4″-methylation producing new peaks predicted to be GX2, JI-20A, GB, and 2′DGX2, respectively, in nearly quantitative yields (Fig. 3a,f,g,h,i). The UPLC retention times and MS/MS fragmentation patterns of these new products were identical to those of commercial GX2 and chemically synthesized JI-20A, GB, and 2′DGX2, respectively (Supplementary Note). These results demonstrate that GB can be generated by a new biosynthetic pathway via two newly identified intermediates 6′A2′DGA2 and 6′A2′DGA. Moreover, while 2′DGX2 is a known synthetic product25, it has not previously been noted as a gentamicin biosynthetic intermediate.
Figure 3. The diverse pathways to the analogs of GA and GX2.
(a) C3″-methylamination steps to GA, 6′AGA, 6′A2′DGA, and 2′DGA catalyzed by GenD2-GenS2-GenN (indicated by green), and C4″ methylation steps to GX2, JI-20A, GB, and 2′DGX2 catalyzed by GenD1 (indicated by pink). (b–e) Chromatograms of GenD2-GenS2-GenN-catalyzed production of GA (selected for m/z = 469.2504) from GA2 (selected for m/z = 456.2188) (b), 6′AGA (selected for m/z = 468.2664) from 6′AGA2 (selected for m/z = 455.2348) (c), 6′A2′DGA (selected for m/z = 469.2504) from 6′A2′DGA2 (selected for m/z = 456.2188) (d), and 2′DGA (selected for m/z = 470.2344) from 2′DGA2 (selected for m/z = 457.2028) (e). (f–i) Chromatograms of GenD1-catalyzed production of GX2 (selected for m/z = 483.2661) from GA (f), JI-20A (selected for m/z = 482.2821) from 6′AGA (g), GB (selected for m/z = 483.2661) from 6′A2′DGA (h), and 2′DGX2 (selected for m/z = 484.2501) from 2′DGA (i). Lower chromatograms show the reactions without enzymes as controls. The dotted line in the upper chromatograms indicates the remaining substrate. Chromatograms show representative results of n>5 independent reactions.
C6′-amination pathway interconnects the intermediates
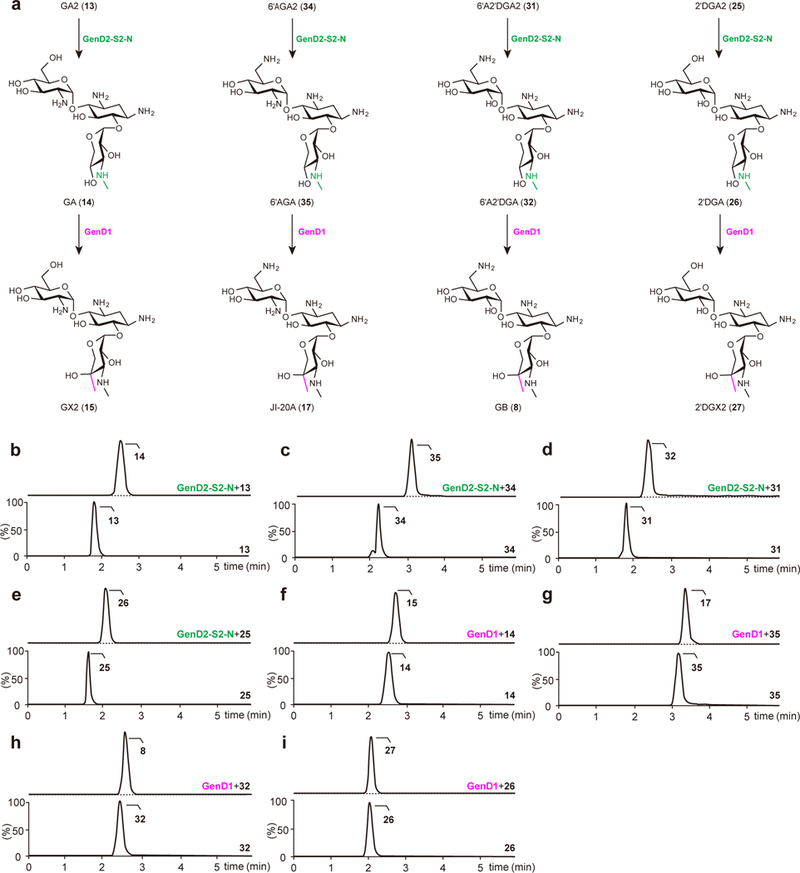
To examine the C6′-amination activity of GenQ-GenB1 toward the two pseudodisaccharides (PM and 2′DPM) and the five pseudotrisaccharides (GA2, GA, 2′DGA2, 2′DGA, and 2′DGX2), the histidine-tagged GenQ and GenB1 were expressed in E. coli and purified (Supplementary Fig. 2). Upon overnight incubation of GenQ-GenB1 with L-glutamine as an amino donor, the natural substrate GX2 was converted into JI-20A in ca. 80% yield. Conversions of GA2, GA, 2′DGA2, 2′DGA, and 2′DGX2 to their C6′-amination products, 6′AGA2, 6′AGA, 6′A2′DGA2, 6′A2′DGA, and GB, by GenQ-GenB1 were also observed (Fig. 4a–g). However, the conversion yield for each substrate was low: GA2, 8%; GA, 14%; 2′DGA2, 2%; 2′DGA, 2%; 2′DGX2, 12%. These results suggest that the presence of the 2′-amino and/or the 4″-methyl group in the pseudotrisaccharides seems to be critical for C6′-amination activity of GenQ-GenB1. A similar trend was also observed upon incubation of PM and 2′DPM with GenQ-GenB1. The yields of NM and 2′DNM were 31% and 5%, respectively (Fig. 4a,h,i). The greater extent of conversion of PM versus 2′DPM is similar to that reported for KanQ-KanB in kanamycin biosynthesis13. Interestingly, amination of the pseudodisaccharide PM was found to be more extensive compared to both GA2 and GA, which lack the 4″-methyl group. Likewise, 2′DPM was also consumed to a greater degree than 2′DGA2 and 2′DGA. These findings again suggest the importance of the 4′′-methyl group for GenQ-GenB1 activity when the third sugar moiety (ring ΙΙΙ) is present. All substrates carrying a 2′-amino group were more completely consumed in the presence of GenQ-GenB1 than their counterparts without a 2′-amino group, implying that the 2′-amino group may also be important for substrate recognition by GenQ-GenB1. Moreover, these results indicate that C6′-amination catalyzed by GenQ-GenB1 can serve to bridge those pathways involving 6′-hydroxylated intermediates with pathways involving 6′-aminated intermediates (Fig. 4a). A case in point is that GB can be derived from 2′DGX2 by GenQ-GenB1-catalyzed C6′-amination.
Figure 4. The interconnecting C6′-amination and C2′-deamination pathways to GB.
(a) C6′-amination and C2′-deamination reactions respectively catalyzed by GenQ-GenB1 (blue arrows) and GenJ-GenK2 (purple arrows) interconnects the intermediates to GB. The second glycosylation catalyzed by GenM2, C3″-methylamination catalyzed by GenD2-GenS2-GenN, and C4″ methylation steps catalyzed by GenD1 are indicated by red, green, and pink arrows, respectively. (b–i) Chromatograms of GenQ-GenB1-catalyzed production of JI-20A from GX2 (b), 6′AGA2 from GA2 (c), 6′AGA from GA (d), 6′A2′DGA2 from 2′DGA2 (e), 6′A2′DGA from 2′DGA (f), GB from 2′DGX2 (g), NM from PM (h), and 2′DNM from 2′DPM (i). Lower chromatograms show the reactions without enzymes as controls. The dotted line in the upper chromatograms indicates the remaining substrate. Chromatograms show representative results of n>5 independent reactions.
The diverse C2′-deamination pathways leading to GB
During the biosynthesis of kanamycin, an α-ketoglutarate-dependent dioxygenase KanJ and a NADPH-dependent reductase KanK act on kanamycin B to produce kanamycin A by C2′-deamination14. Interestingly, heterologous expression of kanJ-kanK in the JI-20A-accumulating M. echinospora mutant resulted in the production of GB26, indicating that GB could also be biosynthesized from JI-20A through C2′-deamination. However, no homologs of the kanJ-kanK genes can be found in the gentamicin gene cluster of M. echinospora. Yet, re-examining the draft genome sequence of M. echinospora led to the identification of two adjacent genes outside of the gentamicin gene cluster that are homologous to kanJ and kanK (55% identity at translated sequence level). They are accordingly designated genJ and genK2 (Supplementary Figs. 3,4). Overnight incubation of the histidine-tagged GenJ and GenK2 (Supplementary Fig. 2) with JI-20A, 6′AGA2, and 6′AGA (along with α-ketoglutarate and (NH4)2Fe(SO4)2·6H2O for GenJ, and NADPH for GenK2) produced GB, 6′A2′DGA2, and 6′A2′DGA, respectively, in nearly quantitative yield (Figs. 4a and 5a–c). GenJ-GenK2 also transformed GA2, GA, and GX2 to their C2′-deaminated products 2′DGA2, 2′DGA, and 2′DGX2 in 10%, 67%, and 78% yield (Figs. 4a and 5d–f). In addition, this enzyme pair could also catalyze C2′-deamination of kanamycin B, which bears a 6′-amino group, to kanamycin A as efficiently as JI-20A is converted to GB (Supplementary Fig. 5). While the 6′-amino group in the substrate appears to be important for efficient turnover by the GenJ-GenK2 pair, this does not seem to be the case for ring ΙΙΙ, particularly when ring Ι has a 6′-amino group. These results demonstrated that GB can be biosynthesized via another independent C2′-deamination pathway from JI-20A by the action of the newly identified GenJ-GenK2.
Figure 5. Analysis of C2′-deamination reactions catalyzed by GenJ-GenK2.
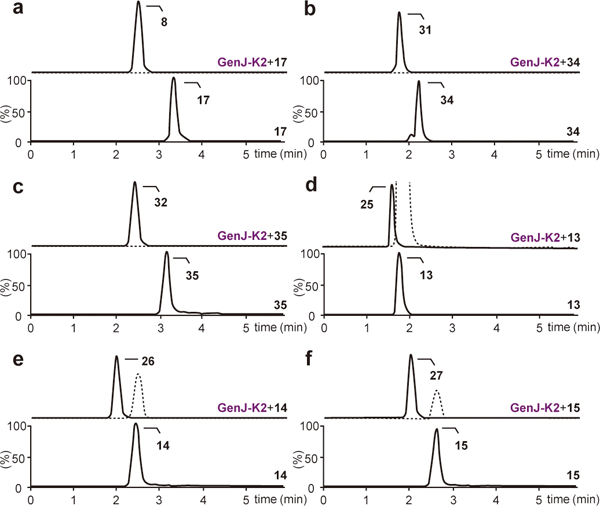
(a–f) Chromatograms of GenJ-GenK2-catalyzed production of GB from JI-20A (a), 6′A2′DGA2 from 6′AGA2 (b), 6′A2′DGA from 6′AGA (c), 2′DGA2 from GA2 (d), 2′DGA from GA (e), and 2′DGX2 from GX2 (f). Lower chromatograms show the reactions without enzymes as controls. The dotted line in the upper chromatograms indicates the remaining substrate. Chromatograms show representative results of n>5 independent reactions.
In the above GenJ-GenK2 coupled reactions, 2′-oxo-products generated from the GenJ reaction were detected based on MS/MS fragmentation patterns (Supplementary Fig. 6). To confirm the identity of these ketone intermediates, the GenJ reaction with JI-20A in the absence of GenK2 was treated with NaBH4 or NaBD4, and the formation of GB or [2′−2H]-GB (36) was observed (Supplementary Fig. 7a,b). Furthermore, the addition of 10 mM phenylhydrazine after the GenJ reaction led to a phenylhydrazone derivative of 2′-oxo-GB (Supplementary Fig. 7c). These results showed that GenJ indeed catalyzes the deamination of JI-20A to form a keto product. To learn more about the GenJ mechanism, [2′−2H]-JI-20A (37) was chemically prepared (Supplementary Note), which upon incubation with GenJ yielded an unlabeled ketone product based on ESI-MS analysis (Supplementary Fig. 8). The oxygen dependency of GenJ was also demonstrated by the incorporation of notable amounts of 18O into JI-20A (32%) under an 18O-enriched atmosphere (Supplementary Fig. 9 and Supplementary Table 1). These findings indicated that GenJ acts as a typical FeII/α-ketoglutarate-dependent dioxygenase and the reaction is initiated by the abstraction of a hydrogen atom at C2′ of JI-20A through the reactive [FeIV=O] species in a mechanism similar to that of KanJ14.
To address why the wild-type M. echinospora is a very poor producer of GB despite the high in vitro C2′-deamination activity of GenJ-GenK2 toward JI-20A, transcriptional analysis was performed using semi-quantitative RT-PCR. The expression levels of genJ-genK2 and six other gentamicin biosynthetic genes located in different operons in the cluster (genM1, genM2, genP, genK, genE, and genN) were compared in M. echinospora. Transcripts of those genes encoded in the cluster were clearly observed at 60 h, increased up to 72‒84 h, and maintained at 96 h. However, transcription of genJ-genK2 was undetectable at 60 h, slightly detected at 72‒84 h, and disappeared at 96 h (Supplementary Fig. 10), indicating that the weak and unsynchronized expression of genJ-genK2 in comparison with other genes in the gentamicin cluster could not support GB biosynthesis in M. echinospora.
Structure of GenB1
A greater substrate specificity for GenM2 and GenQ-GenB1 could explain the limited production of GB in the wild-type M. echinospora strain. As an effort to elucidate the structural basis for C6′-amination efficiency, crystal structures of GenB1 complexed with PLP, PLP/NM, and PLP/JI-20A were solved (Supplementary Table 2). GenB1 is a homo-dimer with monomers consisting of a PLP-binding domain (residues 55‒281) and two flanking domains (residues 9‒59 and 280‒416). The loop (residues 257‒262) between α9 and α10 in one monomer forms an edge of the active site in the other monomer (Fig. 6a; Supplementary Fig. 11). The active site of GenB1 harbors P-, R1-, R2-, and R3-subsites for binding of PLP, ring Ι, ring ΙΙ, and ring ΙΙΙ of the substrates, respectively (Fig. 6b).
Figure 6. Crystal structure of GenB1.

(a) Overall structure of holo-GenB1 in complex with JI-20A. Schematic depiction of the GenB1 primary sequence is shown at the top. Dimeric structure with two flanking domains (green and magenta) and PLP-binding domains (yellow and light pink) are shown in ribbon presentation. The connecting loops between α9 and α10 in each monomer participating in the constitution of the active site in the other monomer are emphasized using thicker lines colored in forest and purple. The N-and C-termini are indicated in circles and secondary structure elements are labeled. Bound PLPs are represented by white sticks. The active site of molecule A is highlighted by a dashed circle. (b) Surface representation of the active site (left) and its schematic diagram with dimensions (right). Bound JI-20A is depicted as a black stick model and its individual sugar rings are marked as Ι, ΙΙ, and ΙΙΙ. (c–e) Close-up views of R1-, R2-, and R3-subsites. Residues constituting subsites are shown as green sticks. (c) Negatively-charged hole of the R1-subsite. The negatively-charged hole is indicated by a dashed circle in surface representation with the electrostatic potential. (d) Aromatic platform of the R2-subsite. The surfaces are colored based on the hydrophobicity of the side chains - yellow to blue representing most hydrophobic to polar side chains. The bound NM is shown as gray sticks. (e) Methyl pocket of the R3-subsite. The moieties of ring ΙΙΙ are indicated by arrows with labels.
In the holo-GenB1, PLP is covalently bound to Lys232, and one magnesium ion in the R1-subsite occupies a negatively charged hole formed by Asp345, Asp395, and the C-terminal end of α11 (Supplementary Fig. 12). Upon binding of NM, an external aldimine formed between PLP and ring Ι of NM (Fig. 6c; Supplementary Fig. 13). Interestingly, the 2′-amino group of ring Ι in NM replaces the magnesium ion to form polar interactions with Asp345, Asp395, and the backbone oxygen of Ala393. Given that GenB1 prefers substrates carrying a 2′-amino group to those with a 2′-hydroxyl group (Fig. 4), the anchoring of the 2′-amino group to the negatively charged hole explains this bias. Tyr132 in the R2-subsite forms C-H/π stacking interactions with the DOS moiety (ring ΙΙ) in NM (Fig. 6d).
The binding modes of PLP and ring Ι of JI-20A in the GenB1/PLP/JI-20A complex are virtually identical to those in the GenB1/PLP/NM complex. However, the GenB1/JI-20A complex structure revealed a semi-sphere-like R3-subsite which accommodates ring ΙΙΙ of JI-20A (Fig. 6e; Supplementary Fig. 14). The methyl groups at C3″ and C4″ of ring ΙΙΙ face the methyl pocket of the R3-subsite lined by Leu156, Trp391, Trp415, and the C-terminal region (Fig. 6e). The R3 methyl pocket may form favorable contacts with hydrophobic groups and is probably responsible for the enhanced amination efficiency of GenB1 towards substrates with methyl substituents including GX2/2′DGX2 over GA2/2′DGA2 (Fig. 4). To study the importance of the R1 negatively charged hole and the R3 methyl pocket, Asp345 and Asp395 were replaced by leucine, and Trp391 and Trp415 were mutated to alanine. Indeed, an approximately 50% decrease in the C6′-amination activity toward GX2 was observed with the four mutant proteins (Supplementary Fig. 15).
Ring ΙΙ in both JI-20A and NM is packed against Tyr132. The 3-amino group of NM forms an ion pair with Asp395, whereas ring ΙΙ in JI-20A is flipped ~180°, losing the favorable interaction (Supplementary Fig. 16). If ring ΙΙ of JI-20A holds the same conformation as that of NM, steric clash would occur between ring ΙΙΙ of JI-20A and the active site. Hence, the presence of ring ΙΙΙ in JI-20A affects the binding mode of ring ΙΙ (Supplementary Fig. 16). The roughly 2–4-fold greater consumption of PM and 2′DPM compared to both GA2/GA and 2′DGA2/2′DGA (Fig. 4) is consistent with the hypothesis that ring ΙΙ of PM and 2′DPM binds to the R2-subsite in the favorable binding mode, while ring ΙΙ of GA2/GA and 2′DGA2/2′DGA is in the less favorable binding mode due to the presence of ring ΙΙΙ. Binding between GenB1 and GA2/GA or 2′DGA2/2′DGA may be further weakened by the absence of methyl groups on ring ΙΙΙ preventing favorable occupancy of the R3 methyl pocket. Together, the R1 negatively-charged hole, the R3 methyl pocket, and the two distinct binding modes of ring ΙΙ in the R2-subsite are all expected to be important for substrate recognition by GenB1.
In vitro toxicity and bioactivity of new intermediates
Because the major therapeutic disadvantages of aminoglycosides are their high nephrotoxicity and ototoxicity27, to assess the therapeutic potential of the newly identified GB derivatives, their toxicities were analyzed using three mammalian kidney cell lines HEK-293, LCC-PK1, and COS-7. G418, which is one of the strongest readthrough inducers among natural aminoglycosides28, was used as a control. The half-maximal lethal concentration (LC50) values for 2′DGA2, 2′DGA, and 2′DGX2 were approximately 1.2–1.3-fold higher than that of G418 in the three cell lines tested. 6′AGA2, 6′AGA, and JI-20A displayed approximately 1.2-fold lower LC50 compared to G418 in human HEK-293 cells (Supplementary Fig. 17). These results display a correlation of reduced toxicity to fewer amino groups, and are consistent with the structure–toxicity relationships observed previously for aminoglycosides29. It was also noted that only the number and not the position of the free amino groups in ring Ι is the key determinant for cell toxicity. The structure of ring ΙΙΙ does not appear to substantially contribute to cell toxicity.
The antibiotic activities of the gentamicin biosynthetic intermediates were tested against four gentamicin-susceptible and -resistant bacteria (Enterococcus faecalis, Pseudomonas aeruginosa, Staphylococcus aureus, and E. coli) (Supplementary Table 3). All compounds tested (compounds 8, 13–15, 17, 25–27, 31, 32, 34, and 35) were inactive against the tested strains except that GB, GX2, JI-20A, 6′A2′DGA, and 6′AGA exhibited weak activity against gentamicin-susceptible S. aureus and E. coli. The level of antibiotic activity follows the order JI-20A>GB>6′AGA>6′A2′DGA>GX2, indicating the importance of the 2′-amino, 6′-amino, 3″-methylamino, and 4″-methyl groups for antibacterial activity.
Next, we examined whether these new intermediates could induce PTC readthrough in human cells. Primary human cystic fibrosis bronchial epithelial cells (cystic fibrosis transmembrane conductance regulator [CFTR]; p.ΔF508/W1282X) were exposed to 25 μM of compounds 13–17, 25–27, 31, 32, 34, and 35 for up to 2 days, and the comparative ratio of full length to truncated CFTR was quantitatively measured using HPLC-ESI-MS. All compounds induced similar levels of PTC readthrough compared to G418 except for JI-20A and GB, which exhibited relatively weak readthrough activity (Supplementary Fig. 18). These results showed that 2′DGA2, 2′DGA, and 2′DGX2 can induce substantial levels of readthrough but with evidently reduced toxicity compared to G418.
Discussion
Our in vitro results now establish that GB can be assembled via three independent biosynthetic routes involving seven newly identified intermediates: 25–27, 31, 32, 34, and 35 (Fig. 4a). The minor biosynthetic intermediates 2′DGA2, 6′A2′DGA2, and 6′AGA2 are generated through xylosylation of the pseudodisaccharides 2′DPM, 2′DNM, and NM, respectively, by the not-so-promiscuous GenM2. These compounds can then be converted to their corresponding new GA analogs, 2′DGA, 6′A2′DGA, and 6′AGA, by the substrate-flexible GenD2-GenS2-GenN system. Subsequent reaction with the GenQ-GenB1 pair can produce small amounts of 6′A2′DGA2, 6′A2′DGA, 6′AGA2, and 6′AGA from 2′DGA2, 2′DGA, GA2, and GA, and the known product JI-20A from GX2 (ref. 20). From this point forward, three pathways can all lead to GB production: C4″-methylation of 6′A2′DGA by the C-methyltransferase GenD1, C6′-amination by GenQ-GenB1 of 2′DGX2, which is derived from 2′DGA by GenD1, and C2′-deamination of JI-20A catalyzed by GenJ-GenK2. In addition, GenJ-GenK2 could also deaminate GA2, GA, GX2, 6′AGA2, and 6′AGA to their 2′-deaminated products. These results uncovered an interwoven GB biosynthetic network showing that all 12 pseudotrisaccharide intermediates are interconnected by GenQ-GenB1, GenD2-GenS2, GenN, GenD1, and GenJ-GenK2 (Fig. 4a).
The present studies also suggest an explanation for the limited in vivo production of GB in the wild-type M. echinospora as being due to the high substrate specificity with respect to both the GenQ-GenB1 pair involved in C6′-amination and the glycosyltransferase GenM2 involved in the attachment of xylose to the pseudodisaccharide acceptor. GenM2 exhibits an increased catalytic preference for PM versus 2′DPM, NM, and 2′DNM, resulting in decreased biosynthesis of the minor pseudotrisaccharides 2′DGA2, 6′AGA2, and 6′A2′DGA2. Furthermore, the C2′-amino and C4″-methyl groups are likely to be important for substrate recognition and subsequent C6′-amination by GenQ-GenB1. This makes GX2 the most abundant and the only productive biosynthetic intermediate for GB (Fig. 4a). The weak and unsynchronized expression of genJ-genK2 located outside the gentamicin cluster also restricts in vivo production of GB, although their translated products show sufficient in vitro 2′-deamination activity toward JI-20A and other 6′-aminated pseudotrisaccharides 6′AGA2 and 6′AGA.
The C6′-amination step is catalyzed by GenQ-GenB1. Although we could not determine the structure of GenQ and did not investigate whether the key determinant of substrate specificity is GenB1, GenQ or both during C6′-amination, the structures of GenB1 provided important insights into the substrate preference of this aminotransferase. BtrR30 from Bacillus circulans and RbmB31 from Streptomyces ribosidificus involved in DOS biosynthesis are PLP-dependent sugar aminotransferases functionally relevant to GenB1. The structure of GenB1 is superimposed onto those of RbmB and BtrR with root-mean-square deviations (rmsd) of 3.6 and 3.5 Å respectively (Supplementary Fig. 19). The large rmsd values reflect the low sequence identities (~12%) between GenB1 and RbmB/BtrR (Supplementary Fig. 20). The active sites of RbmB and BtrR have space only for a single sugar ring (ring ΙΙ) covalently attached to PLP (Supplementary Fig. 21), which is in contrast to the three subsites in GenB1 (Fig. 6). This disparity is related to the fact that the C-terminal extension of RbmB and BtrR covers the region corresponding to the R1- and R3-subsites of GenB1 (Supplementary Fig. 21). Accordingly, the unique subsite organization in the large active site of GenB1 is specifically tailored for its substrate promiscuity and preference. Although we did not perform a mechanistic study of GenB1, structural observations indicate that GenB1 shares a reaction mechanism with other PLP-dependent transaminases (Supplementary Fig. 22).
Compare to G418, the new gentamicin analogues 2′DGA2, 2′DGA, and 2′DGX2 have a reduced toxicity profile while maintaining a similar PTC readthrough activity. It has been shown that installing the (S)-4-amino-2-hydroxybutyric acid (AHBA) moiety at the 1-N position32 and a methyl group at C6′ (refs. 32,33) are effective to improve the termination suppression activity and reduce the toxicity of aminoglycosides. In particular, it was recently reported that the minor component gentamicin B1 (6′-methyl-GB, 38) possesses greater PTC readthrough activity than the gentamicin C series34. Therefore, it is highly likely that these new analogues, such as 2′DGA2, 2′DGA, and 2′DGX2, could be candidates for future development of aminoglycosides with improved readthrough activity and reduced toxicity through the installation of a 1-N-AHBA moiety using the enzymes responsible for its construction in the butirosin biosynthetic pathway13,35–37 and a 6′-methyl group catalyzed by GenK19.
Taken together, results reported herein are important since they provide detailed genetic and molecular insights for enhancing the production of GB. Possible in vivo strategies to increase the biosynthesis of GB include protein engineering of GenB1 (or GenQ) and GenM2 to broaden their substrate specificities, as well as construction of a genK-genP deletion mutant to accumulate the precursor JI-20A and overexpression of genJ-genK2 to increase 2′-deamination activity. In an early report, application of the latter strategy led to the substantially enhanced production of GB in the engineered M. echinospora strain26. Thus far only 150 aminoglycosides, including structurally similar congeners3,38, have been identified since the discovery of the first aminoglycoside in 194339. The scarcity of this class of compounds is partly due to the fact that aminoglycoside biosynthetic gene clusters are not commonly found in the genome of actinomycetes40 and their isolation is particularly challenging compared to many other types of natural products. Our successful efforts at mining the minor biosynthetic intermediates of GB demonstrate the usefulness of this method to discover new aminoglycosides. This alternate approach may be further exploited to expand the diversity of this important class of antibiotics.
Online Methods
Materials, bacterial strains, and culture conditions.
DOS, UDP-GlcNAc, and NM were purchased from Genechem Inc. (Daejeon, Republic of Korea). UDP-Glc, G418, NAD+, NADPH, L-glutamine, SAM, PLP, methylcobalamin, benzyl viologen, α-ketoglutarate, ammonium iron(ΙΙ) sulfate hexahydrate ((NH4)2Fe(SO4)2·6H2O), iron(ΙΙ) sulfate (Fe(ΙΙ)SO4), ampicillin, chloramphenicol, gentamicin, kanamycin, isopropyl β-D-1-thiogalactopyranoside (IPTG), hydrochloric acid (HCl), trifluoroacetic acid (TFA), ammonium hydroxide solution (NH4OH), sulfuric acid, deuterium oxide (D2O), and 5 mm Shigemi advanced NMR microtubes were obtained from Sigma-Aldrich (St Louis, MO, USA). Heptafluorobutyric acid (HFBA) and formic acid were purchased from Fluka (St Louis, MO, USA). UDP-Xyl was supplied by Complex Carbohydrate Research Center (Athens, GA, USA). GA and GX2 were obtained from Toku-E (Bellingham, WA, USA). PM was prepared from paromomycin (Sigma-Aldrich) by hydrolysis as described in the Supplementary Note. HPLC-grade acetonitrile (MeCN), methanol (MeOH), chloroform (CHCl3), and water were acquired from JT Baker (Philipsburg, NJ, USA). Cationic solid-phase exchanger (OASIS MCX SPE, 3 ml/60 mg), Xselect® CSH column XP (2.1×100 mm, 2.5 μm), leucine encephalin, and vacuum manifold were products of Waters Inc. (Milford, MA, USA). Kromasil 1005C18 column (4.6×250 mm, 5 μm) was supplied by Eka Chemicals AB (Bohus, Sweden). Restriction endonucleases and T4 DNA ligase were purchased from New England BioLabs (Ipswich, MA, USA). Polymerase chain reactions were carried out using PrimeSTAR® GXL DNA polymerase from Takara Bio Inc. (Kusatsu, Shiga, Japan). All other chemicals were of the highest purity available.
Bacterial strains and plasmids used in this study are listed in Supplementary Table 4. The gentamicin-producing M. echinospora ATCC 1583541 was obtained from the American Type Culture Collection (Manassas, VA, USA). For genomic DNA isolation and cultivation, M. echinospora was grown for 5 d at 28 °C in ATCC 172 medium (1% glucose, 0.5% yeast extract, 2% soluble starch, 0.5% N-Z amine, and 0.2% CaCO3).
E. coli DH5α42 and plasmid pGEM-T Easy Vector (Promega, Madison, WI, USA) were used for routine subcloning. The pET expression vectors, such as pET15b and pET28a, were purchased from Novagen (Madison, WI, USA). For expression of the recombinant proteins (GenM1, GenD2, GenS2, GenN, GenD1, GenQ, GenB1, GenJ, and GenK2), the E. coli BL21(DE3) (Novagen), E. coli BL21(DE3)pLysS (Novagen), E. coli Rosetta gamiTM2(DE3) (Novagen) and E. coli ArcticExpress(DE3) (Agilent technologies, Santa Clara, CA, USA) were used as heterologous hosts. The E. coli strains were grown in LB liquid medium. Ampicillin (50 μg/ml), chloramphenicol (25 μg/ml), gentamicin (20 μg/ml), and kanamycin (50 μg/ml) were selectively added to the growth media as required. For the recombinant GenM2, the high-copy-number E. coli-Streptomyces shuttle vector pSE34 containing the constitutive ermE* promoter plus a thiostrepton resistance marker was used for expression43. S. venezuelae ATCC 1543944, which was used as a heterologous host for preparing recombinant GenM2, was grown in liquid R2YE45.
Chemical synthesis.
The chemical synthesis and structures of GB, GA2, JI-20A, 2′DGA2, 2′DGX2, 2′DPM, 2′DNM, 6′A2′DGA2, 6′AGA2, and [2′−2H]-JI-20A are described in the Supplementary Note.
UPLC-qTOF-HR-MS analysis and structural identification of gentamicin biosynthetic intermediates.
UPLC-qTOF-HR-MS analysis of the gentamicin biosynthetic intermediates generated by chemical and enzymatic synthesis was performed on a Waters XEVO® G2S Q-TOF mass spectrometer coupled with a Waters Acquity UPLC® system equipped with an Xselect® CSH column XP (2.1×100 mm, 2.5 μm) consisting of an Acquity I-Class system. A gradient elution using solvent A (50 mM ammonium-TFA, pH 2.0) and solvent B (50% aq. acetonitrile with 50 mM ammonium-TFA, pH 2.0) as the mobile phase at a flow rate of 0.2 ml/min at 40 °C was applied. The MS system was operated in ESI with a positive ionization mode. The typical operating parameters were as follows: analyzer, resolution mode; capillary voltage (volt.), 3.0 kV; sampling cone volt., 30 V; source temperature (temp.), 120 °C, source offset temp., 80 °C; desolvation temp., 300 °C; cone gas flow, 10 l/h; desolvation gas flow, 600 l/h: helium collision gas. The analyzer was operated with an extended dynamic range at 60,000 resolution (FWHM at m/z 556) with an acquisition time of 0.1 s. Leucine enkephalin (400 pg/μl, 50% aq. acetonitrile with 0.1% formic acid) as a lockspray was infused at a rate of 5 μl/min for mass correction. Mass spectra were acquired with a scan range of 50−600 amu with scan time 0.1 s.
NMR spectra of GB, PM, GA2, GA, GX2, JI-20A, 2′DGA2, 2′DGA, 2′DGX2, 2′DPM, 2′DNM, 6′A2′DGA2, 6′A2′DGA, 6′AGA2, and 6′AGA were acquired using a Bruker Ascend™ 800 MHz spectrometer or a Varian INOVA 500 MHz spectrometer. Details regarding the characterization of gentamicin biosynthetic intermediates are described in the Supplementary Note. The retention behaviors of compounds 8, 11−15, 17, 25−28, 30−32, 34, and 35 on UPLC-qTOF-HR-MS and data including MS/MS and NMR spectra are described in the Supplementary Note.
Genome sequencing of M. echinospora ATCC 15835.
To determine the genome sequence of strain ATCC 15835, sequencing was performed using the Ion Torrent PGM™ sequencer by GenoTech Corporation (Daejeon, Republic of Korea). The genomic libraries (~200 bp) were constructed by Ion Xpress Plus Fragment Library Kit, and nucleotide sequences with 115-fold coverage of the ATCC 15835 genome were generated. A genome assembly was accomplished by the CLC Genomics Workbench (CLC bio, Inc.). Genome annotation was performed using NewGAS (New Genome Annotation Systems) which is a bioinformatic analysis and database program consisting of automatic genome prediction, annotation pipeline, and NewGAS browser. RNAmmer and tRNAscan-SE were respectively used to detect rRNA and tRNA genes. Protein coding genes were predicted by Glimmer 3.0. AutoFACT was used for automatic functional assignment of the coding sequences with the public databases, GenBank, COG, UniRef90, and KEGG.
Construction of protein expression plasmids.
For recombinant protein expression, the genD2, genS2, genN, genD1, and genB1 genes were codon-optimized for expression in E. coli and synthesized from DNA 2.0 (Menlo Park, CA, USA). Meanwhile, the four genes encoding GenM1, GenQ, GenJ, and GenK2 were obtained by PCR-amplification using the genomic DNA of M. echinospora ATCC 15385 as a template. Each primer sequence contained a restriction site to facilitate cloning of the resulting DNA fragment (see Supplementary Table 5 for oligonucleotide primers). All DNA fragments were cloned into pJExpress404, pET15b, or pET28a to encode a histidine-tagged construct, respectively (see Supplementary Table 4).
To obtain the recombinant GenM2, PCR-amplified DNA fragment (see Supplementary Table 5 for oligonucleotide primers) was cloned into pET28a to generate a histidine-tagged construct, and the digested genM2 fragment was ligated into the complementary sites of pSE3443 (see Supplementary Table 4).
Overexpression of recombinant proteins.
For the expression of GenM1, the expression plasmid pGENM1 was introduced into E. coli ArcticExpress(DE3), and the ArcticExpress(DE3)/pGENM1 strain (see Supplementary Table 4) was grown in LB medium supplemented with 50 μg/ml kanamycin and 20 μg/ml gentamicin. Each liter of culture was inoculated with 10 ml of overnight starter culture. The culture was grown at 37 °C to an optical density (OD600) of 0.6, then expression was induced with 1 mM IPTG. At the same time, the incubation temperature was shifted from 37 to 13 °C and the culture was grown for another 24 h. Recombinant GenM1 was purified as described below. Typically, approximately 0.2 mg of GenM1 was obtained from 1 l of culture (see Supplementary Fig. 2).
For the expression and purification of GenM2, the expression plasmid pGENM2 was transformed into S. venezuelae, yielding the corresponding recombinant strain, S. venezuelae/pGENM2 (see Supplementary Table 4). S. venezuelae protoplast formation and transformation procedures were performed as previously described45. The recombinant strain of S. venezuelae harboring pGENM2 was grown in R2YE medium at 30 °C for 5 d. Recombinant GenM2 was purified as described below. Typically, approximately 0.5 mg of GenM2 was obtained from 1 l of culture (see Supplementary Fig. 2).
For the expression and purification of GenD2, GenS2, and GenN, the synthesized expression plasmids pGEND2 and pGENN were individually introduced into E. coli BL21(DE3), while the pGENS2 was introduced into E. coli BL21(DE3)pLysS. The recombinant BL21(DE3)/pGEND2 and BL21(DE3)/pGENN strains (see Supplementary Table 4) were grown in LB medium supplemented with 50 μg/ml kanamycin. The BL21(DE3)pLysS/pGENS2 strain (see Supplementary Table 4) was grown in the same medium in the presence of 50 μg/ml ampicillin and 25 μg/ml chloramphenicol. Each liter of culture was inoculated with 10 ml of overnight starter culture, and the resulting mixture was grown at 37 °C to an OD600 of 0.6. Overexpression of the desired proteins was induced by 0.5 mM IPTG at 18 °C for another 12 h. Recombinant GenD2, GenS2, and GenN were purified as described below. Typically, approximately 3 mg, 2.5 mg, and 3 mg of purified GenD2, GenS2, and GenN were obtained, respectively, from 1 l of culture (see Supplementary Fig. 2).
For the expression and purification of GenD1, the expression plasmid pGEND1 was introduced into E. coli BL21(DE3)pLysS, and the BL21(DE3)pLysS/pGEND1 strain (see Supplementary Table 4) was grown in LB medium supplemented with 50 μg/ml ampicillin and 25 μg/ml chloramphenicol. Each liter of culture was inoculated with 10 ml of overnight starter culture. The culture was grown at 37 °C to an OD600 of 0.6, then expression was induced with 1 mM IPTG and 2 mM Fe(ΙΙ)SO4. The culture was grown for another 3 h. Recombinant GenD1 was purified and then reconstituted with iron and sulfide in an anaerobic glovebox according to an established procedure18. Typically, approximately 18 mg of GenD1 was obtained from 1 l of culture (see Supplementary Fig. 2).
For the expression and purification of GenQ and GenB1, the expression plasmids pGENQ and pGENB1 were introduced into E. coli Rosetta gamiTM2(DE3) and E. coli BL21(DE3)pLysS, respectively. Each liter of culture was inoculated with 10 ml of overnight starter culture. The Rosetta gamiTM2(DE3)/pGENQ strain (see Supplementary Table 4) was grown in LB medium containing 50 μg/ml kanamycin at 37 °C to an OD600 of 0.6, then expression was induced with 0.5 mM IPTG at 18 °C for another 12 h. Meanwhile, the BL21(DE3)pLysS/pGENB1 strain (see Supplementary Table 4) was cultured in LB medium supplemented with 50 μg/ml ampicillin and 25 μg/ml chloramphenicol until an OD600 of 0.6 was reached. IPTG was then added to the culture to a final concentration of 0.5 mM, and the culture was continued to grow at 18 °C for another 16 h. Recombinant GenQ and GenB1 were also purified as described below. Typically, approximately 1.5 mg and 2 mg of purified GenQ and GenB1 were obtained, respectively, from 1 l of culture (see Supplementary Fig. 2).
For the expression and purification of GenJ and GenK2, the expression plasmids pGENJ and pGENK2 were separately introduced into E. coli BL21(DE3). The recombinant BL21(DE3)/pGENJ and BL21(DE3)/pGENK2 strains (see Supplementary Table 4) were grown in LB medium supplemented with 50 μg/ml kanamycin. Each liter of culture was inoculated with 10 ml of overnight starter culture, and the culture was grown at 37 °C to an OD600 of 0.6. Overexpression of the proteins was induced by 1 mM IPTG at 16 °C for another 20 h. Recombinant GenJ and GenK2 were purified as described below. Typically, approximately 7 mg and 4 mg of purified GenJ and GenK2 were obtained, respectively, from 1 l of culture (see Supplementary Fig. 2).
Protein purification.
Cells were harvested by centrifugation (2 min at 20,000 rpm), re-suspended in lysis buffer (300 mM NaCl, 10 mM imidazole, 50 mM sodium phosphate, pH 8.0), and then lysed by sonication for 5 min using a 2 s on / 2 s off cycle. The lysate was clarified by centrifugation (30 min at 20,000 rpm), and the clarified cell lysate was passed through a column of Ni-nitrilotriacetic acid (NTA) Agarose (Qiagen, Valencia, California, USA). After washing the column with washing buffer (300 mM NaCl, 40 mM imidazole, 50 mM sodium phosphate, pH 8.0), histidine-tagged recombinant proteins were eluted with elution buffer (300 mM NaCl, 500 mM imidazole, 50 mM sodium phosphate, pH 8.0). Imidazole was removed from the purified recombinant protein solution using a PD10 column (GE Healthcare, Piscataway, NJ, USA) with Tris buffer (20 mM Tris-HCl, 250 mM NaCl, 10% glycerol, pH 7.9). The purified proteins were concentrated using an Amicon Ultracel 10K molecular weight cut-off spin filter (Millipore, Bedford, MA, USA), and then stored at -80 °C. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis was employed to ascertain the purity of protein. Protein concentration was determined by the Bradford protein assay using bovine serum albumin as a standard.
Enzyme assays.
First glycosylation by GenM1 was performed in 50 mM sodium phosphate buffer (pH 8.0) containing 0.5 mM DOS, 20 mM glycosyl donor (UDP-Glc or UDP-GlcNAc), 50 μM purified GenM1, and 5 mM MgCl2 at 30 °C for 12 h.
Second glycosylation by GenM2 was performed in 100 mM potassium phosphate buffer (pH 8.0) containing 5 mM glycosyl acceptor (PM, 2′DPM, NM, or 2′DNM), 1.8 mM UDP-Xyl, 0.2 mM purified GenM2, and 10 mM MgCl2 at 37 °C for 12 h.
C3″-methylamination by GenD2-GenS2-GenN was performed in 50 mM Tris-HCl (pH 7.5) containing 0.1 mM substrate (GA2, 2′DGA2, 6′A2′DGA2, or 6′AGA2), 20 μM each enzyme, 2 mM NAD+, 2 mM L-glutamine, and 2 mM SAM at 30 °C for 12 h.
C4″-methylation by GenD1 was performed in 50 mM Tris-HCl (pH 8.0) containing 0.1 mM substrate (GA, 2′DGA, 6′A2′DGA, or 6′AGA), 15 μM purified reconstituted GenD1 (according to an established procedure19), 1 mM methylcobalamin, 1 mM benzyl viologen, 10 mM DTT, 4 mM NADPH, and 4 mM SAM. Assay was carried out in an anaerobic chamber (25 °C) for 12 h.
GenQ-GenB1-mediated C6′-amination was performed in 50 mM Tris-HCl (pH 8.0) containing 0.1 mM GX2, 10 μM each enzyme, and 2 mM L-glutamine at 30 °C for 12 h. For the seven substrates showing the weak GenQ-GenB1-mediated C6′-amination activity (PM, GA2, GA, 2′DGA2, 2′DGA, 2′DGX2, or 2′DPM), reactions were carried out by supplementing 0.1 mM each enzyme.
GenJ-GenK2-mediated C2′-deamination was performed in reaction buffer (40 mM Tris-HCl, 200 mM KCl, 10% glycerol, pH 7.5) containing 0.1 mM substrate (GA2, GA, GX2, JI-20A, 6′AGA2, or 6′AGA), 1.5 μM each enzyme, 1 mM α-ketoglutarate, 1 mM NADPH, and 1 mM (NH4)2Fe(SO4)2·6H2O at 28 °C for 12 h.
The reaction was quenched with chloroform and centrifuged at 13,000 rpm for 10 min. The supernatant containing the product of interest was extracted using OASIS MCX SPE cleanup16, reconstituted with water, and was then analyzed by UPLC-qTOF-HR-MS as described above. Independent experiments were performed 5 or more times. The conversion yield was calculated by dividing the molar equivalent of a product formed by the molar equivalent of the consumption of its reactant. The integrated area under curve (AUC) from the UPLC-qTOF-HR-MS chromatogram was calculated. Calibration curves of each gentamicin intermediate were generated using chemically synthesized, commercially available, or enzymatically synthesized compounds, and were used to convert AUC into the amount of each compound.
Production of 2′DGA, 6′A2′DGA, and 6′AGA by large scale enzyme reactions.
A series of new gentamicin biosynthetic intermediates, 2′DGA, 6′A2′DGA, and 6′AGA were formed from the respective 2′DGA2, 6′A2′DGA2, and 6′AGA2 by one-pot enzymatic reaction. Purified GenD2-GenS2-GenN (40 μM) was incubated with 11 mM substrate (2′DGA2, 6′A2′DGA2, or 6′AGA2), 2 mM NAD+, 2 mM L-glutamine, and 2 mM SAM in 50 mM Tris-HCl (pH 7.5) at 30 °C for 12 h. The reaction was quenched with chloroform and centrifuged at 13,000 rpm for 10 min. The resulting supernatant containing the product of interest was extracted using OASIS MCX SPE cleanup16, and then subjected to UPLC-qTOF-HR-MS analysis. Further reaction was performed until substrate was converted completely.
To obtain these new intermediates (2′DGA, 6′A2′DGA, and 6′AGA), HPLC purification was done using an analytical Kromasil 1005C18 column (4.6×250 mm, 5 μm) on an YL9100 HPLC system (YL Instrument Co. Ltd., Republic of Korea) consisting of a YL9110 gradient pump coupled with a YL9160 photo diode array detector with 1024 channels, and a YL9181 ELSD detector. The fraction containing 2′DGA (3.3 mg), 6′A2′DGA (1.8 mg), and 6′AGA (2.4 mg) was purified by analytical HPLC employing 2% aq. MeCN with 0.05% TFA or 10% aq. MeCN with 10 mM HFBA as the mobile phase with a flow rate of 0.7 ml/min to yield the pure compounds.
GenJ-catalyzed deamination reaction for mechanistic study.
Reactions to determine the activity of GenJ in the deamination of JI-20A to form a keto intermediate were performed by reduction of the product with NaBH4 or NaBD4 for 2 h prior to analysis, or by derivatization of the product with phenylhydrazine. Specifically, 40 μM GenJ was incubated with 1 mM JI-20A, 1 mM α-ketoglutarate, 1 mM (NH4)2Fe(SO4)2∙6H2O in the present of 200 mM KCl, 10% glycerol, and 40 mM Tris-HCl buffer (pH 7.5) at room temperature for 20 h. Excess NaBD4 or NaBH4 was added into the enzyme assay solution and reacted on ice for 45 min. The reaction was quenched by passage through a YM-10 Microcon centrifugal filter and then subjected to ESI-MS analysis. In a separate experiment, the GenJ reaction was treated with 10 mM phenylhydrazine for 2 h. After dinitrophenyl (DNP) derivatization14, the MS measurement was carried out by direct injection into an Agilent 6530 Accurate Mass QTOF-MS at 0.15 ml/min. The results are illustrated in Supplementary Fig. 7.
To determine the mechanism of GenJ-catalyzed deamination and establish the site of the reaction of JI-20A, reactions were carried out with 1 mM [2′−2H]-JI-20A using the same conditions described above (see Supplementary Fig. 8).
The GenJ assays were also conducted under 18O-enriched atmospheres to confirm the oxygen-dependency of GenJ catalysis. Reactions were performed by supplementing 16O2/H216O, 18O2/H216O, 16O2/H218O, or 18O2/H218O under the same conditions as mentioned above. The extend of 18O incorporation in the ketone product under 18O-enriched atmospheres was analyzed as described above, and the results are illustrated in Supplementary Fig. 9 and Table 1.
Transcriptional analysis using semi-quantitative RT-PCR.
Total RNA was isolated from M. echinospora, which had been grown in gentamicin production medium as described previously18. The RNA preparations were treated with DNase I (Qiagen, Hilden, Germany) to eliminate possible chromosomal DNA contamination. Analysis of semi-quantitative RT-PCR was performed with OneStep RT-PCR kit (Qiagen) using 100 ng of the total RNA as a template. The RT-PCR conditions were as follows: cDNA synthesis, 50 °C for 30 min followed by 95 °C for 15 min; amplification, 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min. The cycle number for each gene was optimized in order to obtain sufficient visibility of the RT-PCR band and ensure that amplification was in the linear range and the results were semi-quantitative. The oligonucleotide primers were designed to generate PCR products approximately 500 bp by software Primer3 and summarized in Supplementary Table 5. With each pair of primers, the negative controls were carried out with Taq DNA polymerase (GenoTech Corporation) in the absence of reverse transcriptase to confirm that amplified products were not derived from the chromosomal DNA, whereas the 16S rRNA was used as a positive control.
Purification of GenB1 for its structure determination.
The BL21(DE3)pLysS/pGENB1 recombinant strain was cultured as described above, and cells were harvested by centrifugation. The re-suspended cells in the A-buffer consisting of 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 2 mM β-mercaptoethanol were disrupted by sonication. The crude lysate was centrifuged at 10,000 rpm for 40 min at 4 °C and the cell debris was discarded. The supernatant was loaded onto a nickel sepharose fast flow column (GE Healthcare) and washed with 25 mM imidazole in A-buffer to remove the impurities. Then, retained proteins were eluted isocratically with 500 mM imidazole in A-buffer. Eluted fractions were pooled based on SDS-PAGE analysis. The pooled fractions were concentrated by the concentrator (Milipore, Bedford, MA, USA) and loaded onto a HiLoad 16/60 Superdex-200 prep-grade column (GE Healthcare) equilibrated with the B-buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 2 mM DTT). After the final purification step, the solution was concentrated to ~12 mg/ml and stored at −80 °C. Selenomethionine (Se-Met) substituted GenB1 was produced in the E. coli BL21-CodonPlus (DE3)-RIL-X methionine auxotroph (Novagen) and purified as described above.
Crystallization and data collection of GenB1.
Crystallization experiments with commercially available solution kits from Microlytic, Hampton Research and Emerald Biosystems were performed using the vapor microbatch diffusion crystallization methods at 22 °C: drops of 1 μl protein solution and 1 μl crystallization reagent were mixed under a thin layer of AL’s Oil, a 1:1 mixture of Paraffin (Sigma-Aldrich, St Louis, MO, USA) and Silicone oil (Hampton Research, Aliso Viejo, CA, USA), in 72-well HLA plates (NUNC, Roskilde, Denmark). Initial crystals of holo-GenB1 were grown in two precipitant solutions. One used condition No. 92 of Index from Hampton Research (100 mM magnesium formate and 15% polyethylene glycol (PEG) 3350) and the other used condition No. 67 of SGC from Microlytic (20% PEG 1500, 200 mM MgCl2, 100 mM Tris buffer, pH 8.5). To obtain diffraction quality crystals, the first condition was optimized to 150−200 mM magnesium formate and 15−17% PEG 3350 and the second condition was also changed to 22−25% PEG 1000, 200−150 mM MgCl2, and 100 mM Tris buffer (pH 8.0−8.6). Orthorhombic crystals appeared in a few days. In the case of Se-Met substituted holo-GenB1, crystals were obtained in precipitant solutions consisting of 24% PEG 1000, 200−250 mM MgCl2, and 100 mM Tris buffer (pH 8.0−8.6). For data collection, crystals were cryo-protected using the same precipitant solutions supplemented with 25% glycerol and were flash-frozen in a liquid nitrogen cold stream at -173 °C. Diffraction data were collected at beamlines 5C and 7A of Pohang Light Source (PLS), Republic of Korea. Crystals of holo-GenB1 and Se-Met substituted holo-GenB1 diffracted to 1.7 Å and 1.8 Å resolutions, respectively.
To get crystals of GenB1 complexed with NM or JI-20A, NM or JI-20A dissolved in distilled water was added to holo-GenB1 with a 1:20 molar ratio, and those mixtures were incubated on ice for 1 h. To secure crystals of these complexes, we used the same crystallization solutions for holo-GenB1 and succeeded in collecting diffraction data with crystals grown in precipitation solutions containing 15% polyethylene glycol 3350 and 150 mM magnesium formate. For data collection, Paratone-N or NVH oil was added to the precipitant solutions as a cryo-protectant. Crystals of GenB1 complexed with NM and JI-20A diffracted to 1.76 and 1.4 Å resolutions, respectively, at the beamline 5C and 7A of PLS, Republic of Korea.
All the crystals belonged to the space group P212121 with two molecules in the asymmetric unit, which corresponds to a crystal volume per protein weight of ~2.10 Å3/Da and 41.5% solvent content (Matthews, 1968). All the data were processed and scaled with the program DENZO and SCALEPACK from HKL2000 program suite46. The statistics of data collection are summarized in the Supplementary Table 2.
Structure determination and refinement of GenB1.
The structure of holo-GenB1 was solved using single-wavelength anomalous dispersion (SAD) technique. A reasonable phase was obtained with a mean FOM of 0.371 to a resolution of 1.8 Å and all 10 potential selenium sites were found and refined using PHENIX47. The resulting electron-density maps with a partial model revealed clear main chain density with substantial side-chain details. The GenB1/PLP/NM and GenB1/PLP/JI-20A complex structures were solved by molecular replacement (MR) with holo-GenB1 as a search model. Rounds of manual model building and refinement were performed using COOT48 and PHENIX, respectively. From the beginning of the refinement, 5% of the total reflections were set aside for monitoring Rfree value. The final models of holo-GenB1, GenB1/PLP/NM, and GenB1/PLP/JI-20A include GenB1 residues 9−416 of two copies. The quality of the final models was judged using MolProbity49. The Ramachandran plots indicate 99.0% (GenB1/PLP), 99.0% (GenB1/PLP/NM), and 99.0% (GenB1/PLP/JI-20A) of residues are in the most favored regions. Statistics on data collections and refinements are summarized in Supplementary Table 2. Solvent accessible area and interaction area were calculated by PISA (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html). Sequence alignment was generated using Espript50, and figures were generated using PyMOL51.
Mutational study of GenB1.
For the D345L, D395L, W391A, and W415A substitutions in GenB1, site-directed mutagenesis was carried out using the QuikChange kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s protocols. All primers required for the amplification of DNA fragments containing the mutated GenB1 are listed in Supplementary Table 5. The four mutant proteins were purified, and then examined the C6′-amination activity toward GX2 (Supplementary Fig. 15).
Cytotoxicity of new gentamicin intermediates against mammalian renal cell lines.
To examine the in vitro nephrotoxicity of the newly identified gentamicin intermediates (GA2, GA, and GX2 analogs), a series of cell toxicity assays were carried out using three kidney-derived mammalian cell lines from ATCC, i.e., HEK-293 (human), LLC-PK1 (pig), and COS-7 (monkey). After pre-culturing the cell lines in Dulbecco’s Modified Eagle’s Medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS, 1% glutamine, and 1% penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere, the cells were centrifuged and placed into 96-well microplates (5×103 cells/well). After 1 d, the medium was replaced with a medium without streptomycin. The new gentamicin intermediates were then added to a final concentration of 20−400 μM. The MTT based colorimetric assay was performed after 2 d, according to the manufacturer’s instructions. Cell viability was calculated as the ratio of the numbers of living cells in cultures grown in the treatments, versus those grown without treatments. The concentration of half-maximal lethal dose for cells (LC50) was estimated by fitting concentration response curves to the data obtained from at least two independent experiments, using SigmaPlot software package 10.0.1 (Systat Software Inc., San Jose, CA, USA) (see Supplementary Fig. 17). Data were expressed as means (n=4) ± standard deviations and tested for significance using paired or unpaired two-tailed t-test with analysis of variance as appropriate. Results with P<0.01 were considered significant.
Antibacterial activity of new gentamicin intermediates.
Minimal inhibitory concentration (MIC) test of the gentamicin intermediates (GA2, GA, and GX2 analogs) was performed with the broth dilution method following the Clinical Laboratory Standards Institute (CLSI) recommendation (CLSI, 2018)52,53. Properly diluted Muller Hinton ΙΙ broth (BBL, Sparks, MD, USA) containing each newly synthesized gentamicin intermediates was prepared in a 96 well plate. An aliquot containing each isolate grown at log phase was inoculated in a well containing various concentrations of antimicrobial agents ranging from 0.25 to 128 μg/ml. The final CFUs of bacteria was approximately 5×104 CFU/spot. Then plates were incubated at 35 °C for 18 h under ambient condition. MIC is the lowest concentration of antimicrobial agent that completely inhibits the growth of an organism. A total of 8 test strains were employed to determine the antibacterial spectra of gentamicin biosynthetic intermediates. Four of these strains were type strains: gentamicin-sensitive E. faecalis ATCC 29212, P. aeruginosa ATCC 27853, S. aureus ATCC 29213, and E. coli ATCC 25922. The remaining four strains were clinical isolates obtained from the Culture Collection of Antimicrobial Resistant Microbes (CCARM, Republic of Korea): gentamicin-resistance E. faecalis CCARM 5025, P. aeruginosa CCARM 2002, S. aureus CCARM 3180, and E. coli CCARM 1085. Each MIC test performed in triplicated.
PTC readthrough activity of new gentamicin intermediates.
The readthrough (or suppression of nonsense mutation) activity of the newly synthesized gentamicin intermediates (GA2, GA, and GX2 analogs) was tested ex vivo using primary human cystic fibrosis bronchial epithelial cells (Cystic fibrosis transmembrane conductance regulator [CFTR]; p.ΔF508/W1282X; Asterand Bioscience, Detroit, MI, USA). After pre-culturing in RPMI-1640 medium (Invitrogen) supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere, the cells were separated by centrifugation and cultured for 5 d in T-flasks. On the third day of culturing, the new gentamicin intermediates were added to the culture to a final concentration of 25 μM, and the incubation was continued for another 2 d. Each treatment group from three independent experiments was compared with a positive control group treated by the same method with G418, for which readthrough activity had been reported54. The treated cells were harvested as pellets, and nuclear proteins were prepared using CelLytic NuCLEAR extraction kit (Sigma-Aldrich). After being corrected into an equal concentration, 10 μl of each sample was subjected to HPLC-ESI-MS. The analysis of both truncated and full-length CFTR expressed in the treated cells was performed in denatured state using size exclusion chromatography with LCQ ion trap mass spectrometer (Thermo Fisher Scientific, Sunnyvale, CA, USA); MAbPac SEC-1 column (4×300 mm, 5 μm (Thermo Fisher Scientific)) was eluted with 20% aq. MeCN with 0.05% TFA and 0.1% formic acid at an isocratic flow rate of 0.2 ml/min for 30 min. MS spectra of truncated and full-length CFTR were analyzed using Thermo Scientific™ protein deconvolution 2.0 software, which employs the ReSpect™ algorithm for molecular mass determination. Mass spectra for deconvolution were generated by averaging spectra across the most abundant portion of the elution profile for the target biomolecules. The average spectra were subsequently deconvoluted using an input range of m/z 2000 to 4000 for spectra acquired under denaturation state. On the other hand, an output mass range (160,000 to 180,000 Da for full-length CFTR, whereas 130,000 to 150,000 Da for truncated CFTR) and a minimum of at least five consecutive charge states from the input m/z spectrum were utilized to produce the deconvolution results. The amount of the target protein expressed was estimated based on the peak area shown in the resulting deconvoluted spectrum. Thus, the ratio of full-length CFTR to truncated CFTR was determined as comparative readthrough activity (see Supplementary Fig. 18). Data were expressed as means (n=3) ± standard deviations and tested for significance using paired or unpaired two-tailed t-test with analysis of variance as appropriate. Results with P<0.01 were considered significant.
Statistics and reproducibility.
Information regarding the sample size and number of replicates, along with the statistical test used and statistical significance, is provided in the figure legends.
Supplementary Material
Acknowledgments
This work was supported by the National Research Foundation of Korea grant (2016R1A2A1A05005078) (Y.J.Y.) funded by the Ministry of Science and ICT, Cooperative Research Program for Agriculture Science & Technology Development (PJ01316001) (M.C.S.) and (PJ01317901) (J.W.P.) funded by Rural Development Administration, the project titled “Development of biomedical materials based on marine proteins” funded by the Ministry of Oceans and Fisheries (S.-S.C.), Republic of Korea, and the National Institutes of Health (GM035906) and the Welch Foundation (F-1511), USA (H.-w.L.)
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Data availability.
The sequences of genJ and genK2 genes have been deposited in the GenBank under accession nos: MG879478 (genJ); MG879479 (genK2). Atomic coordinates and structure factors of the reported crystal structures have been deposited in the Protein Data Bank under the accession codes: 5Z83 (GenB1/PLP); 5Z8A (GenB1/PLP/JI-20A); 5Z8K (GenB1/PLP/NM).
References
- 1.Park SR, Park JW, Ban YH, Sohng JK & Yoon YJ 2-Deoxystreptamine-containing aminoglycoside antibiotics: recent advances in the characterization and manipulation of their biosynthetic pathways. Nat. Prod. Rep. 30, 11–20 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Magnet S & Blanchard JS Molecular insights into aminoglycoside action and resistance. Chem. Rev. 105, 477–498 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Kondo S & Hotta K Semisynthetic aminoglycoside antibiotics: development and enzymatic modifications. J. Infect. Chemother. 5, 1–9 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Umezawa H, Umezawa S, Tsuchiya T & Okazaki Y 3’,4’-Dideoxy-kanamycin B active against kanamycin-resistant Escherichia coli and Pseudomonas aeruginosa. J. Antibiot. 24, 485–487 (1971). [DOI] [PubMed] [Google Scholar]
- 5.Kawaguchi H, Naito T, Nakagawa S & Fujisawa K BB-K 8, a new semisynthetic aminoglycoside antibiotic. J. Antibiot. 25, 695–708 (1972). [DOI] [PubMed] [Google Scholar]
- 6.Wright JJ Synthesis of 1-N-ethylsisomicin: a broad-spectrum semisynthetic aminoglycoside antibiotic. J. Chem. Soc., Chem. Commun. 0, 206–208 (1976). [Google Scholar]
- 7.Nagabhushan TL, Cooper AB, Tsai H, Daniels PJL & Miller GH The syntheses and biological properties of 1-N-(S-4-amino-2-hydroxybutyryl)-gentamicin B and 1-N-(S-3-amino-2-hydroxypropionyl)-gentamicin B. J. Antibiot. 31, 681–687 (1978). [DOI] [PubMed] [Google Scholar]
- 8.Kondo S, Iinuma K, Yamamoto H, Maeda K & Umezawa H Syntheses of 1-N-{(S)-4-amino-2-hydroxybutyryl}-kanamycin B and −3’,4’-dideoxykanamycin B active against kanamycin-resistant bacteria. J. Antibiot. 26, 412–415 (1973). [DOI] [PubMed] [Google Scholar]
- 9.Becker B & Cooper MA Aminoglycoside antibiotics in the 21st century. ACS. Chem. Biol. 8, 105–115 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Stelzer AC et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 7, 553–559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prayle A & Smyth AR Aminoglycoside use in cystic fibrosis: therapeutic strategies and toxicity. Curr. Opin. Pulm. Med. 16, 604–610 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Park JW, Ban YH, Nam S-J, Cha S-S & Yoon YJ Biosynthetic pathways of aminoglycosides and their engineering. Curr. Opin. Biotechnol. 48, 33–41 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Park JW et al. Discovery of parallel pathways of kanamycin biosynthesis allows antibiotic manipulation. Nat. Chem. Biol. 7, 843–852 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Sucipto H, Kudo F & Eguchi T The last step of kanamycin biosynthesis: unique deamination reaction catalyzed by the α-ketoglutarate-dependent nonheme iron dioxygenase KanJ and the NADPH-dependent reductase KanK. Angew. Chem. Int. Ed. 51, 3428–3431 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Gao W, Wu Z, Sun J, Ni X & Xia H Modulation of kanamycin B and kanamycin A biosynthesis in Streptomyces kanamyceticus via metabolic engineering. PLoS One. 12, e0181971 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park JW et al. Analytical profiling of biosynthetic intermediates involved in the gentamicin pathway of Micromonospora echinospora by high-performance liquid chromatography using electrospray ionization mass spectrometric detection. Anal. Chem. 79, 4860–4869 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Park JW et al. Genetic dissection of the biosynthetic route to gentamicin A2 by heterologous expression of its minimal gene set. Proc. Natl. Acad. Sci. USA. 105, 8399–8404 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C et al. Delineating the biosynthesis of gentamicin X2, the common precursor of the gentamicin C antibiotic complex. Chem. Biol. 22, 251–261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HJ et al. GenK-catalyzed C-6′ methylation in the biosynthesis of gentamicin: isolation and characterization of a cobalamin-dependent radical SAM enzyme. J. Am. Chem. Soc. 135, 8093–8096 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J et al. Specificity and promiscuity at the branch point in gentamicin biosynthesis. Chem. Biol. 21, 608–618 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu Y et al. Biosynthesis of epimers C2 and C2a in the gentamicin C complex. Chembiochem. 16, 1933–1942 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Li S et al. Methyltransferases of gentamicin biosynthesis. Proc. Natl. Acad. Sci. USA. 115, 1340–1345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagman GH et al. Chromatographic separation of some minor components of the gentamicin complex. J. Chromatogr. A 70, 171–173 (1972). [DOI] [PubMed] [Google Scholar]
- 24.Testa RT & Tilley BC Biotransformation, a new approach to aminoglycoside biosynthesis: II Gentamicin. J. Antibiot. 29, 140–146 (1976). [DOI] [PubMed] [Google Scholar]
- 25.Kim HJ, Liu Y. n., McCarty RM & Liu H.-w. Reaction catalyzed by GenK, a cobalamin-dependent radical S-adenosyl-L-methionine methyltransferase in the biosynthetic pathway of gentamicin, proceeds with retention of configuration. J. Am. Chem. Soc. 139, 16084–16087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni X, Sun Z, Gu Y, Cui H & Xia H Assembly of a novel biosynthetic pathway for gentamicin B production in Micromonospora echinospora. Microb. Cell Fact. 15, 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandrika NT & Garneau-Tsodikova S A review of patents (2011–2015) towards combating resistance to and toxicity of aminoglycosides. Med. Chem. Commun. 7, 50–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howard M, Frizzell RA & Bedwell DM Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 2, 467–469 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Fujisawa K, Hoshiya T & Kawaguchi H Aminoglycoside antibiotics. VII Acute toxicity of aminoglycoside antibiotics. J. Antibiot. 27, 677–681 (1974). [DOI] [PubMed] [Google Scholar]
- 30.Popovic B et al. Crystal structures of the PLP- and PMP-bound forms of BtrR, a dual functional aminotransferase involved in butirosin biosynthesis. Proteins 65, 220–230 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Zachman-Brockmeyer TR, Thoden JB & Holden HM The structure of RbmB from Streptomyces ribosidificus, an aminotransferase involved in the biosynthesis of ribostamycin. Protein Sci. 26, 1886–1892 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nudelman I et al. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 52, 2836–2845 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nudelman I et al. Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 18, 3735–3746 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Baradaran-Heravi A et al. Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proc. Natl. Acad. Sci. USA. 114, 3479–3484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Li Y et al. Biosynthesis of the unique amino acid side chain of butirosin: possible protective-group chemistry in an acyl carrier protein-mediated pathway. Chem. Biol. 12, 665–675 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Llewellyn NM, Li Y & Spencer JB Biosynthesis of butirosin: transfer and deprotection of the unique amino acid side chain. Chem. Biol. 14, 379–386 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Llewellyn NM & Spencer JB Chemoenzymatic acylation of aminoglycoside antibiotics. Chem. Commun. 32, 3786–3788 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Hooper IR The naturally occurring aminoglycoside antibiotics. in Handbook of experimental pharmacology Vol. 62 Aminoglycoside antibiotics. (eds. Umezawa H & Hooper IR) 1–35 (Springer-Verlag, Berlin, Heidelberg, Germany,1982). [Google Scholar]
- 39.Jones D, Metzger HJ, Schatz A & Waksman SA Control of Gram-negative bacteria in experimental animals by streptomycin. Science 100, 103–105 (1944). [DOI] [PubMed] [Google Scholar]
- 40.Doroghazi JR & Metcalf WW Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics 14, 611 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinstein MJ et al. Gentamicin, a new antibiotic complex from Micromonospora. J. Med. Chem. 6, 463–464 (1963). [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF & Maniatis T Molecular Cloning: A Laboratory Manual, 3rd ed. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA,2001). [Google Scholar]
- 43.Smirnova N & Reynolds KA Engineered fatty acid biosynthesis in Streptomyces by altered catalytic function of β-ketoacyl-acyl carrier protein synthase III. J. Bacteriol. 183, 2335–2342 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song JY et al. Complete genome sequence of Streptomyces venezuelae ATCC 15439, a promising cell factory for production of secondary metabolites. J. Biotechnol. 219, 57–58 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Kieser T, Bibb MJ, Buttner MJ, Chater KF & Hopwood DA Practical Streptomyces Genetics, Ch. 10, John Innes Foundation, Norwich, England, (2000). [Google Scholar]
- 46.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Zwart PH et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robert X & Gouet P Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrödinger LLC The PyMOL Molecular Graphics System, Version 1.3r1 (2010). [Google Scholar]
- 52.Weinstein MP et al. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 11th ed. (Clinical Laboratory Standards Institute, Wayne, Pa, USA,2018). [Google Scholar]
- 53.Patel JB et al. Performance standards for antimicrobial susceptibility testing, 27th ed. (Clinical Laboratory Standards Institute, Wayne, Pa, USA,2018). [Google Scholar]
- 54.Murphy GJ, Mostoslavsky G, Kotton DN & Mulligan RC Exogenous control of mammalian gene expression via modulation of translational termination. Nat. Med. 12, 1093–1099 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.