

Summary
The formation of new replication origins (cSDR) and repair of DNA double‐strand breaks (DSBs) in E. coli share a commonality. We find that the two processes require the RNAP‐associated factor, DksA. However, whereas cSDR also relies on (p)ppGpp, the alarmone molecule is dispensable for the repair of topoisomerase type II (Top II) DNA adducts and associated DSBs. The requirement for DksA in repair of nalidixic acid (Nal)‐induced DSBs or for the formation of new origins is not suppressed by a greA deletion mutation, indicating an active role of DksA rather than competition with GreA for insertion into the RNAP secondary channel. Like dksA mutations, transcription termination factor Rho mutations also confer sensitivity to Nal. The rho and dksA mutations are not epistatic, suggesting they involve different repair pathways. The roles of DksA in DSB repair and cSDR differ; certain DksA and RNAP mutants are able to support the first process, but not the latter. We suggest that new origin formation and DNA repair of protein adducts with DSBs may both involve the removal of RNAP without destruction of the RNA:DNA hybrid.
Abbreviations
- cSDR
constitutive stable DNA replication
- RNAP
RNA polymerase
- (p)ppGpp
guanosine penta‐ and tetraphosphate
- DSB
double‐strand break
- dNTPs
deoxyribonucleotide triphosphates
- TEC
transcription elongation complex
- Nal
nalidixic acid
- Top II
type II topoisomerase
Introduction
Escherichia coli utilizes at least three different modes of chromosome replication initiation. In addition to DnaA‐dependent DNA unwinding at oriC, replication can initiate at D‐loops and R‐loops (reviewed in Kogoma (1997)). R‐loops consist of RNA insertions into the DNA double helix, generating a RNA:DNA hybrid and a single‐stranded DNA loop. Replication initiation at R‐loops (constitutive stable DNA replication; cSDR) occurs in cells lacking ribonuclease HI (RNase HI), which degrades R‐loop RNA (Ogawa et al., 1984; Maduike et al., 2014). Mutations in RNase HI suppress dnaA ts mutations and enable the growth of oriC mutants (Kogoma and von Meyenburg, 1983). RecA is required in cSDR to either form or stabilize R‐loops (Kasahara et al., 2000). The RNA within the R‐loop is elongated by DNA polymerase I (Pol I), and subsequent loading of two diverging replisomes by the replication restart machinery creates a new origin of replication (Kogoma and Maldonado, 1997). Replisome reloading proteins PriA and PriB are indispensable for cSDR (Masai et al., 1994; Sandler, 2005). cSDR is also dependent on transcription (von Meyenburg et al., 1987), but the role of the factors associated with RNA polymerase (RNAP) in this reaction has not been studied before. Here we find that DksA, a small RNAP‐binding protein, is required for the formation of new origins in cSDR.
The 17.5 kDa DksA protein shares structural similarity with the two anti‐backtracking factors, GreA and GreB (Perederina et al., 2004). DksA is composed of a globular domain with a zinc‐binding region, a C‐terminal (CT) helix and a coiled‐coil domain that inserts into the secondary channel of RNAP (Perederina et al., 2004; Molodtsov et al., 2018). Unlike the Gre factors, DksA does not induce intrinsic RNA cleavage activity of RNAP (Perederina et al., 2004). DksA, together with the β′ subunit of RNAP, forms (p)ppGpp (guanosine penta‐ and tetraphosphate) binding site 2 (Ross et al., 2016). (p)ppGpp also binds to site 1, located 60 Å away from site 2, on the interface of the ω and β′ RNAP subunits (Ross et al., 2013). (p)ppGpp increases the affinity of DksA to RNAP (Molodtsov et al., 2018). Moreover, the conformation of both DksA and RNAP in the complex changes if (p)ppGpp is present. In the absence of (p)ppGpp, binding of DksA to RNAP bends the β′ rim helix and shifts the βlobe/i4 domain, which is a part of the pincers of the DNA‐binding main channel. This shift might weaken the grip on the non‐template DNA in the transcription bubble and decrease open complex stability. Upon binding of (p)ppGpp to the RNAP–DksA complex, the RNAP is restored to the original apo‐form state. Similarly, DksA conformation reverts to its unbound state in the ternary complex.
DksA, together with (p)ppGpp, participates in the stringent response, a reprogramming of cell metabolism in reaction to environmental stressors, such as nutrient deprivation and heat shock (reviewed in Gaca et al. (2015) and Hauryliuk et al. (2015)). Overall, the stringent response leads to the repression of genes required for rapid growth (such as rRNA and ribosomal protein genes) and the activation of genes involved in amino acid biosynthesis, nutrient acquisition and stress survival. Cellular DksA concentrations are constant under various growth conditions (Paul et al., 2004; Rutherford et al., 2007). DksA acts both on its own and together with (p)ppGpp to regulate transcription initiation (Paul et al., 2004; Paul et al., 2005) and elongation (Tehranchi et al., 2010; Furman et al., 2012). DksA also prevents transcription stalling when translation and transcription are uncoupled (Zhang et al., 2014) and improves transcription fidelity (Roghanian et al., 2015; Satory et al., 2015). For a recent review describing transcriptional responses to DksA and (p)ppGpp see Gourse et al. (2018).
Additionally, both DksA and (p)ppGpp have been implicated in DNA repair. dksA mutants are sensitive to DNA damaging agents, such as UV light, the chemotherapeutic agent mitomycin C and nalidixic acid (Nal, an antibiotic belonging to the quinolone drug class) (Meddows et al., 2005; Trautinger et al., 2005). Similarly, the lack of (p)ppGpp sensitized, whereas increased (p)ppGpp levels provided resistance to several genotoxic agents, such as UV, methyl methanesulphonate (MMS), nitrofurazone (NFZ) and 4‐nitroquinoline‐1‐oxide (4NQO) (McGlynn and Lloyd, 2000; Trautinger et al., 2005; Madison et al., 2014; Kamarthapu et al., 2016). (p)ppGpp was suggested to minimize stalled RNAPs, blocking replication fork progression and promote survival via a mechanism that involves RecA loading on ssDNA and subsequent SOS induction (McGlynn and Lloyd, 2000; Trautinger et al., 2005). More recently, (p)ppGpp was implicated in the Mfd‐independent transcription‐coupled repair (TCR) pathway, facilitating DNA repair by promoting UvrD‐mediated RNAP backtracking (Kamarthapu et al., 2016). A contradicting study, however, demonstrates that genome‐wide TCR is dependent on Mfd but does not require (p)ppGpp (Adebali et al., 2017).
In this study, we focused on Nal‐induced damage, which introduces both DNA–protein adducts and double‐strand breaks (DSBs). In Gram‐negative bacteria, Nal predominantly targets gyrase, a type II topoisomerase. Gyrase introduces negative supercoils into DNA to relieve torsional stress in front of replisomes and transcribing RNAPs (reviewed in Drlica et al. (2008) and Aldred et al. (2014)). Type II topoisomerases induce staggered DNA nicks 4 bp apart on both strands and bind covalently to the 5′ phosphate of the two strands, allowing a second DNA duplex to pass through the DSB. Nal stabilizes the transient gyrase–DNA cleavage complex, preventing DNA religation. The gyrase adduct and the DSB pose a barrier to replication and transcription, which leads to irreversible chromosome fragmentation and cell death (Malik et al., 2006). DksA was proposed to enhance the survival after Nal treatment by destabilizing the transcription complexes, thus clearing the way for recombination and DNA repair (Meddows et al., 2005). Nevertheless, a direct role of DksA in the repair of DSBs or the removal of RNAP has not been shown. Additionally, inactive transcription complexes are removed by Rho helicase, which supports chromosome integrity by suppressing replication fork collisions with stalled RNAPs and subsequent formation of DSBs (Washburn and Gottesman, 2011).
Here we describe an interaction of E. coli DksA with RNAP that creates new replication origins and promotes the repair of Nal‐induced DSBs. We analyzed the role of DksA in E. coli MDS42, an MG1655 derivative lacking ~14% of chromosomal DNA, including non‐essential genes and horizontally acquired sequences (Pósfai et al., 2006). We chose this synthetic E. coli strain to ensure that the observed cellular responses in the absence of transcriptional factor DksA do not stem from the presence of cryptic prophages. It was shown previously that rac prophage present in the MG1655 genome renders it more sensitive than MDS42 to bicyclomycin, an antibiotic targeting transcription termination factor Rho (Cardinale et al., 2008). However, we also tested MG1655 in several experiments.
We find that DksA plays an active role in cSDR and confirm its essentiality in the repair of Nal‐induced DNA damage (Meddows et al., 2005). We assess the roles of other RNAP interacting factors in these DksA‐requiring pathways. Importantly, and in contrast to the repair of phleomycin‐induced DNA lesions, we show that DksA does not act passively to exclude GreA/B from the RNAP secondary channel (Sivaramakrishnan et al., 2017). We propose instead that DksA destabilizes transcription elongation complexes during cSDR and DNA repair but leaves the RNA:DNA hybrid to serve as a primer for new DNA synthesis.
Results
DksA is required for cSDR and repair of DNA DSBs bearing protein adducts
In the absence of RNase HI (ΔrnhA), E. coli cells are capable of replicating using not only the chromosomal origin of replication oriC, but also DnaA‐independent oriK sequences, which fire randomly with respect to the cell cycle (von Meyenburg et al., 1987; Maduike et al., 2014). oriK sites contain R‐loops that are extended by DNA Pol I to form new replication origins. This pathway, named cSDR, enables a strain with a temperature‐sensitive DnaA protein to grow at non‐permissive temperatures. We introduced the dnaA46ts and ΔrnhA mutations into E. coli MDS42 (Fig. 1A). A dnaA46ts mutant cannot grow at 42°C, whereas a dnaA46ts ΔrnhA grows at the highest dilution tested (Fig. 1A i and iii). Since the proteins that extend an RNA primer with dNTPs and reload a replisome have been studied previously, we decided to focus on the possible role of RNAP in the formation of new origins. We studied several RNAP mutants and factors that interact with RNAP, such as transcription factor DksA, anti‐backtracking Gre factors and (p)ppGpp. Fig. 1A shows that the deletion of dksA prevents the growth of dnaA46ts ΔrnhA at 42°C (Fig. 1A iv). DksA expressed from a plasmid reversed this phenotype (Fig. 1B iv). A dnaA + ΔrnhA ΔdksA mutant grew at 42°C, supporting a direct role for DksA in cSDR (Fig. 1A vi). In contrast to WT or ΔrnhA strains, we were unable to introduce ΔdksA into a ΔoriC ΔrnhA strain, which replicates only via cSDR (Fig. 1C). The requirement for DksA in cSDR is not specific to MDS42 background, as the deletion of dksA in MG1655 dnaA46ts ΔrnhA strain also blocked the growth at 42°C (Fig. 7 v and vii). These data indicate that DksA enables the formation of new E. coli origins.
Figure 1.
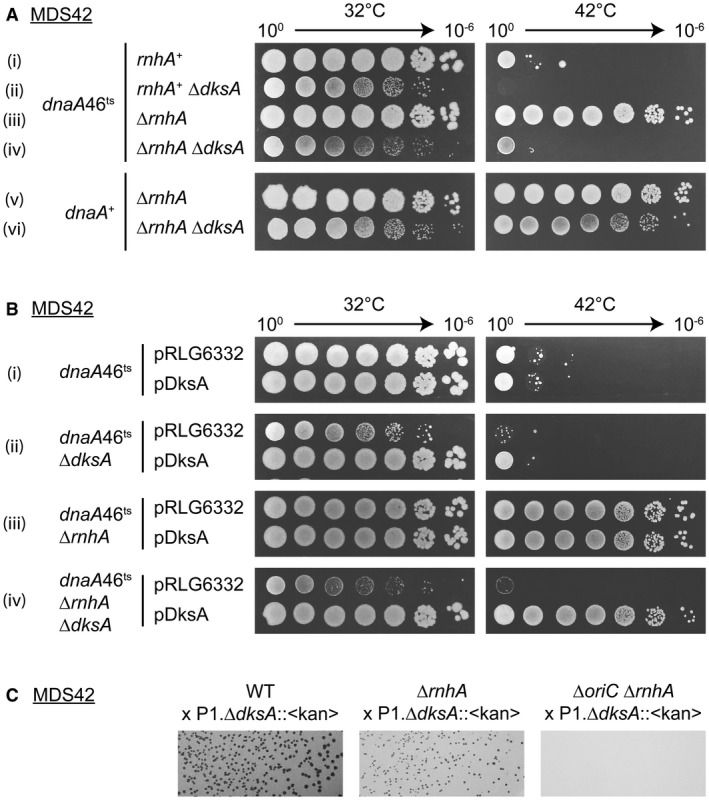
DksA is necessary for oriC‐independent replication. A. Deletion of dksA prevents cSDR and thus growth at 42°C. The strains tested are (i–vi): KM644, KM805, KM650, KM883, 10562, KM777. B. Growth is restored by the expression of a plasmid‐encoded DksA protein. Strains (i–iv): KM644, KM803, KM650, KM882; pDksA = pRLG6333. C. dksA cannot be deleted from a ΔoriC ΔrnhA strain. Strains (left to right): MDS42, 10562, RSW764. P1 was made on JW0141, a Keio collection ΔdksA::kan.
Figure 7.
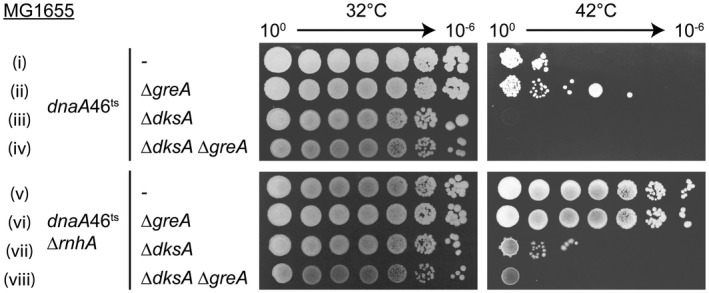
DksA plays an active role in cSDR. Deletion of greA did not restore cSDR to the dnaA46ts ΔrnhA ΔdksA mutant. Strains (i–viii): KM727, KM1058, KM1060, KM1066, KM993, KM1062, KM1064, KM1068.
It has been shown that dksA mutants are sensitive to nalidixic acid (Meddows et al., 2005). Nal inhibits the bacterial DNA gyrase A subunit, creating a DNA DSB with stable 5′ DNA gyrase adducts. This structure creates a lethal barrier for replication and transcription. At present, the repair of Nal lesions is not fully understood. We hypothesized that the repair of Nal‐induced DSBs could require the interaction of DksA with RNAP to create an RNA primer for DNA synthesis, as is the case in cSDR. We decided to compare the requirements for the formation of new origins (cSDR) and DSB repair. We confirmed the sensitivity of ΔdksA mutants to Nal (Fig. 2A ii and B) and showed complementation by plasmid‐encoded DksA (Fig. 2C iv and D).
Figure 2.
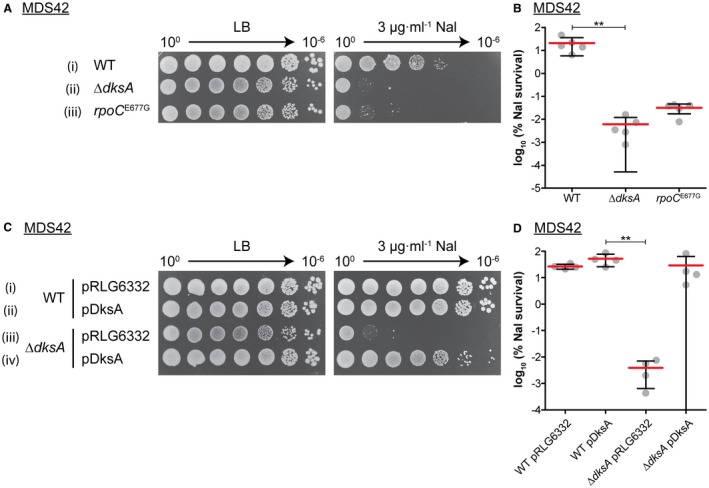
Interaction between RNAP and DksA is required for DNA repair. A. Both ΔdksA and a ‘DksA‐blind’ rpoC E677G mutant are sensitive to Nal. Strains (i–iii): MDS42, KM885, KM807. B. Calculated percentage survival of strains on LB + Nal vs. LB alone. Graph shows mean percentage survival with one standard deviation. Statistical analysis was performed using a nonparametric Kruskal–Wallis test with Dunn’s post test, comparing all data sets. **p < 0.01, n = 5. C. Nalidixic acid sensitivity of ΔdksA is suppressed by the expression of plasmid‐encoded DksA. Strains (i–ii): MDS42, (iii–iv): KM885. pDksA = pRLG6333. D. As in (B), n = 4. [Colour figure can be viewed at wileyonlinelibrary.com].
The ‘DksA‐blind’ RNAP mutant does not support oriC‐independent replication or DSB repair
To confirm that the role of DksA in cSDR and DSB repair involves its interaction with RNAP, we tested a ‘DksA‐blind’ RNAP mutant, rpoC E677G that does not bind to DksA (Satory et al., 2013; Ross et al., 2016). The triple mutant dnaA46ts ΔrnhA rpoC E677G was unable to grow at the non‐permissive temperature, unlike the parental dnaA46ts ΔrnhA (Fig. 3 viii and vii). Control dnaA+ strains displayed a similar colony forming ability at the permissive and non‐permissive temperatures (Fig. 3 i–ii, v–vi). rpoC E677G mutant was also sensitive to Nal (Fig. 2A iii and B). We conclude that the interaction between DksA and RNAP is required for both cSDR and DNA repair.
Figure 3.
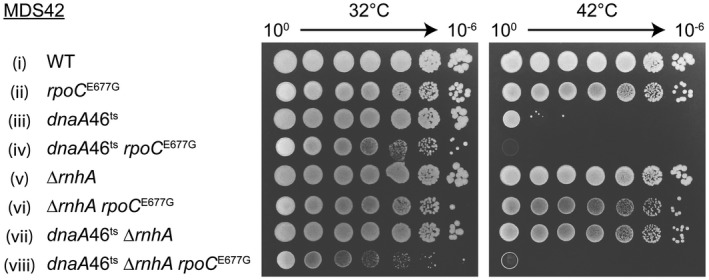
Interaction between DksA protein and RNAP is required for cSDR. A ‘DksA‐blind’ rpoC E677G mutant cannot replicate via cSDR. Strains (i–viii): MDS42, KM807, KM644, KM809, 10562, KM829, KM650, KM801.
(p)ppGpp is required for cSDR but not for the repair of Nal‐induced DNA damage
DksA often acts in concert with (p)ppGpp to regulate transcription initiation. We asked if DksA requires (p)ppGpp to promote new origin formation. First, we deleted relA, which encodes the major (p)ppGpp synthetase in E. coli. The dnaA46ts ΔrnhA ΔrelA strain was able to grow at both low and high temperatures, although colony size at 42°C was decreased relative to the relA + parent (Fig. S1A). Attempts to additionally delete spoT (gene encoding a bifunctional (p)ppGpp synthase/hydrolase) and create a (p)ppGpp0 MDS42 strain were unsuccessful. In E. coli, (p)ppGpp binds to RNAP and also to other protein targets. To determine if the interaction of (p)ppGpp and RNAP is required for cSDR, we investigated RNAP polymerase mutants defective in (p)ppGpp binding. E. coli RNAP carries two (p)ppGpp binding sites. Site 1 is formed by the ω and β′ subunits, whereas site 2 is formed by DksA and β′. The mutations disrupting site 1 (RNAP 1‐) include a deletion of several amino acids from the ω subunit (rpoZ Δ2–5) and three point mutations in the β′ subunit (rpoC R362A R417A K615A) (Ross et al., 2013). The mutations disrupting site 2 (RNAP 2‐) are limited to two substitutions in the β′ subunit (rpoC N680A K681A) that prevent (p)ppGpp but not DksA binding (Ross et al., 2016). We introduced the RNAP 1‐ and 2‐ mutations into the dnaA46ts ΔrnhA strain and tested growth at high temperatures. Mutations in either or both of (p)ppGpp‐binding sites did not affect the growth of the dnaA46ts strains at 32°C (Fig. 4A i–iv). At the non‐permissive temperature, mutations in RNAP binding site 1 reduced colony size, but did not prevent colony formation by dnaA46ts ΔrnhA (Fig. 4A vi). In contrast, site 2 mutations inhibited the growth of dnaA46ts ΔrnhA at 42°C approximately 1000‐fold (Fig. 4A vii). Deletion of both (p)ppGpp‐binding sites was even more inhibitory on the growth of dnaA46ts ΔrnhA at 42°C (Fig. 4A viii).
Figure 4.

Mutations in RNAP (p)ppGpp‐binding sites and the absence of (p)ppGpp inhibit cSDR. A. RNAP (p)ppGpp‐binding site 2 plays a major role in cSDR in MDS42. Strains (i–viii): KK06A, KM899, KK08A, KM911, KK07B, KM901, KK09A, KM913. B. (p)ppGpp is required for oriC‐independent replication. Strains (i–vi): KM712, KM1136, KM1137, KM1171, KM1173, KM1236.
In contrast, deletion of (p)ppGpp‐binding sites had an opposite effect on the sensitivity of strains to Nal. The strains lacking RNAP site 1 grew comparably to the parent, whereas the growth of the RNAP site 2 mutant and the RNAP sites 1 and 2 mutant was significantly improved relative to the wild type (Fig. 5A and B; Fig. S2A and B). Although increased resistance of RNAP 2‐ to Nal was true for both MDS42 and MG1655 background (Figs 5 and S2), the effect of mutations in (p)ppGpp‐binding sites 1 and 2 in cSDR appears to be different depending on the strain background. In MDS42, site 2 plays a bigger role, whereas in MG1655, site 1 seems more important (Fig. S1B). We do not yet have an explanation for this phenomenon.
Figure 5.
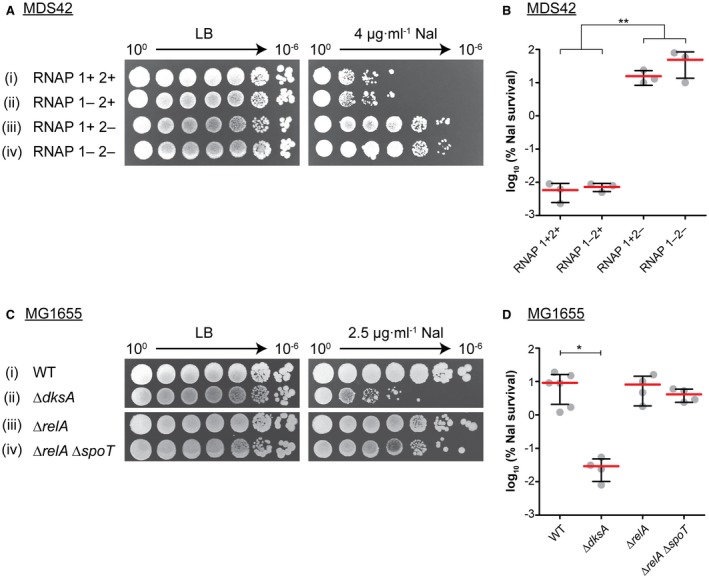
The RNAP (p)ppGpp‐binding site 2 mutation enhances resistance to nalidixic acid, whereas the absence of (p)ppGpp fractionally decreases it. A. The RNAP site 1 mutation did not affect growth on Nal, whereas the RNAP site 2 mutant was more resistant than the wild type. Nalidixic acid concentration was increased to 4 µg ml–1. Strains (i–iv): KK04A, KM915, KK05A, KM917. B. Calculated percentage survival of strains on LB + Nal vs. LB alone. Graph shows mean percentage survival with one standard deviation. Statistical analysis was performed using a nonparametric two‐tailed Mann–Whitney test, comparing combined data for RNAP 2 + vs. RNAP 2‐ mutants. **p < 0.01, n = 3. C. (p)ppGpp is not required for the repair of nalidixic acid‐induced DNA damage. The Nal concentration was decreased to 2.5 µg ml−1 due to higher sensitivity of MG1655‐derived strains. Strains (i–iv): MG1655, KM773, RLG850, RLG847. D. As in (B) but statistical analysis was performed using a nonparametric Kruskal–Wallis test with Dunn’s post test, comparing all data sets. *p < 0.05, n = 6, 4, 4, 4, respectively. [Colour figure can be viewed at wileyonlinelibrary.com].
To eliminate the possibility that the RNAP mutations themselves, rather than the lack of interaction with (p)ppGpp affect R‐loop‐initiated replication and DSB repair, we utilized the MG1655 background. Here, we were able to construct a dnaA46ts ΔrnhA (p)ppGpp0 strain lacking both relA and spoT and test it for growth at permissive and non‐permissive temperatures. We found that the lack of (p)ppGpp prevented cSDR in MG1655 (Fig. 4B vi) to a larger extent than mutations in the (p)ppGpp‐binding sites 1 and 2 (Fig. S1B). However, while RNAP site 2 mutation significantly increased MG1655 resistance to Nal (Fig. S2A and B), the (p)ppGpp0 mutant was ~10‐fold more sensitive to Nal than the parental strain (Fig. 5C and D; Fig. S2C). The discrepancy between the (p)ppGpp0 and RNAP 1‐2‐ phenotypes suggests that the RNAP mutations per se increase resistance to Nal. We confirmed all the (p)ppGpp0 phenotypes by showing that strains failed to grow on a minimal medium and, therefore, had not accumulated suppressors (Figs S1C and S2D). We conclude that (p)ppGpp plays a significant role in cSDR but not in the repair of Nal‐induced DNA damage.
Anti‐backtracking factors are not essential for cSDR
The coiled‐coil domain of DksA protein inserts itself within the RNAP secondary channel. The anti‐backtracking factors GreA and GreB share a similar structure, enter the secondary channel and compete with DksA for binding to RNAP (Vinella et al., 2012). To examine their potential role in cSDR, we deleted each of the genes from the dnaA46ts ΔrnhA strain and assayed the growth at 42°C (Fig. 6 vi–vii). Deletion of greA or greB did not block the growth at non‐permissive temperatures, indicating that the lack of one of the factors does not prevent cSDR. A double greA greB deletion mutant is temperature sensitive in E. coli MG1655 but not in MDS42 (Fig. 6 ix). We were able, therefore, to construct and assay an MDS42 dnaA46ts ΔrnhA ΔgreA ΔgreB mutant. This strain, which lacks both Gre factors, grows at the non‐permissive temperature (Fig. 6 viii). Moreover, a ΔoriC ΔrnhA ΔgreA ΔgreB mutant was also viable (data not shown). Taken together, the data confirm that the anti‐backtracking factors are not required for cSDR.
Figure 6.

GreA and GreB anti‐backtracking factors are not required for cSDR. Deletion of greA or greB does not prevent cSDR in the dnaA46ts ΔrnhA strain. Strains (i–ix): 10583, 12334, 12336, KM1019, KM554, KM586, KM588, KM1021, KM982.
DksA plays an active role in cSDR and DSB repair
It is possible that GreA/GreB blocks cSDR, and that the role of DksA is to reduce the entry of these factors into the RNAP secondary channel (Sivaramakrishnan et al., 2017). To address this question, we attempted to delete greA from dnaA46ts ΔrnhA ΔdksA. We reasoned that if the increased interaction of GreA with RNAP in the absence of DksA inhibited cSDR, then deleting greA should restore the ability of the strain to replicate in an oriC‐independent manner. We were not able to construct the double ΔdksA ΔgreA mutant in MDS42. We could however construct the dnaA46ts ΔrnhA ΔdksA ΔgreA mutant in the MG1655 background (Fig. 7). The mutant was unable to form colonies at the non‐permissive temperature, indicating that DksA exclusion of GreA/B does not account for the DksA requirement for oriC‐independent replication (Fig. 7 viii).
Next, we asked if the deletion of greA rescues the Nal sensitivity of a ΔdksA mutant, as would be predicted if DksA acted passively. dksA mutants are sensitive to the radiomimetic drug, phleomycin (Sivaramakrishnan et al., 2017) (Fig. S3A). A greA deletion not only improved wild‐type growth in phleomycin, but also suppressed the sensitivity of a dksA mutant. This suggested that DksA acts passively to enhance DNA damage repair by excluding GreA from the RNAP secondary channel, thus favoring RNAP backtracking (Sivaramakrishnan et al., 2017). We therefore tested the Nal sensitivity of a MG1655 ΔdksA ΔgreA mutant. Initially, we tested the susceptibility to Nal as in previous experiments, by serially diluting strains and plating them on LB agar containing defined Nal concentrations. We observed that ΔdksA ΔgreA formed colonies on higher dilutions than ΔdksA alone (Fig. S3B). However, unlike WT or ΔgreA strains, ΔdksA ΔgreA formed single colonies starting from the 10−1 dilution. This suggested to us that Nal is bacteriostatic for the double mutant and prompted us to use an additional assay to investigate the effect of Nal on ΔdksA ΔgreA. Growth in the presence of the antibiotic was monitored by the absorbance of cultures at OD600, as well as by counting the viable cells present in the cultures after 3 and 6 h of incubation. The ΔgreA mutant and the wild‐type strain were equally sensitive to Nal and increased in cell mass as well as viability during growth in LB with similar kinetics (Fig. 8). The ΔdksA mutant was very sensitive to Nal showing little increase in culture density and decreasing rapidly in viability with exposure to the inhibitor. After 3 h in Nal, only 3% of the initial ΔdksA culture survived, and by 6 h the viability count was 1% that of the input. The double ΔdksA ΔgreA strain was also sensitive to Nal, showing little increase in OD600 over the 6 h time period (Fig. 8A). However, Nal was bacteriostatic for the double mutant, rather than bactericidal, indicating that ΔgreA has some protective effect in a ΔdksA mutant (Fig. 8B). These findings indicate that DksA plays an active role in the repair of Nal lesions, rather than the passive one of excluding GreA.
Figure 8.

Deletion of greA did not suppress ΔdksA nalidixic acid sensitivity. A. Growth curve in the absence and presence of Nal. Exponential phase cultures were diluted to OD600 = 0.01 and the absorbance was measured hourly. Strains: MG1655, KM1034, KM773, KM1054. B. Quantification of the increase in viable count at 3 and 6 h compared to the viability at t 0 arbitrarily set to 1 for each replicate. Graph represents mean and standard deviation, n = 3. Standard deviation value higher than the mean value results in negative error bars crossing the x‐axis when the y‐axis is in a logarithmic scale.
Rho‐dependent termination is required for the repair of Nal‐induced DNA damage
Inhibition of Rho‐dependent transcription termination leads to chromosomal DSBs (Dutta et al., 2011; Washburn and Gottesman, 2011). This is thought to result from transcription–replication clashes, rather than from failure to repair DSBs. We find that Rho is required to repair DSBs induced by Nal and/or to suppress clashes resulting from such breaks. As shown in Fig. 9, the rho15 missense mutant is highly sensitive to Nal (Fig. 9A iii and C). To determine if Rho and DksA are part of the same repair pathway, we constructed a strain bearing both the ΔdksA and rho15 mutations. To test for epistasis, we lowered the Nal concentration from 3 to 1.5 µg ml−1. At this concentration, both the ΔdksA mutant and the wild type grew (Fig. 9A i–ii), but the rho15 mutant was ~100‐fold more sensitive than the parental strain (Fig. 9A iii and i). The double mutant was more growth‐defective than the rho15 mutant by itself (Fig. 9A iii–iv). We conclude, therefore, that Rho and DksA are involved in different pathways of recovery from Nal‐induced DNA damage.
Figure 9.
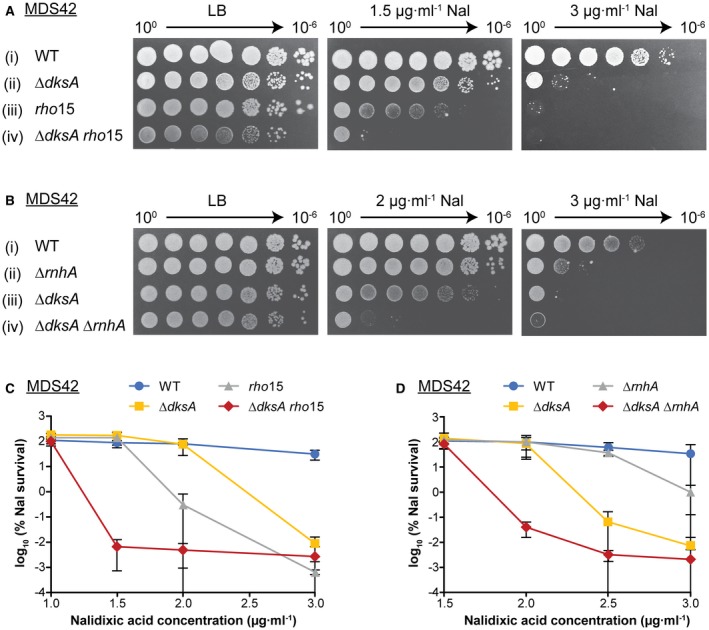
DksA participates in a different DNA repair pathway than Rho and RNase HI. A. The rho15 mutant is more sensitive to Nal than ΔdksA, and together the mutations have an additive inhibitory effect. Strains (i–iv): MDS42, KM885, 10598, 12478. B. The ΔrnhA mutant is more resistant than ΔdksA to Nal and they are not epistatic. Strains (i–iv): MDS42, 10562, KM885, KM777. C. Calculated percentage survival of strains on LB + Nal vs. LB alone. Graph shows mean with standard deviation, n = 3, 2, 3, 4 for Nal concentration 1, 1.5, 2 and 3 µg ml−1, respectively. D. As in (C) n = 3, 3, 3, 6 for Nal concentration 1.5, 2, 2.5 and 3 µg ml−1, respectively.
Roles of RNase HI and DksA in Nal‐induced DNA damage repair
Inactivation of RNase HI is necessary for new DNA origin formation from the resulting persistent R‐loops. On the other hand, R‐loops can initiate DNA breaks (Wimberly et al., 2013). To test if the deletion of rnhA affects Nal sensitivity and if DksA and RNase HI act in the same pathway, we constructed ΔrnhA and ΔdksA ΔrnhA mutants. Abrogation of RNase HI activity exacerbated Nal sensitivity ~100‐fold at 3 µg ml−1 of Nal compared to the wild‐type strain (Fig. 9B ii and D). At lower Nal concentrations, the growth of the rnhA mutant was similar to that of the wild type. Combined, the ΔrnhA and ΔdksA mutations increased Nal sensitivity more than either mutation alone (Fig. 9B and D). We propose that both DksA and RNase HI act to prevent or repair Nal‐induced DNA damage, but that they participate in separate pathways.
A mutation in the RNAP main channel suppresses Nal sensitivity of the dksA mutant and restores new origin formation
To test the hypothesis that DksA might decrease the stability of RNAP and thus contribute to cSDR and DNA repair, we tested several previously isolated RNAP mutants that allow replication in the absence of accessory replicative helicases Rep and UvrD (Baharoglu et al., 2010). One such mutation, rpoB D444G efficiently suppressed the Nal sensitivity of the dksA mutant (Fig. 10A iii–iv and B). Additionally, as shown in Fig. 10C, the rpoB D444G substitution was also able to restore cSDR in the dnaA46ts ΔrnhA ΔdksA strain (Fig. 10C vii–viii). rpoB D444G was shown not only to bypass the need for accessory replicative helicases required to remove transcribing RNAPs, the major obstacle to replication, but also to improve UV resistance of ruvABC, a Holliday junction resolvase mutant (Baharoglu et al., 2010). Based on these observations, it was proposed that the rpoB D444G mutation increases the intrinsic instability of the RNAP–DNA complexes, facilitating both the removal of RNAP upon replication–transcription collisions and replication restart (Baharoglu et al., 2010). Our results are consistent with this model and suggest that destabilization of RNAP is required both for the formation of new origins and for DNA repair.
Figure 10.
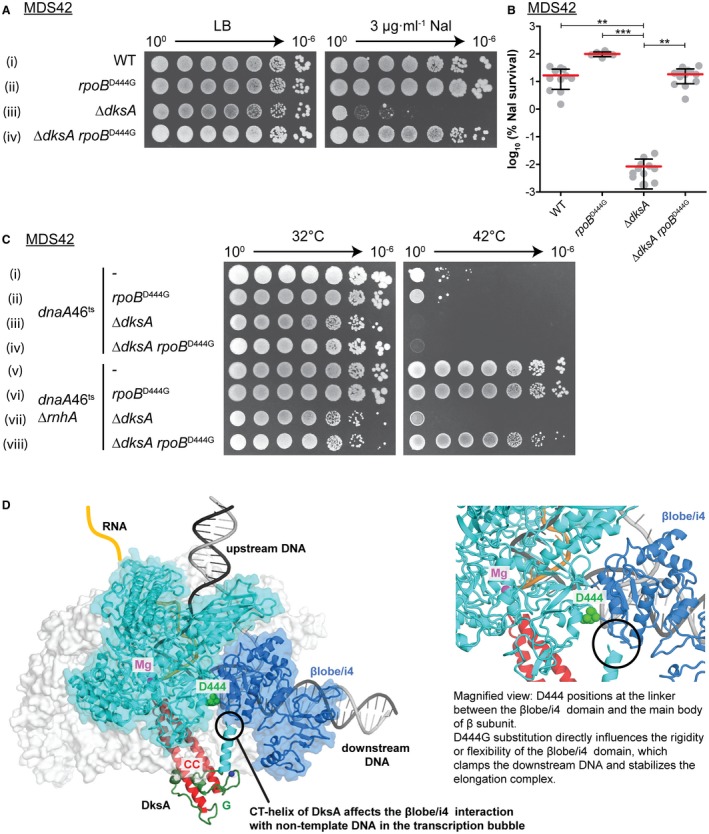
A mutation in the β subunit of RNAP rescues ΔdksA sensitivity to nalidixic acid and allows for replication via cSDR. A. The rpoB D444G mutation enables ΔdksA to grow in the presence of Nal. Strains (i–iv): MDS42, KM1047, KM885, 12481. B. Calculated percentage survival of strains on LB + Nal vs. LB alone. Graph shows the mean percentage survival with one standard deviation. Statistical analysis was performed using a nonparametric Kruskal–Wallis test with Dunn’s post test. **p < 0.01, ***p < 0.001. n = 11, 6, 11, 13. C. rpoB D444G enables cSDR in the ΔdksA mutant. Strains (i–viii): KM644, KM1009, KM803, KM1013, KM650, KM1011, KM882, KM1017. D. Structural model of RNAP in complex with DksA with RNAP residue βD444 annotated.
Separation‐of‐function dksA mutants reveal distinct roles of DksA in cSDR and in DNA repair
To ask if the roles of DksA in cSDR and DNA repair were identical, we tested several DksA mutations previously described as able to complement a dksA deletion. Fortuitously, two DksA point mutants, R91A and D71N/D74N (NN), displayed a separation‐of‐function phenotype. Both were able to complement the sensitivity of ΔdksA to Nal but neither suppressed the temperature sensitivity of dnaA46ts ΔrnhA ΔdksA. This phenotype was seen in both MDS42 (Fig. 11) and MG1655 backgrounds (Fig. S4). The DksA R91A mutation lies in the coiled‐coil domain; DksANN carries two substitutions at the tip of the domain. Both DksA mutants are able to bind to RNAP, but are unable to inhibit transcription from the rrnB P1 promoter in vivo or in vitro (Parshin et al., 2015). When overexpressed from a lac promoter, they can support the growth of ΔdksA on a minimal medium after prolonged incubation (Parshin et al., 2015). These results suggest that the roles of DksA in cSDR and in the repair of Nal‐induced DSBs are not identical.
Figure 11.

DksA coiled‐coil mutant proteins are able to support the repair of nalidixic acid‐induced DNA damage (A) but not cSDR (B). Wild‐type DksA protein and the DksAR91A and DksANN mutants were expressed from pTrc99a plasmids using 1 mM IPTG. Strains used: (A) KM885, (B) KM882.
Discussion
We report here a requirement of the E. coli RNAP‐associated protein, DksA, in the formation of new origins of replication (cSDR) in dnaA46ts ΔrnhA or ΔoriC ΔrnhA mutants (Fig. 1). We also confirm and extend the observation that dksA mutants are sensitive to DNA damage induced by Nal (Meddows et al., 2005). The requirement for DksA for both the formation of new origins and the repair of Nal‐induced DNA damage was demonstrated using a ΔdksA mutation or rpoC E677G, an RNAP β′ subunit mutant that does not bind to DksA (Satory et al., 2013; Ross et al., 2016) (Figs 1, 2, 3).
DksA competes for access to the RNAP secondary channel with the anti‐backtracking factors GreA and GreB. An interplay between the three proteins within cells is complex, involving not only competition for RNAP, but also mutual control of the expression of their genes. Their effects on RNAP activity are in some instances redundant and in others competitive (Vinella et al., 2012). Here, we show that DksA plays an active role both in cSDR and Nal‐induced DSB repair, rather than simply preventing the access of anti‐backtracking factors to the RNAP secondary channel. Thus, the requirement for DksA is not obviated by a greA deletion (Figs 7 and 8). This is in contrast to the mainly passive role of DksA in the repair of phleomycin‐induced DNA damage, which is attributed to the exclusion of GreA and thus to the enhancement of RNAP backtracking (Sivaramakrishnan et al., 2017). The difference in the type of DNA damage inflicted by Nal versus phleomycin might account for this discrepancy. Phleomycin is a glycopeptide antibiotic that cleaves the DNA in the presence of metal cofactors and O2, leaving simple DSBs (Sleigh, 1976). In contrast, Nal‐induced DSBs carry 5′ type II topoisomerase adducts. Repair of such adducts in eukaryotic cells is known to involve different repair functions than simple DSBs (Aparicio et al., 2016).
DksA, together with the RNAP β′ subunit, forms (p)ppGpp‐binding site 2, which is responsible for most of the effects of (p)ppGpp on transcription initiation (Ross et al., 2016). It is conceivable, therefore, that it is not the lack of DksA per se, but the loss of the transcriptional control exerted by (p)ppGpp that is responsible for the inability of ΔdksA and rpoC E677G mutants to carry out cSDR and Nal‐induced DSB repair. Indeed, a lack of (p)ppGpp prevented cSDR in the dnaA46ts ΔrnhA strain (Fig. 4B). However, the (p)ppGpp0 strain was only fractionally more sensitive to Nal than the wild type and more resistant than ΔdksA (Fig. 5C). These results suggest that the effect of ΔdksA on cSDR could be (p)ppGpp‐dependent. DksA plays an active, (p)ppGpp‐independent role in the repair of Nal‐induced DNA damage, consisting of DSBs and Top II DNA adducts. This lack of (p)ppGpp involvement is in contrast to the described role of (p)ppGpp in transcription‐coupled nucleotide excision repair (TC‐NER) (Kamarthapu et al., 2016). However, the main role of (p)ppGpp in TC‐NER is to facilitate RNAP backtracking away from the damage, which allows efficient repair. In the case of Nal‐induced DNA damage, backtracking does not significantly enhance repair since the lack of GreA, an anti‐backtracking factor, did not rescue the sensitivity of dksA mutant (Fig. 8). A precedent for a (p)ppGpp‐independent role of DksA in genome stability exists, since, as previously reported, the suppression of replication–transcription clashes by DksA is likewise independent of (p)ppGpp (Tehranchi et al., 2010).
An analysis of RNAP (p)ppGpp‐binding mutants did not fully clarify the importance of DksA–(p)ppGpp–RNAP interactions for cSDR and DNA repair. Surprisingly, the phenotype of the RNAP mutants was different than the phenotype of cells in the absence (p)ppGpp. Moreover, the two reactions (cSDR and DNA repair) displayed different (p)ppGpp effects. We found that mutations in the RNAP (p)ppGpp‐binding site 2 strongly inhibit cSDR in MDS42, but have less of an effect in the MG1655 background (Figs 4 and S1B). On the other hand, mutations in site 1, which is composed of the ω and β′ RNAP subunits, had little effect on MDS42 but inhibited growth in the MG1655 background. At present, we do not have an explanation for this phenotype. In contrast, mutations in site 2 enhanced the repair of DSBs, whereas site 1 mutations did not affect Nal sensitivity (Figs 5 and S2B). As mentioned above, site 2 accounts for most of the (p)ppGpp effects on transcription initiation (Ross et al., 2016). The discrepancy between the phenotypes of site 2 mutants and ppGpp0 strain in cSDR and upon exposure to Nal was, therefore, unexpected. RNAP is not the only target of (p)ppGpp; perhaps the interaction of (p)ppGpp with other cellular components could explain the divergent phenotypes. However, the in vivo response of a RNAP sites 1 and 2 double mutant to nutritional shifts and amino acid starvation was equivalent to the (p)ppGpp0 strain, confirming that RNAP is the major target of (p)ppGpp (Ross et al., 2016). The opposite effects of RNAP site 2 mutations on the ability to replicate via cSDR and repair Nal‐damaged DNA indicate that the two processes are not identical. Although DksA can bind to RNAP site 2 (Ross et al., 2016), this interaction must be altered compared to the wild‐type RNAP.
Interestingly, two mutations in the coiled‐coil domain of DksA and the RNAP site 2 mutation displayed similar cSDR and DNA repair phenotypes. Both DksAR91A and DksANN, when overexpressed, supported the repair of Nal‐induced DNA damage in ΔdksA, but did not suppress the temperature sensitivity of the dnaA46ts ΔrnhA ΔdksA strain (Fig. 11). The DksA residue R91 is positioned close to the RNAP (p)ppGpp‐binding site 2 residues β′ N680 and K681 and most likely forms salt bridges with the phosphate groups of (p)ppGpp (Molodtsov et al., 2018). The DksAR91A mutant protein binds to RNAP (albeit with reduced affinity) and similarly to β′N680A K681A strongly inhibits (p)ppGpp‐dependent functions (Parshin et al., 2015; Ross et al., 2016). Thus, R91 is proposed to contribute to the formation of RNAP (p)ppGpp‐binding site 2. However, unlike the RNAP site 2 mutant, the DksA R91A substitution also limited DksA inhibition of transcription in the absence of (p)ppGpp (Ross et al., 2016). The RNAP site 2 mutant and DksAR91A both supported growth on minimal media after a prolonged incubation (Parshin et al., 2015; Ross et al., 2016). The DksA R91 residue interaction with the β′ rim helices may stabilize DksA in the secondary channel and aid in the positioning of the tip of the DksA coiled‐coil domain within the active center of RNAP (Parshin et al., 2015). The DksANN mutant with D71N D74N substitutions at the tip of the coiled‐coil domain had similar phenotypes to DksAR91A, enabling DNA repair but not cSDR (Fig. 11). Residue D74 is very well conserved and was previously shown to be required for DksA function alone and together with (p)ppGpp at RNAP site 2 (Parshin et al., 2015; Ross et al., 2016). Residue D74 interacts with the substrate‐binding region of the RNAP active site and is essential for DksA activity (Parshin et al., 2015). Taken together, these data suggest that the correct positioning of the DksA coiled‐coil tip in the RNAP active center is not required for the repair of Nal‐induced DNA damage but is critical for cSDR. Similarly, DksANN suppresses transcriptional pausing and transcription–replication conflicts even though it cannot regulate transcription initiation (Tehranchi et al., 2010). This further supports the notion that the requirement for DksA in repair of Nal‐induced DNA damage involves its role in transcription elongation rather than transcription initiation.
DksA was dispensable for both DNA repair and cSDR in an RNAP mutant with a rpoB D444G substitution. D444 is located in a linker joining the βlobe/i4 domain and the main body of the β subunit (Fig. 10D), and could stabilize the transcription elongation complex (TEC). Several lines of evidence suggest that the rpoB D444G mutation destabilizes RNAP–DNA complexes during transcription initiation and/or elongation. The rpoB D444G mutation allows cells lacking accessory replicative helicases to overcome rich media synthetic lethality, enables their growth in the presence of an inverted rrn operon and facilitates replication restart (Baharoglu et al., 2010). Similarly, the rpoB D444G mutation was also shown to enhance UV survival of ruvABC mutants, which are unable to resolve Holliday junctions, the last step of homologous recombination (Baharoglu et al., 2010). It has been proposed that mutations that destabilize RNAP–DNA complexes facilitate the repair and the removal of obstacles that might otherwise block replication and create the need for RuvABC proteins to promote restart (Trautinger and Lloyd, 2002). In our study, the rpoB D444G mutation rescued the ability of dksA mutants to replicate via cSDR and repair Nal‐induced DNA damage (Fig. 10), which we also attribute to the decreased stability of TECs. A recent report demonstrated that DksA binding to RNAP in the absence of (p)ppGpp distorts both structures as compared to their apo‐forms or when bound in a ternary complex with (p)ppGpp (Molodtsov et al., 2018). In the binary complex, the CT‐helix of DksA rotates the βlobe/i4 domain. The βD444G substitution could, therefore, increase the flexibility of the βlobe/i4 domain, distorting the RNAP pincers, thus phenocopying DksA bound without (p)ppGpp in the secondary binding channel. We speculate that the destabilization of RNAP is required for both cSDR and Nal‐induced DNA repair.
Although no evidence for DksA destabilization of the TEC in vitro has been described (Roghanian et al., 2015; Kamarthapu et al., 2016), it is not ruled out that DksA might promote transcription termination in vivo. Indeed, DksA reduces transcription–replication clashes in vivo, implying that the protein acts on elongating RNAP (Tehranchi et al., 2010). Note that we find that Rho, the transcription termination factor, is essential for recovery from Nal‐induced DNA damage (Fig. 9). Rho maintains genome stability by preventing replisome–TEC clashes that otherwise would induce replication fork arrest and DSBs (Washburn and Gottesman, 2011). rho and dksA mutations are not epistatic, suggesting that they affect different repair pathways, possibly interacting with different states of elongating RNAP.
cSDR and DNA repair presumably share the requirement for the removal of RNAP. For cSDR to occur, RNAP has to be removed to allow DNA Pol I access to the RNA primer. Rho factor removes both the RNAP and RNA:DNA hybrid and thus cannot support cSDR. We suggest that DksA might destabilize the elongating RNAP without unwinding the RNA:DNA hybrid. This notion requires that the 9–10 bp RNA:DNA hybrid in the TEC be sufficiently stable to persist after RNAP removal. Hybrids of this length have been purified (A. Mustaev, personal communication). Furthermore, in vitro construction of a TEC involves the addition of RNAP to an RNA:ssDNA hybrid. The hybrid is then further stabilized by the addition of the complementary DNA strand (Komissarova et al., 2003). In cSDR, the RNA:DNA hybrid might be stabilized by RecA‐dependent formation of an R‐loop that would incorporate the 5′ end of the nascent transcript.
In the case of DNA repair, destabilization of the TEC by DksA could expose the DNA to allow the recombination and assembly of replication forks, as previously suggested (Meddows et al., 2005). If DksA could remove RNAP without disturbing the RNA:DNA hybrid (and possibly the R‐loop upstream), DNA synthesis extending the RNA primer would allow the assembly of replication forks in a manner similar to cSDR. In vitro experiments supporting this notion have been reported. Thus, the E. coli replisome can use an RNA transcript as a primer to continue leading‐strand synthesis after a collision that displaces RNAP from the DNA template (Pomerantz and O'Donnell, 2008). Future experiments with reconstituted replication–transcription systems in vitro will be necessary to establish the precise role of DksA in cSDR and DNA repair.
Experimental procedures
Bacterial strains
All bacterial strains and plasmids used in this study are listed in Supplementary Tables 1 and 2. The strains used in supplementary figures and strains used for construction are in Supplementary Tables 3 and 4.
Viability assays
E. coli strains were grown for 18 h at 37°C with shaking in LB broth. The cultures were then serially diluted 10‐fold in M9 salts. Five‐microliter aliquots were spotted on LB agar plates and incubated at 32°C and 42°C to assess the replication of dnaA46ts strains via cSDR. To test nalidixic acid (Nal) sensitivity, 5 µl aliquots of 10‐fold dilutions were spotted on LB agar plates with and without Nal at a specified concentration. When required, 34 µg ml–1 of chloramphenicol or 100 µg ml–1 of ampicillin was added to the medium for plasmid maintenance. 1 mM IPTG was added to induce gene overexpression, where indicated. Nal sensitivity is presented as the percentage survival on LB + Nal vs. LB. All data points are shown on the graph with the mean marked in red and the standard deviation in black. Statistical analysis was performed using Kruskal–Wallis test with Dunn’s post test, comparing all the data sets. Alternatively, two sets of data were compared using the Mann–Whitney test. All experiments were performed at least twice; representative data sets are shown.
Growth in the presence of Nal
Strains were grown overnight, diluted 100 µl into 5 ml LB in a 50 ml tube and grown at 37°C until cultures reached approx. 108 cfu ml–1, which corresponds to OD600 ~ 0.3–0.5, depending on the strain. Cultures were then diluted to OD600 = 0.01 in 10 ml of LB and split into two 50 ml tubes; one tube was treated with Nal to a final concentration of 3 µg ml−1. The cultures were then incubated, shaking, for 6 h at 37°C. Growth was monitored by measuring absorbance hourly. The viability of the cultures at 0, 3 and 6 h was assessed by serially diluting and spotting on LB plates in triplicates and calculating the cfu ml−1 after overnight incubation. The viability of each culture at t 0 was arbitrarily set to 1 and the viability at t 3 and t 6 was normalized and presented graphically.
Author contributions
The study was designed by KKM and MEG. KKM performed the experiments with the help of KK. KKM, RW and MEG analyzed and interpreted the data. KKM and MEG wrote the manuscript. The authors declare they have no conflict of interest with regard to this study.
Supporting information
Acknowlegements
This work was supported using NIH grant no. GM37219 awarded to MEG. We would like to thank Katsuhiko Murakami for preparing Fig. 10D. We are also indebted to Wilma Ross, Rick Gourse, Bénédicte Michel, Christophe Herman, Bob Lloyd and Sergei Borukhov for providing strains and plasmids. Lastly, we thank everyone mentioned above for their helpful discussions.
References
- Adebali, O. , Sancar, A. and Selby, C.P . (2017) Mfd translocase is necessary and sufficient for transcription‐coupled repair in Escherichia coli . Journal of Biological Chemistry, 292, 18386–18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred, K.J. , Kerns, R.J. and Osheroff, N . (2014) Mechanism of quinolone action and resistance. Biochemistry (Mosc), 53, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio, T. , Baer, R. , Gottesman, M. and Gautier, J . (2016) MRN, CtIP, and BRCA1 mediate repair of topoisomerase II‐DNA adducts. Journal of Cell Biology, 212, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu, Z. , Lestini, R. , Duigou, S. and Michel, B . (2010) RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Molecular Microbiology, 77, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale, C.J. , Washburn, R.S. , Tadigotla, V.R. , Brown, L.M. , Gottesman, M.E. and Nudler, E . (2008) Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli . Science, 320, 935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica, K. , Malik, M. , Kerns, R.J. and Zhao, X . (2008) Quinolone‐mediated bacterial death. Antimicrobial Agents and Chemotherapy, 52, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta, D. , Shatalin, K. , Epshtein, V. , Gottesman, M.E. and Nudler, E . (2011) Linking RNA polymerase backtracking to genome instability in E. coli . Cell, 146, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman, R. , Sevostyanova, A. and Artsimovitch, I . (2012) Transcription initiation factor DksA has diverse effects on RNA chain elongation. Nucleic Acids Research, 40, 3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaca, A.O. , Colomer‐Winter, C. and Lemos, J.A . (2015) Many means to a common end: the intricacies of (p)ppGpp metabolism and its control of bacterial homeostasis. Journal of Bacteriology, 197, 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourse, R.L. , Chen, A.Y. , Gopalkrishnan, S. , Sanchez‐Vazquez, P. , Myers, A. and Ross, W . (2018) Transcriptional responses to ppGpp and DksA. Annual Review of Microbiology, 72, 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauryliuk, V. , Atkinson, G.C. , Murakami, K.S. , Tenson, T. and Gerdes, K . (2015) Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nature Reviews Microbiology, 13, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarthapu, V. , Epshtein, V. , Benjamin, B. , Proshkin, S. , Mironov, A. , Cashel, M. and Nudler, E . (2016) ppGpp couples transcription to DNA repair in E. coli . Science, 352, 993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara, M. , Clikeman, J.A. , Bates, D.B. and Kogoma, T . (2000) RecA protein‐dependent R‐loop formation in vitro . Genes & Development, 14, 360–365. [PMC free article] [PubMed] [Google Scholar]
- Kogoma, T . (1997) Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiology and Molecular Biology Reviews, 61, 212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma, T. and Maldonado, R.R . (1997) DNA polymerase I in constitutive stable DNA replication in Escherichia coli . Journal of Bacteriology, 179, 2109–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma, T. and von Meyenburg, K. (1983) The origin of replication, oriC, and the dnaA protein are dispensable in stable DNA replication (sdrA) mutants of Escherichia coli K‐12. EMBO Journal, 2, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komissarova, N. , Kireeva, M.L. , Becker, J. , Sidorenkov, I. and Kashlev, M . (2003) Engineering of elongation complexes of bacterial and yeast RNA polymerases. Methods in Enzymology, 371, 233–251. [DOI] [PubMed] [Google Scholar]
- Madison, K.E. , Jones‐Foster, E.N. , Vogt, A. , Turner, S.K. , North, S.H. and Nakai, H . (2014) Stringent response processes suppress DNA damage sensitivity caused by deficiency in full‐length translation initiation factor 2 or PriA helicase. Molecular Microbiology, 92, 28–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduike, N.Z. , Tehranchi, A.K. , Wang, J.D. and Kreuzer, K.N . (2014) Replication of the Escherichia coli chromosome in RNase HI‐deficient cells: multiple initiation regions and fork dynamics. Molecular Microbiology, 91, 39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik, M. , Zhao, X. and Drlica, K . (2006) Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Molecular Microbiology, 61, 810–825. [DOI] [PubMed] [Google Scholar]
- Masai, H. , Asai, T. , Kubota, Y. , Arai, K. and Kogoma, T . (1994) Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. EMBO Journal, 13, 5338–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn, P. and Lloyd, R.G . (2000) Modulation of RNA polymerase by (p)ppGpp reveals a RecG‐dependent mechanism for replication fork progression. Cell, 101, 35–45. [DOI] [PubMed] [Google Scholar]
- Meddows, T.R. , Savory, A.P. , Grove, J.I. , Moore, T. and Lloyd, R.G . (2005) RecN protein and transcription factor DksA combine to promote faithful recombinational repair of DNA double‐strand breaks. Molecular Microbiology, 57, 97–110. [DOI] [PubMed] [Google Scholar]
- Molodtsov, V. , Sineva, E. , Zhang, L. , Huang, X. , Cashel, M. , Ades, S.E. and Murakami, K.S . (2018) Allosteric effector ppGpp potentiates the inhibition of transcript initiation by DksA. Molecular Cell, 69, 828–839 e825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, T. , Pickett, G.G. , Kogoma, T. and Kornberg, A . (1984) RNase H confers specificity in the dnaA‐dependent initiation of replication at the unique origin of the Escherichia coli chromosome in vivo and in vitro . Proceedings of the National Academy of Sciences, 81, 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parshin, A. , Shiver, A.L. , Lee, J. , Ozerova, M. , Schneidman‐Duhovny, D. , Gross, C.A. and Borukhov, S . (2015) DksA regulates RNA polymerase in Escherichia coli through a network of interactions in the secondary channel that includes Sequence Insertion 1. Proceedings of the National Academy of Sciences, 112, E6862–6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, B.J. , Barker, M.M. , Ross, W. , Schneider, D.A. , Webb, C. , Foster, J.W. and Gourse, R.L . (2004) DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell, 118, 311–322. [DOI] [PubMed] [Google Scholar]
- Paul, B.J. , Berkmen, M.B. and Gourse, R.L . (2005) DksA potentiates direct activation of amino acid promoters by ppGpp. Proceedings of the National Academy of Sciences, 102, 7823–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina, A. , Svetlov, V. , Vassylyeva, M.N. , Tahirov, T.H. , Yokoyama, S. , Artsimovitch, I. and Vassylyev, D.G . (2004) Regulation through the secondary channel‐structural framework for ppGpp‐DksA synergism during transcription. Cell, 118, 297–309. [DOI] [PubMed] [Google Scholar]
- Pomerantz, R.T. and O'Donnell, M . (2008) The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature, 456, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pósfai, G. , Plunkett, G. 3rd , Fehér, T. , Frisch, D. , Keil, G.M. , Umenhoffer, K. , et al (2006) Emergent properties of reduced‐genome Escherichia coli . Science, 312, 1044–1046. [DOI] [PubMed] [Google Scholar]
- Roghanian, M. , Zenkin, N. and Yuzenkova, Y . (2015) Bacterial global regulators DksA/ppGpp increase fidelity of transcription. Nucleic Acids Research, 43, 1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, W. , Vrentas, C.E. , Sanchez‐Vazquez, P. , Gaal, T. and Gourse, R.L . (2013) The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Molecular Cell, 50, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, W. , Sanchez‐Vazquez, P. , Chen, A.Y. , Lee, J.H. , Burgos, H.L. and Gourse, R.L . (2016) ppGpp binding to a site at the RNAP‐DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Molecular Cell, 62, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, S.T. , Lemke, J.J. , Vrentas, C.E. , Gaal, T. , Ross, W. and Gourse, R.L . (2007) Effects of DksA, GreA, and GreB on transcription initiation: insights into the mechanisms of factors that bind in the secondary channel of RNA polymerase. Journal of Molecular Biology, 366, 1243–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler, S.J . (2005) Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K‐12. Genetics, 169, 1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satory, D. , Halliday, J.A. , Sivaramakrishnan, P. , Lua, R.C. and Herman, C . (2013) Characterization of a novel RNA polymerase mutant that alters DksA activity. Journal of Bacteriology, 195, 4187–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satory, D. , Gordon, A.J. , Wang, M. , Halliday, J.A. , Golding, I. and Herman, C . (2015) DksA involvement in transcription fidelity buffers stochastic epigenetic change. Nucleic Acids Research, 43, 10190–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaramakrishnan, P. , Sepulveda, L.A. , Halliday, J.A. , Liu, J. , Nunez, M.A.B. , Golding, I. , et al (2017) The transcription fidelity factor GreA impedes DNA break repair. Nature, 550, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh, M.J . (1976) The mechanism of DNA breakage by phleomycin in vitro . Nucleic Acids Research, 3, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehranchi, A.K. , Blankschien, M.D. , Zhang, Y. , Halliday, J.A. , Srivatsan, A. , Peng, J. , et al (2010) The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell, 141, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautinger, B.W. and Lloyd, R.G . (2002) Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO Journal, 21, 6944–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautinger, B.W. , Jaktaji, R.P. , Rusakova, E. and Lloyd, R.G . (2005) RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Molecular Cell, 19, 247–258. [DOI] [PubMed] [Google Scholar]
- Vinella, D. , Potrykus, K. , Murphy, H. and Cashel, M . (2012) Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli . Journal of Bacteriology, 194, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenburg, K. , Boye, E. , Skarstad, K. , Koppes, L. and Kogoma, T . (1987) Mode of initiation of constitutive stable DNA replication in RNase H‐defective mutants of Escherichia coli K‐12. Journal of Bacteriology, 169, 2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn, R.S. and Gottesman, M.E . (2011) Transcription termination maintains chromosome integrity. Proceedings of the National Academy of Sciences, 108, 792–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly, H. , Shee, C. , Thornton, P.C. , Sivaramakrishnan, P. , Rosenberg, S.M. and Hastings, P.J . (2013) R‐loops and nicks initiate DNA breakage and genome instability in non‐growing Escherichia coli . Nature Communications, 4 Article number: 2115. https://www.nature.com/articles/ncomms3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Mooney, R.A. , Grass, J.A. , Sivaramakrishnan, P. , Herman, C. , Landick, R. and Wang, J.D . (2014) DksA guards elongating RNA polymerase against ribosome‐stalling‐induced arrest. Molecular Cell, 53, 766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials