

Summary:
Detection of cytosolic DNA by the enzyme cGAS triggers the production of cGAMP, a second messenger that binds and activates the adaptor protein STING, which leads to interferon (IFN) production. Here we found that in vivo, natural killer (NK) cell killing of tumor cells, but not normal cells, depended on STING expression in non-tumor cells. Experiments using transplantable tumor models in STING and cGAS-deficient mice revealed that cGAS expression by tumor cells was critical for tumor rejection by NK cells. In contrast, cGAS expression by host cells was dispensable, suggesting that tumor-derived cGAMP is transferred to non-tumor cells where it activates STING. cGAMP administration triggered STING activation and interferon-β production in myeloid cells and B cells but not NK cells. Our results revealed that the anti-tumor response of NK cells critically depended on the cytosolic DNA sensing pathway similarly to its role in defense against pathogens, and identified tumor-derived cGAMP as a major determinant of tumor immunogenicity with implications for cancer immunotherapy.
Keywords: Natural Killer cells, NK cells, tumor immunity, STING, cGAS, 2'3'-cGAMP, DNA sensor, cancer immunology, cancer immunotherapy
Graphical Abstract
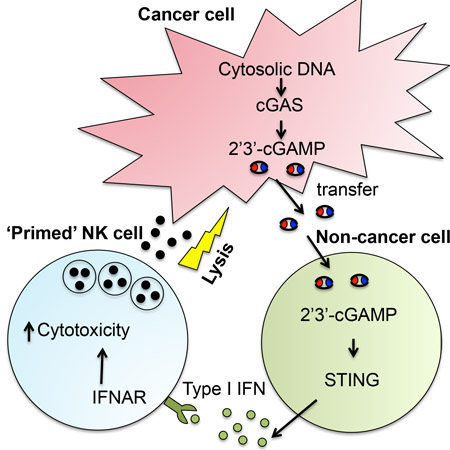
In Brief
Marcus et al. find that cGAMP produced by tumor cells triggers the activation of the STING pathway in immune cells within the tumor microenvironment, leading to interferon production by these cells, which in turn activates NK cell anti-tumor immunity.
Introduction
Natural Killer (NK) cells are cytotoxic lymphocytes that can directly kill infected and transformed cells, and shape adaptive immune responses by secretion of cytokines. NK recognition of target cells is mediated by the balance of signaling conveyed by germline-encoded activating receptors for stress-induced or virus-encoded ligands on target cells (Lanier, 2005; Marcus et al., 2014; Yokoyama and Plougastel, 2003), and inhibitory receptors that engage MHC I molecules (Karre, 2008).
In vivo, NK cells can reject sensitive tumor cells efficiently, but ex vivo, resting NK cells obtained from healthy animals or donors often exhibit relatively low cytotoxicity (Diefenbach et al., 2001; Glas et al., 2000; Raulet, 2004). The mechanisms whereby NK cells acquire strong effector activity in vivo against tumor cells are not well defined. Acquisition of strong effector activity (‘priming’) can be conferred by infections, cytokines (e.g. type I interferon (IFN), IL-15 and IL-12), ligands for pattern recognition receptors (e.g. double-stranded RNA) and irradiated tumor cells (Chaix et al., 2008; Diefenbach et al., 2001; Glas et al., 2000; Guia et al., 2008; Mortier et al., 2009). Whether these pathways are relevant in priming NK cell activity in the tumor setting is unclear.
Because IFNs can prime strong effector activity in NK cells, the cGAS-STING pathway is an attractive candidate when considering the activation of NK cells to exert anti-tumor activity, The cGAS-STING pathway mediates cellular immune responses to cytosolic DNA (Chen et al., 2016b; Ishii et al., 2006; Stetson and Medzhitov, 2006). The cGAS enzyme, when bound by cytosolic DNA, catalyzes the synthesis of a cyclic-GMP-AMP dinucleotide called 2'3'-cGAMP (Ablasser et al., 2013a; Diner et al., 2013; Gao et al., 2013b; Wu et al., 2013; Zhang et al., 2013). cGAMP binds and activates the ER-resident adaptor protein STING (stimulator of interferon genes protein) (Ablasser et al., 2013a; Diner et al., 2013; Gao et al., 2013c; Ishikawa et al., 2009; Zhang et al., 2013), which leads to the downstream activation of the transcription factors interferon regulatory factor 3 (IRF3) and nuclear factor κ (NF-κB) (Chen et al., 2016b) and the expression of type I IFN, IFN responsive genes, and various other chemokines and cytokines (e.g., CCL5). The CGAS-STING pathway plays an important role in immune responses to viral infections (Chen et al., 2016b; Ishii et al., 2006; Stetson and Medzhitov, 2006) and emerging evidence in both tumor transfer models and autochthonous models of cancer suggests a role for this pathway in anti-tumor immunity as well (Brzostek-Racine et al., 2011; Gasser and Raulet, 2006a; Hartlova et al., 2015; Lam et al., 2014; Ohkuri et al., 2014; Woo et al., 2014; Zhu et al., 2014).
It has been suggested that DNA leaking from tumor cell nuclei or from dying tumor cells can activate STING in host cells and induce T cell-mediated anti-tumor responses (Klarquist et al., 2014; Ohkuri et al., 2014; Woo et al., 2014). The model suggests that tumor derived-DNA accesses the cytosol of host antigen presenting cells (APC) by some unknown mechanism, where it triggers the cGAS-STING pathway and causes production of IFN. IFN causes maturation of APC and enhances priming of T cells against the tumor. Because STING activation and IFN production can potentially prime strong effector activity in NK cells, the cGAS-STING pathway could be important in the activation of intra-tumoral NK cells responses.
Here we found that spontaneous in vivo NK cell rejection of tumor cells, but not untransformed cells, depends critically on the cGAS-STING pathway. pathway. cGAS in tumor cells was active under steady-state conditions, and could elicit spontaneous NK responses to tumor cells via activation of STING in host cells and subsequent IFN-mediated priming. Our findings provide insight into the mechanisms activating NK cell anti-tumor activity in vivo, and have implications as to the activation of T cells and other immune cells in the tumor.
Results
Stinggt/gt mice are susceptible to tumors independently of effects on T and B cells
STING is important for inducing T cell responses against tumors (Woo et al., 2014). To test whether STING plays a role in anti-tumor responses against tumors that are poorly recognized by T cells, we challenged mice with the TAP2-deficient RMA-S lymphoma and the poorly immunogenic B16-BL6 melanoma. RMA-S lymphoma cells were rejected by WT mice but grew progressively in STING-deficient (Stinggt/gt) mice (Fig. 1A). Rejection of RMA-S cells was also impaired in mice deficient for IRF3, which acts downstream of STING (Fig. 1B). B16-BL6 melanoma tumors also grew more rapidly in Stinggt/gt mice and Irf3−/− mice than in WT mice (Fig. 1C, D). To rule out a contribution of T cells or B cells to these anti-tumor responses, we bred Stinggt/gt mice with Rag2−/− mice, which lack T and B cells. Rag2−/− Stinggt/gt mice were significantly more susceptible to RMA-S and B16-BL6 tumor challenge than Rag2−/− mice (Fig. 1E,F). In responses to other transplanted tumors, T cells play an important role, but if the cells are NK-sensitive, NK cells may also participate in rejection. For example, T cells play an important role in rejecting the MC38 colon carcinoma, but these cells are also NK-sensitive due to expression of ligands for NKG2D and other activating receptors on NK cells. In order to circumvent the T cell response and more clearly reveal the NK cell response, we once again employed mice on the Rag2 background. Rag2−/− Stinggt/gt mice were significantly more susceptible than Rag2−/− mice to challenge with MC38, thereby establishing the relevance of host STING in yet another tumor model (Fig 1G).
Figure 1. Stinggt/gt mice are susceptible to tumors independently of effects on T and B cells.

Tumor cells were injected s.c. into mice (n=4-6). Tumor growth was assessed by caliper measurements, and statistical significance was assessed by 2-way ANOVA. Bars represent +/− means SEM. Results are representative of two to four independent experiments. Tumors were injected into WT or Stinggt/gt mice (A, C), into WT or Irf3−/− mice (B, D), or into Rag2−/− or Rag2−/− Stinggt/gt mice (E,F,G). The mice were injected with the following tumor doses: (A, B, E) 2 × 105 RMA-S or RMA cells; (C, D, F) 2 × 104 B16-BL6 cells; (F) 5 × 104 RMA-RAE-1ε cells; (G) 105 MC38 cells
STING induces NK cell-mediated anti-tumor responses
Both RMA-S and B16-BL6 are sensitive to NK cell rejection, suggesting that STING can induce NK cell responses. STING-mediated protection against RMA-S and B16-BL6 tumors (Fig. 2A,C) was abolished by antibody-mediated depletion of NK cells (Fig. 2B,D). These analyses confirmed that spontaneous STING-mediated protection against RMA-S and B16-BL6 tumors required NK cells but not T cells or B cells. Furthermore, host STING played no role in tumor growth when tumors were NK-insensitive, as in the case of RMA, the MHC I+ counterpart of RMA-S (Fig. 2E). Thus, the impact of STING in these responses is mediated through NK cells and not through the cytostatic effects of IFN or some other NK-independent mechanism.
Figure 2. STING induces NK cell-mediated anti-tumor responses.

Tumor cells were injected s.c. into mice (n=4-6). Tumor growth was assessed by caliper measurements, and statistical significance was assessed by 2-way ANOVA. Bars represent means +/− SEM. Results are representative of two to four independent experiments. In some groups (B, D), NK cells were depleted with PK136 antibody. Tumor cells were injected into WT or Stinggt/gt mice. The mice were injected with the following tumor doses: (A,B) 2 × 105 RMA-S; (C,D) 2 × 104 B16-BL6 cells; (E) 105 RMA cells; (F) 5 × 104 RMA-RAE-1ε cells.
Despite expression of MHC I, RMA cells are rendered NK-sensitive when transduced with an NK activating ligand, RAE-1ε, a ligand for the NKG2D receptor, (Diefenbach et al., 2001). Notably, rejection of RMA-RAE-1ε cells was also dependent on host STING expression (Fig. 2F). Therefore, STING was required for rejection of tumor cells that were sensitive to NK cells due to MHC I-deficiency or expression of activating ligands. These findings suggest a role for STING against many tumor types, as NKG2D ligand expression and NK sensitivity are common features of tumors (Raulet et al., 2013).
Stinggt/gt mice have functional NK cells and are capable of rejecting MHC I-deficient bone marrow grafts
We asked whether STING was required for the normal development of NK cells. WT and Stinggt/gt mice contained comparable numbers of splenic NK cells and showed similar expression of phenotypic markers such as CD11b, Ly6C, and NKG2D (Fig 3A-E), suggesting that this is not the case. Moreover, NK cells in Stinggt/gt mice responded normally with respect to cytokine induction when stimulated ex vivo with plate-bound antibodies against activating receptors NKp46 or NKG2D (Fig 3F). NK cells in Stinggt/gt mice were also functional in vivo in rejecting bone marrow grafts from MHC I-deficient B2m−/− mice, whereas negative control NK cell-deficient NK-DTA mice were unable to reject B2m−/− bone marrow cells, as expected (Fig. 3G). These data suggest that STING is essential for NK rejection responses against tumor cells in vivo, but not for in vivo rejection of untransformed MHC I-deficient cells.
Figure 3. Stinggt/gt mice have functional NK cells, and are capable of rejecting MHC I-deficient cells.
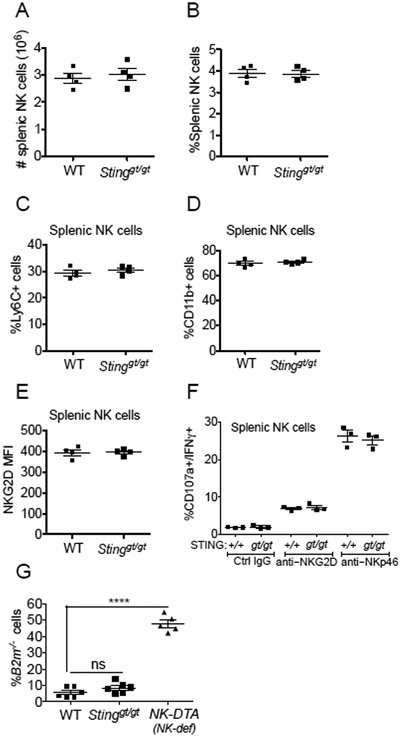
Splenocytes from WT or Stinggt/gt mice (n=4) were analyzed by flow cytometry for (A, B) absolute number (A), or percentage (B), of NK cells; (C, D) percentages of Ly6C+ (C) or CD11b+ (D) NK cells; (E) Mean Fluorescence Intensity (MFI) of NKG2D staining of NK cells. (F) Splenocytes (n=3) were stimulated with plate-bound antibodies (for NKG2D or NKp46, or control IgG). The percentages of CD107a+/IFN-γ+ NK cells were assessed by flow cytometry. (G) Rejection of B2m−/− bone marrow cells by WT and Stinggt/gt mice but not NK cell-deficient NK-DTA mice (n=5-6). A 50:50 mixture of CFSE-labeled B2m−/− and WT bone marrow cells was injected intravenously, and recovery of B2m−/− cells was assessed by flow cytometry three days later. Results are representative of two to four independent experiments.Bars represent means +/− SEM. Statistical significance was assessed using 2-tailed t tests in panels (A-F), where no significant differences were noted, or 1-way-ANOVA with Bonferroni’s correction for multiple comparisons (G).
Host cGAS is dispensable for tumor rejection
It has been previously suggested that, via an unknown mechanism, DNA from dying tumor cells accesses the cytosol of host cells where it triggers the cGAS-STING pathway and the production of IFN, thus inducing T cell-mediated anti-tumor responses (Klarquist et al., 2014; Ohkuri et al., 2014; Woo et al., 2014). In light of the requirement for STING for NK-dependent rejection of tumors, we assessed the requirement for host cGAS, which acts upstream of STING. We used CRISPR/Cas9 technology to generate mice lacking expression of Cgas gene, (see Methods). When tested for the anti-tumor NK response, Cgas−/− mice rejected RMA-S and B16-BL6 tumors as well as WT mice did, demonstrating that host cGAS is not required for these STING-dependent antitumor responses (Fig. 4A, B). As before, Stinggt/gt mice tested in parallel were defective in rejecting both of these tumors. As expected, splenocytes from Cgas−/− mice failed to respond to stimulation with DNA (Fig. 4C), but did respond to stimulation with 2'3'-cGAMP (Fig. 4D). Stinggt/gt mice, in contrast, failed to respond to stimulation with DNA or with 2'3'-cGAMP (Fig. 4C-D). In addition, we verified that the numbers and phenotype of NK cells were similar in Cgas−/− and WT mice (Fig. 4E-H). Finally, we verified that both the Cgas−/− mice and Stinggt/gt mice exhibited the expected sensitivity to infections with DNA viruses, such as HSV-1 (Fig. S1).
Figure 4. Host cGAS is dispensable tumor rejection.

(A,B) Tumor cells were injected s.c. into WT, Stinggt/gt, or Cgas−/− mice (n=4-6). Analysis was as in Fig. 1 legend. Results are representative of two independent experiments. Mice were injected with 2 × 105 RMA-S cells (A) or 2 × 104 B16-BL6 cells (B). (C,D) Splenocytes from WT, Stinggt/gt or Cgas−/− mice were transfected with either Vaccinia Virus dsDNA 70mer (C), or 2'3'-cGAMP (D), and secreted type I IFN in culture supernatants was measured using an IFN bioassay. Splenocytes from WT or Cgas−/− mice (n=6) were analyzed by flow cytometry for the absolute number (E) or percentage (F) of NK cells, or the percentages of NK cells expressing CD11b (G) or Ly6C (H). Results are representative of two to four independent experiments. Bars represent means +/− SEM. Statistical significance was assessed using 2-tailed t tests (E-H) or 1-way-ANOVA with Bonferroni’s correction for multiple comparisons (C-D).
Exogenous cGAMP can activate NK cells extrinsically
The requirement for host STING but not host cGAS for anti-tumor NK responses raised the possibility that the 2'3'-cGAMP cyclic dinucleotide necessary to activate STING originates not in host cells, but in tumor cells. Consistent with the possibility that exogenously supplied cGAMP can activate NK cells, intraperitoneal injections of 2'3'-cGAMP caused NK activation, as shown by increased expression of CD69 and CD137 (4-1BB), and NK cell recruitment to the peritoneum (Fig 5A-F). NK cell activation and peritoneal recruitment induced by 2'3'-cGAMP were both dependent on STING-expression in the recipient mice (Fig 5A-F). We hypothesized that NK cell activation occurs downstream of STING-induced type I IFN. To test that possibility we injected 2'3'-cGAMP into WT, Stinggt/gt, or Ifnar−/− mice. NK cell activation, assessed by CD69 activation, was abolished in both Stinggt/gt, and Ifnar−/− mice, demonstrating that type I IFN acts downstream of STING to activate NK cells (Fig 5G). We further tested whether type I IFN acts directly on NK cells by transferring Ifnar−/− splenocytes into congenic CD45.1 mice, allowing for the co-existence of donor and host NK cells in the same animal, and then challenging the mice with 2'3'-cGAMP. Both WT and Ifnar−/− NK cells were activated, suggesting an indirect effect of IFN on NK cells, but Ifnar−/− NK cells were not activated as well as WT NK cells, indicating an additional direct effect of IFN on NK cells (Fig 5H). Thus, type I IFN activates NK cells both directly and indirectly.
Figure 5. Exogenous 2'3'-cGAMP activates NK cells in a cell extrinsic fashion.

(A-G) 200 ηmol 2'3'-cGAMP was injected i.p. into WT, Stinggt/gt mice or Ifnar−/− mice (n=3-4), and 18 hours later splenocytes were analyzed by flow cytometry. (A,C,G) Percentages of CD69+ or 4-1BB+ splenic NK cells are shown. (B,D) Representative CD69 (B) and 4-1BB (D) staining profiles. (E,F) Analysis of peritoneal wash cells collected from 2'3'-cGAMP-injected mice, showing percentages (E) and absolute numbers (F) of peritoneal NK cells. (H,J) 30 × 106 Ifnar−/− (H) or Stinggt/gt (J) splenocytes were transferred i.v. into CD45.1 mice, and the mice were challenged i.p with 200 ηmol 2'3'-cGAMP. 12 hours later, splenocytes were harvested and donor and host NK cells were analyzed for CD69 expression. (I) Gated NK cells from WT and Stinggt/gt mice were stained intracellularly for STING expression. (K-L) 500 μg (696 ηmol) 2'3'-cGAMP was injected into 7-day established RMA-S tumors (K) or ip in non-tumor bearing mice (L), and one hour later the tumors (K), or peritoneal wash cells (L), were harvested for analysis. Cells were incubated with brefeldin/monesin for five hours prior to intracellular IFN-β staining. Shown are the results for CD11b+ tumor myeloid cells (negative for CD3, CD19, NKp46, Ly6G) (K) and peritoneal B cells (L). Results are representative of two to four independent experiments. Bars represent means +/− SEM. For cGAMP injections, data was analyzed using 1-way ANOVA with Bonferroni’s correction for multiple comparisons.
We stained NK cells for STING expression and found that a subset of them did express STING (Fig. 5I), raising the possibility that STING acts intrinsically in NK cells. We therefore tested whether NK cell-intrinsic STING was necessary for NK cell activation induced by 2'3'-cGAMP. We adoptively transferred splenocytes from Stinggt/gt mice into CD45.1 mice, and challenged the mice with 2'3'-cGAMP. 2'3'-cGAMP injections resulted in equivalent activation of WT and Stinggt/gt NK cells as assessed by CD69 expression (Fig. 5J), demonstrating that NK cell-intrinsic STING signaling is dispensable for NK cell activation and STING must act via other cell type(s).
To address which cells respond to 2'3'-cGAMP, the cyclic dinucleotides were injected directly into RMA-S tumors, and one hour later tumors were harvested and incubated for an additional five hours in the presence of Golgi transport inhibitors allowing for cytokines to accumulate. Cells from tumor dissociates were tested for intracellular accumulation of IFN-β by intracellular cytokine staining. IFN-β was detected in CD11b+ cells (negative for CD3, CD19, NKp46, Ly6G), but not in other infiltrating leukocytes such as neutrophils, T cells or NK cells (Fig. 5K), suggesting that these cells play a role when 2'3'-cGAMP is injected into tumors.
B cells were largely absent from these tumors but could play a role in different tumors in responses to 2'3'-cGAMP. Indeed, when 2'3'-cGAMP was injected intraperitoneally, intracellular IFN-β was detected in peritoneal B cells when examined ex vivo. Taken together, our results suggest that multiple cell types are capable of responding to 2’3’ cGAMP and that the relevant cells for a given response may be context-dependent (Fig. 5L).
cGAS is specifically active in tumor cells
To test directly whether cGAS in tumor cells was required for tumor rejection mediated by host STING, we used CRISPR/Cas9 technology and two gRNAs concurrently to delete the first exon of the Cgas gene in B16-BL6 tumor cells, without stably introducing any other marker proteins in the cells. cGAS-deficient B16-BL6 cells failed to respond to DNA stimulation and did not increase expression of Ifnb, Ccl5, and Ifit1 (Fig. S2). In the absence of DNA stimulation, WT cells showed significant steady-state expression of cGAS/STING target genes Ccl5 and the interferon-inducible Ifit1 gene, and this expression was significantly reduced in cGAS-deficient B16-BL6 cells, arguing that cGAS is partially active without purposeful induction in the WT tumor cells (Fig 6A,B). Ccl5 and Ifit1 expression were restored when the mutant tumor cells were transduced with wildtype cGAS, but not enzymatically inactive cGAS (G198A/S199A). Transduced cGAS, both wildtype and mutant, were expressed at similar levels to each other, but had increased expression compared with endogenous cGAS. Compared to non-transduced cells, the increased expression of wildtype cGAS, but not of mutant CGAS, augmented Ccl5 and Ifit1 expression. Steady-state activation of the cGAS-STING pathway was also observed in human monocytic cell line THP1 with THP1 TMEM173−/− cells expressing lower levels of IFIT1 and CXCL10 compared with THP1 WT cells (Fig S3A-B). These data suggest that cGAS is active in both tumor cell lines in the absence of exogenous DNA stimulation, resulting in low but detectable constitutive expression of IFN-inducible genes. In contrast to the results with the tumor cells, splenocytes, bone marrow cells, lung cells, and liver cells from WT and cGAS-deficient mice expressed similar levels of Ccl5 and Ifit1l (Fig 6C,D, Fig S3C-H). These data indicate that constitutively active cGAS is a distinguishing feature of tumor cells. One possible explanation for the constitutive activation of cGAS-STING pathway is that it is activated by DNA damage in the tumor cells, as was previously suggested (Ho et al., 2016; Lam et al., 2014; Shen et al., 2015). Indeed, induction of DNA damage in B16 cells by chemotherapy drug ARA-C led to increased cGAS-dependent secretion of type I IFN (Fig S3I).
Figure 6. Constitutive cGAS activation in tumor cells leads to tumor rejection dependent on host STING.

(A, B) QRT-PCR analysis of Ccl5 (A) and Ifit1 (B) expression levels in B16-BL6-Cgas+/+, B16-Cgas−/− or B16-Cgas−/− tumor cells, or B16-Cgas−/− cells transduced with either active (Cgaswt) or inactive (Cgasmut) CGAS expression vector. (C,D) QRT-PCR analysis of Ccl5 (C) and Ifit1 (D) expression levels in splenocytes isolated from WT or Cgas−/− mice. Results are representative of three to six independent experiments, and data consists of three technical replicates. Fold expression is shown relative to mutant cells. Statistical significance was assessed using 2-tailed t tests. (E-H) Tumor cells (105) were injected s.c. into WT or Stinggt/gt mice and tumor growth monitored as in Fig. 1 legend. Each group contained 4-6 mice, and results are representative of two independent experiments. Injected tumor cells were: B16-BL6-CGAS+/+ cells (E); B16-BL6-Cgas−/− cells (F); B16-BL6-Cgas−/− cells transduced with active (Cgaswt, G), or inactive (Cgasmut, H) cGAS expression vector. Bars represent means +/− SEM. Statistical significance was assessed as in Fig 1.
Expression of cGAS by tumor cells is crucial for tumor rejection
We tested the impact of cGAS expression on tumor rejection in vivo. Remarkably, cGAS-deficient tumor cells were not rejected in a STING-dependent fashion in vivo, whereas WT tumor cells were partially rejected, as before (Fig 6E-F). Restoration with WT cGAS restored the STING-dependent tumor rejection response, whereas restoration with catalytically inactive cGAS did not (Fig. 6G-H, and Fig S3J-K), confirming that the defect was due to differences in cGAS enzymatic activity and not to other clonally variable properties of the tumor cells. Taken together, these data indicate that the NK-mediated rejection of B16-BL6 tumors requires active cGAS in the tumor cells and STING expression in host cells.
CGAS expression correlates with immune activation and improved survival in melanoma
We sought to evaluate the clinical relevance of our findings by analyzing cGAS gene expression in tumors using publicly available data from The Cancer Genome Atlas (TCGA). Since our findings suggest a role for expression of CGAS in tumors in inducing anti-tumor responses, we focused on melanoma, which tends to be a relatively immunogenic tumor type.
We proceeded to look for gene expression relationships consistent with immune activation being induced by cGAS expression. As a control comparison, we examined pairs of genes that are known to be co-expressed, such as CD3E and CD3D, which did indeed display a significant although imperfect correlation (Fig. 7A). As CGAS-induced genes are also induced by other upstream sensors, we opted to use cGAS expression as a surrogate marker for cGAS activation, based on the reasoning that higher cGAS expression leads to a stronger activation of the pathway. Immune activation was quantified based on expression of several genes including CD69, IFNG, TNF, GZMA, GZMB, and IFIT1. We found strong correlations between cGAS expression levels and expression of immune activation genes (Fig 7B, Fig S4). We could not assess NK cell activation directly because there are no immune activation markers that are specific to NK cells. We could, however, quantify NK cell infiltration by examining expression of several genes that are preferentially expressed by NK cells including: KIR2DL4, NCR1, KLRD1, KLRC1, KLRC2, KLRC3, KLRC4, KLRB1, KLRK1. There was a significant correlation between cGAS and expression levels of the various NK cell receptors (Fig. 7C-D, Fig. S5).
Figure 7. Clinical correlations with cGAS and NK cells in human melanoma.

All the data were obtained from The Cancer Genome Atlas (TCGA). (A) Plot of expression of CD3E vs. expression of CD3D across all cancers. (B) Plot of expression of cGAS vs. expression of CD69 in melanoma. (C) Plot of expression of cGAS vs. expression of KIR2DL4 in melanoma. (D) Plot of expression of cGAS vs. expression of NCR1 in melanoma. (E) Plot of expression of cGAS vs. expression of ULBP1 in melanoma. (F) Plot of expression of cGAS vs. expression of ULBP3 in melanoma. (G) Kaplan-Meier plot of melanoma patient survival; patients are segmented by cGAS expression (highest and lowest thirds). (H) Kaplan-Meier plot of melanoma patient survival; patients are segmented by KIR2DL4 expression (highest and lowest thirds). For correlations of expression, statistical significance was assessed using the Spearman coefficient. For survival analysis, statistical significance was assessed using the log-rank test.
We then asked whether increased cGAS expression is associated with expression of ligands that render cells NK-sensitive. There was a significant correlation between cGAS expression and expression of the NKG2D ligands ULBP1 and ULBP3 (Fig 7E-F), in accordance with an earlier report that showed that cGAS-STING activation can lead to expression of NKG2D ligands (Lam et al., 2014). Finally, in melanoma patients we demonstrated a significant correlation between cGAS expression levels and survival, and between NK cell receptor expression levels (including NCR1) and survival (Fig 7G-H, Fig S6). These data are consistent with a role of cGAS in the induction of anti-tumor responses in melanoma patients in accordance with our model.
Discussion
Two major conclusions can be derived from our findings. First, NK-dependent rejection of tumor cells is largely reliant on activation of STING in non-tumor cells, at least in some cases. Second, steady state activation of cGAS in tumor cells rather than cGAS activation in host cells is required for NK cell responses against tumors, suggesting that tumor-derived cGAMP is responsible for STING activation, and may be transferred to non-cancerous cells to activate the response. Thus, aberrant cGAS activation in tumors boosts anti-tumor immune responses. Notably, rejection of normal MHC I-deficient bone marrow cells by NK cells did not require host STING expression, nor does T cell-mediated skin allograft rejection (Woo et al., 2014). These findings support the conclusion that the immunogenicity of tumor cells in both the NK and T cell responses may be amplified by the cGAS-STING pathway, though it remains possible that other differences between these tumor cells and normal cells or in the experimental protocols may account for the different outcomes.
The evidence that the cGAS-STING pathway underlies NK cell responses to tumors provides a new foundation to the decades-old field of natural cytotoxicity to tumors (Herberman et al., 1975; Kiessling et al., 1975). The findings provide a mechanism for amplifying NK cell responses to aberrant tumor cells, analogous to the role of pattern recognition receptors, including cGAS, in amplifying other immune responses to pathogens. In viral infections, cGAS, RNA sensors and Toll-like receptors play an important role in promoting anti-viral responses, but the role of such receptors in triggering spontaneous NK responses to tumors was not previously demonstrated. STING, which is downstream of cGAS, is important in CD8 T cell responses to tumors (Woo et al., 2014), and it will be interesting to address whether tumor or host cGAS is similarly necessary for such T cell responses.
It has previously been proposed that tumor cells activate the cGAS-STING pathway in vivo through transfer of tumor cell DNA into host cells (Klarquist et al., 2014; Ohkuri et al., 2014; Woo et al., 2014), although the mechanism whereby tumor DNA might access the cytosol and the causal relationship between DNA transfer and STING activation were not established. The DNA transfer model predicts that cGAS, like STING, is required in host cells, contrary to our results. Instead, our results suggest that cGAMP is produced in tumor cells and may be transferred to host cells to trigger STING, resulting in production of the cytokines, such as type I IFN, which are known to enhance NK cell cytotoxicity. cGAMP transfer can occur in the context of viral infection, at least in an in vitro setting, through a variety of mechanisms including: transfer through gap junctions, transfer through viral particles, and transfer via membrane fusion (Ablasser et al., 2013b; Gentili et al., 2015; Xu et al., 2016). In vivo, cGAMP transfer through gap junctions between tumor cells and astrocytes promotes brain metastases, although the underlying mechanisms are not clear (Chen et al., 2016a), but evidence supporting a role for cGAMP transfer in vivo in inducing immune responses is lacking.
Our data suggest that cGAMP is transferred from tumor cells to host cells to initiate the response. Injected 2’3’-cGAMP induced IFNβ expression in CD11b+ cells within tumors, but not in neutrophils, NK cells or T cells, suggesting that CD11b+ cells are candidates for receiving 2’3’-cGAMP during anti-tumor responses. However, it remains possible that other cell types participate, such as B cells, which have not been previously suggested to act as 2’3’-cGAMP sensors but are capable of responding to it. More generally, it is plausible that different cell types play roles in different tumor microenvironments. While we have demonstrated that extracellular cGAMP can trigger host STING and initiate an NK response, additional studies will be necessary to establish if and how cGAMP is transferred from tumor cells to host cells.
STING can be activated in response to DNA damage, which may explain why cGAMP is produced in tumor cells. DNA damage and activation of the DDR are a hallmark of cancer (Gasser and Raulet, 2006b; Hanahan and Weinberg, 2000), and some tumor cell lines such as YAC-1, EμM1, TRAMPC2, DU145, and PC-3 contain detectable cytosolic DNA (Ho et al., 2016; Lam et al., 2014; Shen et al., 2015), and spontaneously produce cytokines in culture in a STING-dependent manner. Together with our finding in this report that the cGAS-STING pathway is constitutively active in B16 and THP1 cells, these data suggest that constitutive activation of the cGAS-STING pathway is fairly common and likely occurs in many tumors. We propose that genomic abnormalities and the consequent activation of the DNA sensing pathway helps to mark these cells as abnormal, and triggers cGAS in the tumor cells. This leads in turn to STING activation in host cells, and eventually mobilizes a sufficiently potent NK cell response to reject the tumors (completely or partially, depending on the system). This proposal would establish cGAS activation as a distinguishing characteristic of tumor cells, and would explain why rejection of non-transformed MHC I-deficient bone marrow cells did not require host STING. It is plausible that rejection of non-transformed cell grafts requires only weak NK activity, that is independent of host STING activation, whereas rejection of growing tumor cells requires a more potent or sustained response that depends on factors downstream of host STING.
Our model positions cGAS expression in tumors as a major determinant of tumor immunogenicity. Interestingly, cGAS is inactivated in certain tumors (Xia et al., 2016a; Xia et al., 2016b). Loss of cGAS from tumor cells might be a mechanism by which tumors evolve to escape the cGAS-STING dependent immune response we describe here. It is difficult to envisage, however, how loss of cGAMP production in one cell would provide a fitness advantage to that cell if tumor cells nearby continued to produce cGAMP. Alternatively, loss of cGAS expression by tumor cells may provide cell-intrinsic benefits to tumor cells, e.g., permit the tumor cell to circumvent senescence (Gluck et al., 2017; Yang et al., 2017), or prevent immunostimulatory NKG2D ligand expression (Lam et al., 2014). Regardless, our results imply that heterogeneity in cGAS activity across tumors may be an important predictor of cancer prognosis and response to treatment. Indeed, our results reinforce the rationale for the use of exogenous cyclic-dinucleotides for tumor immunotherapy (Corrales et al., 2015), and suggest that NK cells may play an important role in mediating the anti-tumor effects of the treatment.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be delivered to and will be fulfilled by the Lead Contact David H. Raulet (raulet@berkeley.edu). Completed of Material Transfer Agreements may be required for obtaining mutant mice or cell lines generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
All experiments were carried out with mice on the C57BL/6J background. C57BL/6J breeder mice were obtained from Jackson Laboratories. Goldenticket Stinggt/gt mutant mice were previously described (Sauer et al., 2011). Rag2−/− mice were crossed to Stinggt/gt mice to generate Rag2−/− Stinggt/gt mice. Irf3−/− mice were kindly provided by T. Taniguchi (University of Tokyo, Tokyo, Japan). B2m−/− and B2m-Ly5.1 mice were bred in our facility. Sex and age-matched (6 to 14 weeks old) mice were used in experiments. Nkp46iCre mice, which express improved CRE recombinase in NK cells (Narni-Mancinelli et al., 2011), were generously provided by Eric Vivier. Nkp46iCre mice were crossed to Rosa26-LSL-DTA (Jackson Laboratories) to generate NK-DTA mice, such that diphtheria toxin is expressed in nascent NK cells, resulting in NK cell-deficiency. Cgas−/− mice were generated in house using CRISPR/Cas9 technology (see below for details). All experiments were approved by the UC Berkeley Animal Care and Use Committee.
Cell lines and culture conditions
RMA-S, B16-BL6, RMA, RMA-RAE-1ε, MC38, THP1, and L929-ISRE cells were cultured in 5% CO2 in RPMI containing 5% FBS (Omega scientific), 0.2 mg/ml glutamine (Sigma-Aldrich), 100 u/ml penicillin (Thermo Fisher Scientific), 100 μg/ml streptomycin (Thermo Fisher Scientific), 10 μg/ml gentamicin sulfate (Lonza), 50 μM β-mercaptoethanol (EMD Biosciences), and 20 mM HEPES (Thermo Fisher Scientific). B16-BL6-Cgas−/− cells were generated using CRISPR/Cas9 technology (see below for details). THP1 and THP1 TMEM173−/− cells were obtained from Invivogen. MC38-GFP/Luc cells were kindly provided by Dr. Michel Dupage (UC Berkeley). All cell lines tested negative for mycoplasma contamination.
METHOD DETAILS
In Vivo Tumor Models
Cells were resuspended in 100 μl of PBS, and injected subcutaneously. Tumor development and growth were monitored by caliper measurements. Tumor experiments typically included 4-6 mice per group. In vivo depletions of NK cells were done by intraperitoneal injections of 200 μg of PK136 antibody (recognizing NKR-P1C, also known as NK1.1) on day −1 prior to tumor injections, and once weekly thereafter. PK136 was purified and validated in our laboratory. NK cell-depletion was verified by flow cytometry.
In vivo stimulation with cGAMP
200 ηmol of 2'3'-cGAMP (made in-house), as well as 2'3'-cGAMP provided by Aduro Biotech, was injected intraperitoneally. 12 or 18 hours later (depending on the experiment), splenocytes and peritoneal wash cells were harvested for analysis.
For intracellular cytokine staining studies, 500 μg (696 ηmol) was injected either intratumorally or intraperitoneally. Tumor dissociates or peritoneal wash cells were isolated one hour later, and incubated for an additional five hours in the presence of brefeldin and monensin prior to intracellular cytokine staining.
In vivo rejection assay
Bone marrow cells from CD45.1 B2m−/− and CD45.1 WT mice were labeled with 10 μM CFSE or 1 μM CFSE, respectively. A mixture of 5 × 106 cells of each type was injected intravenously into recipient mice. Donor cell rejection was assessed 72 hours later by harvesting spleens and analyzing the percentages of CFSEhigh and CFSElow cells by flow cytometry.
HSV1 infections
Age and sex-matched mice were infected intravenously with HSV-1 (ATCC), and monitored thereafter for paralysis and mortality. Moribund mice were euthanized in accordance with animal care guidelines.
Cgas mutant mice
Cgas mutant mice were generated using the CRISPR/Cas9 system. A gRNA was chosen to target the sequence 5’- TGACTCAGCGGATTTCCTCGTGG-3’ in the second exon. A gRNA targeting the tyrosinase gene was also included, so that a coat color change would serve to indicate mice derived from embryos in which targeting was successful. The gRNAs were in vitro transcribed and injected together with Cas9 mRNA (Trilink) into single cell embryos as previously described (Wang et al., 2013). Several founder mice carrying frameshift mutations in the Cgas gene were identified, and a mouse carrying a 31 bp deletion was chosen. The mutation deleted the following sequence: CAAAAGAATTCCACGAGGAAATCCGCTGAGT. The mouse was back-crossed for eight generations to C57BL/6J mice in order to eliminate any tyrosinase mutations or other variants, before intercrossing to generate homozygous mutant mice. Whole genome SNP analysis (UC Davis, Mouse Biology Program) was used to confirm the mice were on a pure C57BL/6 background.
Cgas−/− tumor cells
Cgas−/− B16-BL6 cells were generated using the CRISPR/Cas9 system. Two gRNA flanking the first exon, including the first ATG codon, were selected. The gRNA were transfected together with Cas9 mRNA (Trilink) using lipofectamine2000 (Thermo Fisher Scientific). Cells were single cell-cloned, and mutant cells were identified using PCR. The genomic target sequences used for targeting were: 5’- GTCAGATGTCGATTGATGCC -3’ 5’- GGTGACCTTAAAGTAGTCGC -3’
Plasmids, Mutagenesis, and transduction
The retroviral Cgas expression plasmid, based on the MSCV2.2-IRES-EGFP backbone containing the complete open reading frame of mouse Cgas cDNA, was previously described (Diner et al., 2013). The plasmid was mutagenized to generate inactive Cgas (G198A/S199A) using QuikChange Site-directed mutagenesis kit according to the manufacturer’s instructions. Retroviral supernatants were generated, and transductions were performed, as previously described (Deng et al., 2015). Briefly, 293T cells were co-transfected with plasmids encoding VSV gag/pol, Env, and pMSCV vectors using lipofectamine2000 (Thermo Fisher Scientific). Culture supernatants were collected 48 hours post-transfection, and added to pre-plated cells together with 8μg/ml polybrene. Transduced cells were selected based on GFP expression using an Influx cell sorter.
Flow cytometry
Flow cytometry was performed using standard protocols. Briefly, cells were stained in 50 μl FACS buffer (2% BSA, 0.02% sodium azide, 1mM EDTA). Dead cells were excluded using Live-Dead fixable stain kit using the manufacturer protocols. Cells were incubated for 20 minutes with 2.4G2 hybridoma supernatant (prepared in the lab) to block FcγRII/III receptors. For intracellular staining cells were fixed using Cytofix/Cytoperm (BD). Multicolor flow cytometry was performed on one of the following machines: LSR II, or LSR Fortessa or LSR X20 (BD). Data were analyzed using FlowJo software (Tree Star). NK cells were identified as CD3−, CD19−, NKp46+ cells.
DNA transfections and DNA damage induction
All transfections were carried out with lipofectamine2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. B16 cells were pre-plated at 5 × 105/well in 6-well plates, and transfected with HT-DNA at a final concentration of 100 μg/ml, and cultured for 6 hours, before the cells were harvested for RNA isolation. Splenocytes were plated at 106/well in 96-well plates, and transfected with Vaccina 70mer at a final concentration of 0.5 μg/ml for 4 hours. Afterwards, the media was replaced with fresh media and the cells were incubated overnight. The following day media was harvested for the type I IFN bioassay. Splenocytes were also transfected with 2'3'-cGAMP in digitonin buffer for 30 minutes. Following transfection, the medium was replaced with fresh medium, and the cells were incubated for 7 hours before harvesting the culture supernatant for the type I IFN bioassay. DNA damage was induced by incubation with 50 μM ARA-C, and after 48 hours secreted type I IFN was measured using an IFN bioassay.
NK cell responsiveness assay
High protein-binding flat bottom plates were pre-coated with 5 μg/ml NKG2D (MI-6) or 5μg/ml NKp46 (29A1.4) antibody, or an isotype control antibody. Splenocytes were then incubated in the well for 5 hours in the presence of 1 μg/ml GolgiPlug (BD), 1 μg/ml GolgiStop (BD), 1000 u/ml IL-2 (National Cancer Institute) and CD107a antibody. Following stimulation, the cells were stained for surface markers in order to identify NK cells, and intracellular IFN-γ.
Quantitative RT-PCR
Total RNA was isolated RNAeasy kit (Qiagen) and reverse transcribed using iScript (Bio-Rad Laboratories) according to the manufacturer’s protocol. Q-PCR was performed on a CFX96 thermocycler (Bio-Rad Laboratories) using SSO-Fast EvaGreen Supermix (Bio-Rad Laboratories). Rpl19, and Actin were used as references.
Type I IFN bioassay
L929-ISRE IFN reporter cells have been previously described (Sauer et al., 2011). Briefly, L929-ISRE cells were pre-plated at 5 × 104 per well in flat bottom 96 well plates and incubated in medium for 5 hours. Following incubation, the cells were lysed in passive lysis buffer (Promega) for 5 minutes at room temperature. Cell lysates were incubated with firefly luciferase substrate, and luminescence was measured using SpectraMax Luminescence microplate reader (Molecular Devices).
QUANTIFICATION AND STATISTICAL ANALYSIS
Group sizes, number of replications, and explanation of the mean and error bars are provided in the figure legends. Statistical analysis was done using Prism software (GraphPad software). Tumor growth experiments were analyzed with repeated measures 2-way ANOVA. Flow cytometry, QPCR, and Stimulation experiments were analyzed using 2-tailed t tests or 1-way ANOVA. P-values less than 0.05 were considered significant. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Clinical data analysis
All data were obtained from TCGA using the cbioportal website (Cerami et al., 2012; Gao et al., 2013a). Correlation of gene expression was assessed using the Spearman coefficient. For survival analysis Kaplan Meier curves were plotted comparing the patient with the highest level (upper 33%) and lowest level of particular gene (lower 33%). The curves were compared using the log-rank test.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NK1.1 (PK136) – BV711 | Biolegend | 108745; RRID:AB_2563286 |
| CD3ε (145-2C11) – PE/Cy5 | Biolegend | 100309, RRID:AB_312674 |
| CD19 (6D5) – BV605 | Biolegend | 115540, RRID:AB_2563067 |
| CD45.1 (A20) – Percp/Cy5.5 | Biolegend | 110728, RRID:AB_893346) |
| CD45.2 (104) – BUV395 | BD | 564616 |
| IFN-γ (XMG1.2) – PE | Biolegend | 505808, RRID:AB_315402 |
| CD107a (1D4B) – A647 | Biolegend | 121609, RRID:AB_571990 |
| NKG2D (CX5) – PE | Biolegend | 130207, RRID:AB_1227713 |
| NKp46 (29A1.4) – FITC | Biolegend | 137606, RRID:AB_2298210 |
| CD11b (M1/70) – PE/Cy7 | Biolegend | 101216, RRID:AB_312799 |
| CD69 (H1.2F3) – PE/Dazzle594 | Biolegend | 104536, RRID:AB_2565583 |
| 4-1BB (17B5) – PE | Biolegend | 106106, RRID:AB_2287565 |
| STING (clone 41) | Millipore | MABF213 |
| Bacterial and Virus Strains | ||
| HSV1 | ATCC | ATCC VR-1487 (KOS) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2’3’-cGAMP | Produced in-house | N/A |
| Vaccinia 70mer | Invivogen | tlrl-vav70n |
| ISD DNA | Invivogen | tlrl-isdn |
| Herring Testes HT-DNA | Sigma-Aldrich | D6898 |
| Poybrene | Sigma-Aldrich | TR-1003 |
| Cytofix/Cytoperm | BD | 554722 |
| GolgiPlug | BD | 555029 |
| GolgiStop | BD | 554724 |
| CFSE | ThermoFisher Scientific | C34554 |
| Cytarabine ARA-C | Cayman Chemical | 16069 |
| Critical Commercial Assays | ||
| QuikChange Site Mutagenesis kit | Agilent technologies | 210514 |
| RNAeasy Mini kit | Qiagen | 74104 |
| iScript reverse transcripase | Bio-Rad | 1708841 |
| SSO-Fast Eva Green Supermix | Bio-Rad | 1725203 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | CRL-3216 |
| B16-BL6 | Laboratory of James P. Allison | N/A |
| B16-BL6-Cgas−/− | This paper | N/A |
| RMA-S | Laboratory of Klas Karre | N/A |
| RMA | Laboratory of Klas Karre | N/A |
| RMA-RAE-1ε | Produced in house | N/A |
| THP1 | Invivogen | thpd-nfis |
| THP1-TMEM173−/− | Invivogen | thpd-kostg |
| MC38-GFP/Luc | Laboratory of Dr. Michel Dupage | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | 000664 |
| C57BL/6J Tmem173−/− | Jackson Laboratory | 017537 |
| C57BL/6J Cgas−/− | This paper | N/A |
| C57BL/6J Ifnar1−/− | Jackson Laboratory | 028228 |
| C57BL/6J Irf3−/− | Laboratory of Dr. T. Taniguchi | N/A |
| C57BL/6J Ly5.1 | Jackson Laboratory | 002014 |
| C57BL/6J B2m−/− | Jackson Laboratory | 002087 |
| C57BL/6J Rag2−/− | Jackson Laboratory | 008449 |
| C57BL/6J NKp46iCRE | Laboratory of Dr. Eric Vivier | N/A |
| C57BL/6J R26-LSL-DTA | Jackson Laboratory | 009669 |
| Oligonucleotides | ||
| Synthetic DNA sequences see Table S1 | ||
| Recombinant DNA | ||
| Cas9 mRNA | Trilink | L-7606 |
| MSCV2.2 | Addgene | 60206 |
| Software and Algorithms | ||
| Prism | GraphPrism software | Version 7.0 |
| Flowjo | TreeStar software | Version 10 |
Highlights:
Sting−/− mice but not Cgas Cgas−/− mice fail to mount optimal NK cell anti-tumor responses
Cgas−/− tumor cells fail are defective in inducing anti-tumor responses by NK cells
cGAS is constitutively active in tumor cells but not in untransformed cells
cGAMP is transferred from tumor cells to other cells in the TME to activate STING
Acknowledgements
We thank L. Zhang and T. Trevino for their assistance in the lab, and members of the Raulet as well as Vance labs member for discussions. Hector Nolla and Alma Valeros provided assistance with cell sorting. We acknowledge the technical support of Chulho Kang and the Cancer Research Laboratory Gene Targeting Facility in the generation of Cgas-deficient mice. This research was supported by NIH grant R01-AI0113041 to D.H.R. R.E.V. is supported by an Investigator Award from the HHMI, and by NIH grants AI075039 and AI063302.
Footnotes
Lead Contact: David H Raulet, raulet@berkeley.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary information including six figures can be found online
Declaration of Interests
DHR is a co-founder of Dragonfly Therapeutics, and served or serves on the Scientific Advisory Boards of Dragonfly, Aduro Biotech, Innate Pharma, and Ignite Immunotherapy; he has a financial interest in all four companies and may benefit from commercialization of the results of this research. REV is an inventor on US patent US 9,724,408, Compositions and Methods for Activating Stimulator of Interferon Gene-Dependent Signaling.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, and Hornung V (2013a). cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, and Hornung V (2013b). Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzostek-Racine S, Gordon C, Van Scoy S, and Reich NC (2011). The DNA damage response induces IFN. J Immunol 187, 5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaix J, Tessmer MS, Hoebe K, Fuseri N, Ryffel B, Dalod M, Alexopoulou L, Beutler B, Brossay L, Vivier E, et al. (2008). Cutting edge: Priming of NK cells by IL-18. J Immunol 181, 1627–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu K, et al. (2016a). Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, and Chen ZJ (2016b). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. (2015). Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11, 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, and Raulet DH (2015). Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 348, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbach A, Jensen ER, Jamieson AM, and Raulet DH (2001). Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, and Vance RE (2013). The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, and Chen ZJ (2013a). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. (2013b). Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. (2013c). Structure-function analysis of STING activation by c[G(2',5')pA(3',5')p] and targeting by antiviral DMXAA. Cell 154, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser S, and Raulet D(2006a). The DNA damage response, immunity and cancer. Semin Cancer Biol 16, 344–347. [DOI] [PubMed] [Google Scholar]
- Gasser S, and Raulet DH (2006b). The DNA damage response arouses the immune system. Cancer Res 66, 3959–3962. [DOI] [PubMed] [Google Scholar]
- Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C, Boyron M, Lombard B, Durand S, Kroemer G, et al. (2015). Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 349, 1232–1236. [DOI] [PubMed] [Google Scholar]
- Glas R, Franksson L, Une C, Eloranta ML, Ohlen C, Orn A, and Karre K(2000). Recruitment and activation of natural killer (NK) cells in vivo determined by the target cell phenotype: An adaptive component of NK cell-mediated responses. Journal of Experimental Medicine 191, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasser A (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guia S, Cognet C, de Beaucoudrey L, Tessmer MS, Jouanguy E, Berger C, Filipe-Santos O, Feinberg J, Camcioglu Y, Levy J, et al. (2008). A role for interleukin-12/23 in the maturation of human Natural Killer and CD56+ T cells in vivo. Blood. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA, et al. (2015). DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote antimicrobial innate immunity. Immunity 42, 332–343. [DOI] [PubMed] [Google Scholar]
- Herberman RB, Nunn ME, and Lavrin DH (1975). Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. I. Distribution of reactivity and specificity. International Journal of Cancer 16, 216–229. [DOI] [PubMed] [Google Scholar]
- Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, Cheung FS, Lim WK, Tan PH, Ngeow J, et al. (2016). The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 44, 1177–1189. [DOI] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, et al. (2006). A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 7, 40–48. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, and Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karre K (2008). Natural killer cell recognition of missing self. Nat Immunol 9, 477–480. [DOI] [PubMed] [Google Scholar]
- Kiessling R, Klein E, and Wigzell H (1975). "Natural" killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol 5, 112–117. [DOI] [PubMed] [Google Scholar]
- Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, and Janssen EM (2014). STING-Mediated DNA Sensing Promotes Antitumor and Autoimmune Responses to Dying Cells. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AR, Le Bert N, Ho SS, Shen YJ, Tang ML, Xiong GM, Croxford JL, Koo CX, Ishii KJ, Akira S, et al. (2014). RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res 74, 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL (2005). NK cell recognition. Annu Rev Immunol 23, 225–274. [DOI] [PubMed] [Google Scholar]
- Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, Wang L, Shifrin N, and Raulet DH (2014). Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol 122, 91–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortier E, Advincula R, Kim L, Chmura S, Barrera J, Reizis B, Malynn BA, and Ma A (2009). Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity 31, 811–822. [DOI] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, et al. (2011). Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci U S A 108, 18324–18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, Watkins SC, Sarkar SN, and Okada H (2014). STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res 2, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulet DH (2004). Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol 5, 996–1002. [DOI] [PubMed] [Google Scholar]
- Raulet DH, Gasser S, Gowen BG, Deng W, and Jung H (2013). Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol 31, 413–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, et al. (2011). The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 79, 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SS, Khatoo M, Tan NY, Ishii KJ, and Gasser S (2015). Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep 11, 460–473. [DOI] [PubMed] [Google Scholar]
- Stetson DB, and Medzhitov R (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, Ahn J, and Barber GN (2016a). Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 14, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, and Barber GN (2016b). Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res 76, 6747–6759. [DOI] [PubMed] [Google Scholar]
- Xu S, Ducroux A, Ponnurangam A, Vieyres G, Franz S, Musken M, Zillinger T, Malassa A, Ewald E, Hornung V, et al. (2016). cGAS-Mediated Innate Immunity Spreads Intercellularly through HIV-1 Env-Induced Membrane Fusion Sites. Cell Host Microbe 20, 443–457. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama WM, and Plougastel BF (2003). Immune functions encoded by the natural killer gene complex. Nat Rev Immunol 3, 304–316. [DOI] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Sun L, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 51, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Man SM, Gurung P, Liu Z, Vogel P, Lamkanfi M, and Kanneganti TD (2014). Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J Immunol 193, 4779–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.