

Abstract
Growing evidence links extremes of self-reported sleep duration with higher circulating markers of inflammatory disease risk, although not all findings are consistent. Extremes of sleep duration also associate with activation of the hypothalamic-pituitary-adrenocortical (HPA) system and the peripheral release of cortisol, a glucocorticoid (GC) important in downregulating transcription of pro-inflammatory molecules. Polymorphic variation in the gene encoding the GC receptor (GR; NR3C1) modulates cellular sensitivity to GC-mediated anti-inflammatory signaling, thereby affecting levels of pro-inflammatory molecules. Thus, we hypothesized that extremes of self-reported sleep duration may covary with circulating levels of inflammatory markers as a function of allelic variation in NR3C1. Specifically, we examine the possibility that a single nucleotide polymorphism of the GR gene—(rs6198), the minor (G) allele of which confers reduced GR sensitivity—moderates an association of sleep duration with interleukin (IL)-6 and C-reactive protein (CRP) among a large sample (IL-6: N = 857; CRP: N = 929) of midlife community volunteers of European ancestry. Findings showed that sleep duration varied inversely with IL-6 (β = −.087, p = .012), and this association was stronger among individuals homozygous for the rs6198 G-allele compared to alternate genotypes (β = −.071 , p = .039). We also found that sleep duration showed a U-shaped association with CRP (polynomial term: β = .093, p = .006), which was not moderated by rs6198 genotype. In conclusion, we show that a common genetic variant in the GR moderates an inverse association of self-reported sleep duration with circulating IL-6, possibly contributing to the increased disease risk observed among some short sleepers.
Keywords: Sleep, Genetics, Inflammation, Hypothalamic-Pituitary-Adrenal Axis, Gene-Environment Interaction, Glucocorticoid Receptor, Polymorphism, Interleukin-6, rs6198
Introduction
Mounting evidence suggests that both short and long sleep duration associate with all-cause mortality and acceleration of many diseases of aging, including cardiovascular disease (Cappuccio et al., 2010; Cappuccio et al., 2011). To shed light on these associations, the psychobiological processes linking sleep and health have received recent attention, with accumulating evidence that inflammatory mechanisms may play a role (Hall et al., 2015; Irwin et al., 2016; Irwin, 2015). Indeed, meta-analytic evidence shows that both short and long sleep are independently associated with inflammatory mediators such as interleukin (IL)-6 and C-reactive protein (CRP), although not all associations are consistent (Irwin & Carroll, 2016). In addition, at least one study has shown that inflammatory markers account for a significant portion of the association between self-reported sleep duration and mortality risk, although the magnitude of the effect was greater for short sleepers than for long sleepers (Hall et al., 2015). Together, this evidence suggests that inflammatory mechanisms may play a role in linking sleep to health-related outcomes.
Activation of the hypothalamic-pituitary-adrenal (HPA)-axis and the peripheral release of glucocorticoids (GCs), primarily cortisol in humans, functions to down-regulate inflammatory processes via binding of glucocorticoid receptors (GRs) in immune cells (Barnes, 2011; Kadmiel & Cidlowski, 2013). Accumulating evidence suggests that individuals reporting habitually short sleep display modestly elevated HPA-activation. For example, short sleep has been shown to predict higher evening cortisol levels and an attenuation of the normal decline in cortisol release over the day (e.g. Abell et al., 2016; Castro-Diehl et al., 2015; Steiger et al., 1997), although findings vary across studies (e.g. Vgontzas et al., 2004; Guyon et al., 2014; Reynolds et al., 2012; Pejovic et al., 2013). Little is known about mechanisms underlying the health consequences of long sleep duration. Nevertheless, habitually extended sleep has been linked to conditions associated with both HPA-axis activation and inflammatory mediators such as depression, obesity, and diabetes (Patel et al., 2006; Grandner & Drummond, 2007). Together, these observations raise the possibility that recruitment of the HPA-axis may be a prominent physiological effect of extremes of sleep duration, whether short or long. While one might expect cortisol to mediate a homeostatic down-regulation of immune activation, the efficacy of GC-mediated signaling is ultimately effected at the level of the GR. In this regard, individual differences in GR sensitivity have been observed as a consequence of several molecular processes—including reduced expression, weakened ligand affinity, decreased nuclear translocation, and altered interaction with other regulatory transcription proteins (Kino, Su, & Chrousos, 2009; Oakley & Cidlowski, 2011, 2013; Kadmiel & Cidlowski, 2013). Regardless of cause, variability in GR sensitivity may moderate the impact of cortisol on immune cell activity, thereby helping to explain individual differences in the magnitude of association observed between sleep duration and inflammatory disease risk.
Accumulating evidence suggests that polymorphic variation in the gene encoding the GR (NR3C1) can modulate inflammatory responses to endogenous cortisol and the propensity to develop diseases with inflammatory pathophysiology, including cardiovascular disease (Koper et al., 2014; Otte et al., 2010; van den Akker et al., 2008; Koeijvoets et al., 2008). In this regard, the A3669G (rs6198) single nucleotide polymorphism (SNP) has received particular attention, given its reported association with circulating levels of CRP and IL-6 (Otte et al., 2010; van den Akker et al., 2008), as well as risk for cardiovascular disease, hypertension, and depression (Chung et al., 2009; Koeijvoets et al., 2008; Otte et al., 2010; Szczepankiewicz et al., 2011; van den Akker et al., 2008). Specifically, the minor G-allele of rs6198 promotes an alternative NR3C1 mRNA splicing that results in synthesis of the GR-9β isoform of the GR protein, which has previously been found associated with reduced sensitivity of immune cells to GC-mediated anti-inflammatory signaling (DeRijk & deKloet, 2008; Kumsta et al., 2007; Lewis-Tuffin & Cidlowski, 2006). Ordinarily, the more frequent GR-α isoform binds cortisol in the cytoplasm and translocates to the nucleus. The primary mechanisms by which GR-α downregulates inflammation are two-fold: 1) downregulation of NFkB and AP-1 stimulated production of pro-inflammatory cytokines, and 2) enhanced transcription of certain anti-inflammatory factors (e.g. Inhibitor of κβ (Iκβ)) by binding of the GR-α complex to glucocorticoid response elements (GREs) in promotor regions of genes encoding these anti-inflammatory proteins. In contrast, molecular studies show that the GR-9β protein isoform does not bind cortisol, resides constitutively in the nucleus, and by itself is transcriptionally inactive at GRE promotor sites (reviewed by Kino et al., 2009; Oakley & Cidlowski, 2011, 2013; Kadmiel & Cidlowski, 2013; Lewis-Tuffin & Cidlowski, 2006). This suggests that allelic variation in rs6198 may contribute to individual differences in GR sensitivity, such that persons carrying the G-allele may exhibit elevated markers of systemic inflammation and increased risk for inflammatory disease.
Taken together, converging evidence suggests that extremes of sleep duration associate with HPA-axis activation and elevated markers of inflammation, albeit imperfectly and likely of varying impact among individuals. This raises the possibility that differences in the magnitude of the association between sleep and inflammation could be moderated by allelic variation in rs6198, the minor G-allele of which associates with decreased GR sensitivity and elevated circulating markers of inflammation. In the current study, we examine for the first time whether sleep duration covaries with plasma IL-6 and CRP as a function of polymorphic variation in rs6198. Specifically, we explore (a) whether circulating levels of IL-6 and CRP covary with self-reported sleep duration and/or rs6198 genotype, and (b) whether associations between sleep duration and circulating levels of IL-6 and CRP vary as a function of rs6198 genotype. We also hypothesized that sleep duration and circulating levels of IL-6 and CRP would show a non-linear association, with the highest levels of circulating inflammatory markers seen in individuals reporting the shortest and longest sleep. We expected that relative to the A allele of rs6198, presence of the less frequent G allele would associate with higher levels of circulating IL-6 and CRP, and consistent with Otte et al. (2010) and van den Akker et al. (2008), possibly only among individuals homozygous for the G allele. Further, we posited that any effect of rs6198 genotype on IL-6 and CRP levels would be magnified among persons who reported habitually short or long sleep duration. In exploratory analyses, we examined whether lifestyle factors (physical activity, over the counter medication use, BMI, smoking status, and alcohol use) that covary with both systemic markers of inflammation (O’Connor et al., 2009) and sleep duration (Ferrie et al., 2007) contribute to any effects of sleep duration on levels of inflammation and/or any moderating effects of rs6198 genotype.
Methods
Participants
Study data were derived from the University of Pittsburgh Adult Health and Behavior (AHAB) project and were collected between 2001 and 2005. The AHAB project provides a registry of behavioral and biological measurements, plus DNA for genetic analysis of registry phenotypes, on 1295 midlife community volunteers recruited via mass-mail solicitation from communities of southwestern Pennsylvania (principally Allegheny County; Halder et al., 2007, 2010; Manuck et al., 2011; Sweitzer et al., 2013; Erickson et al., 2013). AHAB participants were 30-54 years of age, with no clinical history of atherosclerotic cardiovascular disease, chronic kidney or liver disease, cancer treatment in the preceding year or major neurologic disorders, schizophrenia or other psychotic illness. Other AHAB study exclusions included pregnancy and use of insulin, glucocorticoid, antiarrhythmic, psychotropic or prescription weight-loss medications (Sweitzer et al., 2013). Data collection occurred over multiple laboratory sessions and informed consent was obtained in accordance with approved protocols and guidelines of the University of Pittsburgh Institutional Review Board.
Procedure
All blood samples were drawn between the hours of 7:30 and 10:30 AM. Participants were asked to fast overnight for 8 hours, to avoid exercise for 12 hours, and to avoid alcohol for 24 hours prior to the blood draw. At the same research visit, a nurse conducted a medical history and medication use interview -and obtained measures of height and weight.
Demographic Factors
The following variables were self-reported by participants: age at study entry (years), sex (M/F), and years of education (an indicator of socioeconomic status). Sex was coded as follows: 0 = male, 1 = female.
Sleep Duration
As described previously (Hall et al., 2008; Wong et al., 2015), self-reported sleep duration was obtained from administration of the Stanford Five-City Physical Activity Interview (Sallis et al., 1985). Participants were asked separately about the number of hours per night they had slept over the prior 5 weekday nights (Sunday-Thursday) and the previous 2 weekend nights (Friday and Saturday). Consistent with other studies that have reported on sleep duration using this measure and indices of cardiometabolic risk (Knutson et al., 2006; Taheri et al., 2004), average daily reported sleep duration was calculated as the weighted average of weeknight and weekend values [(5 × weekday sleep duration) + (2 × weekend sleep duration) / 7]. Short sleep duration using this self-reported sleep measure predicted both aggregated cardiometabolic risk (metabolic syndrome) and lower insulin sensitivity in prior studies of the AHAB cohort (Hall et al., 2008; Wong et al., 2015).
DNA isolation and genotyping
Genomic DNA was isolated from peripheral blood mononuclear cells using the PureGene kit (Gentra Systems, Minneapolis, MN). The rs6198 SNP was amplified using unique sequence primers and genotyped by the fluorescent polarization method of Chen et al. (1999). Forward and reverse primer sequences were F-5’-CCTACGCAGTGAAATGTC-3 and R-5’-CAGATTGGACAATCGGAAC-3’ respectively. Genotypes were assigned using the program AlleleCaller by comparison to controls of known genotype. A 5% resample was genotyped independently to check for reproducibility, and all genotypes were confirmed.
Although the AHAB registry includes measurements on a small proportion of African Americans, study analyses were limited to the larger non-Hispanic Caucasian cohort (N=1081) to mitigate effects of sample heterogeneity, race/ethnic differences in allele frequencies at rs6198 (NCBI, 2017), and unknown extent and variability of European genetic admixture among African American participants (Wang et al., 2004; Haider et al., 2009). Of this sample, 97% were successfully genotyped (N = 1050). Frequency of the minor (G) allele was 18%, and similar to other European American samples (e.g. Otte et al., 2010; van den Akker et al., 2008). The distribution of genotypes (GG: N = 40, GA: N = 295, AA: N = 715) conformed to Hardy-Weinberg equilibrium (χ2(1) = 1.88, p = .17)1. For analysis, genotypes were coded 1 (GG), 0 (AG), and −1 (AA). Owing to prior reports that levels of IL-6 or CRP were elevated only in association with homozygosity for the G allele (van den Akker et al., 2008, Otte et al., 2010), further tests contrasted GG genotype (coded 1) with participants carrying any A allele (i.e., AG/AA; coded 0).
Markers of peripheral inflammation (IL-6, CRP).
Blood was collected in citrate treated tubes and plasma was stored at −80°C until batch-analysis. IL-6 levels were determined using a high sensitivity quantitative sandwich enzyme immunoassay kit (R&D Systems, Cat # HS600B) run according to manufacturer’s directions. The range of detection for the assay was 0.156 to 10pg/mL. Samples were run in duplicate, and the average intra-assay CV was 5%. The inter-assay variation was < 3%. As described previously (Haider et al., 2010), plasma CRP was assayed using a BN II nephelometer (Dade-Behring, Inc., Deerfield, Illinois, USA). This assay has a detection range of 0.175–1100 mg/L. Intra-assay CVs range from 2.3-4.4% and inter-assay CVs range from 2.1- 5.7%.
Among participants who were successfully genotyped (N = 1050), 50 individuals were excluded because they were missing both CRP and IL-6 values. Because AHAB exclusions did not include common acute illnesses, such as colds, we also excluded individuals with a history of hepatitis B or C, rheumatoid arthritis, current use of steroids (including inhaled corticosteroids), immunosuppressants, or cold medications (including sedating anti-histamines and antibiotics), individuals with CRP > 10 mg/L (N = 30), and individuals with IL-6 >10 pg/mL, but only if their BMI was < 30 kg/m2 (N = 66). There were 3 individuals missing IL-6 values only, and 3 outliers with IL-6 values > 3 SD above the mean (outlier mean = 103.1 pg/mL, range = 30.90–175.88 pg/mL). This resulted in a final analytic sample of N = 857 for IL-6 and N = 929 for CRP. Individuals included in the analyses were not significantly different from the parent sample on age, sex, educational attainment, sleep duration, or genotype distribution (p’s > .10). The final analytic sample for IL-6 was also not significantly different than the sample for CRP on any of these variables. Finally, raw score distributions for IL-6 and CRP demonstrated abnormal positive skew and were transformed using a natural log (x’ = ln(x)), resulting in normal distribution of the data.
Lifestyle Factors
Physical Activity.
Physical activity was measured using the Paffenbarger Physical Activity Questionnaire (Paffenbarger et al., 1978). This measure asks about the daily distance walked, pace of walking, flights of stairs climbed, and the type, duration, and frequency of exercise, sports, and other recreational activities in the past year. From these activities, weekly total energy expenditure (total kilocalories) is estimated (Paffenbarger et al., 1978). The Paffenbarger questionnaire has adequate test-retest reliability (r = .72) and validity compared to other physical activity measures (Albanes et al., 1990), and predicts risk for all-cause mortality (Paffenbarger et al., 1993). Raw scores for physical activity were transformed using a natural log (x’ = ln(x)) to correct for positive skew.
Body Mass Index (BMI).
Participants’ weight and height was taken before each blood draw for the determination of BMI [(weight in kg)/height in meters squared (kg/m2)]. BMI was assessed on two different days of the study and averaged for these analyses.
Smoking.
Smoking status was self-reported by participants and coded as follows: 0 = never smoker, tried it, ex-smoker; 1 = current cigarette smoker.
Alcohol use.
Average weekly alcohol use was derived from a brief interview in which an open-ended question assessed how many alcoholic drinks each participant drank in a typical week in the past 6 months. Raw scores for alcohol use were transformed using a natural log (x’ = ln(x)) to correct for positive skew.
Over the counter medications.
Over the counter (OTC) medications and dietary supplements were assessed in the medical history interview. Participants reported taking a wide range of OTC products, including: anti-ulcer medications (IL-6 sample: 2.8%; CRP sample: 3.2%), antacids (IL-6 sample: 1.4%; CRP sample: 1.3%), laxatives (IL-6 sample: 0.9%; CRP sample: 0.9%), aspirin (IL-6 sample: 7.2%; CRP sample: 7.2%), non-steroidal anti-inflammatories (IL-6 sample: 7.0%; CRP sample: 7.2%), acetaminophen (IL-6 sample: 1.4%; CRP sample: 1.5%), antioxidants (Vit E, A, C, Selenium, CoQlO, OPC3, pycogenol, pinepark, folic acid, querceti, isoflavomide, alpha lipoic acid) (IL-6 sample: 18.6%; CRP sample: 18.6%), other vitamins (Vit B, Dermavit, PrimeVit, MVI) (IL-6 sample: 43.1%; CRP sample: 45.2%), dietary supplements (Ginko, ginseng, 5-HTP, SAMe, Melatonin, Acetyl-L-Carnitine, Kava Kava, Inositol, Phenylalanine, DMEA) (IL-6 sample: 28.5%; CRP sample: 27.8%), St. John’s Wart (IL-6 sample: 0.9%; CRP sample: 1.1%), niacin (Nicomide, Niaspan) (IL-6 sample: 0.5%; CRP sample: 0.4%), fish oil (cod liver oil, salmon oil, omega 3 oil) (IL-6 sample: 2.3%; CRP sample: 2.5%), short-chain omega-3 oils (flaxseed oil, essential fatty acids, pycnogenol, GLA/gammalinolenic, evening primrose oil, borage oil) (IL-6 sample: 2.3%; CRP sample: 2.5%). OTC medication categories were coded as 1 if a participant reported taking ≥ 1 medication or supplement. All categories were then summed, with values ranging from 0-13.
Statistical analyses
Statistical analyses were conducted using SPSS v22 (IBM Corporation, Armonk, NY) and STATA/SE 14.2 for Windows (StataCorp LP, College Station, TX). Dropout analyses and group comparisons were performed using independent sample t-tests and one-way analysis of variance for continuous variables, and χ2-tests for categorical and dichotomous variables. Preliminary analyses examined relationships among the demographic variables (age, sex, education) and variables of interest (IL-6, CRP, sleep duration, genotype) using Pearson correlations for continuous variables and one-way analysis of variance and χ2-tests for categorical variables.
Multiple regression (MRE) analyses were used to test the association between sleep duration, genotype, and markers of inflammation. Initial models examined the effect of sleep duration and rs6198 genotype on IL-6 and CRP, adjusting for age, sex, and years of education. To test quadratic relationships between sleep duration and markers of inflammation, sleep duration was mean centered and squared, with the squared term entered hierarchically following demographic covariates and the linear term. A significant change in R2 for the quadratic term indicated a significant prediction of the quadratic term above and beyond the linear term. As recommended by Aiken & West (1991), in such instance we performed follow-up analyses to determine the duration of sleep (in hours) at which the inflammatory marker differed significantly from its minimal value along the quadratic curve.
To test the association between sleep duration and markers of inflammation as a function of rs6198 genotype, sleep duration was mean centered and multiplied by the coded genotype model(s). For linear models, sleep duration, genotype, and the interaction term were entered hierarchically, after age, sex, and years of education. For quadratic models, both the linear and quadratic terms were multiplied by the coded genotype and entered into the model with the individual linear and quadratic terms, adjusting for age, sex, and years of education. A significant change in R2 for the linear and quadratic interaction terms together indicated a significant moderation of sleep duration by genotype. As suggested by Keller (2014), we also controlled for covariate × sleep and covariate × genotype interactions to account for any additional confounding effects of the covariates (age, sex, or years of education) on the interaction term.
To investigate whether lifestyle factors could account for any sleep and genotype dependent associations or interactions, we first examined relationships between lifestyle factors (physical activity, OTC medication use, alcohol use, smoking status, and BMI) and the main variables of interest (IL-6, CRP, sleep duration, genotype). We then entered the lifestyle factors into relevant MREs following the demographic covariates. For IL-6, we compared the change in effect size for an observed linear interaction term with and without the lifestyle factors in the model, using an independent samples t-test, accounting for the difference in standard error across models (standard error of the difference; Cohen, Cohen, West, & Aiken, 2003). For a quadratic relationship observed between sleep and CRP, we compared the 95% Confidence Interval (CI) on the R2 term for the linear and polynomial terms together, with and without the lifestyle factors in the model (Cumming & Fidler, 2010).
Results
Subject Characteristics
Subjects were on average 45 years of age, 51% female, and reported an average 16 years of education (Table 1). With respect to relationships between the demographic factors (age, sex, education) and variables of interest (IL-6, CRP, sleep duration, and genotype), age was positively associated with levels of IL-6 (r =.072, p = .034) and negatively associated with sleep duration (IL-6 sample: r = −.117, p = .001; CRP sample: r = −.126, p < .001). Comparison between sexes revealed that males had higher levels of IL-6 than females (M: 1.96 ± 2.15, F: 1.67 ± 1.78, t = 3.118, p = .002), and females reported longer sleep duration than males (IL-6 sample: M: 6.73 hr. ± 1.03, F: 7.04 hr. ± 0.95, t = −4.572, p < .001; CRP sample: M: 6.74 hr. ± 1.02, F: 7.04 hr. ± 0.95, t = −4.54, p < .001). More education was associated with lower levels of IL-6 (r = −.092, p = .007) and lower levels of CRP (r = −.098, p = .003). IL-6 and CRP were moderately correlated (r = .336, p < .001).
Table 1.
Subject Characteristics by subsample
| IL-6 sample (N = 857) mean ± SD |
CRP sample (N = 929) mean ± SD |
|
|---|---|---|
| Age at 1st visit (yrs) | 44.9 ± 6.8 | 44.8 ± 6.8 |
| Sex (%F, M/F) | 51, 421/436 | 51, 460/469 |
| Education (yrs school) | 16.0 ± 2.8 | 16.0 ± 2.8 |
| Sleep duration, average (hrs) | 6.89 ± 1.00 | 6.89 ± 1.00 |
| rs6198 genotype (G/G, G/A, A/A) | 33/236/566 | 34/262/609 |
| IL-6 (pg/mL)d,e | 1.81 ± 1.98 | 1.81 ± 1.96 |
| CRP (μg/mL)d,e | 1.60 ± 1.60 | 1.59 ± 1.84 |
| Weekly Physical activity (kcal)f | 2482.2 ± 1774.4 | 2502.9 ± 1779.9 |
| BMI (kg/m2) | 26.98 ± 5.28 | 26.86 ± 5.21 |
| Smoking status (%Y, N/Y)g | 16, 723/133 | 16, 780/148 |
| Weekly Alcohol use (drinks/week) | 4.9 ± 7.6 | 4.9 ± 7.6 |
| OTC medications | 1.2 ± 1.3 | 1.2 ± 1.3 |
Notes:
mean concentration;
circulating value;
Based on Paffenbarger;
Y: current smoker; N: never smoker, tried it, ex-smoker; BMI: Body Mass Index; OTC: Over the counter; IL-6: Interleukin-6; CRP: C-reactive protein; rs6198: Glucocorticoid Receptor Single Nucleotide Polymorphism (SNP), (G) minor allele
Sleep duration and markers of inflammation (IL-6, CRP)
The relationship between IL-6 and sleep duration was best represented as a linear association, while the relationship between CRP and sleep duration was quadratic (Table 2, Figure 1). More specifically, sleep duration was inversely related to circulating IL-6 after adjusting for age, sex, and education (β = −0.087 (SE = 0.025, p = .012; Table 2, Model 2; Figure 1A). After adjusting for demographic factors, the quadratic relationship between sleep duration and CRP conformed to an upward-U shape (Table 2, Model 6; Figure 1B). The lowest levels of CRP were observed at an average sleep duration of 7.24 hours (CRP = 0.55 mg/L), and the simple slope was significantly greater than zero when sleep duration was less than 6.87 hours (t(x = 6.86) = −1.98, p < .05) and greater than 7.86 hours (t(x =7.87) = 1.98, p < .05). That is, sleep duration was associated with a significant increase in CRP when individuals reported that they slept less than ~7 hours and greater than ~8 hours, on average.
Table 2.
Main Effect of Sleep Duration on Markers of Inflammation
| IL-6 |
Model 1 |
Model 2 |
Model 3 |
|||
|---|---|---|---|---|---|---|
| Variables | β | p | β | p | β | p |
| Age | .072 | .034 | .062 | .071 | .066 | .053 |
| Sex | −.114 | .001 | −.100 | .004 | −.100 | .004 |
| Education | −.096 | .005 | −.092 | .007 | −.089 | .009 |
| Avg Sleep | −.087 | .012 | −.072 | .045 | ||
| (Avg Sleep)2 | .048 | .174 | ||||
| F | 7.580 | < .001 | 7.31 | <.001 | 6.22 | < .001 |
| R2 | 0.026 | 0.033 | 0.035 | |||
| ΔR2 | 0.007 | .012 | 0.002 | .174 | ||
| CRP |
Model 4 |
Model 5 |
Model 6 |
|||
| Variables | β | p | β | p | β | p |
| Age | −.004 | .895 | −.016 | .626 | −.006 | .849 |
| Sex | −.006 | .867 | .009 | .791 | .009 | .794 |
| Education | −.097 | .003 | −.093 | .005 | −.088 | .008 |
| Avg Sleep | −.092 | .006 | −.065 | .059 | ||
| (Avg Sleep)2 | .093 | .006 | ||||
| F | 2.910 | .034 | 4.09 | .003 | 4.79 | < .001 |
| R2 | 0.009 | 0.018 | 0.025 | |||
| ΔR2 | 0.008 | .006 | 0.008 | .006 | ||
Notes: IL-6: Interleukin-6; CRP: C-reactive protein; Age & Education in years; Sex: 0 = male, 1 = female; Avg Sleep = weighted average of weeknight and weekend sleep duration (hours); Avg sleep is mean centered in quadratic models (Models 3 & 6).
Figure 1:
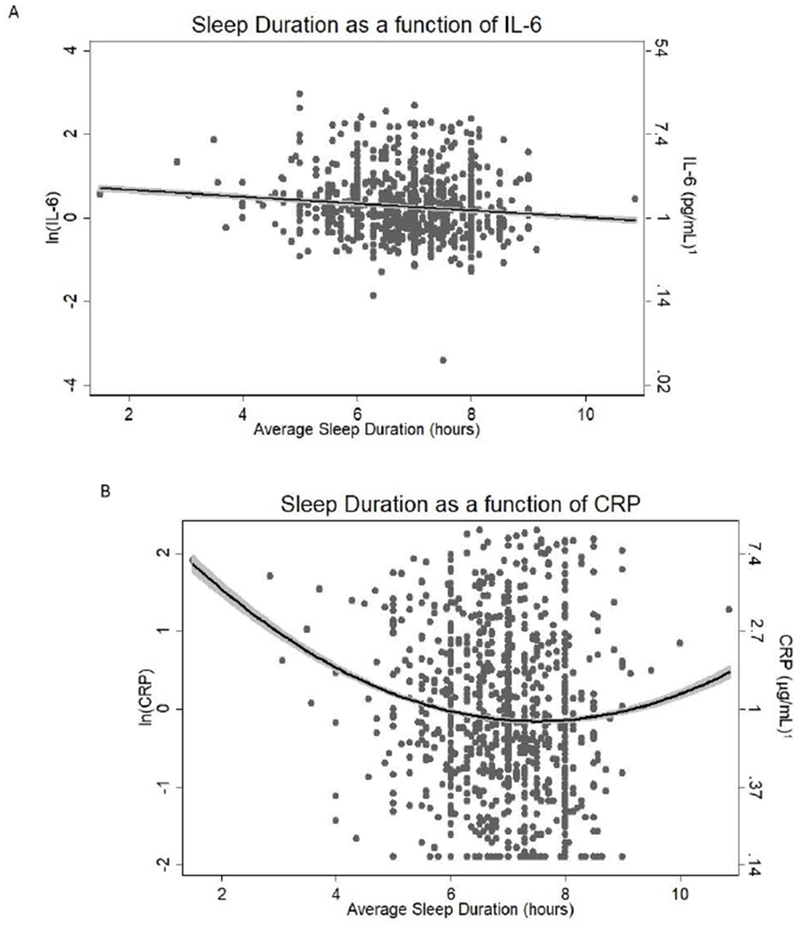
Association between sleep duration and markers of inflammation; A. IL-6 = Interleukin(IL)-6; B. CRP = C-reactive protein. Dark line: Line of best fit. Shading in grey: 95% CI. 1Mathematical transformation of natural log (ln) transformed values (i.e. eŶ)
rs6198 genotype and markers of inflammation (IL-6, CRP)
There was no significant difference in mean levels of IL-6 or CRP between rs6198 genotypes with or without adjustment for age, sex, and education (Table 3, Models 1&3). When the association was examined comparing individuals homozygous for the G-allele (GG) against those carrying any A allele (i.e., AG/AA), there was a marginal effect for IL-6, such that persons homozygous for the G-allele exhibited higher levels of IL-6 compared to those with any A-allele (Table 3, Model 2: β = 0.064 (SE = 0.130), p = .063). CRP was unrelated to rs6198 genotype (Table 3, Models 4).
Table 3.
Main Effect of Genotype on Markers of Inflammation
| IL-6 |
CRP |
|||||||
|---|---|---|---|---|---|---|---|---|
| Model 1 |
Model 2 |
Model 3 |
Model 4 |
|||||
| Variables | β | p | β | p | β | p | β | p |
| Age | .063 | .068 | .062 | .071 | −.002 | .949 | −.003 | .938 |
| Sex | −.113 | .001 | −.114 | .001 | −.005 | .888 | −.005 | .878 |
| Education | −.098 | .004 | −.100 | .004 | −.098 | .003 | −.099 | .003 |
| rs6198 | .054 | .119 | .021 | .530 | ||||
| rs6198 GG | .064 | .063 | .018 | .589 | ||||
| F | 5.940 | < .001 | 6.20 | < .001 | 2.24 | .063 | 2.22 | .065 |
| R2 | .028 | .029 | .010 | .010 | ||||
Notes: Age & Education in years; Sex: 0 = male, 1 = female; rs6198: a glucocorticoid receptor splice variant with an A to G substitution. ‘rs6198’ is coded as 1 (GG), 0 (AG), and −1 (AA). ‘rs6198 GG’ is coded 0 (AG/AA), 1 (GG).
Moderation of sleep duration and systemic levels of inflammation by rs6198
For IL-6, the additive model showed a significant linear interaction with sleep duration, after adjusting for age, sex, and education (β = −0.123 (SE = 0.049), p = .033; Table 4, Model 2; Figure 2A). Follow-up analyses showed that the inverse association of IL-6 with sleep duration was stronger among GG homozygotes than among individuals homozygous for the A allele, but did not differ from heterozygotes (Figure 2A; GG vs. AA: β = −0.078 (SE = 0.159), p = .025; GG vs. GA: β = −0.116 (SE = 0.165), p = .067; GA vs. AA: β = −0.047 (SE = 0.058), p = .234). A model in which individuals homozygous for the G-allele (GG) were compared against a combined group of individuals with any A allele (i.e., AG/AA) also revealed a significant linear interaction with sleep duration in the prediction of IL-6 (β = −0.074 (SE = 0.158), p = .033, Table 4, Model 3; Figure 2B). Additional adjustment for covariate × sleep and covariate × genotype interactions did not substantially alter the results.
Table 4.
Moderation of the Association Between Average Sleep Duration and Markers of Inflammation by rs6198 Genotype
| IL-6 |
CRP |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model 1 |
Model 2 |
Model 3 |
Model 4 |
Model 5 |
||||||
| Variables | β | p | β | p | β | p | β | p | β | p |
| Age | .053 | .126 | .051 | .138 | .048 | .162 | −.022 | .533 | −.022 | .535 |
| Sex | −.098 | .005 | −.099 | .004 | −.097 | .005 | .011 | .757 | .010 | .763 |
| Education | −.095 | .006 | −.095 | .006 | −.095 | .006 | −.072 | .037 | −.072 | .036 |
| Avg Sleep | −.092 | .009 | −.190 | .001 | −.080 | .023 | −.052 | .150 | −.083 | .162 |
| rs6198 | .056 | .104 | .057 | .094 | .026 | .448 | .034 | .414 | ||
| rs6198 GG | .068 | .047 | ||||||||
| rs6198*Avg Sleep | −.123 | .033 | −.039 | .508 | ||||||
| rs6198 GG*Avg Sleep | −.074 | .033 | ||||||||
| (Avg Sleep)2 | .085 | .016 | .064 | .401 | ||||||
| rs6198*(Avg Sleep)2 | −.016 | .745 | ||||||||
| F | 6.14 | < .001 | 5.91 | < .001 | 6.06 | < .001 | 2.69 | .014 | 2.07 | .036 |
| R2 | .036 | .041 | .042 | .019 | .019 | |||||
| ΔR2 | .005 | .033 | .005 | .033 | .001 | .791 | ||||
Notes: Age & Education in years; Sex: 0 = male, 1 = female; Avg Sleep = weighted average of weeknight and weekend sleep duration (hours); Avg sleep is mean centered in all models; rs6198: a glucocorticoid receptor SNP with an A to G substitution. ‘rs6198’ is coded as 1 (GG), 0 (AG), and −1 (AA). ‘rs6198 GG’ is coded 0 (AG/AA), 1 (GG).
Figure 2:
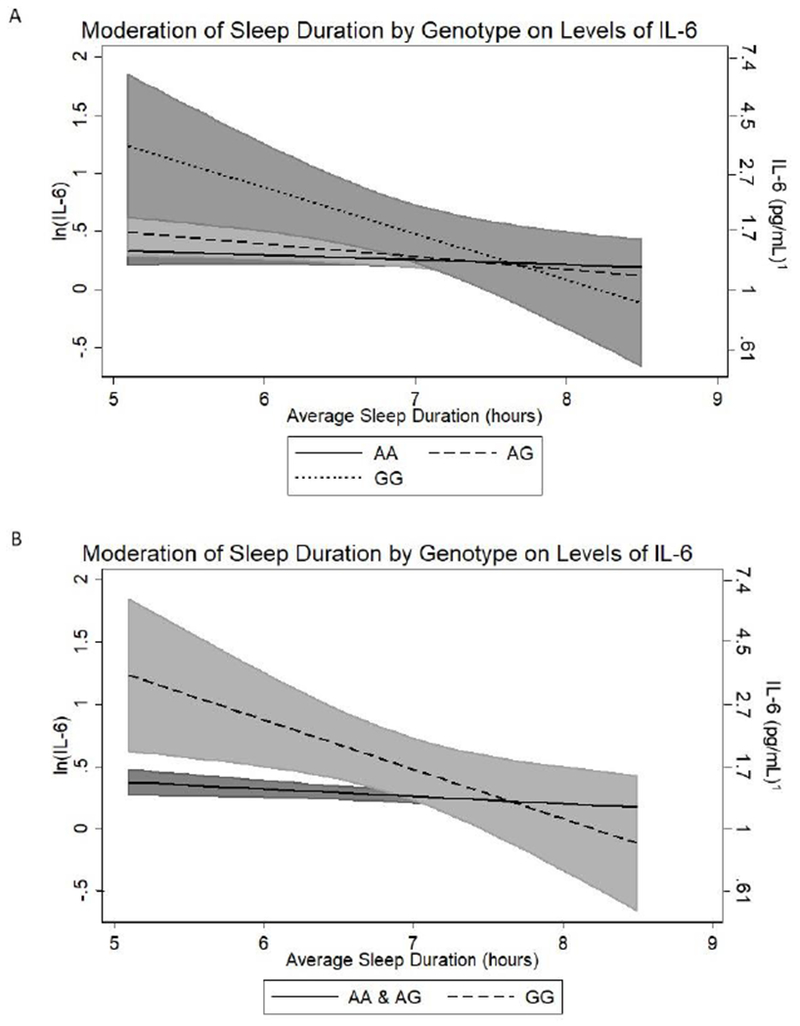
Association between sleep duration and Interleukin(IL)-6 as a function of genotype; A. rs6198 additive model; B. rs6198 GG vs AG/AA model. Shading in grey: 95% CI. 1Mathematical transformation of natural log (ln) transformed values (i.e. eŶ)
For CRP, there was no significant interaction between sleep duration and rs6198 genotype in models that adjusted for age, sex, and education (ΔR2 = .001, p = .791; Table 4, Model 5).
Role of Lifestyle Factors
We examined the association x’ of lifestyle factors (physical activity, OTC medication use, alcohol use, smoking status, and BMI) with IL-6, CRP, sleep duration, and genotype. Physical activity level was inversely related to IL-6 (r = −.101, p < .01) and CRP (r = −.125, p < .01), but was not related to sleep duration (IL-6 sample: r = −.044, CRP sample: r = −.036, p’s > .05) or genotype (p’s > .7). Individuals who reported greater OTC medication use had lower levels of CRP (r = −. 104, p < .01), but not IL-6 (r = −.047, p >.05). OTC medication use was not significantly associated with sleep (IL-6 sample: r= .013, p >.05; CRP sample: r = .011, p > .05), or genotype (p’s > .7). Alcohol use was not related to levels of IL-6 (r = .059, p = .139), CRP (r = −.003, p = .932), sleep duration (IL-6 sample: r = −.051, p = .202; CRP sample: r = −.050, p = .191), or genotype (IL-6 sample: F = 1.65, p = .193; CRP sample: F = 1.56, p = .210). Compared to non-smokers, smokers had significantly higher levels of IL-6 (t = −2.56, p = .011), but not CRP (t = −0.446, p = .656). Smokers did not differ from non-smokers in average sleep duration (IL-6 sample: t = 1.206, p = .228; CRP sample: t = 1.177, p = .240) or genotype distribution. Finally, BMI was positively correlated with IL-6 (r = .301, p < .001) and CRP (r = .489, p < .001), and inversely correlated with sleep duration (IL-6 sample: r = −.159, p < .001; CRP sample: r = −.160, p < .001); BMI did not differ by genotype (p’s > .7). Given that OTC medication use and alcohol use were not related to sleep duration, genotype, or IL-6, we considered only physical activity, smoking status, and BMI as potential confounders of the interaction of sleep duration and rs6198 genotype on IL-6. Similarly, we considered only physical activity, OTC medication use, and BMI as potential confounders of the quadratic relationship between sleep and CRP.
Together, physical activity, BMI, and smoking status accounted for 8.6% of the variance in IL-6 (F(3,819) = 26.28, p < .001), after adjusting for age, sex, and education. Although these lifestyle factors accounted for significant variation in levels of IL-6, they did not individually or together significantly erode the magnitude of the interaction of sleep duration and rs6198 genotype on levels of IL-6 (Table 5, tdiff, Models 2-5). Additional adjustment for lifestyle factor × sleep and lifestyle factor × genotype interactions did not substantially change the results.
Table 5.
Relationship between and IL-6 and average sleep duration by rs6198 genotype, controlling for lifestyle factors
| IL-6 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model 1 |
Model 2 |
Model 3 |
Model 4 |
Model 5 |
||||||
| Variables | β | p | β | p | β | p | β | p | β | p |
| Age | .051 | .138 | .050 | .150 | .043 | .198 | .053 | .124 | .044 | .184 |
| Sex | −.099 | .004 | −.109 | .002 | −.056 | .097 | −.092 | .008 | −.053 | .121 |
| Education | −.095 | .006 | −.086 | .013 | −.074 | .026 | −.089 | .009 | −.061 | .067 |
| Physical Activity | −.106 | .002 | −.054 | .110 | ||||||
| BMI | .271 | < .001 | .270 | .000 | ||||||
| Smoking | .058 | .096 | .083 | .013 | ||||||
| Avg Sleep | −.190 | .001 | −.194 | .001 | −.142 | .011 | −.186 | .001 | −.138 | .014 |
| rs6198 | .057 | .094 | .054 | .113 | .060 | .068 | .058 | .089 | .059 | .072 |
| rs6198*Avg Sleep | −.123 | .033 | −.118 | .038 | −.105 | .059 | −.119 | .037 | −.098 | .074 |
| F | 5.91 | < .001 | 6.55 | < .001 | 14.58 | < .001 | 5.57 | < .001 | 12.44 | < .001 |
| R2 | 0.041 | 0.053 | 0.110 | 0.045 | 0.121 | |||||
| tdiff | 0.043 | .965 | 0.221 | .825 | 0.044 | .965 | 0.25 | .802 | ||
Notes: Age & Education in years; Sex: 0 = male, 1 = female; Physical Activity (kCal/week); BMI (kg/m2); Smoking (Y: current smoker; N: never smoker, tried it, ex-smoker); Avg Sleep = weighted average of weeknight and weekend sleep duration (hours); Avg sleep is mean centered in all models; rs6198: a glucocorticoid receptor SNP with an A to G substitution. ‘rs6198’ is coded as 1 (GG), 0 (AG), and −1 (AA).
After controlling for age, sex, and education, 24.7% of the variance in CRP was accounted for by physical activity, OTC medication use, and BMI together (F(3, 912) = 100.84, p < .001). The magnitude of the effect of sleep duration on CRP was not significantly reduced when controlling for these lifestyle factors (Table 6, 95%CI for Models 2-5 are contained within the 95%CI for Model 1).
Table 6.
Relationship between sleep duration and CRP, controlling for lifestyle factors
| CRP |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model 1 |
Model 2 |
Model 3 |
Model 4 |
Model 5 |
||||||
| Variables | β | p | β | p | β | p | β | p | β | p |
| Age | −.006 | .849 | −0.012 | .722 | −0.022 | .452 | 0.013 | .693 | −0.011 | .727 |
| Sex | .009 | .794 | −0.001 | .989 | 0.089 | .003 | 0.023 | .479 | 0.096 | .001 |
| Education | −.088 | .008 | −0.078 | .018 | −0.052 | .073 | −0.079 | .017 | −0.042 | .149 |
| Physical Activity | −0.117 | < .001 | −0.027 | .365 | ||||||
| BMI | 0.492 | < .001 | 0.485 | < .001 | ||||||
| OTC meds | −0.099 | .003 | −0.076 | .010 | ||||||
| Avg Sleep Duration | −.065 | .059 | −0.072 | .037 | −0.01 | 0.751 | −0.064 | .060 | −0.010 | .754 |
| (Avg Sleep Duration)2 | .093 | .006 | 0.082 | .016 | 0.059 | 0.050 | 0.091 | .007 | 0.057 | .060 |
| F | 4.79 | < .001 | 6.07 | < .001 | 51.58 | < .001 | 5.57 | < .001 | 39.82 | < .001 |
| R2 | 0.025 | 0.038 | 0.253 | 0.035 | 0.260 | |||||
| ΔR2 | 0.016 | < .001 | 0.0149 | < .001 | 0.0038 | < .001 | 0.0155 | < .001 | 0.0035 | .119 |
| 95% CI | [.001, .032] | [.000, .030] | [.000, .012] | [.000, .031] | [.000, .011] | |||||
Notes: Age & Education in years; Sex: 0 = male, 1 = female; Physical Activity (kCal/week); BMI (kg/m2); OTC meds (over the counter medications); Avg Sleep = weighted average of weeknight and weekend sleep duration (hours); Avg sleep is mean centered in all models
Discussion
The current study investigated whether self-reported sleep duration covaried with circulating levels of IL-6 and CRP as a function of rs6198, the G-allele of which has been associated with elevated levels of these markers, as well as increased risk for diseases with inflammatory pathophysiology (Koper et al., 2014; Otte et al., 2010; van den Akker et al., 2008; Koeijvoets et al., 2008). In a relatively large sample of healthy midlife adults, we found an inverse relationship between sleep duration and IL-6, with a larger association among individuals homozygous for the rs6198 G-allele, compared to other rs6198 genotypes. In addition, individuals who reported habitual short (< 7 hours) or long (> 8 hours) sleep duration showed higher levels of CRP than individuals who slept between 7 and 8 hours. Unlike IL-6, these associations were not moderated by rs6198 genotype. Our findings suggest that alterations in the GR associated with the rs6198 G-allele, specifically via production of the GR-9β isoform, modulate effects of short sleep duration on IL-6.
Consistent with some prior studies (e.g. Ferrie et al., 2013; Marsland et al., 2008; Taveras et al., 2011), we observed that individuals who report habitually short sleep have elevated systemic IL-6. Given that short sleep duration also associates with recruitment of the HPA-axis (Abell et al., 2016; Castro-Diehl et al., 2015) and that cortisol normally downregulates inflammatory activity through binding to the GR (Kadmiel & Cidlowski, 2013), we hypothesized that individual differences in GR sensitivity may moderate associations between short sleep and heightened levels of IL-6. Consistent with our hypothesis, we found that individuals homozygous for the G-allele of rs6198 had significantly higher levels of IL-6 for the same duration of short sleep as individuals homozygous for the alternate A-allele. In explanation, the rs6198 G-allele has been associated with elevated levels of GR-9β, a GR protein isoform that does not bind cortisol, resides constitutively in the nucleus, and has been shown to attenuate typical GC-mediated anti-inflammatory signaling (Kino et al., 2009; Oakley & Cidlowski, 2011, 2013; Kadmiel & Cidlowski, 2013; Lewis-Tuffin & Cidlowski, 2006). Indeed, while normally the cortisol-bound GR-α protein isoform binds to the transcription factors NF-kB and AP-1 to down-regulate transcription of genes encoding pro-inflammatory cytokines (e.g. IL-6), it has been posited that GR-9β may compete for these binding sites, thereby promoting production of pro-inflammatory cytokines in the context of HPA-activation (Oakley & Cidlowski, 2011, 2013; Kadmiel & Cidlowski, 2013). In support of this mechanism as related to our findings, sleep loss has been associated with upregulation of pro-inflammatory genes regulated by NFkB and AP-1 (Irwin et al. 2006, 2008; Irwin, 2015). In addition, some studies have found that levels of GR-9β can be selectively increased by cellular exposure to pro-inflammatory cytokines (Hauk et al., 2000; Tliba et al., 2006; Webster et al., 2001). Thus, it could be that in the context of short sleep, individuals homozygous for the rs6198 G-allele show greater activation of pro-inflammatory pathways compared to those carrying any A-allele due to heightened levels of GR-9β; this process is then is exacerbated by further increases in levels of GR-9β due to a pro-inflammatory microenvironment. In sum, our findings suggest that among relatively healthy midlife adults, short sleep duration associates with heightened circulating levels of IL-6, and that individuals homozygous for the rs6198 G-allele may be particularly susceptible to this effect.
In contrast to prior studies, we did not find a main effect of rs6198 genotype on peripheral levels of IL-6. Notably, the two studies to date reporting higher levels of inflammatory markers among individuals homozygous for the rs6198 G-allele, compared to those with the alternate A-allele, were in a population of aging individuals (55 years and older; van den Akker et al., 2008) and individuals with existing cardiovascular disease (CVD) (Otte et al., 2010). Indeed, ours is the first study to examine associations between the rs6198 G-allele and levels of IL-6 among a relatively healthy sample of midlife adults. Thus, one possible explanation for the difference between studies could be that the impact of the rs6198 G-allele on peripheral levels of IL-6 in younger, healthy study participants is revealed only among individuals exposed to environmental factors that promote inflammatory conditions. Specifically, both aging and CVD have been associated with HPA-axis dysregulation (Van Cauter et al., 1996; Walker, 2007) and elevated markers of inflammation (Kovacic et al., 2011). Thus, in keeping with the proposed mechanism explaining our findings among individuals with habitually short sleep, elevated levels of GR-9β among rs6198 G-allele homozygotes could heighten inflammation associated with aging or CVD. In support of this idea, both Otte et al., (2010) and van den Akker et al. (2008) reported higher mean levels of IL-6 than found in our study, with the highest levels again among those homozygous for the G-allele. This conjecture, together with observation that the rs6198 G-allele predicted higher IL-6 levels only among short sleepers, suggests that in younger and well populations effects of the rs6198 minor G-allele on IL-6 may be conditioned by environmental factors to which some, but not all, individuals are exposed.
Given that a number of lifestyle factors associate with both habitually short sleep duration and elevated levels of systemic IL-6 (Ferrie et al., 2013; O’Connor et al., 2009), it is possible that individuals homozygous for the rs6198 G-allele engage in lifestyle choices that increase their susceptibility to both elevated levels of IL-6 and shortened sleep duration. However, we found that the moderation of short sleep duration by rs6198 genotype on levels of peripheral IL-6 was independent of a number of related health behaviors, including presumed caloric intake in excess of energy expenditure (represented by BMI), smoking, and physical activity. While modification of any of these factors (e.g. losing weight, increasing levels of physical activity) may improve sleep or reduce levels of systemic IL-6 independently (Chennaoui et al., 2015; Martin et al., 2016; Nicklas et al., 2005; Selvin et al., 2007), it appears that these pathways do not influence the interactive effects of sleep duration and rs6198 genotype on IL-6 concentrations observed here.
In contrast to IL-6, we found the association between subjectively reported sleep duration and CRP followed an upward U-shaped curve, whereby individuals who reported sleeping less than about 7 hours or greater than 8 hours showed higher levels of CRP than participants sleeping 7-8 hours. These findings are consistent with at least one other study reporting a U-shaped association between self-reported sleep duration and CRP among non-Hispanic white individuals (Grandner et al., 2013). In that study, individuals reporting less than 5 hours or greater than 9 hours of sleep showed higher levels of CRP. Our study replicates and extends these findings, suggesting that individuals sleeping between 5 to 7 hours and between 8 to 9 hours may also exhibit elevated levels of CRP. These findings were independent of related lifestyle factors, including presumed caloric intake in excess of energy expenditure (represented by BMI), physical activity levels, and over the counter medication use that could impact inflammation. Notably, elsewhere we have reported that in this same sample and by the same self-reported measure of sleep duration, individuals sleeping less than 7 or greater than 8 hours per night were also more likely than others to meet criteria for presence of the metabolic syndrome (Hall et al., 2008). Here, we did not find that levels of CRP varied by rs6198 genotype, nor did we observe that rs6198 moderated associations between sleep duration and CRP. Given that CRP is an acute phase protein that is released by the liver as part of the inflammatory cascade (Gruys et al., 2005), it may be understandable that CRP was not moderated in the same way as IL-6 by this polymorphism. That is, the rs6198 polymorphism increases levels of GR-9β, a GR protein known to be permissive of pro-inflammatory cytokine transcription in immune cells. While the release of CRP from hepatocytes is stimulated by IL-6, it is not directly regulated by GR signaling at the level of the immune cell. Thus, it could be that direct effects of rs6198 genotype found in previous studies (e.g. van den Akker et al., 2008; Otte et al., 2008) are a down-stream consequence of elevations in IL-6 and/or or other pro-inflammatory cytokines known to stimulate release of CRP (e.g. TNF-α, IL-1β; Gruys et al., 2005). In sum, the current findings add to a growing literature showing an association between self-reported sleep duration and levels of CRP, a clinical marker of cardiovascular disease risk (Ridker et al., 1999), and suggest that at least among relatively healthy midlife individuals, rs6198 does not modulate CRP levels in the context of extremes of sleep duration.
To our knowledge, this is the first study showing that the association between sleep duration and IL-6 is moderated by polymorphic variation in the glucocorticoid receptor gene. Although our findings raise the possibility that decrements in GR sensitivity associated with the rs6198 G-allele may heighten systemic inflammation associated with short sleep, several limitations warrant consideration. First, the cross-sectional design of the current study was limited to single concurrent assessments of sleep duration and IL-6 levels, precluding causal inference. Second, this study is limited by the use of self-report measures for both sleep duration and physical activity. With regard to sleep duration, meta-analysis suggests that associations between subjective assessments of sleep and the inflammatory markers IL-6 and CRP tend to be smaller than in studies employing instrumented measures of sleep (Carroll et al., 2016). Thus, it may be that our findings underestimate an association between sleep and inflammation. The current study was also limited by including individuals of European-American ancestry only, which leaves the generalizability of our findings to other ethnic/racial ancestries uncertain. Additionally, while our findings highlight the HPA-system in the regulation of inflammatory cytokines, other candidate pathways may also play a role in the association between sleep and systemic inflammation. For example, the sympathetic nervous system (SNS) is known to upregulate pro-inflammatory gene expression (Cole et al., 2010), and several studies have shown that sleep deprivation activates SNS pathways (Irwin, 2015). Thus, future studies should consider the role of additional molecular pathways linking the central nervous and peripheral immune systems, as well as their interaction with the HPA-system in the context of habitually shortened sleep. Finally, although not a limitation per se, we note that the G allele of rs6198 is infrequent in Caucasian populations and that GG homozygosity was present here in only 4% of study participants. Thus, only a small portion of the overall variance in IL-6 can be explained by the interaction of sleep duration and GR allelic variation, when the latter is indexed to this single polymorphism. Although we focused on rs6198 here because of its well-characterized effects on glucocorticoid sensitivity, it is possible that other DNA sequence variation in NR3C1, if similarly functional, might also influence these inflammatory markers. Nonetheless, our findings are novel and raise the possibility that genetically modulated individual differences in counter-regulatory effects of HPA activation on inflammation. may contribute to the elevations in chronic disease risk that accompany short sleep duration.
Highlights.
Higher circulating IL-6 is associated with short sleep duration in midlife adults
Short sleep on IL-6 levels is moderated by variation in the GR gene (NR3C1)
CRP levels are elevated in association with both short and long sleep duration
The effect of sleep on CRP levels was not moderated by NR3C1 variation
Acknowledgments
Disclosure Statement: This work was supported by NIH Grant PO1 HL040962 (SBM),- and NIH Training Grant 4T32HL007560 (CPW). All authors declare that there are no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
In prior work (Erickson et al., 2013), we tested for possible genetic substructure in this sample using a panel of genome-spanning SNPs and the program STRUCTURE (Falush, Stephens & Pritchard, 2003). Because that analysis showed the data to best fit a model specifying a single population, as opposed to either two or three subpopulations, no further adjustments were made for stratification.
References
- Abell JG, Shipley MJ, Ferrie JE, Kivimäki M, & Kumari M (2016). Recurrent short sleep, chronic insomnia symptoms and salivary cortisol: A 10-year follow-up in the Whitehall II study. Psychoneuroendocrinology, 68, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanes D, Conway JM, Taylor PR, Moe PW, Judd J. Validation and comparison of eight physical activity questionnaires. Epidemiology 1990;1:65–71. [DOI] [PubMed] [Google Scholar]
- Ferrie JE, Kivimaki M, Akbaraly TN, Singh-Manoux A, Miller MA, Gimeno D, … Shipley MJ (2013). Associations between change in sleep duration and inflammation: findings on C-reactive protein and interleukin 6 in the Whitehall II Study. Am J Epidemiol, 178(6), 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2011). Glucocorticosteroids: current and future directions. British Journal of Pharmacology, 163(1), 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappuccio FP, D’Elia L, Strazzullo P, & Miller MA (2010). Sleep duration and all-cause mortality: A systematic review and meta-analysis of prospective studies. Sleep, 33(5), 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappuccio FP, Cooper D, D’elia L, Strazzullo P, & Miller MA (2011). Sleep duration predicts cardiovascular outcomes: a systematic review and meta-analysis of prospective studies. European heart journal, 32(12), 1484–1492. [DOI] [PubMed] [Google Scholar]
- Castro-Diehl C, Roux AVD, Redline S, Seeman T, Shrager SE, & Shea S (2015). Association of sleep duration and quality with alterations in the hypothalamic-pituitary adrenocortical axis: The multi-ethnic study of atherosclerosis (MESA). Journal of Clinical Endocrinology and Metabolism, 100(8), 3149–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, Cohen P, West SG, & Aiken LS (2013). Applied multiple regression/correlation analysis for the behavioral sciences. New York, NY: Routledge. [Google Scholar]
- Cowen PJ (2010). Not fade away: The HPA axis and depression. Psychological Medicine, 40(1), 1–4. [DOI] [PubMed] [Google Scholar]
- Chen X, Levine L, & Kwok PY (1999). Fluorescence polarization in homogeneous nucleic acid analysis. Genome research, 9(5), 492–498. [PMC free article] [PubMed] [Google Scholar]
- Chennaoui M, Arnal PJ, Sauvet F, & Léger D (2015). Sleep and exercise: a reciprocal issue?. Sleep medicine reviews, 20, 59–72. [DOI] [PubMed] [Google Scholar]
- Chung CC, Shimmin L, Natarajan S, Hanis CL, Boerwinkle E, & Hixson JE (2009). Glucocorticoid receptor gene variant in the 3’ untranslated region is associated with multiple measures of blood pressure. J Clin Endocrinol Metab, 94(1), 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming G and Fidler F (2010). Effect Sizes and Confidence Intervals In Hancock GR, Mueller RO, & Stapleton LM (Eds.), The Reviewer’s Guide to Quantitative Methods in the Social Sciences (pp. 79–91). New York, NY: Routledge. [Google Scholar]
- Derijk RH, de Kloet ER. (2008). Corticosteroid receptor polymorphisms: determinants of vulnerability and resilience. Eur J Pharmacol 583:303–311 [DOI] [PubMed] [Google Scholar]
- Erickson KI, Banducci SE, Weinstein AM, MacDonald III AW, Ferrell RE, Halder I, … & Manuck SB (2013). The brain-derived neurotrophic factor Val66Met polymorphism moderates an effect of physical activity on working memory performance. Psychological science, 24(9), 1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, & Pritchard JK (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics, 164(4), 1567–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrie JE, Shipley MJ, Cappuccio FP, et al. (2007). A prospective study of change in sleep duration: associations with mortality in the Whitehall II cohort. Sleep, 30, 1659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CF, & Nemeroff CB (2005). Hypercortisolemia and depression. Psychosomatic Medicine, 67(SUPPL. 1), 28–30. [DOI] [PubMed] [Google Scholar]
- Grandner MA, & Drummond SP (2007). Who are the long sleepers? Towards an understanding of the mortality relationship. Sleep medicine reviews, 11(5), 341–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruys E, Toussaint MJM, Niewold TA, & Koopmans SJ (2005). Acute phase reaction and acute phase proteins. Journal of Zhejiang University. Science. B, 6(11), 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon A, Balbo M, Morselli LL, et al. (2014). Adverse effects of two nights of sleep restriction on the hypothalamic-pituitary-adrenal axis in healthy men. J Clin Endocrinol Metab, 99, 2861–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder I, Muldoon MF, Ferrell RE, & Manuck SB (2007). Serotonin receptor 2A (HTR2A) gene polymorphisms are associated with blood pressure, central adiposity, and the metabolic syndrome. Metabolic syndrome and related disorders, 5(4), 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder I, Yang BZ, Kranzler HR, et al. (2009). Measurement of admixture proportions and description of admixture structure in different US populations. Human Mutation, 30(9), 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MH, Smagula SF, Boudreau RM, Ayonayon HN, Goldman SE, Harris TB, … & Stone KL (2015). Association between sleep duration and mortality is mediated by markers of inflammation and health in older adults: the health, aging and body composition study. Sleep, 38(2), 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MH, Muldoon MF, Jennings JR, Buysse DJ, Flory JD, & Manuck SB (2008). Self-reported sleep duration is associated with the metabolic syndrome in midlife adults. Sleep, 31(5), 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk PJ, Hamid QA, Chrousos GP, Leung DY. (2000) Induction of corticosteroid insensitivity in human PBMCs by microbial superantigens. J Allergy Clin Immunol, 105(4), 782–7. [DOI] [PubMed] [Google Scholar]
- Irwin MR, Olmstead R, & Carroll JE (2016). Archival Report Sleep Disturbance, Sleep Duration, and Inflammation : A Systematic Review and Meta-Analysis of Cohort Studies and Experimental Sleep Deprivation. Biological Psychiatry, 80(1), 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MR (2015). Why sleep is important for health: a psychoneuroimmunology perspective. Annual review of psychology, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, & Cole S (2006). Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Archives of internal medicine, 166(16), 1756–1762. [DOI] [PubMed] [Google Scholar]
- Irwin MR, Wang M, Ribeiro D, Cho HJ, Olmstead R, et al. (2008). Sleep loss activates cellular inflammatory signaling. Biol. Psychiatry 64:538–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmiel M, & Cidlowski JA (2013). Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci, 34(9), 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MC (2014). Gene×environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biological psychiatry, 75(1), 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, Su YA, & Chrousos GP (2009). Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci, 66(21), 3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson KL, Ryden AM, Mander BA, et al. (2006). Role of sleep duration and quality in the risk and severity of type 2 diabetes mellitus. Arch Intern Med, 166, 1768–1774. [DOI] [PubMed] [Google Scholar]
- Kovacic JC, Moreno P, Nabel EG, Hachinski V, & Fuster V (2011). Cellular senescence, vascular disease, and aging: part 2 of a 2-part review: clinical vascular disease in the elderly. Circulation, 123(17), 1900–1910. [DOI] [PubMed] [Google Scholar]
- Koeijvoets KC, van der Net JB, van Rossum EF, Steyerberg EW, Defesche JC, Kastelein JJ, Lamberts SWJ, Sijbrands EJ. (2008). Two common haplotypes of the glucocorticoid receptor gene are associated with increased susceptibility to cardiovascular disease in men with familial hypercholesterolemia. J Clin Endocrinol Metab, 93, 4902–4908. [DOI] [PubMed] [Google Scholar]
- Koper JW, van Rossum EF, & van den Akker EL (2014). Glucocorticoid receptor polymorphisms and haplotypes and their expression in health and disease. Steroids, 92, 62–73. [DOI] [PubMed] [Google Scholar]
- Kumsta R, Entringer S, Koper JW, van Rossum EFC, Hellhammer DH, Lewis-Tuffin LJ, & Cidlowski JA (2006). The physiology of human glucocorticoid receptor β (hGRβ) and glucocorticoid resistance. Annals of the New York Academy of Sciences, 1069(1), 1–9. [DOI] [PubMed] [Google Scholar]
- Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR. (2008). Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry, 64, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuck SB, Craig AE, Flory JD, Halder I, & Ferrell RE (2011). Reported early family environment covaries with menarcheal age as a function of polymorphic variation in estrogen receptor-α. Development and psychopathology, 23(1), 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CK, Bhapkar M, Pittas AG, Pieper CF, Das SK, Williamson DA, … & Stewart T (2016). Effect of calorie restriction on mood, quality of life, sleep, and sexual function in healthy nonobese adults: the CALERIE 2 randomized clinical trial. JAMA internal medicine, 176(6), 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI (2017, October 6). Reference SNP Cluster Report: rs6198. Retrieved from https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=6198
- Nicklas BJ, You T, & Pahor M (2005). Behavioural treatments for chronic systemic inflammation: effects of dietary weight loss and exercise training. Canadian Medical Association Journal, 172(9), 1199–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, & Cidlowski JA (2011). Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem, 286(5), 3177–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, & Cidlowski JA (2013). The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. Journal of Allergy and Clinical Immunology, 132(5), 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte C, Wüst S, Zhao S, Pawlikowska L, Kwok PY, & Whooley MA (2010). Glucocorticoid receptor gene, low-grade inflammation, and heart failure: the Heart and Soul study. The Journal of Clinical Endocrinology & Metabolism, 95(6), 2885–2891. [DOI] [PubMed] [Google Scholar]
- Paffenbarger RS Jr, Wing AL, Hyde RT. (1978) Physical activity as an index of heart attack risk in college alumni. Am J Epidemiol, 108, 161–75. [DOI] [PubMed] [Google Scholar]
- Paffenbarger RS Jr, Hyde RT, Wing AL, Lee IM, Jung DL, Kampert JB. (1993). The association of changes in physical-activity level and other lifestyle characteristics with mortality among men. N Engl J Med, 328, 538–45. [DOI] [PubMed] [Google Scholar]
- Patel SR, Malhotra A, Gottlieb DJ, White DP, Hu FB. (2006). Correlates of long sleep duration. Sleep, 29, 881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejovic S, Basta M, Vgontzas AN, et al. (2013). Effects of recovery sleep after one work week of mild sleep restriction on interleukin-6 and cortisol secretion and daytime sleepiness and performance. Am J Physiol Endocrinol Metab, 305, E890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax R. a, Manenschijn L, Koper JW, Hazes JM, Lamberts SWJ, van Rossum EFC, & Feelders R. a. (2013). Glucocorticoid sensitivity in health and disease. Nature Reviews. Endocrinology, 9(11), 670–86. [DOI] [PubMed] [Google Scholar]
- Reynolds AC, Dorrian J, Liu PY, et al. (2012). Impact of five nights of sleep restriction on glucose metabolism, leptin and testosterone in young adult men. PLoS One, 7, e41218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallis JF, Haskell WL, Wood PD, Fortmann SP, Rogers T, Blair SN, & Paffenbarger RS Jr (1985). Physical activity assessment methodology in the Five-City Project. American journal of epidemiology, 121(1), 91–106. [DOI] [PubMed] [Google Scholar]
- Selvin E, Paynter NP, & Erlinger TP (2007). The effect of weight loss on C-reactive protein: a systematic review. Archives of internal medicine, 167(1), 31–39. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz A, Leszczynska-Rodziewicz A, Pawlak J, Rajewska-Rager A, Dmitrzak-Weglarz M, Wilkosc M, … Hauser J (2011). Glucocorticoid receptor polymorphism is associated with major depression and predominance of depression in the course of bipolar disorder. J Affect Disord, 134(1–3), 138–144. [DOI] [PubMed] [Google Scholar]
- Steiger A, Holsboer F. (1997). Neuropeptides and human sleep. Sleep, 20, 1038–1052. [PubMed] [Google Scholar]
- Sweitzer MM, Halder I, Flory JD, Craig AE, Gianaros PJ, Ferrell RE, & Manuck SB (2013). Polymorphic variation in the dopamine D4 receptor predicts delay discounting as a function of childhood socioeconomic status: evidence for differential susceptibility. Social cognitive and affective neuroscience, 8(5), 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri S, Lin L, Austin D, et al. (2004). Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med, 1, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveras EM, Rifas-Shiman SL, Rich-Edwards JW, Mantzoros CS. (2011). Maternal short sleep duration is associated with increased levels of inflammatory markers at 3 years postpartum. Metabolism, 60, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tliba O, Cidlowski JA, Amrani Y. (2006). CD38 expression is insensitive to steroid action in cells treated with tumor necrosis factor-alpha and interferon-gamma by a mechanism involving the upregulation of the glucocorticoid receptor beta isoform. Mol Pharmacol, 69(2), 588–96. [DOI] [PubMed] [Google Scholar]
- Van Cauter E, Leproult R, & Kupfer DJ (1996). Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. The Journal of Clinical Endocrinology & Metabolism, 81(7), 2468–2473. [DOI] [PubMed] [Google Scholar]
- van den Akker EL, Koper JW, van Rossum EF, Dekker MJ, Russcher H, de Jong FH, … & Lamberts SW (2008). Glucocorticoid receptor gene and risk of cardiovascular disease. Archives of internal medicine, 168(1), 33–39. [DOI] [PubMed] [Google Scholar]
- Vgontzas AN, Zoumakis E, Bixler EO, et al. (2004). Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J Clin Endocrinol Metab, 89, 2119–2126. [DOI] [PubMed] [Google Scholar]
- Walker BR (2007). Glucocorticoids and cardiovascular disease. European Journal of Endocrinology, 157(5), 545–559. [DOI] [PubMed] [Google Scholar]
- Wang E, Ding YC, Flodman P, Kidd JR, Kidd KK, Grady DL, … & Moyzis RK (2004). The genetic architecture of selection at the human dopamine receptor D4 (DRD4) gene locus. The American Journal of Human Genetics, 74(5), 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JC, Oakley RH, Jewell CM, Cidlowski JA. (2001). Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci, 98(12), 6865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PM, Manuck SB, DiNardo MM, Korytkowski M, & Muldoon MF (2015). Shorter sleep duration is associated with decreased insulin sensitivity in healthy white men. Sleep, 38(2), 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüst S (2007). Sex specific associations between common glucocorticoid receptor gene variants and hypothalamus-pituitary-adrenal axis responses to psychological stress. Biol Psychiatry, 62, 863–869. [DOI] [PubMed] [Google Scholar]