

Abstract
Circular RNAs (circRNAs) are a large class of noncoding RNAs that have potential regulatory roles in disease pathogenesis and progression. Recently, circRNAs have been found to be expressed in hepatocellular carcinoma (HCC) tissues and involved in the development and metastasis of HCC. However, the significance of circRNAs in peripheral blood mononuclear cells (PBMCs) of HCC patients remains unclear. In this study, RNA sequencing analysis was performed to identify circRNAs from four HCC patients and three healthy controls to determine the expression pattern of circRNAs in the PBMCs and the circRNAs’ molecular regulatory networks in HCC pathogenesis. A total of 58 circRNAs were found to be significantly changed (≥2 or ≤0.5‐fold) in the PBMCs of HCC patients compared with those of the healthy cases. Six random representative circRNAs (three up‐ and three down‐regulated) were further validated by real‐time RT‐PCR in 72 samples of PBMCs from HCC patients and 30 control subjects. Chi‐square test indicated that one of the up‐regulated circRNA candidates—circ_0000798—was correlated with clinical variables. Highly expressed circ_0000798 was associated with poor overall survival of HCC patients. Receiver operating characteristic curve analysis further revealed that circ_0000798 was discriminating HCC patients from healthy controls. Finally, the predicted competing endogenous RNA network of circ_0000798 showed that it might act as a “sponge” of target microRNAs, that would subsequently regulate the expression of target genes in PBMCs. In summary, this is the first study to comprehensively identify dysregulated circRNAs in PBMCs of HCC patients, and its findings suggest that dysregulated circ_0000798 in PBMCs has potential as a convenient biomarker for diagnosing or prognosticating HCC.
Keywords: biomarker, circRNAs, hepatocellular carcinoma, high‐throughput sequencing
1. INTRODUCTION
Among the most common malignancies worldwide, hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver and exhibits one of the highest mortality rates.1 More than 750 000 people are newly diagnosed with HCC every year globally.2 Although advances in diagnosis, surgical techniques, and liver transplantation have been achieved, the long‐term survival of patients with HCC remains poor to date. Moreover, the prognosis after curative resection of HCC has remained unsatisfactory because of a high incidence of postoperative recurrence.3 Identifying early diagnostic and prognostic biomarkers for metastatic HCC is of paramount importance to overcome these ongoing challenges to human health.
Circular RNAs (circRNAs) are a type of endogenous RNA with a stable structure that is widely expressed in mammal genomes.4 The circRNAs were initially observed in RNA viruses in the 1970s5 and were considered mere by‐products of splicing in the 1990s.6 However, the advent of RNA sequencing technology and bioinformatics in the recent decades have led to the novel and extraordinary findings that circRNAs are abundant, conserved, and stable, have a tissue‐specific expression pattern in mammalian cells,4, 7 and are unique noncoding RNA (ncRNA) molecules, with sizes ranging from hundreds to thousands of nucleotides. Although most of the circRNAs discovered to date appear to not encode proteins, the circRNAs in general have been shown to play crucial regulatory roles in a variety of cellular processes and biological pathways,8 similar to other ncRNAs such as microRNAs (miRNAs) and long noncoding RNAs (lncRNAs). The regulation mechanisms implicated to date include cell proliferation, apoptosis, differentiation, and angiogenesis.9, 10 A number of studies have also shown a close relationship between circRNAs and cancers, wherein circRNAs could regulate the mRNA activity of target genes by acting as miRNA sponges, promoting or inhibiting the development and metastasis of various cancer types.11, 12, 13 Collectively, these lines of evidence provide a new direction for exploring circRNAs as targets for the diagnosis and prognosis of diseases.
It is well known that under normal physiologic conditions immune cells monitor and clear mutated cells from the body. However, the immune cells can also establish conditions supportive of tumor growth and invasion, such as the proinflammatory environment that develops during chronic inflammation.14 The peripheral blood mononuclear cells (PBMCs) are composed of various leukocyte subpopulations, including T lymphocytes, B lymphocytes, monocytes, natural killer cells, and dendritic cells. Studies of the key genes in PBMCs have found associations between altered gene expression and diseases, such as cancer, and disease states, such as cancer‐related immunity.15 Thus, altered gene expression in PBMCs has been proposed as a potential clinical tool, as it may reflect a disease's diagnosis or prognosis. Moreover, since, as described above, circRNAs are capable of regulating gene expression and the dysregulated gene expression in PBMCs might, at least partially, attribute to the altered circRNAs profile observed in PBMCs, the expression profile of circRNAs in PBMCs might represent a promising tool for discovering biomarkers associated with physiological or pathological events. For example, determining how the circRNAs profile is changed in PBMCs of HCC patients and whether those differential expression profiles could be useful for the diagnosis or prognosis of HCC.
In this study, we used high‐throughput sequencing to identify aberrantly expressed circRNAs in the PBMCs of HCC patients, as compared to healthy controls. The sequencing efficiency was validated by the selected dysregulated circRNAs by quantitative reverse transcription (qRT)‐PCR, and the putative circRNA‐miRNA‐mRNA interaction was predicted for the selected dysregulated circ_0000798. Finally, a receiver operating characteristic (ROC) curve was used to analyze the potential diagnostic and clinical significance of circ_0000798 for HCC.
2. MATERIALS AND METHODS
2.1. Sample collection
A total of 102 peripheral blood samples were collected during surgery (72 patients with HCC and 30 healthy controls) between 2016 and 2018 in the Southwest Hospital of the Third Military Medical University (Chongqing, China). The patients had not been treated with radiotherapy or chemotherapy before surgery. Healthy subjects who received a regular physical examination at the Department of Health from the same hospital were recruited as normal control subjects. Fresh peripheral blood samples (10 mL) were collected from all of the subjects in ethylenediaminetetraacetic acid (EDTA) tubes. This study was approved and supervised by the ethical committee of the Southwest Hospital. Written informed consent was obtained from all subjects in this study.
2.2. PBMC preparation and total RNA extraction
Fresh 10‐mL peripheral blood samples were collected in EDTA tubes before use in the study procedures. Within 4 hours, PBMCs from each donor were isolated using density centrifugation (700 g for 20 minutes) with the total 10‐mL blood sample layered on 5 mL Ficoll‐Paque PLUS (Cat. No. 17‐1440‐02; GE Healthcare, Uppsala, Sweden) at room temperature. The cells were subsequently frozen in TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and stored in liquid nitrogen (−180°C). Total RNA was extracted from the isolated PMBC samples using TRIzol reagent according to the manufacturer's instruction. The quantity of the isolated RNA was measured using a NanoDrop spectrophotometer (Agilent Technologies, Santa Clara, CA, USA).
2.3. Whole transcriptome sequencing
The RNA extracted by TRIzol reagent and having an RNA integrity number of >8.0 was utilized to construct an rRNA depletion library (VAHTSTM Total RNA‐seq [H/M/R]; Agilent Technologies) according to the manufacturer's instructions. Whole transcriptome sequencing was performed by NovelBio Corp. Laboratory (Shanghai, China) using a Hiseq™ XTEN Sequencer (Illumina, San Diego, CA, USA). The sequencing data were first filtered utilizing domestic Java code (removing the adaptor sequences, reads with >5% ambiguous bases (noted as N), and low‐quality reads containing more than 20% of bases with qualities of <20) and then mapped to the human genome (human genome version: GRCH38.p7 National Center for Biotechnology Information [NCBI]) utilizing HISAT2.16
2.4. Data analysis and identification of differentially expressed circRNAs
First, low‐quality reads were removed from the raw sequence data. The Unmapped Reads Collection (HISAT2)17 was then used as the method of mapping to the reference genome (GRCH38.p7 NCBI; utilizing the parameter ‐hisat2 ‐p 8 ‐5 5 ‐3 5 –min‐intronlen 20‐max‐intronlen 500000 ‐k 3 –phred33.). Next, the pipeline “acfs2” (publicly available at https://code.google.com/p/acfs2/) was used to identify candidate circRNA in each sample, according to the procedures described in the published literature.18
2.5. qRT‐PCR validation of differentially expressed circRNAs
The dysregulated expressions of six randomly selected circRNAs from our RNA sequencing data were validated by qRT‐PCR. For this, cDNA was first synthesized from 1 μg total RNA using a Prime‐Script RT reagent kit with gDNA Eraser (TaKaRa, Shiga, Japan). Then, specific divergent primers for each circRNA were designed based on circPrimer1.2 (http://www.bioinf.com.cn/) and according to the “spliced sequence” of circRNA originated from our sequence data (consistent with circBase [http://www.circbase.org/]) (Table 1). The real‐time PCR was performed using SYBR qPCR Super Mix (Novoprotein, Shanghai, China) with the Stratagene Mx3000P Real‐Time PCR System (Agilent Technologies) and following the manufacturer's instructions. The PCR reactions were: denaturation at 95°C for 2 minutes, 40 cycles of 95°C for 30 seconds, 46‐55°C for 30 seconds, and 72°C for 30 seconds. The human gene GAPDH was used as an internal control. The relative gene expression levels were analyzed by the 2−∆∆Ct method. Three independent replicates of each experiment were performed.
Table 1.
Sequences of the primer pairs used to analyze the genes
| ID | Primer sequence (5′‐3′) | Tm (°C) | Ct |
|---|---|---|---|
| hsa_circ_0005505 | R: GCCAGCTGCTTGAAAGTCTC F: TGCAGTGTAAGAAGCATTGGA | 53 | 25‐32 |
| hsa_circ_0001394 | R: TCCATCAGTCATCTTGGTCCA F: TTCATGAAGCTGAGGAGGGG | 55 | 25‐27 |
| hsa_circ_0000798 | R: GTACCTGCATCTGGGGTGAC F: ACTCCTGGACAAGGATCTGC | 55 | 23‐25 |
| hsa_circ_0004771 | R: AGCTCACAATCCAAACACTTCC F: ACTTTTCAACAGCCTTCTCAAT | 48 | 28‐32 |
| hsa_circ_0001074 | R: TTACTCTTTGATTTACGACTGC F: GTAACTCTGTCCTTATTATCGG | 46 | 31‐35 |
| hsa_circ_0067735 | R: TCTCTGCACTCTTCACATTCCA F: TGACTTGTGCCTATTATTCTGC | 48 | 26‐32 |
| GAPDH | R: GGTCTGGGAGCCTGGAAAA F: TTCGCTCCTGGAAGATGGTAAT | 46‐55 | 16‐24 |
Ct, cycle threshold; F, forward; R, reverse; Tm, annealing temperature.
2.6. Prediction of circRNA‐miRNA‐mRNA interactions
circRNA binding to a miRNA allows for its indirect regulation of the translation of an mRNA.4 We utilized miRanda v3.3a (http://www.microrna.org/microrna/home.do) and RNAhybrid 2.1 (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/) as tools to predict the related target miRNAs and mRNAs for circ_0000798. Ultimately, the graphs of circRNA‐miRNA‐mRNA sharing meaningful correlation interaction networks were drawn using Cytoscape 3.3.0 (https://cytoscape.org/).
2.7. Statistical analysis
In this study, after tested by the one‐sample Kolmogorov‐Smirnov test (Table S1), the data in Figure 2a,b were analyzed by t‐test with Welch's correction in case of the normal distribution among groups; otherwise, the data were analyzed by Mann‐Whitney U‐test among groups (Table S2). The relationship between the differentially regulated circRNA and clinical features were analyzed using the chi‐square test. Survival analysis was carried out by Kaplan‐Meier analysis, and statistical analysis was performed through log‐rank test. A ROC curve was computed and the specificity and sensitivity of predictive power was assessed by the area under the curve (AUC) to indicate the diagnostic value of the selected dysregulated circRNA in the PBMCs of HCC patients compared to healthy controls. P < 0.05 was considered statistically significant. All the data were analyzed using SPSS statistical software (version 17.0; SPSS, Chicago, IL, USA).
3. RESULTS
3.1. circRNA expression profiling in PBMCs from HCC patients and healthy controls
Based on the result of ACFS circRNA prediction and identification pipeline, the supporting reads for each circRNA was calculated, representing the confidence of the predicted circRNA. Global transcriptome expression analysis detected 5609 distinct circRNA candidates (Figure 1a). The size of the circRNAs ranged from 100 to 2100 nt (Figure 1b). Expression of the circRNAs in HCC patients and healthy controls was measured based on RPKM (mapped back‐splicing junction reads per million mapped reads), which demonstrated the normalized intensities from the seven samples (Figure 1c). The variations in circRNA expression were illustrated according to the volcano plot (Figure 1d). Hierarchical clustering heatmap analysis displayed a distinguishable circRNA expression profile among the samples (Figure 1e). These results showed that there was a markedly distinguishable circRNA expression profile in PBMCs between the HCC and healthy controls. Specifically, 58 circRNAs were significantly changed in the HCC patients compared with the healthy controls, of which 21 were up‐regulated and 37 were down‐regulated in the HCC group (Table S3). Significant differences in circRNAs between HCC patients and healthy controls were classified as fold change ≥2 or fold change ≤0.5, P‐value <0.05, and false discovery rate <0.05, as determined by having to satisfy both of the above conditions. The RNA‐seq data have been submitted to the NCBI Gene Expression Omnibus (GEO), under GEO accession number GSE120663 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120663).
Figure 1.
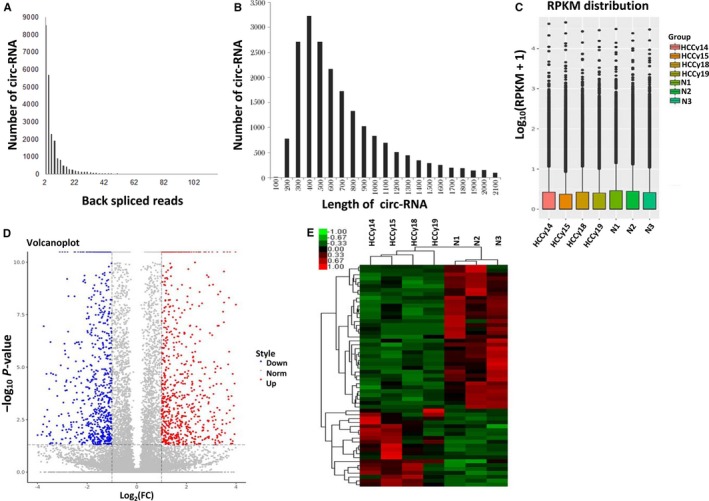
Dysregulated Circular RNA (circRNA) expression profiles in hepatocellular carcinoma (HCC) samples compared with normal samples from healthy controls. (a) The number of circRNAs in HCC samples compared with normal samples. The Y axis representing the circRNA supported reads number and the X axis representing the circRNA number with such reads supported were used to display the predicted circRNA and the circRNA prediction quality. (b) Length distribution of circRNAs. (c) A box plot showed the normalized intensities from the four HCC examples and three control examples. The box plots represent the interquartile range (25%‐75%, IQR) with whiskers corresponding to 1.5 × IQR and points—to outliers. (d) A volcano plot showing the dysregulated circRNA expression in the samples. Red and blue represent the up‐regulated and down‐regulated circRNAs in HCC, respectively. (e) Heat map of circRNAs showing hierarchical clustering of circRNAs with altered expression in HCC samples compared with normal samples. Each row represents one circRNA, and each column represents each sample. Cluster plot was done utilizing Cluster 3.0 software (open source) based on the normalized circRNA expression data using the parameter: 1. average linkage; 2. Pearson correlation. The up‐ and down‐regulated genes are colored in red and green, respectively. N1, N2, and N3 represent healthy controls, and HCCy15, HCCy19, HCCy14, and HCCy18 represent HCC patients
3.2. Validation of differentially expressed circRNAs by qRT‐PCR
To validate the sequencing data, six circRNAs, representing three of the up‐regulated circRNAs (circ_0005505, circ_0001394, and circRNA_0000798) and three of the down‐regulated circRNAs (circ_0004771, circ_0001074, and circRNA_0067735) (Table 2), were randomly selected from the 58 significantly dysregulated circRNAs (Table S1) for validation by qRT‐PCR using PBMCs of 72 HCC patients and 30 healthy controls. The results showed that the expression profiles of these six circRNAs were consistent with those from the sequencing results (Figure 2a,b). Among the six selected circRNAs, circRNA_0000798 showed the greatest stability among all subjects, as compared with the other five circRNAs (Figure 2a,b). Accordingly, the qRT‐PCR product of circRNA_0000798 was then subjected to agarose gel electrophoresis and Sanger sequencing, which demonstrated the expected 86‐bp size and correct sequence of the product (Figure 2c,d). Collectively, these data suggest the accuracy and reproducibility of the RNA sequencing findings.
Table 2.
The six selected circRNAs in HCC samples compared with normal samples
| circRNA ID | Log2FC | FDR | Regulation | Chr | Strand | Gene symbol | Genomic length, bp | Spliced length, bp |
|---|---|---|---|---|---|---|---|---|
| hsa_circ_0005505 | 1.172 122 | 0.019 256 | Up | Chr12 | + | IRAK3 | 24 660 | 754 |
| hsa_circ_0000798 | 3.945 709 | 4.43E‐10 | Up | Chr17 | + | BPTF | 2898 | 1226 |
| hsa_circ_0001394 | 2.932 023 | 5.46E‐05 | Up | Chr4 | + | TBC1D14 | 739 | 739 |
| hsa_circ_0001074 | −4.708 075 | 0.004 433 | Down | Chr2 | − | ORC4 | 3237 | 242 |
| hsa_circ_0004771 | −2.045 652 | 5.71E‐13 | Down | Chr21 | − | NRIP1 | 29 231 | 203 |
| hsa_circ_0067735 | −1.838 783 | 0.003 161 | Down | Chr3 | + | MED12L | 11 647 | 457 |
The six selected circRNAs from sequencing.
Chr, chromosome; circRNA, circular RNA; FC, fold change; FDR, false discovery rate; HCC, hepatocellular carcinoma.
Figure 2.
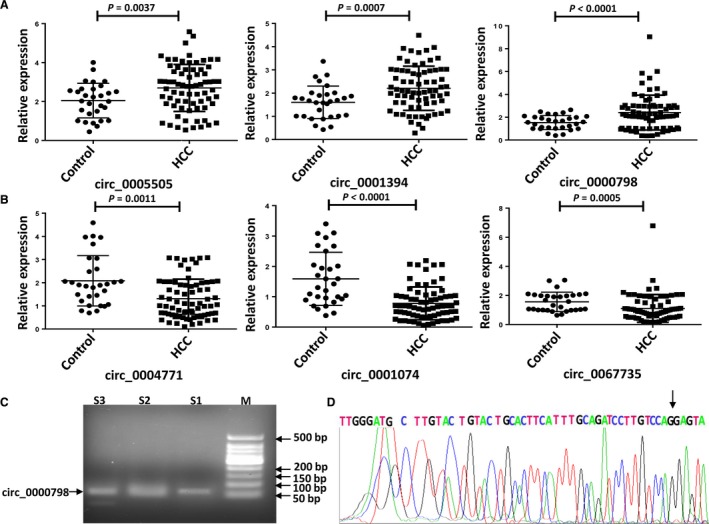
Validation of the expression of selected Circular RNAs (circRNAs) by qRT‐PCR. The relative expression of specific circRNAs between the peripheral blood mononuclear cells of hepatocellular carcinoma (HCC) patients and healthy individuals (controls). Relative expression of the three up‐regulated (a) and the three down‐regulated (b) circRNAs were determined by qRT‐PCR. The differences in the expression of circRNA candidates between HCC group and healthy group were analyzed by t‐test with Welch's correction, except circ_0067735, of which the expression difference was calculated by Mann‐Whitney U‐test. The data are presented as the means ± SD of three independent experiments. (c) The circRNA_0000798 expression was analyzed by qRT‐PCR, followed by 2.5% agarose gel electrophoresis. Lane M is the DL0501 Marker (GENERAY, Shanghai, China); Lanes S1‐S3 are the qRT‐PCR products of three repeated samples. (d) Validation of the circRNA_0000798 splicing junction site through sequencing of its qRT‐PCR product. The 2−ΔΔCt method was used to calculate the circRNAs expression level relative to the GAPDH housekeeping control. Three independent replicates of each experiment were performed
3.3. circ_0000798 up‐regulation is associated with clinical features of HCC
We further selected a representative circRNA, circ_0000798, to investigate its clinical significance, because this circRNA was more stably up‐regulated among the 72 patients, as compared with the healthy controls (Figure 2a). The results demonstrated that there were no significant correlations between circ_0000798 expression in the PBMCs of HCC patients and the patients’ sex, age, family history, diabetes, α‐fetoprotein, tumor differentiation, lymph node status, and distant metastasis; however, circ_0000798 expression was markedly correlated with tumor size and cirrhosis (P < 0.05) (Table 3). Moreover, to evaluate the prognostic significance of circ_0000798 expression in HCC, the HCC patients were divided into high‐ and low‐expression groups based on the expression levels of circ_0000798 detected in their PBMCs by qRT‐PCR. The results showed that HCC patients with high circ_0000798 expression level had a significantly lower survival rate than those with low circ_0000798 expression (median survival of 10 months vs 28 months; P < 0.0001; Figure 3).
Table 3.
The association between circ_0000798 expression and clinicopathological characteristics of 72 hepatocellular carcinoma patients
| Characteristic | Total, n = 72 | circ_0000798 expression | χ2 | P | |
|---|---|---|---|---|---|
| Low, n = 24 (%) | High, n = 48 (%) | ||||
| Sex | |||||
| Male | 16 | 7 (43.7) | 9 (56.3) | ||
| Female | 56 | 17 (30.4) | 39 (69.6) | 1.004 | 0.316 |
| Age, years | |||||
| <50 | 24 | 9 (37.5) | 15 (62.5) | ||
| ≥50 | 48 | 15 (31.3) | 33 (68.7) | 0.281 | 0.596 |
| Family history | |||||
| Yes | 20 | 7 (35.0) | 13 (65.0) | ||
| No | 52 | 17 (32.7) | 35 (67.3) | 0.035 | 0.852 |
| Tumor size, cm | |||||
| <5 | 28 | 22 (78.6) | 6 (21.4) | ||
| ≥5 | 44 | 2 (4.5) | 42 (95.5) | 42.195 | <0.0001 * |
| Tumor differentiation | |||||
| Well/moderate | 56 | 16 (28.6) | 40 (71.4) | ||
| Poor | 16 | 8 (50.0) | 8 (50.0) | 2.571 | 0.109 |
| Diabetes | |||||
| Yes | 8 | 2 (25.0) | 6 (75.0) | ||
| No | 64 | 22 (34.4) | 42 (65.6) | 0.018 | 0.895 |
| Cirrhosis | |||||
| Yes | 42 | 3 (7.1) | 39 (92.9) | ||
| No | 30 | 21 (70.0) | 9 (30.0) | 31.114 | <0.0001 * |
| AFP | |||||
| ≤20 | 21 | 8 (38.1) | 13 (61.9) | ||
| >20 | 51 | 16 (31.4) | 35 (68.6) | 0.303 | 0.582 |
| Lymph node status | |||||
| Yes | 22 | 5 (22.7) | 17 (77.3) | ||
| No | 50 | 19 (38.0) | 31 (62.0) | 1.604 | 0.205 |
| Distant metastasis | |||||
| M0 | 41 | 17 (41.5) | 24 (58.5) | ||
| M1 | 31 | 7 (22.6) | 24 (77.4) | 2.832 | 0.092 |
AFP, α‐fetoprotein.
Statistically significant (P < 0.05); the P‐value was from the χ2.
Figure 3.
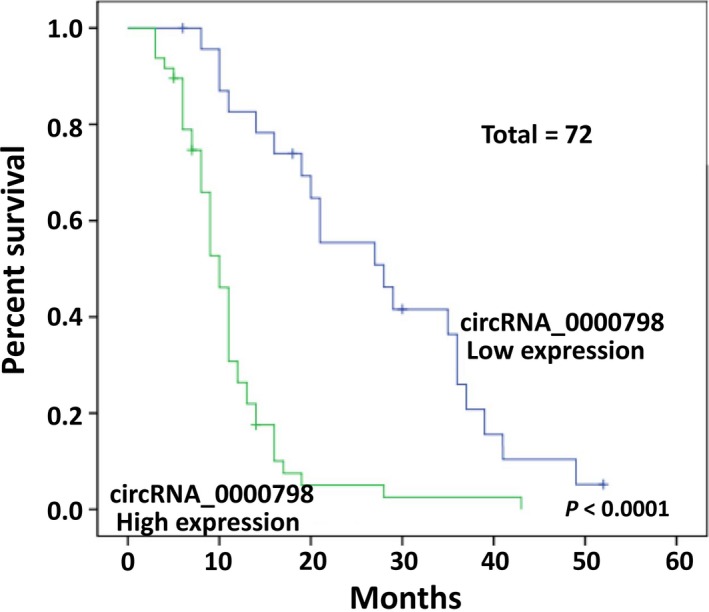
The correlation between circ_0000798 expression and hepatocellular carcinoma patients’ survival rate was analyzed using Kaplan‐Meier survival curves and log‐rank tests
3.4. Diagnostic values of circ_0000798 for HCC patients
To assess the potential value of significantly and differentially expressed circ_0000798 for HCC diagnosis, a ROC curve was computed to describe the circ_0000798 expression in discriminating HCC patients from healthy controls according the expression of fold changes of circ_0000798 in all samples. The results showed that the AUC of circ_0000798 reached 0.703 (95% confidence interval [CI]: 0.604‐0.803; P = 0.001), indicating that circ_0000798 in PBMCs could separate the patients with HCC from the healthy controls, and the circ_0000798 expression level might have potential clinical significance as a tumor marker (Figure 4).
Figure 4.
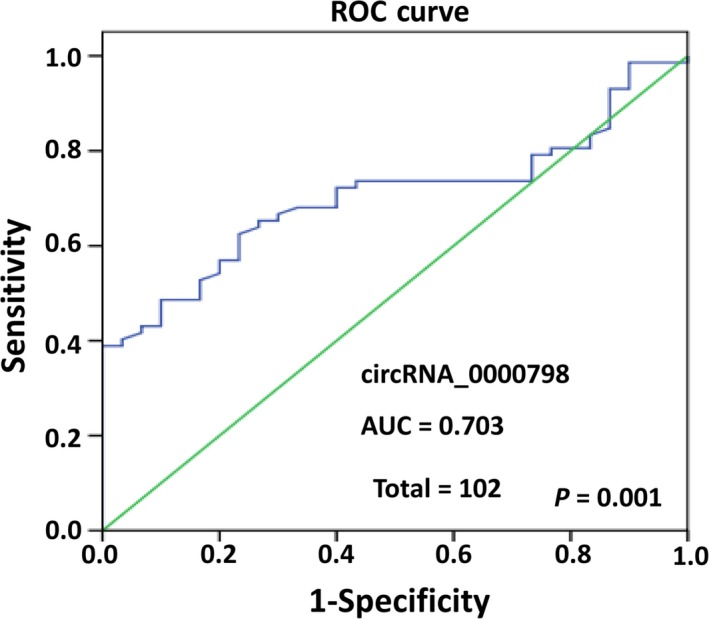
Analysis of the sensitivity and specificity of circ_0000798 as a novel hepatocellular carcinoma (HCC) marker by receiver operating characteristic (ROC) curve. The ROC curve was computed using the expression of fold changes of circ_0000798 in all samples to describe the circ_0000798 expression in discriminating HCC patients from healthy controls
3.5. Predicted circRNA‐miRNA‐mRNA regulatory network
In order to further explore the potential functions of circ_0000798 in PBMCs of HCC patients, we performed a circRNA‐miRNA‐mRNA network analysis in which the immune‐related mRNAs from our RNA‐seq data were selected (Figure 5). We found that there were several potential target mRNAs of circ_0000798, mediated by the predicted miRNAs through “sponging” mechanisms. The predicted target mRNAs include the well‐known effector molecules of immune cells, such as IFNG, IFNGR2, IL17RA, TNFAIP, and ICAM1. In addition, the predicted target mRNAs also include the regulatory molecules in immune cells, mainly the transcription factors involved in the regulation of the proliferation, activation, and apoptosis, such as FOS, BCL3, BCL2A1, BCL6, SOCS, and SGK1.
Figure 5.
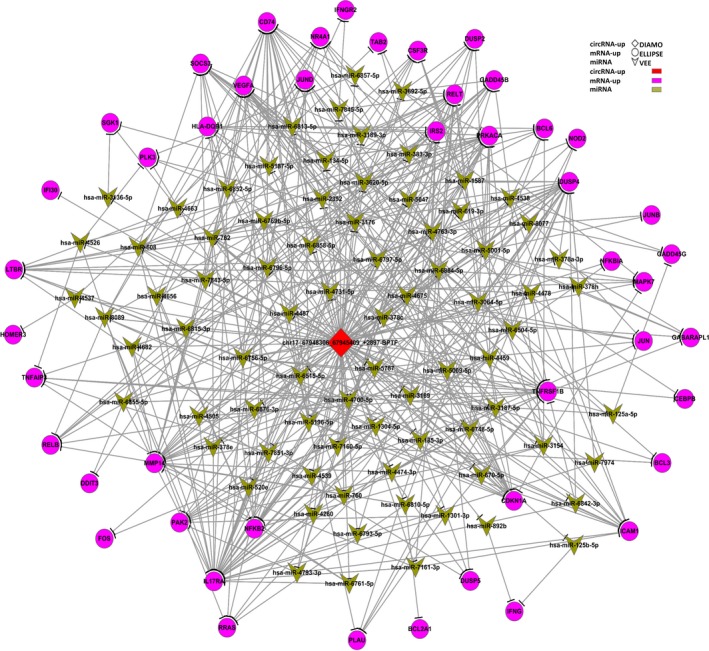
Competing endogenous RNA network in peripheral blood mononuclear cells from hepatocellular carcinoma
4. DISCUSSION
Hepatocellular carcinoma is a high‐incidence malignant tumor, and most HCC patients are diagnosed at advanced stages of the disease, when there is tumor cell metastasis or diffusion. This advanced stage of disease at diagnosis causes a delay in treatment and supports poor prognosis.19 Recently, studies have found that circRNAs are important members of the ncRNA family in different species.20, 21 The development of high‐throughput sequencing technology has increased our understanding of the role and the molecular mechanism of circRNAs in human diseases, especially in cancer,22 and has promoted the development of these RNAs as a novel biomarker for cancer diagnosis.23, 24 In this study, circRNA sequencing was used to identify differences in the circRNA expression profile of PBMCs between HCC patients and healthy controls and has demonstrated the possible involvement of differentially expressed circRNAs in HCC pathology.
Compared with miRNAs and lncRNAs, circRNAs are widely expressed in human cells and more stable in mammalian cells,9 which endows circRNAs with the potential to be ideal biomarkers for human diseases. In the present study, to explore novel biomarkers of HCC, we first explored the expression profiles of circRNAs in PBMCs between four HCC patients and three healthy individuals (controls) by high‐throughput RNA sequencing. A total of 58 circRNAs were found to be significantly dysregulated (21 up‐regulated and 37 down‐regulated) in the HCC patients compared with the healthy controls. The expression of six randomly selected circRNAs (circ_0005505, circ_0001394, circRNA_0000798, circ_0004771, circ_0001074 and circRNA_0067735) were further analyzed by qRT‐PCR and confirmed as being consistent with the sequencing data.
Early diagnosis is the key to successful treatment and improved prognosis for HCC patients.25 circRNAs might be novel biomarkers, and have already been reported as helpful biomarkers for several diseases, including cancers such as gastric cancer, acute myeloid leukemia, pancreatic ductal adenocarcinoma, and lung cancer.15, 16, 26, 27 As such, they should be helpful for the diagnosis of HCC as well. Shang et al.25 found that circ_0005075 was up‐regulated in HCC tissues and its expression was positively related to the tumor size in HCC. In the current study, we found that several circRNAs were significantly up‐ or down‐regulated in PBMCs from HCC patients, among which circ_0000798 was selected for further investigation. As expected, circ_0000798 was confirmed as being markedly up‐regulated in the PBMCs of 72 HCC patients by qRT‐PCR. Prognostic analysis of circ_0000798 in these 72 HCC patients using Kaplan‐Meier analysis indicated that the up‐regulated circ_0000798 was always associated with larger tumor size and malignant cirrhosis. Furthermore, the survival rate of HCC patients with high circ_0000798 expression was significantly lower than those with low circ_0000798 expression. The AUC indicated that circ_0000798 had the potential to assist in the diagnosis for HCC. Together, these data indicate that circ_0000798 might be markedly correlated with poor prognosis in HCC.
Recently, many studies have indicated that circRNAs could function as a tumor regulator in HCC. For instance, Yu et al.28 showed that the expression level of Cdr1as was up‐regulated in HCC tissues, silencing of which could suppress the proliferation of HCC cells (SMMC‐7721 and HepG2). Qin et al.12, 29 found that down‐regulation of hsa_circ_0001649 in HCC cells significantly increased the mRNA level of matrix metallopeptidases 9, 10, and 13, promoting the metastasis of HCC. Zhu and colleagues30 demonstrated that the circRNA, circ_0067934, promotes tumor growth and metastasis in HCC through regulation of the miR‐1324/FZD5/Wnt/b‐catenin axis. All these findings strongly support the notion that circRNAs play important roles in tumor progression.
In our latest study, presented herein, we used RNA‐seq data to construct a circRNA‐miRNA‐mRNA network in order to predict the potential functions of circ_0000798 in PBMCs of HCC patients. We found that the potential target mRNAs of circ_0000798 include the well‐known effector molecules of immune cells, such as IFNG, IFNGR2, IL17RA, TNFAIP, and ICAM1. Moreover, the predicted target molecules also include molecules that have been verified to function in immune cells, such as FOS, SOCS, SGK1, and BCL family members. For instance, an earlier study found that after activation by mTORC2, SGK1 promoted T helper type 2 differentiation by negatively regulating degradation of the transcription factor JunB mediated by the E3 ligase Nedd4‐2, and that mice with selective deletion of SGK1 in T cells were resistant to experimentally induced asthma, as a result of a substantial IFN‐γ increase in response to viral infection and more readily rejected tumors.31 In addition, Li et al.32 reported that the mRNAs of SOCS family genes could be marked by m6A, increasing the levels of mRNAs and proteins of SOCS family members in Mettl3‐deficient naive T cells. The up‐regulated SOCS family activity subsequently inhibited IL‐7‐mediated STAT5 activation and T‐cell homeostatic proliferation and differentiation.
5. CONCLUSION
This study is the first to assess circRNAs expression in PBMCs from HCC patients and compare the data with that from healthy controls. Bioinformatics analysis indicated that the up‐regulated expression of circ_0000798 was closely related to the development of HCC. Furthermore, circ_0000798 may be helpful for HCC diagnosis or acts as an independent prognostic biomarker for HCC patients. These observations and verifications have potential clinical significance, as dysregulated circRNAs such as circ_0000798 are potential diagnostic markers in HCC patients. In addition, bioinformatic analysis revealed that circ_0000798 could be involved in the pathophysiology of HCC by regulating the functions of peripheral immune cells and the consequent occurrence and progression of HCC. The predicted functions of this study's identified circ_0000798 in PBMCs of HCC patients should be further characterized with loss‐ and gain‐of‐function experiments in the future, which are expected to give further insight into how circ_0000798 exerts its functions on HCC by acting as a miRNA sponge.33
CONFLICT OF INTEREST
The authors declare no conflicts of interest related to this publication.
Supporting information
ACKNOWLEDGMENTS
This work was supported by grants from the National Key Research and Development Project (No. 2016YFA0502203), the National Foundation of China (Nos. 81502728 and 81670534), and the Anhui Provincial Natural Science Foundation (No. 1408085MH149). The funders had no role in the study design, data analysis, or decision to publish.
Lei B, Zhou J, Xuan X, et al. Circular RNA expression profiles of peripheral blood mononuclear cells in hepatocellular carcinoma patients by sequence analysis. Cancer Med. 2019;8:1423‐1433. 10.1002/cam4.2010
Bo Lei and Jian Zhou contributed equally to this work.
Contributor Information
Bing Ni, Email: nibing@tmmu.edu.cn.
Hui Pang, Email: 617990023@qq.com.
Weiping Fan, Email: fanweiping26418@126.com.
REFERENCES
- 1. Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271‐289. [DOI] [PubMed] [Google Scholar]
- 3. Poon RT. Differentiating early and late recurrences after resection of HCC in cirrhotic patients: implications on surveillance, prevention, and treatment strategies. Ann Surg Oncol. 2009;16:792‐794. [DOI] [PubMed] [Google Scholar]
- 4. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333‐338. [DOI] [PubMed] [Google Scholar]
- 5. Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single‐stranded covalently closed circular RNA molecules existing as highly base‐paired rod‐like structures. Proc Natl Acad Sci USA. 1976;73:3852‐3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205‐211. [DOI] [PubMed] [Google Scholar]
- 7. Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: identification, biogenesis and function. Biochem Biophys Acta. 2016;1859:163‐168. [DOI] [PubMed] [Google Scholar]
- 9. Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014;28:2233‐2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Starke S, Jost I, Rossbach O, et al. Exon circularization requires canonical splice signals. Cell Rep. 2015;10:103‐111. [DOI] [PubMed] [Google Scholar]
- 11. Zhong Z, Huang M, Lv M, et al. Circular RNA MYLK as a competing endogenous RNA promotes bladder cancer progression through modulating VEGFA/VEGFR2 signaling pathway. Cancer Lett. 2017;403:305‐317. [DOI] [PubMed] [Google Scholar]
- 12. Qin M, Liu G, Huo X, et al. Hsa_circ_0001649: a circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomarkers. 2016;16:161‐169. [DOI] [PubMed] [Google Scholar]
- 13. Li J, Zhen L, Zhang Y, et al. Circ‐104916 is downregulated in gastric cancer and suppresses migration and invasion of gastric cancer cells. Onco Targets Ther. 2017;10:3521‐3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meirow Y, Baniyash M. Immune biomarkers for chronic inflammation related complications in non‐cancerous and cancerous diseases. Cancer Immunol Immunother. 2017;66:1089‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li X, Wang Y, Han C, Li P, Zhang H. Colorectal cancer progression is associated with accumulation of Th17 lymphocytes in tumor tissues and increased serum levels of interleukin‐6. Tohoku J Exp Med. 2014;233:175‐182. [DOI] [PubMed] [Google Scholar]
- 16. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods. 2012;9:357‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. You X, Vlatkovic I, Babic A, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015;18:603‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu K, Wu X, Zang X, et al. TRAF4 regulates migration, invasion, and epithelial‐mesenchymal transition via PI3K/AKT signaling in hepatocellular carcinoma. Oncol Res. 2017;25:1329‐1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han J, Gao L, Dong J, Bai J, Zhang M, Zheng J. The expression profile of developmental stage‐dependent circular RNA in the immature rat retina. Mol Vis. 2017;23:457‐469. [PMC free article] [PubMed] [Google Scholar]
- 21. Fu L, Yao T, Chen Q, Mo X, Hu Y, Guo J. Screening differential circular RNA expression profiles reveals hsa_circ_0004018 is associated with hepatocellular carcinoma. Oncotarget. 2017;8:58405‐58416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsiao KY, Sun HS, Tsai SJ. Circular RNA—new member of noncoding RNA with novel functions. Exp Biol Med. 2017;242:1136‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang MM, Mai ZT, Wan SZ, et al. Microarray profiles reveal that circular RNA hsa_circ_0007385 functions as an oncogene in non‐small cell lung cancer tumorigenesis. J Cancer Res Clin Oncol. 2018;144:667‐674. [DOI] [PubMed] [Google Scholar]
- 24. Meng S, Zhou H, Feng Z, et al. CircRNA: functions and properties of a novel potential biomarker for cancer. Mol Cancer. 2017;16:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shang X, Li G, Liu H, et al. Comprehensive circular RNA profiling reveals that hsa_circ_0005075, a new circular RNA biomarker, is involved in hepatocellular crcinoma development. Medicine. 2016;95:e3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ouyang Q, Wu J, Jiang Z, et al. Microarray expression profile of circular RNAs in peripheral blood mononuclear cells from rheumatoid arthritis patients. Cell Physiol Biochem. 2017;42:651‐659. [DOI] [PubMed] [Google Scholar]
- 27. Tout I, Gomes M, Ainouze M, et al. Hepatitis B virus blocks the CRE/CREB complex and prevents TLR9 transcription and function in human B cells. J Immunol. 2018;201:2331‐2344. [DOI] [PubMed] [Google Scholar]
- 28. Yu L, Gong X, Sun L, Zhou Q, Lu B, Zhu L. The circular RNA Cdr1as act as an oncogene in hepatocellular carcinoma through targeting miR‐7 expression. PLoS ONE. 2016;11:e0158347. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Heldin CH, Moustakas A. Role of Smads in TGFbeta signaling. Cell Tissue Res. 2012;347:21‐36. [DOI] [PubMed] [Google Scholar]
- 30. Zhu Q, Lu G, Luo Z, et al. CircRNA circ_0067934 promotes tumor growth and metastasis in hepatocellular carcinoma through regulation of miR‐1324/FZD5/Wnt/beta‐catenin axis. Biochem Biophys Res Comm. 2018;497:626‐632. [DOI] [PubMed] [Google Scholar]
- 31. Heikamp EB, Patel CH, Collins S, et al. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat Immunol. 2014;15:457‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li HB, Tong J, Zhu S, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL‐7/STAT5/SOCS pathways. Nature. 2017;548:338‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Won C, Kim BH, Yi EH, et al. Signal transducer and activator of transcription 3‐mediated CD133 up‐regulation contributes to promotion of hepatocellular carcinoma. Hepatology. 2015;62:1160‐1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials