

Abstract
Liver interstitial dendritic cells (DC) have been implicated in the control of ischemia/reperfusion injury (IRI) and host immune responses following liver transplantation. Mechanisms underlying these regulatory functions of hepatic DC remain unclear. We have shown recently that the transmembrane immunoadaptor DNAX-activating protein of 12 kDa (DAP12) negatively regulates mouse liver DC maturation and their pro-inflammatory and immune stimulatory functions. Here, we used PCR analysis and flow cytometry to characterize expression of DAP12 and its associated triggering receptor, triggering receptor expressed on myeloid cells 2 (TREM2) by mouse and human liver DC and other immune cells compared with DC in other tissues. We also examined the roles of DAP12 and TREM2 and their expression by liver DC in regulation of liver IRI. Injury was induced in DAP12−/−, TREM2−/− or WT mice by 1 hour of 70% clamping and quantified following 6 hours reperfusion. Both DAP12 and TREM2 were co-expressed at comparatively high levels by liver DC. Mouse liver DC lacking DAP12 or TREM2 displayed enhanced levels of nuclear factor κB and co-stimulatory molecule expression. Unlike normal WT liver DC, DAP12−/− liver DC failed to inhibit proliferative responses of activated T cells. In vivo, DAP12−/− and TREM2−/− mice exhibited enhanced IRI accompanied by augmented liver DC activation. Elevated alanine aminotransferase levels and tissue injury were markedly reduced by infusion of WT but not DAP12−/− DC. Conclusions: our data reveal a close association between DAP12 and TREM2 expression by liver DC and suggest that, by negatively regulating liver DC stimulatory function, DAP12 promotes their control of hepatic inflammatory responses. The DAP12/TREM2 signaling complex may represent a novel therapeutic target for control of acute liver injury/liver inflammatory disorders.
Keywords: Innate immunity, T cells, tissue injury, inflammation
Introduction
The liver microenvironment is a site of immune regulation and tolerance induction (1), with a unique constituency of innate and adaptive immune cells (1–3). Liver-resident antigen (Ag)-presenting cells (APC) of hematopoietic and non-hematopoietic origin, that have been shown to regulate inflammatory and T cell-mediated immune responses, include dendritic cells (DC), Kupffer cells, sinusoid-lining endothelial cells and hepatic stellate cells. In particular, liver DC, that are regarded as critical instigators and regulators of innate and immune-mediated inflammatory responses have been implicated in control of acute liver injury (4, 5), T cell-mediated (6) and autoimmune hepatitis (7), and in the promotion of oral (8) and transplant tolerance (9, 10).
Several molecular mechanisms have been reported to regulate the function of hepatic APC. These include the expression by liver conventional myeloid DC of CD39(5), programmed death ligand 1 (PD-L1) = B7 homologue1 (B7-H1) (11), IL-10 (12) and DNAX activating protein of 12kDa (DAP12)(13) that negatively regulate their proinflammatory/T cell stimulatory functions. DAP12 is a homodimeric, immunoreceptor tyrosine-based activation motif (ITAM)-bearing transmembrane adaptor molecule. It integrates signals through multiple receptors, including triggering receptor expressed by myeloid cells (TREM) 1 and 2, NKG2D, Ly49, myeloid DAP12-associated lectin 1 and CD200R (14). By associating with distinct receptors, DAP12 can enhance or suppress leukocyte activation, the outcome being determined by the avidity of receptor-ligand interaction (15). This is in keeping with divergent immunological outcomes in DAP12−/− mice that exhibit enhanced Toll-like receptor (TLR) responses (16), accelerated diabetes development in NOD mice (15) or, by contrast, resistance to endotoxemia and septic peritonitis (17) or the induction of experimental autoimmune disease (18).
There is evidence that signaling through the TREM2/DAP12 receptor complex in response to extracellular glycoproteins and lipids negatively regulates TLR responses in myeloid cells (19) and that DAP12 stabilizes the C-terminal fragment of TREM2 and protects against lipopolysaccharide (LPS)-induced pro-inflammatory responses (20) through reducing mitogen-activated protein kinase (MAPK) activation (19). Conventional DC generated from the bone marrow (BM) of DAP12−/− mice have enhanced TLR responses and costimulatory molecule expression (21, 22) and CD11c+ lung APC of DAP12−/− mice display increased nuclear factor (NF) κB activation and enhanced TLR responses (23). Further, we have shown (13, 24) that DAP12 is a negative regulator of mouse liver DC maturation in vitro. However, our understanding of the role of TREM2/DAP12 in regulation of inflammatory responses is not well understood. In humans, defective TREM2/DAP12 functions within macrophages (microglial cells) of the central nervous system enhances their production of inflammatory cytokines and leads to chronic neurodegenerative disease (25, 26). We have shown recently (24) that DAP12 deficiency in mouse liver allografts can enhance host T cell responses to donor and abrogate ‘spontaneous’ transplant tolerance. Here, we have examined the expression of both DAP12 and TREM2 by mouse and human liver DC, the role of DAP12 in control of the activation and regulatory function of these cells, and the significance of TREM2/DAP12 expression in control of liver IRI.
Materials and Methods
MICE
Male C57BL/6 (B6; H2b), C3H/HeJ (H2k) and TREM2−/− (B6 background) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Male DAP12−/− mice (B6 background) were bred from pairs kindly provided by Dr. Marco Colonna, Washington University School of Medicine, St. Louis, MO. Animals were maintained in the specific pathogen-free Central Animal Facility of the University Pittsburgh School of Medicine. Experiments were conducted under an Institutional Animal Care and Use Committee-approved protocol and in accordance with criteria outlined in the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Animals were fed a diet of Purina rodent chow (Ralston Purina, St. Louis, MO) and received tap water ad libitum. Mice (4-5 per cage) were housed in Optimice® (Animal Care Systems, Inc., Centennial, CO). All surgeries were performed during the light cycle, without fasting.
LIVER I/R INJURY
Male WT B6, DAP12−/− and TREM2−/− mice (8-12 weeks old) were subjected to 70 % warm I/R as described (27), with minor modifications. Sham-operated animals underwent the same procedures without liver ischemia. Animals were euthanized after 6 hours. Serum alanine aminotransferase (ALT) was quantified as described (11). Liver tissue was fixed in 10% vol/vol (v/v) formalin, embedded in paraffin, then sectioned and stained with hematoxylin and eosin (H&E). Areas of liver necrosis were quantified by Image J (NIH, MD).
ISOLATION OF MOUSE AND HUMAN LIVER NON-PARENCHYMAL CELLS (NPC)
Mouse and human liver non-parenchymal cells (NPC) were isolated by collagenase digestion and centrifugal elutriation as described (10) with minor modifications. Histologically normal human liver tissue was obtained following liver tumor resection in the absence of chemotherapy under a University of Pittsburgh IRB protocol (# 12100076). Written informed consent were received from all participants prior to inclusion in the study.
GENERATION AND INTRAVENOUS ADMINISTRATION OF DC
Conventional myeloid DC were generated from freshly-isolated WT or DAP12−/− BM cells (BM-DC), as described(28) using recombinant (r) mouse granulocyte-macrophage colony-stimulating factor and r mouse IL-4 (both 1000 U/ml; R & D Systems, Minneapolis, MN). In some experiments, mice were injected with Fms-like tyrosine kinase 3 ligand (Amgen, Thousand Oaks, CA; 10 μg/day for 10 days) to expand endogenous DC. On day 7 of BM-DC culture, the cells were harvested, counted and CD11c+ cells isolated by anti-CD11c immune-magnetic bead purification (Miltenyi Biotec, San Diego, CA). For retro-orbital venous plexus injection, a 28G needle was used and 1-2 × 106 DC administered in 100μl. For in vivo cell trafficking studies, 4-5 × 106 DC were labeled with Violet Proliferation Dye 460 (VPD450; BD Bioscience, San Jose, CA) and infused systemically, as described (29), immediately after clam removal.
FLOW CYTOMETRY
Cells were stained with Zombie Aqua™ dye (Zombie; Biolegend, San Diego, CA) for 30 min at room temperature (RT), then incubated with FcγR-blocking rat α-mouse CD16/32 antibody (Ab) (clone #: 93; Biolegend) for 20 min at RT to prevent non-specific reactions. For cell surface staining, the cells were incubated for 20 min at 4 °C with different combinations of fluorochrome-conjugated Abs against mouse CD3 (clone #: 17A2), CD4 (H129.19), CD8α (53-6.7), CD11c (HL3), B220/CD45R (RA3-6B2), NK1.1 (PK136), CD86 (GL1), CD80 (16-10A1), I-Ab (AF6-120.1), IA/IE (M5/114.15.2), nuclear factor (NF)κB (D14E12), TREM1 (174031) and TREM2 (78.18) or human CD1c (F10/21A3), CD3 (SP34-2), CD4 (L200), CD8α (RPA-T8), CD11c (B-ly6), CD16 (3G8), CD19 (SJ25C1), CD20 (2H7), CD56 (B159), CD66b (G10F5), CD123 (6H6), CD141 (JAA17), HLA-DR (G46-6), DAP12 (406288), TREM1 (6B1) or TREM2 (237920) (eBioscience, San Diego, CA; BD Biolegend; R&D Systems)). After staining, cells were fixed with 4% v/v paraformaldehyde. Flow data were acquired on a LSR Fortessa flow cytometer (BD) and analyzed using FlowJo version 10 software (Tree Star, San Carlos, CA).
CYTOKINE QUANTITATION
Cytokine levels in cell culture supernatants and in mouse serum were determined using mouse or human Th1, Th2 and Th17 cytokine bead array kits (BD).
STATISTICAL ANALYSES
GraphPad Prism (version 7.00; Graphpad Software Inc., San Diego, CA) was used for statistical analyses. Results are expressed as means ± SEM. Significances of differences between means were determined using Student’s ‘t’ test. Multiple comparisons on a single data set were performed by ANOVA. In all experiments, ‘P’ <0.05 was considered significant.
ADDITIONAL METHODS
Details of additional reagents and methods are provided in the Supporting Information.
Results
DAP12 AND ITS CO-RECEPTOR TREM2 ARE EXPRESSED AT MUCH HIGHER LEVELS BY LIVER NPC THAN PC AND AT HIGHER LEVELS THAN IN OTHER TISSUES
We first examined the expression of DAP12 and its co-receptors TREM1 and TREM2 by wild-type (WT) B6 mouse liver, kidney and lung parenchymal cells (PC) and NPC and by human liver PC and NPC. As shown in Fig. 1A, RT-PCR analysis revealed that NPC from each mouse tissue examined expressed higher levels of DAP12, TREM1 and TREM2 mRNA than PC from the same tissue. There was a striking similarity between the overall pattern and level of expression of DAP12 and TREM2, but not DAP12 and TREM1 for the various tissues. DAP12 and TREM2 expression was higher for mouse liver and lung NPC than for kidney NPC or total spleen cells. Mouse BM cells also expressed comparatively high levels of DAP12 and TREM2 mRNA. As in the mouse, human liver NPC expressed much higher levels of DAP12 and TREM2 than liver PC (Fig. 1B).
FIG. 1.
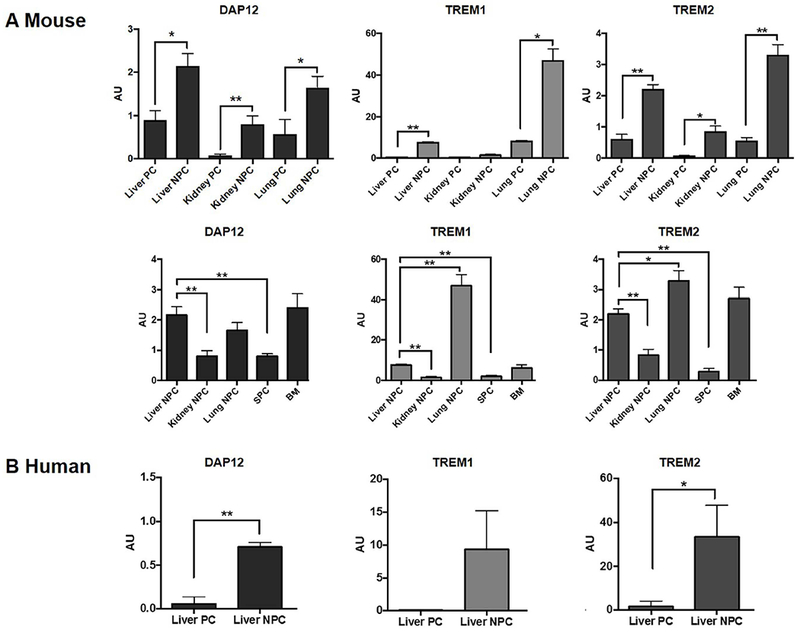
Expression of the transmembrane immunoadaptor DAP12 and its co-receptors, TREM1 and TREM2 by parenchymal (PC) and non-parenchymal cells (NPC) of various normal mouse tissues and histologically normal human liver tissue was quantified by RT-PCR. (A) Liver, kidney and lung PC and NPC, total spleen cells (SPC) and bone marrow (BM) cells were isolated from wild-type (WT) B6 mice. (B) Human liver PC and NPC were isolated from histologically normal liver surgical resection specimens. Data shown are means + SEM obtained from n = 4 mice and n = 3 human subjects in independent experiments. *p<0.05; **p<0.001.
EXPRESSION OF DAP12 AND TREM2 BY LIVER IMMUNE CELL POPULATIONS
Next, we determined the levels of DAP12 and TREM2 protein expression by liver immune cell populations using flow cytometry. An Ab against mouse DAP12 was not available and thus DAP12 protein expression by mouse immune cells could not be determined. Mouse liver conventional CD11c+ DC expressed the highest levels of TREM2, compared with lung, kidney and especially spleen DC (Fig. 2A). Moreover, mouse liver DC expressed much higher levels of TREM2 than liver T cells, B cells, NK cells or macrophages (Fig. 2B). TREM1 expression patterns on mouse tissue DC and liver immune cells were similar to those of TREM2, whereas those on human liver immune cells were dissimilar to those of TREM2 (Suppl. Fig. 1). The expression of DAP12 and TREM2 by human liver immune cells is shown in Fig. 2C and 2D. Thus, whereas virtually all DC and macrophages expressed DAP12, a lower incidence of NK cells and virtually no T or B cells expressed DAP12 (Fig. 2C). Human liver DC and macrophages expressed the highest levels of DAP12 MFI, with a similar overall pattern of staining intensity being observed for TREM2 (Fig. 2C). By contrast, no significant differences in expression between liver immune cell populations was observed for TREM1 (Suppl. Fig. 1). Furthermore, co-expression of DAP12 and TREM2 was significantly (about 2-fold higher) than that of DAP12 and TREM1 (Fig. 2D).
FIG. 2.
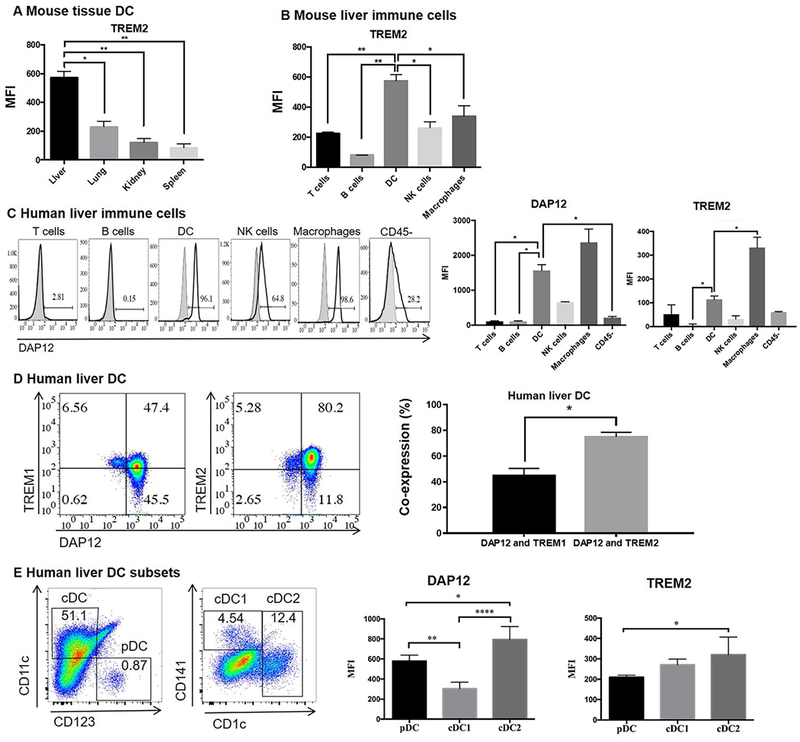
Expression of DAP12 and TREM2 by liver immune cell populations. (A) Freshly-isolated WT B6 mouse liver, lung, kidney NPC and splenocytes were stained for TREM2 by flow cytometry, as described in the Materials and Methods. (B) Expression of TREM2 by various mouse liver immune cell populations. Data are means + SEM obtained from n = 4 mice. *p<0.05; **p<0.01. (C) Left; representative staining of various human liver immune cell populations for DAP12. Figures on histograms denote % positive cells. Gray profiles indicate isotype controls. *p<0.05. Right; aggregate data showing mean fluorescence intensity (MFI) of DAP12 and TREM2 expression by human liver immune cells. (D) Co-expression of DAP12 and TREM1 or TREM2 by human liver DC (CD3− CD19− CD14− HLA-DR+ CD11c+ cells). Representative (left) and aggregate data (right) are shown. Data in (C) and (D) are means + SEM obtained from 3 human subjects. *p<0.05. (E) Expression of DAP12 and TREM2 by human liver conventional (c) DC and plasmacytoid (p)DC subsets (cDC1: lin−HLA-DR+ CD11c+ CD141+; cDC2: lin− HLA-DR+ CD11c+ CD1c+; pDC: lin− HLA-DR+ CD11c+ CD123+). Representative flow data (left) and aggregate data (right) showing MFI of DAP12 and TREM2 expression. Data are means + SEM obtained from n=4 human subjects. *<0.05; **p<0.01; ****p<0.0001.
Human liver DC comprise both conventional (c) myeloid DC (cDC) and plasmacytoid (pDC) subsets (30). Fig. 2E shows DAP12 expression by human liver cDC1 (lin− HLA-DR+ CD11c+ CD141+), cDC2 (lin− HLA-DR+ CD11c+ CD1c+) and pDC (lin− HLA-DR+ CD11c− CD123 [IL-3R]+). DAP12 MFI was highest on cDC2, while TREM2 expression was also expressed at higher levels on cDC2 compared with the other two DC subsets examined.
DAP12 REGULATES NFκB AND BOTH MHC CLASS II AND COSTIMULATORY MOLECULE EXPRESSION BY LPS-STIMULATED LIVER DC IN VIVO
Activation of the transcription factor NFκB is well-recognized to promote DC maturation and activation(31). We therefore investigated the role of DAP12 in regulation of NFκB expression in liver DC after TLR4 ligation in vivo. WT or DAP12−/− mice were injected intravenously (i.v.) with 100ng LPS or PBS and euthanized 2 hours later. NFκB expression in freshly-isolated liver and spleen CD11c+ DC and other immune cells was then evaluated by flow cytometry. While as expected, NFκB expression in WT splenic DC was increased in response to LPS exposure in vivo, by contrast, liver DC exhibited decreased NFκB expression (Fig. 3 A and 3B). In DAP12−/− mice given LPS, a similar fold increase in splenic DC NFκB expression was observed to that found in WT mice, while the extent of reduction in NFκB expression in liver DC was significantly less marked than in those of WT mice (Fig. 3C).
FIG. 3.
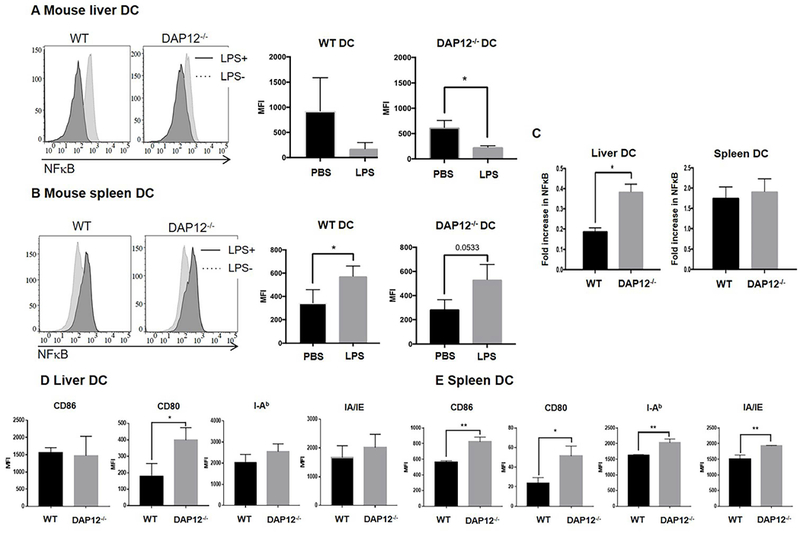
DAP12 negatively regulates NFκB and cell surface costimulatory molecule expression by liver DC. (A, B) WT or and DAP12−/− B6 mice were injected with 100ng E.coli LPS or PBS (control) via the portal vein and euthanized 2 hours later. (A, B) NFκB expression in freshly-isolated liver DC (A) and spleen DC (B) was assessed by flow cytometry. Left-hand histograms show representative flow data. Right hand panels show aggregate data from n=3 independent experiments. Compared with PBS injection, NFκB expression was reduced in liver DC 2 hours after LPS injection, but increased in splenic DC. (C) Whereas the fold increase in NFκB in spleen DC was similar for WT and DAP12−/− spleen DC after LPS injection, there was significantly less reduction in DAP12−/− liver DC compared with WT liver DC. Data are means + SEM obtained from n = 3 independent experiments. (D, E) Expression of MHC class II (IAb) and costimulatory molecules by WT and DAP12−/− liver DC (D) and spleen DC (E) (following LPS stimulation for 2 hours in vivo). *p<0.05; **p<0.001.
We reported previously(13, 24) that, in vitro, DAP12−/− or DAP12 small interfering RNA-treated liver DC exhibited a more activated/mature phenotype and enhanced production of pro-inflammatory cytokines compared with WT liver DC(24). When we examined the extent to which DAP12 regulated both MHC Ⅱ (I-Ab and IA/IE) and costimulatory molecule expression by liver and spleen DC following LPS stimulation in vivo, we found that the MFI of these molecules expressed by both liver and spleen DC from LPS-injected DAP12−/− mice was increased compared with WT controls (Fig. 3D and 3E). Taken together, these data show for the first time, that DAP12 negatively regulates the response of liver DC to TLR4 ligation/activation in vivo.
LIVER DC INHIBIT T CELL PROLIFERATION AND CYTOKINE PRODUCTION IN A DAP12-DEPENDENT MANNER
We next examined the influence of mouse or human liver DC on alloAg (DC)- or αCD3/CD28-induced T cell proliferation and cytokine production (Fig. 4 A). Liver DC from both mouse and human inhibited CD4 and CD8 T cell proliferative responses (Fig. 4A and 4C, accompanied in mouse MLR cultures, by significant reductions in pro-inflammatory cytokine (TNFα, IFNγ, IL-6, IL-4) production and in human MLR, by reductions in IL-2 production (Fig. 4B and 4D). Further analysis showed that, in the absence of DAP12, mouse liver DC had no regulatory capacity (Fig. 5A and 5B) consistent with their markedly diminished expression of the T cell suppressive factors indoleamine dioxygenase (IDO) and arginase-1 (Fig. 5C). These data show that, in addition to regulating their activation, DAP12 expression by liver DC controls their ability to suppress T cell responses induced by αCD3/CD28 or alloAg stimulation.
FIG. 4.
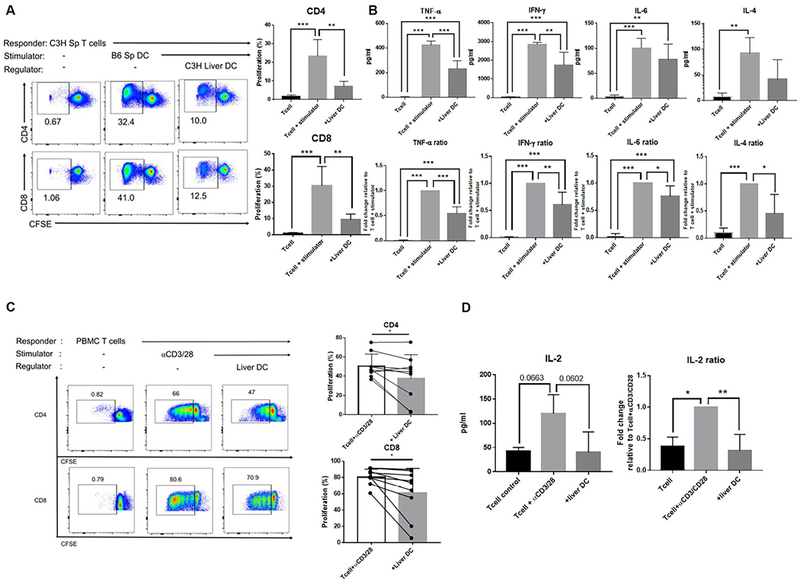
Liver DC inhibit alloAg- or αCD3/CD28-induced T cell proliferation and cytokine production. (A) Left; representative flow data showing the ability of freshly-isolated WT mouse liver DC to inhibit alloAg-induced splenic CD4 and CD8 T cell proliferation in 5-day CFSE-MLR. Freshly-isolated C3H spleen T cells were plated as responders and γ-irradiated, B6 spleen DC used as stimulators at a ratio of 10 T cells:1 DC. C3H liver DC were tested as regulators. Right; histograms show means + SEM from n=3 independent experiments. (B) Cytokine levels in MLR supernatants (day 5) and fold change relative to control value were determined using mouse CBA kits. (C) Left; representative flow data (left) showing the ability of freshly-isolated human liver DC to suppress T cell proliferation induced by αCD3/CD28 stimulation. Human PMBC were plated as responders, α human CD3/CD28 Ab beads were used as stimulators, and human liver DC tested as regulators. The ratio of T cells: DC was 10:1. Cultures were harvested at day 5. Right, cumulative results from n=7 independent experiments. (D) IL-2 levels and fold change relative to control values. Data are means + SEM. *p<0.05; **p<0.001; ***p<0.0001.
FIG. 5.
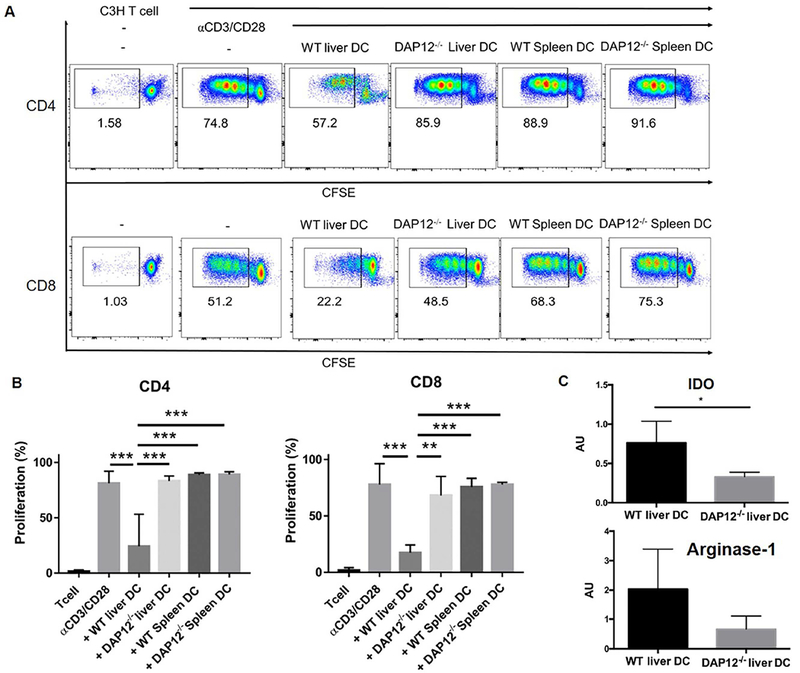
DAP12 controls the negative regulatory effect of liver DC on CD4 and CD8 T cell proliferation. Freshly-isolated, CFSE-labeled mouse C3H spleen T cells were stimulated with αCD3/CD28 beads and either γ-irradiated (30Gy) B6 WT spleen or liver DC, or DAP12−/− spleen or liver DC added at the start of culture. The ratio of T cells: DC was 10:1. Cultures were harvested at day 5 and proliferation determined by CFSE dilution. (A) Representative data from n=4 independent experiments. (B) Aggregate data showing means + SEM obtained from n=4 independent experiments. (C) Absence of DAP12 diminishes expression of the T cell inhibitory factors indoleamine dioxygenase (IDO) and arginase-1 by mouse liver DC. Aggregate RT-PCR data showing means + SEM obtained from liver DC isolated from n=5 WT mice and n=4 DAP12−/− mice in independent experiments. *p<0.05; **p<0.01; ***p<0.001.
DAP12 AND TREM2 EXPRESSION NEGATIVELY REGULATES LIVER IRI
To investigate the in vivo functional significance of DAP12 and TREM2 expression by the liver in relation to the host response to acute liver injury, we first subjected WT, DAP12−/− or TREM2−/− mice to warm IRI. As shown in Fig. 6 A-6C, using the 70% clamp model, we observed significant increases in serum ALT and histological evidence of tissue injury in DAP12−/− and TREM2−/− livers compared with WT livers. At the same time, there were increases in circulating levels of TNFα and IL-6 in both DAP12−/− and TREM2−/− animals (Fig. 6D). Concomitant with the increased levels of liver injury was evidence of enhanced activation (upregulated costimulatory CD86 expression) by interstitial liver DC in DAP12−/− and TREM2−/− animals (Fig. 6E).
FIG. 6.
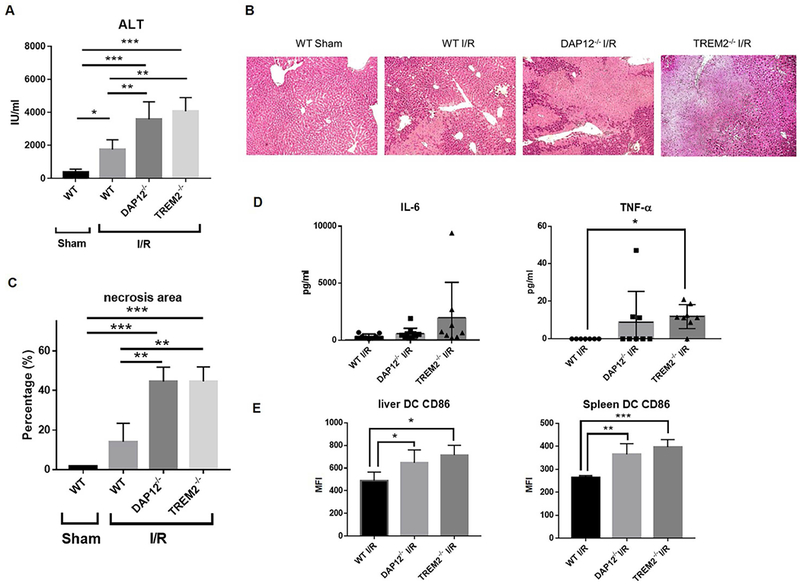
DAP12 and TREM2 regulate the severity of mouse liver IRI. Injury was induced in sham-operated B6 WT, DAP12−/− or TREM2−/− mice by 70% clamping, as described in the Materials and Methods. IRI was determined 6 hours after clamp release. (A) Serum ALT levels in 5-8 mice per group. Data shown are means + SEM. (B) H&E staining of injured liver tissue. (C) Areas of necrosis were quantified by Image J. Data shown are means + SEM obtained from n = 3 animals in each group. (D)Serum IL-6 and TNFα levels evaluated 6 hours after clamp release. Data are means ± SEM obtained from n = 5-8 mice per group. (E) Cell surface costimulatory molecule (CD86) expression by liver and spleen DC 6 hours after liver IR. Data are means + SEM obtained from n = 3 mice in each group. *p<0.05; **p<0.01; ***p<0.001.
INFUSION OF WT DC BUT NOT DAP12−/− DC REVERSES LIVER IRI
To investigate the role of DAP12 expression by DC in regulation of liver IRI, we performed adoptive (i.v.) transfer of 1-2 × 106 WT or DAP12−/− DC to DAP12−/− mice that were subjected to liver IRI. The cells were infused at the time of clamp removal. WT DC but not DAP12−/− DC (that displayed enhanced cell surface MHC II and CD86 expression in response to TLR4 ligation compared with WT DC; Fig. 7A), showed the ability to significantly reduce elevated ALT levels, systemic levels of IL-6 and the extent of tissue injury (Fig. 7B-7E), indicating an important role for DAP12 expression by DC in regulation of liver IRI. Moreover, when we examined the trafficking of adoptively-transferred, VPD450-labeled WT DC to the liver and other tissues (kidney and spleen) of injured DAP12−/− mice, we observed that a much greater proportion of total CD11c+ cells in the host injured liver comprised infused DC compared with those in the kidney or secondary lymphoid tissue (Fig. 7F). However, no significant difference in the intensity of TREM2 expression by the infused DC was detected between the tissues in which they were located (Fig. 7F).
FIG. 7.
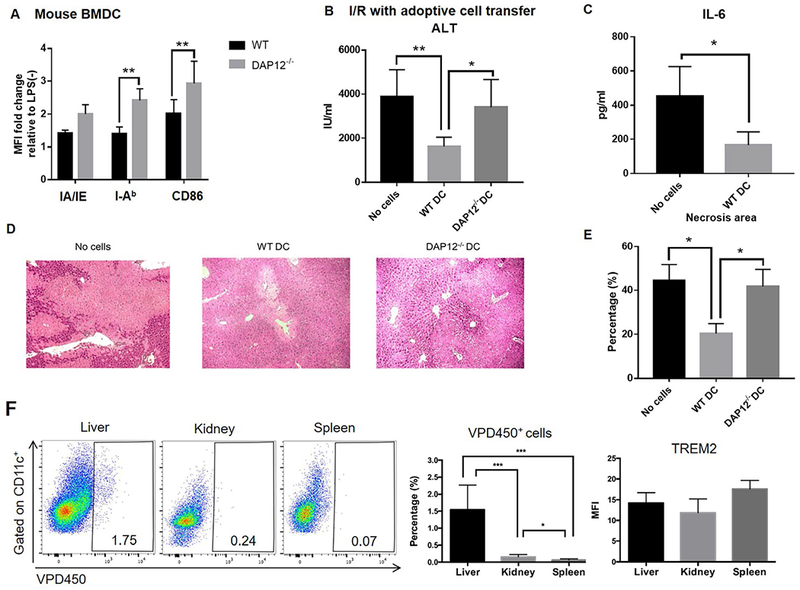
WT but not DAP12−/− DC reduce the severity of liver IRI in DAP12−/− mice. (A) Phenotype of WT or DAP12−/− DC that were infused (1-2 × 106 i.v.) into DAP12−/− mice immediately after clamp removal. (B) Serum ALT levels evaluated 6 hours after I/R. Data shown are means + SEM obtained from n = 4-12 mice per group. (C) Circulating levels of IL-6. (D) Extent of liver tissue injury observed in H&E-stained sections. (E) Quantitation of areas of necrosis. Data shown are means + SEM obtained from n = 3 mice per group. *p<0.05; **p<0.01. (F) Analysis of the in vivo migration of VPD450-labeled WT DC (infused systemically immediately after clamp removal) to the injured liver, kidney and spleen of DAP12−/− recipients performed 6 hours after I/R. Left, representative flow data from n=6 mice. Middle, aggregate data from n=6 mice denote percentages of migrated cells in the total CD11c+ population for each tissue. Right, aggregate data from n=3 mice showing the mean fluorescence intensity (MFI) of TREM2 staining on migrated DC in each tissue. *p<0.05; **p<0.01; ***p<0.001.
Discussion
The present findings offer several new insights into the immunobiology of hepatic DC, their refractory behavior in response to TLR ligation and their role in regulation of inflammatory responses and acute liver injury/IRI. We have shown previously (13) that the transmembrane immunoadapter protein DAP12, signaling through which inhibits TLR responses in myeloid cells (19), is expressed constitutively at comparatively high levels by primary mouse liver cDC. These levels of DAP12, and as we have shown here, liver DC-expressed TREM2, are higher than those expressed by their DC counterparts freshly-isolated from the lung, kidney or spleen. This may be linked to the phenomena of endotoxin tolerance and cross tolerance exhibited by myeloid cells after primary TLR4 ligation (32, 33), that occurs in APC of the liver (13, 34, 35), an organ that is exposed continually to microbial products that reach the liver directly via the portal vein. Signaling pathways such as TREM2/DAP12, that negatively regulate TLR signaling in myeloid cells (19, 36) and promote endotoxin tolerance in liver DC (13) may serve to protect the liver from acute and chronic inflammation in the healthy steady state. Indeed, our earlier work (13) shows that DAP12 promotes the expression of IL-1 receptor-associated kinase-M (IRAK-M), a negative regulator of TLR signaling and IL-10 production by mouse liver cDC. Moreover, new data that we report herein suggest that, in the absence of DAP12, liver DC express reduced levels of the suppressive factors IDO and arginase-1 that may also contribute to the anti-inflammatory/tolerogenic function of these cells.
In addition to DC, liver macrophages (Kupffer cells; KC) have been implicated in immune regulation within the liver microenvironment and in the promotion of tolerance (3), including allograft tolerance (37). Since, in this study, we have also observed comparatively high levels of DAP12 and TREM2 expression by liver macrophages (Fig. 2), these molecules may be important in mediating anti-inflammatory/immune regulatory functions of these cells, as well as of liver DC.
We show in this study that both DAP12 and its co-receptor TREM2 are expressed at higher levels by liver-resident mouse cDC than by liver NK cells or intrahepatic T or B lymphocytes. Consistent with this observation, and confirming the translational relevance of our findings, we have also noted that human liver DC (including cDC1, cDC2 and pDC subsets) display comparatively high levels of DAP12 and TREM2/DAP12 co-expression. Furthermore, while in contrast to spleen cDC that displayed enhanced activation following LPS stimulation, WT mouse liver DC showed reductions in NFκB activation. Mechanisms by which TREM2/DAP12 signaling might lead to inhibition of NFκB activation have been discussed (19, 38). Absence of DAP12 in liver DC however, significantly reversed this deficiency.
Our investigations also show, for the first time to our knowledge, that DAP12 plays an important role in controlling the capacity of liver DC to negatively regulate responses of adaptive immune cells (proliferative responses of T cells) to stimulation that can be triggered by IRI (39). This function of DAP12 may be an important factor in the control of T cell-mediated immune responses induced by allogeneic DC in vivo (24), or by liver allografts that are accepted in mice without immunosuppressive therapy but rejected acutely when the grafts are from DAP12-deficient donors (24). We found that, in vivo, the absence of DAP12 or TREM2 on liver cells in our established IRI model enhanced intrahepatic DC activation and exacerbated acute tissue injury. The extent of damage was however, reversed by the adoptive transfer of WT DC that migrated to the liver, but not by DAP12−/− DC at the time of injury. Taken together, these observations strengthen evidence for a key role of the TREM2/DAP12 complex in regulation of liver DC activation/function and the inflammatory response to liver IRI.
TREM2 signals via the ITAM motif of DAP12 to attenuate TLR-driven macrophage activation/cytokine production/inflammation (40). Recently, liver non-parenchymal TREM2 has been shown to protect the liver from immune-mediated hepatocellular damage (41) and appears to act as a natural brake on inflammation during hepatocellular injury (following carbon tetrachloride and acetaminophen intoxication). Conceivably, there may be active TREM2/DAP12-dependent regulation of inflammation/immunity in the liver. Further, while there is evidence that mice lacking the endogenous TLR2/4 ligand high mobility group box 1 (HMGB1) specifically in hepatocytes are more susceptible to liver IRI (27), the extent to which TREM2/DAP12 and HMGB1 expression/function might be co-regulated is unknown, but may be worthy of investigation.
There is evidence that DAP12-associated receptors such as TREM2 recognize host-encoded ligands and those encoded by microbial products, such as LPS (42, 43) and it appears likely that, as with DAP12, TREM2 plays an important role in regulation of both innate and adaptive immune responses. Thus, TREM2-deficient mouse BM-derived DC exhibit a phenotype similar to that of DC lacking DAP12 (44) and, as with DC that lack DAP12, are hyper-responsive to TLR stimulation, as determined by cell maturation and inflammatory cytokine production. As we have shown previously for DAP12-deficient liver DC (13), there is evidence that TREM2-deficient DC can induce enhanced levels of Ag-specific T cell proliferation (44).
Our data reveal that in DAP12−/− or TREM2−/− livers subjected to IRI, interstitial DC express a more activated phenotype, concomitant with functional and histological evidence of enhanced tissue injury. Moreover, adoptive transfer of WT DC expressing DAP12 that could be tracked to the injured liver, but not transfer of DC lacking DAP12, reversed elevated ALT levels and markedly reduced the severity of tissue injury. WT DC also suppressed systemic pro-inflammatory (IL-6) cytokine levels. These data provide direct evidence of a key role of DAP12 in the protection afforded by DC. Previously, we and others have shown that liver DC can attenuate hepatic inflammation and fibrosis (45–47) and regulate liver warm (4) and transplant-induced IRI (5).
As discussed in detail elsewhere (48), the pathogenesis of liver IRI involves sequential phases of ischemic organ damage and inflammation-related reperfusion injury in which a dialogue between cells of the innate and adaptive immune systems (DC, KC, NK cells and T cells) plays a key role. These cells use distinct immune receptors to recognize and trigger both endogenous and endogenous molecules/molecular complexes that instigate and regulate innate immune activation pathways and parenchymal cell death. Improved understanding of this complex regulatory network is essential for the design of improved strategies to alleviate IRI in the clinic. The present findings suggest that the TREM2/DAP12 complex plays a key role in active regulation of liver IRI and that DC that express this complex at comparatively high levels within the liver may thus exert a protective effect. Moreover, a protective effect of WT DC against liver IRI was achieved after their adoptive transfer. This finding could be relevant for certain clinical indications, such as prophylaxis of liver IRI in recipients of extended criteria organs. On the other hand, overexpression of TREM2/DAP12 could potentially be achieved in donor livers by adenoviral/adeno-associated viral gene delivery, as has been described for other gene products that inhibit liver ischemic injury (49, 50).
Supplementary Material
Acknowledgments:
This work was supported by National Institutes of Health grants R56 and R01 AI118777 (to AWT). We thank Alan F. Zahorchak, Nicole Martik-Hays, Heather Waring, and Yawen Zheng for technical support and advice, and Miriam Freeman for administrative support.
Abbreviations:
- Ab
antibody
- Ag
antigen
- ALT
alanine aminotransferase
- APC
antigen-presenting cell(s)
- BM
bone marrow
- (c)DC
(conventional) dendritic cell(s)
- DAP12
DNAX activating protein of 12kDa
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IRI
ischemia reperfusion injury
- LPS
lipopolysaccharide
- MFI
mean fluorescence intensity
- MHC
major histocompatibility complex
- MLR
mixed leukocyte reaction
- mRNA
messenger RNA
- NFκB
nuclear factor κB
- NPC
non-parenchymal cell(s)
- PBMC
peripheral blood mononuclear cell(s)
- PC
parenchymal cell(s)
- TREM
triggering receptor expressed on myeloid cells
- RT
room temperature
- VPD450
violet proliferation dye 450
- WT
wild-type
Footnotes
Conflict of interest: The authors declare that they have no conflict of interests.
Authors’ names in bold designate shared co-authorship.
Contributor Information
Toshimasa Nakao, Email: toshimasanakao19830205@gmail.com.
Yoshihiro Ono, Email: yoshi_hiro18@hotmail.com.
Helong Dai, Email: helong68888@163.com.
Ryosuke Nakano, Email: nakanor@upmc.edu.
Angelica Perez-Gutierrez, Email: angelyk.pg@gmail.com.
Geoffrey Camirand, Email: camirandg@upmc.edu.
Hai Huang, Email: huangh2@upmc.edu.
David A. Geller, Email: gellerda@upmc.edu.
Angus W. Thomson, Email: thomsonaw@upmc.edu.
References
- 1.Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev 2000;174:21–34. [DOI] [PubMed] [Google Scholar]
- 2.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol 2009;27:147–163. [DOI] [PubMed] [Google Scholar]
- 3.Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol 2010;10:753–766. [DOI] [PubMed] [Google Scholar]
- 4.Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest 2010;120:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida O, Kimura S, Jackson EK, Robson SC, Geller DA, Murase N, Thomson AW. CD39 expression by hepatic myeloid dendritic cells attenuates inflammation in liver transplant ischemia-reperfusion injury in mice. Hepatology 2013;58:2163–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe T, Katsukura H, Chiba T, Kita T, Wakatsuki Y. Periportal and sinusoidal liver dendritic cells suppressing T helper type 1-mediated hepatitis. Gut 2007;56:1445–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia S, Guo Z, Xu X, Yi H, Wang Q, Cao X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood 2008;112:3175–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, Trinchieri G, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity 2008;29:464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yokota S, Yoshida O, Ono Y, Geller DA, Thomson AW. Liver transplantation in the mouse: Insights into liver immunobiology, tissue injury, and allograft tolerance. Liver Transpl 2016;22:536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ono Y, Perez-Gutierrez A, Nakao T, Dai H, Camirand G, Yoshida O, Yokota S, et al. Graft-infiltrating PD-L1(hi) cross-dressed dendritic cells regulate antidonor T cell responses in mouse liver transplant tolerance. Hepatology 2018;67:1499–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ueki S, Castellaneta A, Yoshida O, Ozaki K, Zhang M, Kimura S, Isse K, et al. Hepatic B7 homolog 1 expression is essential for controlling cold ischemia/reperfusion injury after mouse liver transplantation. Hepatology 2011;54:216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, Gonen M, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol 2009;182:1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sumpter TL, Packiam V, Turnquist HR, Castellaneta A, Yoshida O, Thomson AW. DAP12 promotes IRAK-M expression and IL-10 production by liver myeloid dendritic cells and restrains their T cell allostimulatory ability. J Immunol 2011;186:1970–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev 2009;227:150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aoki N, Kimura S, Takiyama Y, Atsuta Y, Abe A, Sato K, Katagiri M. The role of the DAP12 signal in mouse myeloid differentiation. J Immunol 2000;165:3790–3796. [DOI] [PubMed] [Google Scholar]
- 16.Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol 2005;6:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnbull IR, McDunn JE, Takai T, Townsend RR, Cobb JP, Colonna M. DAP12 (KARAP) amplifies inflammation and increases mortality from endotoxemia and septic peritonitis. J Exp Med 2005;202:363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakker AB, Hoek RM, Cerwenka A, Blom B, Lucian L, McNeil T, Murray R, et al. DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity 2000;13:345–353. [DOI] [PubMed] [Google Scholar]
- 19.Hamerman JA, Pottle J, Ni M, He Y, Zhang ZY, Buckner JH. Negative regulation of TLR signaling in myeloid cells--implications for autoimmune diseases. Immunol Rev 2016;269:212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong L, Chen XF, Zhang ZL, Wang Z, Shi XZ, Xu K, Zhang YW, et al. DAP12 Stabilizes the C-terminal Fragment of the Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) and Protects against LPS-induced Pro-inflammatory Response. J Biol Chem 2015;290:15866–15877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomasello E, Desmoulins PO, Chemin K, Guia S, Cremer H, Ortaldo J, Love P, et al. Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity 2000;13:355–364. [DOI] [PubMed] [Google Scholar]
- 22.Chu CL, Yu YL, Shen KY, Lowell CA, Lanier LL, Hamerman JA. Increased TLR responses in dendritic cells lacking the ITAM-containing adapters DAP12 and FcRgamma. Eur J Immunol 2008;38:166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Divangahi M, Yang T, Kugathasan K, McCormick S, Takenaka S, Gaschler G, Ashkar A, et al. Critical negative regulation of type 1 T cell immunity and immunopathology by signaling adaptor DAP12 during intracellular infection. J Immunol 2007;179:4015–4026. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida O, Kimura S, Dou L, Matta BM, Yokota S, Ross MA, Geller DA, et al. DAP12 deficiency in liver allografts results in enhanced donor DC migration, augmented effector T cell responses and abrogation of transplant tolerance. Am J Transplant 2014;14:1791–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neumann H, Takahashi K. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 2007;184:92–99. [DOI] [PubMed] [Google Scholar]
- 26.Yeh FL, Hansen DV, Sheng M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol Med 2017;23:512–533. [DOI] [PubMed] [Google Scholar]
- 27.Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR, Ding Q, et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology 2014;59:1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raich-Regue D, Rosborough BR, Watson AR, McGeachy MJ, Turnquist HR, Thomson AW. mTORC2 Deficiency in Myeloid Dendritic Cells Enhances Their Allogeneic Th1 and Th17 Stimulatory Ability after TLR4 Ligation In Vitro and In Vivo. J Immunol 2015;194:4767–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai H, Watson AR, Fantus D, Peng L, Thomson AW, Rogers NM. Rictor deficiency in dendritic cells exacerbates acute kidney injury. Kidney International;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly A, Fahey R, Fletcher JM, Keogh C, Carroll AG, Siddachari R, Geoghegan J, et al. CD141(+) myeloid dendritic cells are enriched in healthy human liver. J Hepatol 2014;60:135–142. [DOI] [PubMed] [Google Scholar]
- 31.Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med 1998;188:2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe T, Kudo M, Chiba T, Wakatsuki Y. Molecular mechanisms of portal vein tolerance. Hepatol Res 2008;38:441–449. [DOI] [PubMed] [Google Scholar]
- 33.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol 2009;30:475–487. [DOI] [PubMed] [Google Scholar]
- 34.Uhrig A, Banafsche R, Kremer M, Hegenbarth S, Hamann A, Neurath M, Gerken G, et al. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J Leukoc Biol 2005;77:626–633. [DOI] [PubMed] [Google Scholar]
- 35.Liu ZJ, Yan LN, Li XH, Xu FL, Chen XF, You HB, Gong JP. Up-regulation of IRAK-M is essential for endotoxin tolerance induced by a low dose of lipopolysaccharide in Kupffer cells. J Surg Res 2008;150:34–39. [DOI] [PubMed] [Google Scholar]
- 36.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol 2005;5:446–458. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Liu Z, Liang S, Luan X, Long F, Chen J, Peng Y, et al. Role of Kupffer cells in the induction of tolerance of orthotopic liver transplantation in rats. Liver Transpl 2008;14:823–836. [DOI] [PubMed] [Google Scholar]
- 38.Turnbull IR, Colonna M. Activating and inhibitory functions of DAP12. Nat Rev Immunol 2007;7:155–161. [DOI] [PubMed] [Google Scholar]
- 39.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant 2011;11:1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 2006;177:3520–3524. [DOI] [PubMed] [Google Scholar]
- 41.Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, Jais A, Mann J, et al. Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.N’Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, Hamerman JA, et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol 2009;184:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quan DN, Cooper MD, Potter JL, Roberts MH, Cheng H, Jarvis GA. TREM-2 binds to lipooligosaccharides of Neisseria gonorrhoeae and is expressed on reproductive tract epithelial cells. Mucosal Immunol 2008;1:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ito H, Hamerman JA. TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol 2012;42:176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang M, Ueki S, Kimura S, Yoshida O, Castellaneta A, Ozaki KS, Demetris AJ, et al. Roles of dendritic cells in murine hepatic warm and liver transplantation-induced cold ischemia/reperfusion injury. Hepatology 2013;57:1585–1596. [DOI] [PubMed] [Google Scholar]
- 46.Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, Barilla R, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013;58:589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiao J, Sastre D, Fiel MI, Lee UE, Ghiassi-Nejad Z, Ginhoux F, Vivier E, et al. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology 2012;55:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu L, Zhou H, Ni M, Wang X, Busuttil R, Kupiec-Weglinski J, Zhai Y. Innate Immune Regulations and Liver Ischemia-Reperfusion Injury. Transplantation 2016;100:2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bilbao G, Contreras JL, Eckhoff DE, Mikheeva G, Krasnykh V, Douglas JT, Thomas FT, et al. Reduction of ischemia-reperfusion injury of the liver by in vivo adenovirus-mediated gene transfer of the antiapoptotic Bcl-2 gene. Ann Surg 1999;230:185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klune JR, Dhupar R, Kimura S, Ueki S, Cardinal J, Nakao A, Nace G, et al. Interferon regulatory factor-2 is protective against hepatic ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 2012;303:G666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.