

Abstract
Hyoscyamine 6β-hydroxylase (H6H) is an α-ketoglutarate dependent mononuclear nonheme iron enzyme that catalyzes C6-hydroxylation of hyoscyamine and oxidative cyclization of the resulting product to give the oxirane natural product scopolamine. Herein, the chemistry of H6H is investigated using hyoscyamine derivatives with modifications at the C6 or C7 position as well as substrate analogues possessing a 9-azabicyclo[3.3.1]-nonane core. Results indicate that hydroxyl rebound is unlikely to take place during the cyclization reaction and that the hydroxylase versus oxidative cyclase activity of H6H is correlated with the presence of an exohydroxy group having syn-periplanar geometry with respect to the adjacent H atom to be abstracted.
The mononuclear nonheme iron dependent oxidases are an important class of enzymes that catalyze a diverse array of reaction types including hydroxylation, desaturation, epimerization, halogenation and epoxidation.1–4 Members of this enzyme family typically require α-ketoglutarate (α-KG) as a cosubstrate for catalysis and possess a highly conserved His/His/Asp(Glu) facial triad that coordinates the catalytic iron center.1–4 Despite significant progress in understanding this class of enzymes, questions remain regarding how these enzymes are able to catalyze specific transformations thereby preventing alternative reaction outcomes.
An atypical member of this enzyme family is hyoscyamine 6β-hydroxylase (H6H), which is involved in the biosynthesis of the anticholinergic alkaloid scopolamine (1) in the Solanaceae family of plants.5–13 What makes H6H unusual is its versatility, because it can catalyze hydroxylation (2 → 3), dehydrogenation (3 → 1) and in vitro epoxidation (4 → 1) reactions as shown in Scheme 1.7–13 Two other enzymes with similar catalytic properties are clavaminate synthase (CAS)14 and LolO,15,16 which catalyze the conversion of deoxyguanidino proclavaminate (5) to clavaminate (8) and exo-1-acetamidopyrrolizidine (9) to N-acetylnorloline (11), respectively (Scheme 1). Particularly notable are the cyclization reactions (3 → 1, 6 → 7 and 10 → 11), which are also dehydrogenations. 9,13,17 The conversion of (S)-2-hydroxypropylphosphonate (HPP, 12) to fosfomycin (13) is another example of this chemistry also catalyzed by a mononuclear nonheme iron enzyme, HPP epoxidase (HppE), which uses H2O2 rather than α-KG as the oxidant.18,19
Scheme 1.
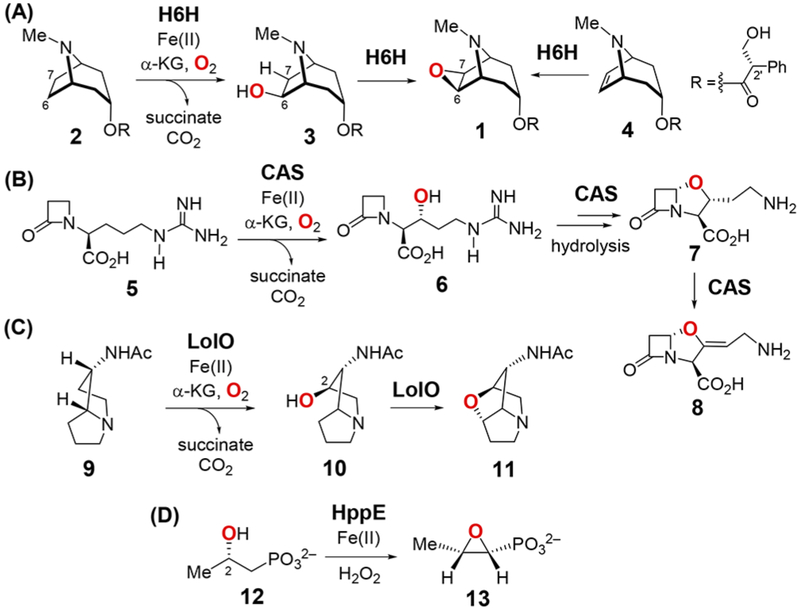
Reactions Catalyzed by (A) H6H, (B) CAS, (C) LolO and (D) HppE
Similar to other α-KG-dependent nonheme iron enzymes,1–4 the hydroxylation reaction catalyzed by H6H is predicted to involve H atom abstraction from C6 of hyoscyamine (2) to give 15 by a high valent iron-oxo species (14) that is generated via the reaction of the α-KG-Fe(II)-substrate complex with O2 (see Scheme 2). Subsequent rebound of the metal-coordinated hydroxide then yields the exo-6-hydroxylated product (15 → 3). A similar iron-oxo intermediary species could also play a key role in formation of the oxirane ring (3 → 1). However, the reasons behind the conversion of H6H to an oxidative cyclase rather than a hydroxylase when 3 is the substrate have yet to be established. We report here the investigation of four substrate analogues as mechanistic probes to delineate those features that determine the outcome of H6H-catalyzed reactions. The results indicate that cyclization does not involve a diol intermediate and that hydroxylation versus cyclization correlates with the degree to which the abstracted exo-hydrogen eclipses the adjacent hydroxy group in the unbound substrate.
Scheme 2.
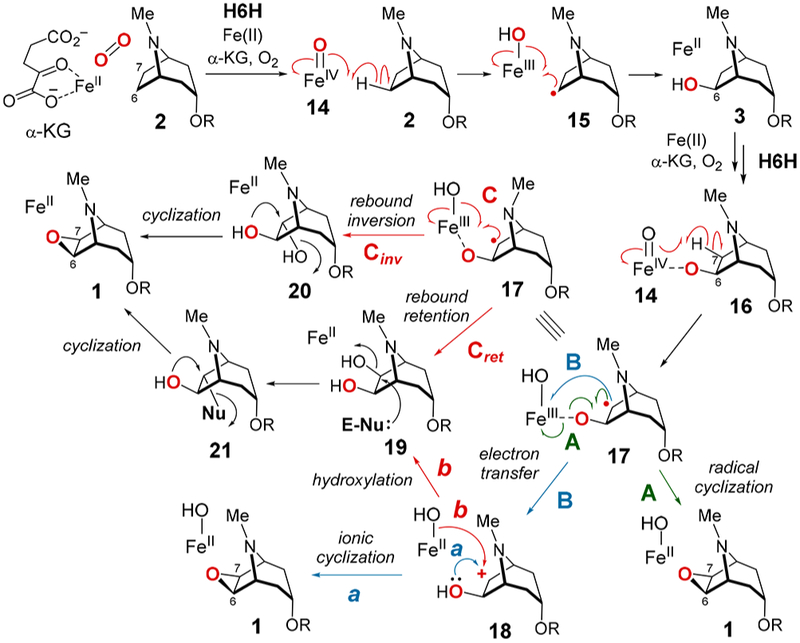
Possible Mechanisms for the Epoxidation of 2
Several possible mechanisms for the conversion of 3 to 1 are shown in Scheme 2. The 6-OH of 3 may coordinate the iron center in the active site of H6H similar to substrate binding in the HppE-catalyzed epoxidation reaction (12 → 13, Scheme 1) 20–23 Such an interaction could facilitate H atom abstraction from the exo-C7 position in 3 by the Fe(IV)═O complex (14) to generate the radical intermediate 1710 prior to radical-mediated cyclization (route A). Ring formation may also result from intramolecular nucleophilic addition of the exo-C6-OH to the C7 carbocation in 18, which would be produced via electron transfer from 17 to the Fe(III)OH complex (route B-a). Alternatively, hydroxyl rebound to 17 could yield a 1,2-diol (19 or 20), which can then be converted to 1 through nucleophilic displacement of the C7 hydroxy group (route C). The dihydroxylation could also occur via a cation intermediate (18, route B-b).
To test whether a 1,2-diol intermediate is formed during the catalytic cycle, 19 and 20 were synthesized (the latter as a 3:2 mixture of 2′S and 2′R diastereomers) and incubated with H6H from Hyoscyamus niger heterologously expressed and purified using Escherichia coli (see Supporting Information). No consumption of 19 (1 mM) was detected under standard assay conditions (aerobic, 68 μM H6H, 0.40 mM FeSO4, 5.0 mM α-KG, 4.0 mM ascorbate, 50 mM tris(hydroxymethyl)-aminomethane (Tris) buffer, pH 7.4). Replacing α-KG with succinate, which is the byproduct derived from α-KG upon formation of the iron-oxo species,1–4 did not change the outcome (Figure S2). These results indicated that 19 is an unlikely intermediate in the conversion of 3 to 1 (Scheme 3).
Scheme 3.
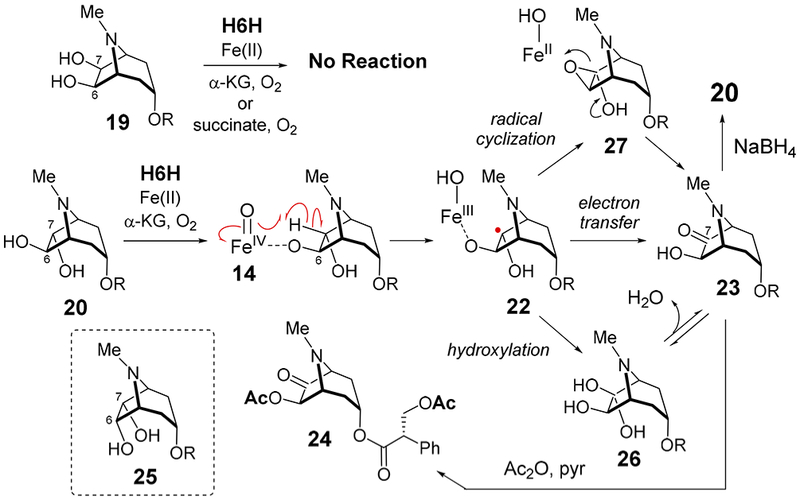
H6H-Catalzyed Oxidation of 20
On the contrary, consumption of (2′S)-20 was observed under the standard assay conditions with α-KG, and a new peak was detected by HPLC different from that of 1; however, no reaction was observed with succinate or (2′R)-20 (see Figure S3). ESI-MS analysis of the reaction product suggested that one of the hydroxy groups in 20 had been oxidized to a carbonyl (calcd m/z for C17H22NO5+ [M + H]+, 320.1492; obsd, 320.1500, see Figure S12). Isolation and ESI-MS analysis of the reaction product following treatment with Ac2O in pyridine (80 °C, 15 min) was consistent with formation of a diacetylated species (e.g., 24, calcd m/z for C21H26NO7+ [M + H]+, 404.1704; obsd, 404.1717, see Figure S12). Furthermore, reduction of the reaction product with sodium borohydride gave a compound that coeluted with (2′S)-20, but not with 6α,7α-dihydroxyhyoscyamine (25, Figure S4). This implies that the keto functionality of the reaction product is at C7 rather than C6, because reduction of the keto group of 23 with NaBH4 should occur from the less hindered exo face to regenerate 20.24 Based on these observations, the reaction product of 20 with H6H was assigned as 7-keto-6β- hydroxyhyoscyamine (23).
As shown in Scheme 3, the 7-keto product 23 could be produced either by rebound of the hydroxyl group from Fe(III)–OH to the radical intermediate (22 → 26), the direct oxidation of 22 via electron transfer to the ferric iron (22 → 23), or oxidative cyclization of 22 to 27 followed by ring opening. When the reaction of 20 (as a 2′S/2′R mixture) was conducted under 18O2, no 18O incorporation was found in 23 (Figure S5). Incorporation of 18O into 23 was observed when the reaction was run in H218O; however, this could be fully explained by hydration of the resulting ketone (Figure S5). Though these results do not rule out the possibility for stereoselective elimination of the exo-OH from 26, they do suggest a mechanism involving either electron transfer (22 → 23) or an oxirane intermediate (22 → 27 → 23). In either case, neither 19 nor 20 appears to be an intermediate during the cyclization of 3 to 1 arguing against the formation of a 1,2-diol intermediate and effectively ruling out routes C and B-b (Scheme 2).
In contrast to 2, which is hydroxylated in the presence of H6H, compounds 3 and 20 undergo dehydrogenation with the former known to result in an oxirane. These observations raised the question as to what determines hydroxylation versus dehydrogenation. One possibility is that the presence of an exo-hydroxy functionality vicinal to the site of H atom abstraction plays a role in redirecting reaction flux to dehydrogenation. As a test of this hypothesis, 7β-hydroxyhyoscyamine (28)25,26 was prepared and incubated with H6H (Scheme 4). In this experiment, formation of the C6-hydroxylated product 19 from 28 was expected assuming the presence of the exo C7-OH leaves the H6H catalytic cycle essentially unperturbed compared to that for 2. Instead, however, 28 was slowly cyclized to 1 in the presence of H6H under standard conditions (Scheme 4), which was confirmed by ESI-MS spectroscopy (calcd m/z for C17H22NO4+ [M + H]+, 304.1543; obsd, 304.1554) and coelution with a standard (Figures S6 and S12). No dihydroxylated product (e.g., 19) could be detected. Hence, the presence of an exo-hydroxy group at either C6 or C7 indeed influences the course of the reaction. Consistent with previous reports,10 the observation that 3, 20 and 28 are all substrates for H6H whereas 19 is not implies that H atom abstraction can occur from either the exo-C6 or exo-C7 position but not from the corresponding endo positions.
Scheme 4.

H6H-Catalyzed Oxidation of 28
To explore the relationship between substrate structure and reaction course, an analogue with a 9-azabicyclo[3.3.1]nonane core (30) was synthesized (Scheme 5A). Two new products in an approximately 3:2 ratio were detected by HPLC when 30 was incubated with H6H for 10 min under standard conditions (Figure S9). These were assigned as the monohydroxylated species 31a (or its isomer 31b) and 34 based on NMR and ESI-MS (calcd m/z for C18H26NO4+ [M + H]+, 320.1856; obsd, 320.1848 and 320.1849 for 31 and 34, respectively, see Supporting Information). The identity of the major product was confirmed to be 34 by coelution with a synthetic standard (Figure S10). Upon further incubation, these products are consumed with concomitant formation of two new species in a roughly 3:1 ratio both having masses consistent with dihydroxylated derivatives of 30 (calcd m/z for C18H26NO5+ [M + H]+, 336.1805; obsd, 336.1799 and 336.1795, see Figures S9 and S12). The major dihydroxylated species was identified as the 6β,7β-dihydroxylated compound 33a (or its isomer 33b) by HPLC coelution with a mixed synthetic standard of 33a and 33b (Figure S11). Purification of the monohydroxylated products (31 and 34) followed by reincubation with H6H showed that both could be converted to 33 (Figure S10). Though the structure of the minor dihydroxylated species could not be determined, it does not coelute with the mixed standard of 33a and 33b by HPLC. The only evidence for cyclization was an LCMS signal consistent with 32 or a C6/C8-keto derivative of 30 (calcd m/z for C18H24NO4+ [M + H]+, 318.1700; obsd, 318.1695, see Figure S12) when purified 30 was incubated with H6H. However, the MS signal intensity of this species was less than 5% relative to that of the major dihydroxylation product (33). Therefore, if cyclization does take place, then it is only a very minor side reaction.
Scheme 5.
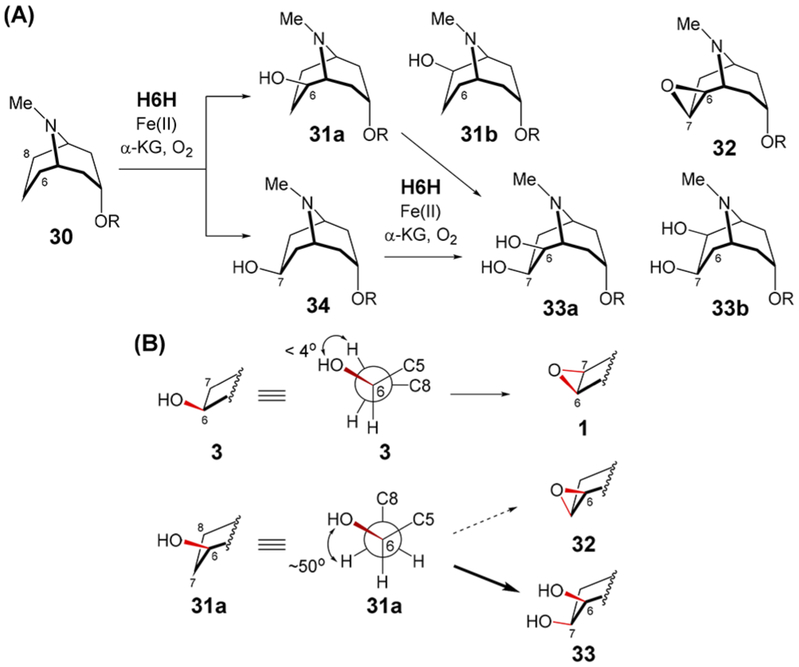
(A) H6H-Catalyzed Oxidation of 30; (B) Correlation of H6H-Catalyzed Reaction Outcome with Substrate Conformation
The observation that H6H catalyzes cyclization of hydroxy-azabicyclo[3.2.1]octanes (3 and 28) and hydroxylation of hydroxy-azabicyclo[3.3.1]nonanes (31 and 34) suggests that the orientation of the exo-C—H bond to be broken versus the adjacent exo-C—O bond is correlated with the subsequent reaction course. The X-ray crystal structure of 30 shows that it adopts a staggered conformation with an H—C6—C7—H dihedral angle of 49° (Figure S8). Furthermore, gas phase RB3LYP/6-31G* computations of models of 31a (with R = CH3) implied that a similar HO—C6—C7—H dihedral angle is retained in 31a (see Scheme 5B and Figure S42), which is consistent with characterization by NMR (see Supporting Information). Likewise, computational models of 34 (R = CH3) show H—C6—C7—OH dihedral angles ranging from 35° to 52° with respect to the exo-C6—H bond (see Figure S40). This continues a general trend of staggered substrate conformations observed among other α-KG-dependent nonheme iron enzymes catalyzing the β-hydroxylation of alcohols to produce vicinal diols (e.g., OrfP,27 PolL,28 GA 2β oxidase,29 KdoO,30 BcmG31,32 and RbtG33). In contrast, gas phase models of 3 (R = CH3) exhibit dihedral angles less than 4° versus the exo-C7—H bond (see Scheme 5B and Figure S38).
One possible explanation for this correlation is that a similar geometric arrangement is maintained between the exo-C—OH bond and the adjacent partially filled p-orbital in the radical intermediate resulting from H atom abstraction. This would then facilitate cyclization of radicals derived from 3 and 28 (i.e., 17 and 29) as opposed to oxygen rebound for radicals derived from 31 and 34. However, though gas phase UB3LYP/6-31G* computations indicated that the HO—C6—C7 p angle increases to no more than 22° in the modeled radicals derived from 3 (R = CH3, see Figure S39), angles less than 15° (i.e., periplanar) could also be found among optimized conformers of radicals derived from 31 and 34 (see Figures S41 and S43). Therefore, interactions between the enzyme and bound substrate may be important to maintain the initial substrate geometry in order to explain the observed correlation. Alternatively, a discrete radical intermediate susceptible to rebound may only form from substrates with staggered (31 and 34) rather than eclipsed alignments (3 and 28). In the latter case, cyclization may proceed in concert with H atom abstraction resulting in a one-step-two-electron reduction of the Fe(IV)═O complex. Though enzymatic Fe(IV)═O complexes are typically modeled as stepwise one-electron acceptors, Fe(IV)═O complexes have been implicated in two-electron processes,34,35 and a similar chemistry may be at work in the catalytic cycle of H6H.
H6H represents an excellent system for studying the partitioning of radical-mediated catalytic cycles among different reaction paths. Herein, evidence is provided that the oxidative cyclization catalyzed by H6H does not involve rebound of the hydroxyl group following H atom abstraction. Furthermore, cyclization versus rebound appears to require that an exo-OH not only be adjacent to the site of H atom abstraction but also have the correct syn-periplanar configuration. Future investigation of these properties and the chemistry underlying them will provide new insights into the mechanics of nonheme iron biocatalysts.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Takashi Hashimoto at Nara Institute of Science and Technology for generously providing the clone of h6h. This work was supported by grants from the National Institutes of Health (GM113106 and GM040541) and the Welch Foundation (F-1511).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b03729.
Details regarding H6H assays along with HPLC protocols, chemical synthesis of compounds and computational methods and results (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Hausinger RP Biochemical diversity of 2-oxoglutarate dependent oxygenases In 2-Oxoglutarate-dependent oxygenases; Schofield CJ, Hausinger RP, Eds.; The Royal Society of Chemistry: Cambridge, U. K, 2015; pp 1–58. [Google Scholar]
- (2).Kovaleva EG; Lipscomb JD Nat. Chem. Biol 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Costas M; Mehn MP; Jensen MP; Que L Jr. Chem. Rev. 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- (4).Krebs C; Fujimori DG; Walsh CT; Bollinger MJ Jr. Acc. Chem. Res 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Klinkenberg I; Blokland A Neurosci. Biobehav. Rev 2010, 34, 1307–1350. [DOI] [PubMed] [Google Scholar]
- (6).Fodor G; Romeike A; Janzsó G; Koczor I Tetrahedron Lett. 1959, 1, 19–23. [Google Scholar]
- (7).Hashimoto T; Yamada Y Plant Physiol. 1986, 81, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hashimoto T; Yamada Y Eur. J. Biochem 1987, 164, 277–285. [DOI] [PubMed] [Google Scholar]
- (9).Hashimoto T; Kohno J; Yamada Y Plant Physiol. 1987, 84, 144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hashimoto T; Kohno J; Yamada Y Phytochemistry 1989, 28, 1077–1082. [Google Scholar]
- (11).Matsuda J; Okabe S; Hashimoto T; Yamada Y J. Biol. Chem 1991, 266, 9460–9464. [PubMed] [Google Scholar]
- (12).Hashimoto T; Matsuda J; Yamada Y FEBS Lett. 1993, 329, 35–39. [DOI] [PubMed] [Google Scholar]
- (13).Li J; van Belkum MJ; Vederas JC Bioorg. Med. Chem 2012, 20, 4356–4363. [DOI] [PubMed] [Google Scholar]
- (14).Elson SW; Baggaley KH; Gillett J; Holland S; Nicholson NH; Sime JT; Woroniecki SR J. Chem. Soc., Chem. Commun 1987, 1736–1738. [Google Scholar]
- (15).Pan J; Bhardwaj M; Faulkner JR; Nagabhyru P; Charlton ND; Higashi RM; Miller A-F; Young CA; Grossman RB; Schardl CL Phytochemistry 2014, 98, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pan J; Bhardwaj M; Zhang B; Chang W.-c.; Schardl CL; Krebs C; Grossman RB; Bollinger JM Jr. Biochemistry 2018, 57, 2074–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Krol WJ; Basak A; Salowe SP; Townsend CA J. Am. Chem. Soc 1989, 111, 7625–7627. [Google Scholar]
- (18).Liu P; Murakami K; Seki T; He X; Yeung S-M; Kuzuyama T; Seto H; Liu H.-w.-w. J. Am. Chem. Soc 2001, 123, 4619–4620. [DOI] [PubMed] [Google Scholar]
- (19).Wang C; Chang W.-c.; Guo Y; Huang H; Peck SC; Pandelia ME; Lin G.-m.; Liu H.-w.; Krebs C; Bollinger JM Jr. Science 2013, 342, 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Higgins LJ; Yan F; Liu P; Liu H.-w.; Drennan CL Nature 2005, 437, 838–844. [DOI] [PubMed] [Google Scholar]
- (21).Chang W.-c.; Dey M; Liu P; Mansoorabadi SO; Moon S-J; Zhao BK; Drennan CL; Liu H.-w. Nature 2013, 496, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yun D; Dey M; Higgins LJ; Yan F; Liu H.-w.; Drennan CL J. Am. Chem. Soc 2011, 133, 11262–11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yan F; Moon S-J; Liu P; Zhao Z; Lipscomb JD; Liu A; Liu H.-w. Biochemistry 2007, 46, 12628–12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Grundt P; Kopajtic TA; Katz JL; Newman AH Bioorg. Med. Chem. Lett 2004, 14, 3295–3298. [DOI] [PubMed] [Google Scholar]
- (25).Ishimaru K; Shimomura K Phytochemistry 1989, 28, 3507–3509. [Google Scholar]
- (26).Muñoz MA; Muñoz O; Joseph-Nathan PJ Nat. Prod 2006, 69, 1335–1340. [DOI] [PubMed] [Google Scholar]
- (27).Chang C-Y; Lyu S-Y; Liu Y-C; Hsu N-S; Wu C-C; Tang C-F; Lin K-H; Ho J-Y; Wu C-J; Tsai M-D; Li T-L Angew. Chem., Int. Ed 2014, 53, 1943–1948. [DOI] [PubMed] [Google Scholar]
- (28).Qi J; Wan D; Ma H; Liu Y; Gong R; Qu X; Sun Y; Deng Z; Chen W Cell. Chem. Biol 2016, 23, 935–944. [DOI] [PubMed] [Google Scholar]
- (29).Thomas SG; Phillips AL; Hedden P Proc. Natl. Acad. Sci. U. S. A 1999, 96, 4698–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chung HS; Raetz CRH Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Patteson JB; Cai W; Johnson RA; Santa Maria KC; Li B Biochemistry 2018, 57, 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Meng S; Han W; Zhao J; Jian X-H; Pan H-X; Tang G-L Angew. Chem., Int. Ed 2018, 57, 719–723. [DOI] [PubMed] [Google Scholar]
- (33).Bai J; Yan D; Zhang T; Guo Y; Liu Y; Zou Y; Tang M; Liu B; Wu Q; Yu S; Tang Y; Hu Y Angew. Chem., Int. Ed 2017, 56, 4782–478. [DOI] [PubMed] [Google Scholar]
- (34).Pestovsky O; Bakac A J. Am. Chem. Soc 2004, 126, 13757–13764. [DOI] [PubMed] [Google Scholar]
- (35).Oh NY; Suh Y; Park MJ; Seo MS; Kim J; Nam W Angew. Chem 2005, 117, 4307–4311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.