

Abstract
Mercury (Hg) exposure remains a major public health concern due to its widespread distribution in the environment. Organic mercurials, such as MeHg, have been extensively investigated especially because of their congenital effects. In this context, studies on the molecular mechanism of MeHg-induced neurotoxicity are pivotal to the understanding of its toxic effects and the development of preventive measures. Post-translational modifications (PTMs) of proteins, such as phosphorylation, ubiquitination, and acetylation are essential for the proper function of proteins and play important roles in the regulation of cellular homeostasis. The rapid and transient nature of many PTMs allows efficient signal transduction in response to stress. This review summarizes the current knowledge of PTMs in MeHg-induced neurotoxicity, including the most commonly PTMs, as well as PTMs induced by oxidative stress and PTMs of antioxidant proteins. Though PTMs represent an important molecular mechanism for maintaining cellular homeostasis and are involved in the neurotoxic effects of MeHg, we are far from understanding the complete picture on their role, and further research is warranted to increase our knowledge of PTMs in MeHg-induced neurotoxicity.
Keywords: post-translational modification, methylmercury, neurotoxicity
1. Introduction
Mercury (Hg) exposure remains a major public health concern as the metal is present in the environment due to both natural and anthropogenic sources. Within the environment, Hg occurs as both inorganic (elemental or Hg0, Hg+ or Hg2+) and organic compounds, such as methylmercury (MeHg) [1]. Organic mercurials, such as MeHg, have received extensive attention due to their ability to cause congenital effects, leading to the development of characteristic neurological symptoms, including mental retardation, cerebellar ataxia and cerebral palsy, commonly characterized as Fetal Minamata disease (FMD) [2, 3]. In adults, toxic effects of environmental level of MeHg are characterized by a long latency period before the appearance of neurotoxic symptoms, which include weight loss, blurred vision and paresthesia, followed by visual field constriction and ataxia [4].
MeHg is produced by biomethylation of inorganic mercury present in aquatic sediments, a reaction catalyzed primarily by aquatic microorganisms [5]. It accumulates up the aquatic food chain, and reaches maximal concentrations in long-lived, predatory fish such as tuna, swordfish, shark and whale [5]. As a consequence, MeHg toxicity represents an ongoing environmental problem to human health, especially in susceptible populations whose diet consists largely of fish and other seafood products [1, 6]. As mentioned previously, seafood is the main source of MeHg in the human diet, and about 95% of that ingested is absorbed in the gastrointestinal tract [7]. After absorption, MeHg is ubiquitously distributed, and readily penetrates the central nervous system (CNS) [8]. It can distribute to all brain regions by crossing the blood-brain barrier via the neutral amino acid transport system l as a complex with L-cysteine [9]. The brain has high affinity for MeHg, and concentrations can be 3–6 times greater than those found in blood [10]. In fact, it is well documented that the brain is particularly vulnerable to MeHg, especially the developing CNS [11–13]. In this regard, the most susceptible brain regions to MeHg-mediated injury are the brain cortex and the cerebellum, with particular susceptibility exhibited by cerebellar granule cells (CGC) [2]. The neurotoxic effects of MeHg are largely related to its pro-oxidative properties. MeHg is a soft electrophile that has extremely high affinity for nucleophilic thiol (-SH) and selenol (-SeH) groups, which plays a fundamental role in MeHg-induced toxicity. Indeed, the high affinity of MeHg for -SH and -SeH groups on amino acids such as cysteine and selenocysteine may block critical (catalytic) functional groups and/or alter the structure of a large number of proteins [14], leading to disruption of various intracellular functions, including elevations of reactive oxygen species (ROS) [15–17].
The toxic consequences of MeHg exposure are triggered by its ability to interrupt intracellular homeostasis, such as redox status, glutamine recycling and calcium homeostasis, to name a few. Indeed, the electrophilic properties of MeHg entails that a large pool of cellular molecules could be its candidate targets. Given the pivotal role of post-translational modifications (PTMs) of proteins in the regulation of cellular environment, the understanding of its roles in MeHg-induced neurotoxicity could be fundamental to the knowledge of this metal, facilitating targeted therapies or interventions for MeHg-related injuries of the CNS, including neurodevelopmental disorders early in life and neurodegenerative diseases associated with aging. This review summarizes the most current knowledge regarding the role of PTMs in MeHg-induced neurotoxicity. Considerable attention has been directed toward the most commonly occurring PTMs (protein phosphorylation, protein acetylation, and protein ubiquitination), as well as PTMs induced by oxidative stress and PTMs of antioxidant proteins.
2. Mechanism of MeHg-induced neurotoxicity
2.1. Astrocytes, Ca+2 and neurotransmitters
As mentioned earlier, MeHg is a highly and selectively neurotoxic compound, leading to neurological and developmental deficits in the CNS, both in humans and experimental animals [5, 18, 19]. While MeHg can adversely affect many cell types, a central role of astrocytes in mediating MeHg-induced neurotoxicity had been characterized (Fig. 1) [20]. Indeed, a number of experimental findings suggest that astrocytes represent a preferential site for MeHg accumulation [21–23]. Astrocytes have an essential role in control of extracellular pH and ionic balance, neurotrophic factor secretion, and most importantly, astrocytes are primarily responsible for glutamate uptake [24]. In addition to the inhibition of glutamate uptake by astrocytes, increase in spontaneous release of glutamate from presynaptic terminals, and inhibition of vesicular glutamate uptake, have also been considered critical phenomena linked to MeHg-mediated excitotoxicity [20, 25, 26]. These evidences are in line with the increased levels of extracellular glutamate in microdialysis probes implanted in the frontal cortex of adult Wistar rats exposed to MeHg [27]. Earlier studies have demonstrated that MeHg-induced toxic effects can be attenuated by glutamate receptor antagonists [28]. Importantly, during critical CNS developmental windows, MeHg exposure induced disruption of the glutamate homeostasis [29]. Although the molecular mechanisms mediating increased glutamate release and decreased glutamate uptake in MeHg toxicity are not yet fully understood, evidence shows that hydrogen peroxide, whose levels are increased during MeHg exposure, may have a role in the inhibition of astrocyte glutamate transport [30].
Fig. 1. General mechanisms of MeHg-induced neurotoxicity.
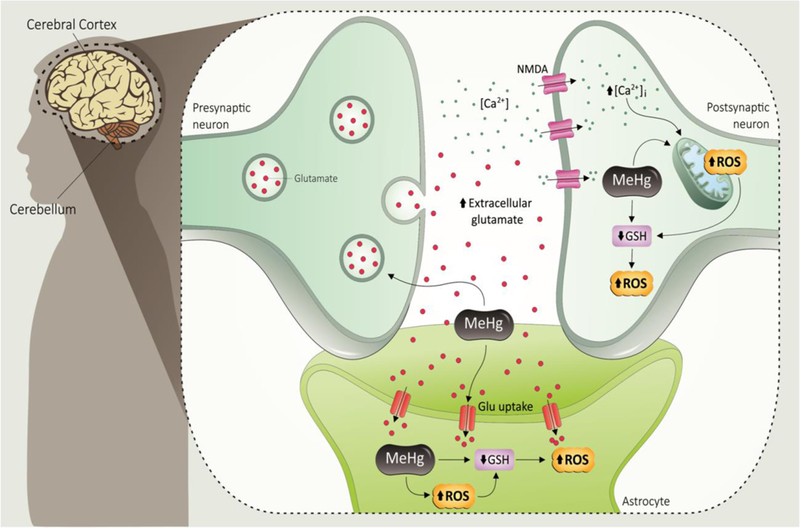
MeHg inhibits astrocytic glutamate uptake and enhances glutamate release from presynaptic terminals. The increased extracellular glutamate (GLU) levels lead to overactivation of N-methyl D-aspartate (NMDA)-type glutamate receptor, increasing the influx of Ca2+ into postsynaptic neurons. The increased levels of intracellular Ca2+ may cause mitochondrial dysfunction and increased reactive oxygen species (ROS) formation. MeHg can affect directly the mitochondrial electron transfer chain (mainly at the level of complexes II-III), also leading to increased ROS generation. MeHg disrupts the redox state of the cells that indirectly depletes GSH, which will further increase ROS production in both astrocytes and neurons.
The increase of the extracellular glutamate levels with consequent overactivation of N-methyl D-aspartate (NMDA) receptors increases Ca2+ influx into neurons, therefore leading to activation of important pathways involved in cell death. In many instances, such effects are mediated by increased generation of ROS in the mitochondria or other intracellular compartments [31, 32]. Indeed, studies have shown that MeHg at low micromolar concentrations causes prolonged increases in intracellular Ca2+ concentrations [33, 34], which may be secondary to increases in extracellular glutamate [23]. Sustained elevated Ca2+ levels have been found in various cell types after MeHg exposure, and protective effects of Ca2+-chelators or Ca2+-channel blockers have been reported in vitro and in vivo [33–36]. Furthermore, studies with acutely prepared cerebellar slices showed that multiple sources of Ca2+ contributes to the elevation of intracellular Ca2+ [37], which is developmentally-dependent [38], and contributes to MeHg-induced stimulation of spontaneous GABA release in cerebellar neurons [37]. Blockage of GABA receptors in hippocampal CA1 neurons is responsible for MeHg’s early stimulatory effect on excitatory synaptic transmission [39]. Other experiments have demonstrated that the sensitivity of GABA receptors to MeHg may explain vulnerability of CGC to this organometal [40] (see review [41] on the mechanisms of MeHg-induced alteration of chemical neurotransmission).
Dopaminergic neuron and dopamine metabolism are also important targets of MeHg in various models. In vivo study using microdialysis coupled to high-performance liquid chromatography demonstrated that 6 ppm MeHg induced significant increases in the striatal release of dopamine (DA) and the acidic dopamine (DA) catabolites homovanillic acid (HVA) and 3,4-dihydroxyphenylacetic acid (DOPAC) [42]. The DA release by MeHg in striatum can be prevented by administration of -SH containing compounds [43]. Intrastriatal infusion of 400 μM MeHg markedly increased the extracellular DA levels [44]. MeHg (0.5 ppm) exposure for 14 days during perinatal period caused reduced binding activity of dopamine D2 receptor in the caudate putamen in rats, which correlates with retarded learning ability in adult stage [45]. In vitro study using PC12 cell suggests that the DA release is partly due to the increased synthesis of DA [46] and indirect inhibition of aldehyde dehydrogenase (ALDH) by MeHg [47]. Comparative studies on MeHg and MPP(+) demonstrated that MeHg acts like MPP(+) impairing DA metabolism [48, 49]. The researches on MeHg exposure and serotonin (5-HT) system revealed that this is a highly rewarded area, as serotonin levels have been linked to psychiatric disorders. 5-HT uptake in astrocytes was inhibited by treatment of 10 μM MeHg for 30 min [50]. Perinatal exposure of MeHg was associated with decreased monoamine oxidase (MAO) activity in female offspring [51]. A decreased level of extracellular 5-HT was found in MeHg treated zebrafish, which was associated with anxiogenic-like behaviors [52].
2.2. Mitochondrial damage and ROS
The mitochondrion is very well known as the central core of energy metabolism within the cell, producing the major source of cellular ATP through the process of oxidative phosphorylation (OXPHOS). This multifunctional organelle not only maintain and regulate nutrient flow through metabolic pathways, such as fatty acid and glucose oxidation and urea cycle, but also is crucial to thermogenesis, calcium homeostasis, heme and phospholipid biosynthesis, redox signaling and apoptosis [53, 54].
Mitochondrial respiration is the main source of constitutive cellular ROS [55], and its significance as a target in the MeHg-induced neurotoxicity is primarily linked to this effect [56–58]. In fact, several studies have shown that the mitochondria are actively involved in the MeHg-induced neurotoxic damage [20]. In this sense, a study from our group using primary astrocytes culture demonstrated that MeHg induces a concentration-dependent impairment in the mitochondrial membrane potential (ΔΨm), and this event might be related to increased Ca2+ levels triggering ROS formation and increased oxidative stress [20]. Consistent with these observations, MeHg has been shown to promote a concentration/dose- and time-dependent increase in lipid peroxidation and ROS generation, both in vitro [25, 26] and in vivo [27].
It is noteworthy that mitochondria have a large number of redox centers, potentially prompt to react with xenobiotics and metals, which could in turn produce reactive metabolites and ROS [59]. In this context, one key site for both ROS production and ROS damage is the mitochondrial electron transport chain (ETC). The OXPHOS site takes place in the inner mitochondrial membrane and consists of five multi-subunit protein complexes named complex I to V (CI-CV) [60]. Several in vivo and in vitro studies have shown mitochondrial effects related to MeHg toxicity, which includes alterations in complexes II and III of the mitochondrial ETC [61], depression of respiration and ATP production [62] and swelling of the mitochondrial matrix [57, 63, 64]. In mice, it was demonstrated that MeHg can disrupt complexes I and II, and reduce the number of mitochondria [65, 66]. Corroborating these findings, using C. elegans as an experimental model, our group has also demonstrated that MeHg induces depletion in cellular NAD+ levels, in addition to oxidative stress and mitochondrial dysfunction. These latter effects were prevented by NAD+ supplementation prior to MeHg exposure, which is an essential cofactor required for electron transfer reactions. This effect demonstrates the importance of NAD+ cofactor in the MeHg-induced toxicity [67].
Besides the pivotal role of mitochondria in energy metabolism, these organelles are also directly involved in the regulation of cell death pathways. Neuronal death via apoptosis and/or necrosis is commonly a consequence of oxidative damage. Not only the antioxidant response, but also the repair processes to the damaged biomolecules are triggered in response to oxidative injury [68]. It is well known that mitochondria can release several apoptotic mediators, including cytochrome C (cyt C), apoptosis protease-activating factor 1 (Apaf-1) and apoptosis inducing factor (AIF) [69], in addition, mitochondria can be the primary target for several other molecules, especially proteins related to the Bcl-2 family [70]. In this context, it has been demonstrated that MeHg is able to induce apoptosis via a classical mitochondrial pathway, where the loss of mitochondrial membrane potential [71] is followed by the release of the pro-apoptotic mediator cyt C [72] and Bax activation [15, 73].
The brain has been drawn much attention in MeHg studies due to its high susceptibility to oxidative stress, secondary to its high energy demand and redox-activity [74]. To illustrate that, a study has compared the effects of MeHg on regular murine embryonal carcinoma cells (undifferentiated), as well as in the differentiated neuronal state (after exposition to retinoic acid), evaluating the susceptibility of these cells to oxidative stress and apoptotic cell death. MeHg treatment induced mitochondrial depolarization, cyt C release, and significantly higher levels of ROS production in differentiated carcinoma cells when compared to undifferentiated cells [75]. In general, it seems that the magnitude of MeHg neurotoxicity and specific consequences of mitochondrial dysfunction likely depend on the dose, cell type and, particularly, the stage of development [75].
3. PTMs- a mechanism to diversify protein functions
The function, structure and stability of proteins in cells are frequently modulated by chemical modifications introduced after translation from RNA. PTMs are important steps in the biosynthesis of proteins, and are known to be essential mechanisms used by eukaryotic cells to diversify their protein functions and dynamically coordinate their signaling networks [76]. Most proteins do not perform their molecular function as unmodified folded polypeptides. In fact, while cognate genes encode the basic biological functions of proteins, the real-time dynamics and regulation of protein structure and function are generally carried out by over 200 protein PTMs that have been described, including phosphorylation, glycosylation, ubiquitination, nitrosylation, methylation, acetylation, lipidation and proteolysis [77, 78]. PTMs are found in all types of proteins, from nuclear transcription factors to metabolic enzymes, structural proteins and plasma membrane receptors [77]. These regulatory processes involve an alteration of the original chemical composition of a protein, usually through the covalent addition of a small molecule to one of the amino-acid residues in proteins, usually in the context of a particular sequence motif [79]. By the covalent addition of functional groups or proteins, proteolytic cleavage of regulatory subunits, or degradation of entire proteins, PTMs have been shown to play prominent roles in protein alteration for destruction [80], general regulation [81] and stress response [82], without involvement of genomic, transcriptomic or translational regulation [83].
Protein phosphorylation is one of the most important, prevalent and best-studied PTMs, which is involved in almost every cellular processes including cell cycle, growth and apoptosis, as well as participating in signal transduction pathways [84]. Virtually all types of extracellular signals exert their physiological effects by modulating phosphorylation of specific proteins in target cells [85]. This reversible mechanism is mediated by protein kinases, and consists of the addition of a phosphate group (PO4) donated from ATP to the polar group R of various amino acids. A complementary group of enzymes, the phosphatases, can remove the attached PO4 group from a phosphorylated protein substrate, reverting the action of the protein kinases. In eukaryotic cells, this occurs most commonly on serine, followed by threonine and tyrosine residues [86]. It is important to note that an imbalance in this equilibrium may produce hyper- or hypo-phosphorylation of target proteins, resulting in cellular dyshomeostasis and, ultimately, in disease conditions, including Alzheimer’s disease (AD) and post-encephalitic Parkinsonism [85].
Protein acetylation occurs on the lysine residue of histone proteins at the N-terminal tail of lysine and is regulated by the enzymes, histone acetylases (HATs), or histone deacetylases (HDACs), and thus is dynamic and reversible [87]. Due to the consumption of Ac-CoA during acetylation and NAD+ during deacetylation, acetylation interferes with metabolic processes and energy homeostasis. Recent studies not only have linked the process of protein acetylation with a number of diseases but also have shown that amino acid acetylation significantly contributes to the overall pathophysiology of the diseases. Indeed, malfunctioning acetylation machinery can lead to disorders including cancer, neurodegenerative and cardiovascular diseases [88–90].
The covalent attachment of the small, highly conserved, and abundant protein ubiquitin to a variety of cellular proteins (ubiquitination) is one of the most common PTMs in eukaryotic cells. Ubiquitination was initially recognized as a signal for proteasome-mediated protein degradation. Besides regulating protein longevity, ubiquitination is also be involved in the control of many cellular processes, including protein activity, localization, and interactions [91]. Ubiquitin has a flexible C-terminus, terminating with a glycine residue that via an isopeptide bond can be attached covalently with ε-amino groups of lysine residues on substrates [92]. Although the most commonly described sites for this ligation are lysine residues, ubiquitination is also known to occur on the side chains of other amino acids, such as cysteine, serine, and threonine [93]. Alterations of protein ubiquitination have been linked to many diseases including cancer, neurodegenerative, cardiovascular and inflammatory diseases [94].
Although transcriptional induction of stress genes constitutes a major cellular defense program against a variety of stressors, posttranslational control directly regulating the activities of preexisting stress proteins provides a faster-acting alternative response [95]. In fact, to add functional diversity and adaption to their alternative environments, proteins may respond to stresses by a transformation of structure and, then, function [96]. The rapid and transient nature of many PTMs allows efficient signal transmission in response to internal and environmental stimuli. On the other hand, the excess of reactive chemical species may be able to post-translationally modify proteins, resulting in the disturbance of cellular redox systems and the incidence of oxidative stress. For instance, cysteine oxidation in cells is an emerging class of redox protein PTMs that creates a range of modifications, which can regulate signal transduction and biological functions [97, 98].
4. PTMs in MeHg-induced neurotoxicity
4.1. Phosphorylation
The phosphorylation process is regulated by the activity of different protein kinases and phosphatases. Previous studies have shown that MeHg can modulate protein phosphorylation in the brain cytosol fraction of rats exposed to 10 ppm MeHg for 7 days, and in cultures of cerebellar granule neurons (CGNs) exposed to 5 μM MeHg for 24 h [99–101]. These alterations are developmentally dependent and cell-type specific. Moreover, it was recently shown that MeHg (1 μM, 24 h) can decrease protein tyrosine phosphatase 1B activity in human brain microvascular endothelial cells and pericytes [102], and in vivo data have indicated an increase in cerebellar PP2B expression following gestational exposure to 2 ppm MeHg [103]. Overall, MeHg exposure can induce alteration of protein phosphorylation in a concentration, developmental stage- and cell-type dependent manner. In this review, we are focusing on the modulation of mitogen-activated protein kinases (MAPKs), protein kinase A (PKA) and protein kinase C (PKC) following MeHg exposure. Those effects are summarized in the table 1.
Table 1.
Effects of MeHg on different kinases in the CNS.
| Protein Kinase | Experimental Model | Effect | Reference |
|---|---|---|---|
| ERK1/2 | PC12 cells | Attenuated NGF-induced ERK1/2 phosphorylation | [104, 105] |
| In vivo exposure during gestational period in rats | Decreased ERK1/2 phosphorylation in the cerebellum and hippocampus of young rats | [103, 106] | |
| Primary cortical astrocyte culture | Increase ERK1/2 phosphorylation | [107] | |
| Primary cerebellar neurons | ERK1/2 inhibition attenuated MeHg-induced cell death | [108] | |
| Neuro-2A cells | Increase ERK1/2 phosphorylation ERK1/2 inhibition attenuated MeHg-induced cell death |
[109] [110] |
|
| Repeated administration in adult mice | Increase ERK1/2 phosphorylation in the cerebral cortex | [111] | |
| JNK | Primary cortical astrocyte culture | Increase on JNK phosphorylation | [112] |
| Repeated administration in adult mice | Increase on JNK phosphorylation in the cerebral cortex | [103] | |
| In vivo exposure during gestational period in rats | Decreased JNK phosphorylation in the cerebellum of young rats | [106] | |
| p38MAPK | In vivo exposure during gestational period in rats | Increase hippocampal p38MAPK phosphorylation | [110] |
| Repeated administration in adult mice | Increase p38MAPK phosphorylation in the cerebral cortex | [109] | |
| Neuro-2A cells | Increase p38MAPK phosphorylation p38 inhibition attenuated MeHg-induced cell death | [113, 114] [102] | |
| SH-SY5Y cells | Increase p38MAPK phosphorylation | [115] | |
| Human brain microvascular endothelial cells | Increase p38MAPK phosphorylation | [116] | |
| Primary cortical astrocyte culture | Increase p38MAPK phosphorylation | [117] | |
| Primary microglia culture | Increase p38MAPK phosphorylation | [118] | |
| PKC | Repeated administration in adult mice | Decrease PKC activity | [103, 106] |
| In vivo exposure during gestational period in rats | Decrease PKC activity Impairment of PKCa and e in the brain |
[107] | |
| PC12 cells | Decrease PKC activity | [105] | |
| Recombinant PKC | Decrease PKCd and z activity and increase PKCa | [105] | |
| PKA | In vivo exposure during gestational period in rats | Increase PKAc expression | [103] |
| Repeated administration in adult mice | Increase PKAc activity | [110] | |
Abbreviations: ERK1/2: extracellular signal-regulated kinase 1/2; JNK: c-Jun N-terminal kinases; PKC: protein kinase C; PKA: Protein kinase A; PKAc: Protein kinase A catalytic subunit.
In mammalian cells, 14 MAPKs have been characterized. Among them, extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinases (JNKs), and p38MAPK isoforms are the foremost studied MAPKs [119, 120]. ERKs are important components of the Ras-Raf-MEK signaling pathway that mediate the transduction of intracellular stimuli and gene expression. As a member of the MAPKs family, the phosphorylation of a serine and a threonine residue (in this case: Thr202 and Tyr204) are essential to ERK1/2 activity. Regulation of ERK1/2 phosphorylation by MeHg is contextually dependent. Current evidence supports a notion that MeHg modulates ERK1/2 phosphorylation indirectly rather than directly. Treatment of PC12 cells with nerve growth factor (NGF) for 5 min induced a marked increase of ERK1/2 phosphorylation, which was believed a mechanism for NGF-induced neurite outgrowth [121]. Cotreatment with 0.01~0.30 μM MeHg inhibited NGF-stimulated ERK1/2 phosphorylation with an EC50 of 0.018 μM [121]. The down-regulated ERK1/2 was supposed to be an indirect effect of MeHg on the upstream pathway that regulates ERK1/2 phosphorylation [121]. ERK1/2 phosphorylation in the hippocampus of gestationally exposed young rats (2 ppm MeHg) was down regulated with concomitant hippocampus-mediated behavior abnormity [122]. Furthermore, a time-dependent increase of ERK1/2 phosphorylation was observed in primary astrocyte cultures treated with low level of MeHg [123]. In primary culture of rat cerebellar granular cells, inhibition of ERK1/2 phosphorylation ameliorated cell death caused by 30 nM MeHg alone or a combination of 30 nM MeHg and 10ng/ml BDNF, and the later was shown to accelerate MeHg-induced cell death [124]. In Neuro-2a cells, ERK1/2 phosphorylation was elevated after 0.5 h post treatment of 3μM MeHg, and ERK1/2 inhibitor attenuated MeHg-induced cytotoxicity [125].
JNKs are a member of the MAPKs kinase family responsible for phosphorylating serine or threonine residues that are flanked by a carboxy-terminal proline. The JNK family proteins were initially discovered for being strongly activated by cellular stress and for its key role in apoptosis. For this reason, these kinases have also been referred to as stress-activated protein kinases (SAPKs). Indeed, JNK activity is higher in brain tissue when compared to other tissues, which may suggest a pivotal role of this kinase family in CNS function. In the human brain, three JNK genes are expressed and are associated with at least ten splice variants of 46 kDa or 54 kDa JNK proteins. All of these isoforms require a double phosphorylation in a serine and a tyrosine residue for their kinase activity [126, 127]. It was recently demonstrated treatment with 3 μM MeHg for 24 h induced increase in JNK phosphorylation as well as TNF-α level in primary astrocytes, which is associated with cytoskeleton remodeling and necrotic death of astrocytes [111]. In other cell models like HepG2 cell, 8 μM MeHg induced a peak elevation of JNK phosphorylation at 8 h of treatment, which was inhibited by cotreatment of 10 μM selenocystine (SeCys), an organoselenium compound that can ameliorate the oxidative damage caused by MeHg [128],while in mouse embryonic fibroblast (MEF) cells, 1 μM MeHg induced a peak elevation of JNK phosphorylation at 4 h of treatment, which is associated with the expression of p62, an autophagy marker which level was increased by MeHg exposure [129]. Upregulation of JNK phosphorylation in the cerebral cortex was found in adult mice exposed to 30 ppm MeHg for 8 weeks, which is associated with an increase in tau-hyperphosphorylation and characteristic neuropathological changes in the deep layer of primary motor cortex and prelimbic cortex [112]. JNK also plays an important role during brain development, such as morphogenesis and neural migration [126, 130]. Recently, a decrease in the cerebellar JNK phosphorylation in young rats gestationally exposed to 2 ppm MeHg was shown to be associated with the misregulated redox equilibrium of developing cerebellar cells [103]. Due the complex regulation among all the JNK isoforms and their function in different phases of neural development, further studies are warranted to elucidate the role of JNKs in response to MeHg-induced neurotoxicity.
p38MAPK is a member of the MAPKs family responsive to stress stimuli such as inflammatory cytokines and oxidative stress. There are 4 isoforms of this kinase named α, β, γ and δ. The p38α and β isoforms share 70% of homology, which are ubiquitously expressed in most tissues including the CNS [131]. In vivo models including C57BL/6NJcl mice exposed to 30 ppm MeHg for 8 weeks and postnatal 30-day wistar rats gestationally exposed to 2 ppm MeHg, have shown that MeHg exposure increased p38MAPK phosphorylation [106, 110]. However, the consequences of the MeHg-induced p38MAPK phosphorylation seem to be dependent on the cell type and MeHg concentration. The activation of p38MAPK in human brain endothelial cells could mediate a pro-inflammatory response after MeHg treatment [102]. On the other hand, exposure of astrocytes to low concentrations of MeHg is able to increase p38MAPK phosphorylation, leading to release of ATP and interleukin-6, and affording neuronal protection against this metal [115, 116]. In Neuro-2a cells [125] and cortical neurons [132], the MeHg-induced increase in p38MAPK phosphorylation and consequent kinase activation, is commonly associated with cell death.
PKC represents an important family of serine/threonine kinase that plays a role in cell growth, differentiation, pos-synaptic transport, neuronal long-term potentiation (LTP), synaptic plasticity and cell survival. The PKC family comprises 12 enzymes that can be divided into three subclasses: the conventional PKC that are sensitive to Ca2+ and diacylglycerol, the novel PKCs (also named non-conventional PKC) that are only activated by DAG, and atypical PKCs that are not regulated by Ca2+ or DAG [133, 134]. In vivo data have demonstrated that repeated MeHg administrations (10 ppm) can induce a decrease in overall PKC activity in the mouse brain [117]. Similarly, Haykal-Coates et al. [118] have shown that MeHg exposure during gestational period decreases the cortical PKC activity on the postnatal day 1 and 4, and also impairs the postnatal ontogeny of PKCα and PKCε in the rat brain. Furthermore, PKC activity in vitro was also decreased after MeHg treatment in cerebellar granular neurons, PC12 cells, and rat brain homogenates [105, 135]. Further studies using recombinant PKC enzymes have demonstrated an in vitro inhibition of the PKC isoforms δ and ζ kinase activity. On the other hand, the PKCα activity was increased after the treatment with a low concentration of MeHg. Since PKCa contains cysteine-rich calcium binding domains and cysteine residues in the catalytic domain of the enzyme, it is speculated that MeHg in low concentrations can mimic the effect of a divalent cation after binding to cysteine (mimicking calcium activation) and stimulate PKCα activity [105].
5’-cyclic-adenosine monophosphate (cAMP) is an intracellular signaling molecule that regulates many processes in the CNS, such as apoptosis, establishment of neuronal circuitry and plasticity, and other cognitive functions. The cAMP synthesis in the brain is related to the activity of transmembrane adenylyl cyclases (ACs) and soluble ACs which are expressed in both glia and neurons. PKA is positively regulated by cAMP and is one of the main downstream effectors of cAMP signaling. The PKA inactive structure consists of two catalytic subunits (PKAc) and two regulatory subunits (PKAr I and II), which bind together as a heterotetramer. The increase in AC activity upregulates cAMP levels in the intracellular compartment, which binds to PKAr I or PKAr II causing dissociation of PKAc subunit and promotes the kinase activity [136, 137]. An increase in the PKAc subunit content in the cerebellum of mice exposed to MeHg during the gestational period has been documented [103]. The study from Fujimura et al. [110] suggested an upregulation in PKA activity after MeHg treatment, and an increase in ser-499 phosphorylation of RAP1-GAP (a specific marker of PKA activity) and ser-133 CREB phosphorylation (a downstream PKA target). This PKA activation could be associated with neural hyperactivity that precedes MeHg-induced neuronal degeneration.
4.2. Ubiquitination and Acetylation
As mentioned before, ubiquitin is a 76-amino acid protein that can be conjugated to other proteins, either singly or as polyubiquitin chains. Ubiquitination can target proteins for degradation, but can also modify the protein structure and function. This process is associated with complex cellular machinery that involves the activation of E1 ubiquitin-activating enzymes, E2 ubiquitin conjugates enzymes and E3 ubiquitin ligases. On the other hand, ubiquitin is removed from substrate proteins by a class of enzymes called deubiquitinases (DUBs). The ubiquitin system is central to the regulation of a wide range of cellular processes due to its ability to control protein activity and abundance [138, 139]. Interestingly, in an early attempt to search for genes that confer resistance to MeHg, a gene, CDC34, that encodes an ubiquitin-conjugating enzyme (E2) was identified to play a protective role in yeast cells following incubation of 0~1.0 μM MeHg for 48 h. CDC34 contains one cysteine residue that is essential for its function and might be a direct target of MeHg, and overexpression of other ubiquitin-conjugating enzymes might confer resistance to MeHg. The later was confirmed by the experiments showed that Ubc4 and Ubc 7 also play a protective role in MeHg’s toxicity. Moreover, treatment of yeast cells (W303B) with 25 nM MeHg for 90 min could induce increased transcription levels of CDC34, Ubc4 and Ubc7 [140]. A further exploration found another ubiquitin system enzyme, Rad23, confer resistance to MeHg by its appropriate domain function that binds to the multiubiquitin chain of ubiquitinated proteins [141]. The Skp1/Cullin/F-box protein (SCF) complex contains a F-box protein that binds directly to the target protein for degradation. Genetic screenings found that F-box containing proteins Hrt3, Ylr224w, and Ymr258c confer protection against MeHg’s toxicity in yeast cells that incubated with 0~140 nM MeHg for 48 h [142, 143]. In the human derived cell line model, Toyama et al. [144] have demonstrated that the treatment of the cell line SH-SY5Y with MeHg (1 μM, 9 h) induced inhibition of one of the enzymes in the DUB family, specifically binding to Cys152 residue of the ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), and inhibiting the enzyme activity. Furthermore, proteasomic and genomic analyses of MN9D cells exposed to MeHg (1~5 μM, 48 h) revealed an increase in ubiquitin conjugating enzyme E2L3 (UBE2L3) [145]. It has been noted that symptoms of MeHg toxicity share some similarity with those of Parkinson’s disease (PD) [146, 147]. PD is characterized by the loss of dopaminergic neurons and the presence of inclusion bodies (Lewy body) in brain tissue that is associated with the aging process. Pathological investigation revealed that malfunction of ubiquitin system components or overload of the system implicates in the formation of toxic protein aggregation [148]. A genomic and proteomic study suggests that MeHg and MPP(+) share similar signaling pathways that are involved in the pathogenesis of PD [49]. Thus, further investigations on the role of environmental factors, such as MeHg exposure, in the aberrations of protein ubiquitination system and its relation to the development of neurodegenerative disease are warranted. In addition, animal studies showed that exposure to environmental level of MeHg (0.50~0.59 ppm, 14 days) [149–151] predisposed animals to depression, a debilitating disease that is highly prevalent among US adults [152]. It has been known that MeHg (10 μM, 30 min) treatment of astrocytes significantly inhibited the fluoxetine-sensitive 5-HT uptake [50]. Perinatal MeHg exposure sensitized behavioral response to amphetamine in adult stage [153]. Treatment of MeHg induced anxiogenic behavior in zebrafish, which is associated with decreased level of extracellular 5-HT [52]. Although many drugs and behavior therapies have been developed, there is still a proportion of patients don’t respond to either of them, which caused a great number of economic loss and social burdens [154]. A clear understanding and elucidation of the causal relationship between environmental factors, like MeHg exposure, and pathogenesis of depression will improve the prevention and treatment measures of the disease.
The development of an organism is controlled by sequentially happened programming events, among which epigenomic remodeling plays a key role. MeHg are a known neurodevelopmental toxicant, and early life stage demonstrates greater sensitivity to MeHg. As to epigenetic effects, MeHg exposure caused global DNA hypomethylation as well as methylation level changes at specific regions of genes in various models (see review [155]). In a prospective pre-birth cohort study, it was found that methylation at paraoxonase 1 gene (PON1) that is relative to prenatal mercury exposure, is associated with cognitive performance in childhood [156]. Experimental studies support that MeHg-induced epigenetic modification might explain the latent neurobehavioral effects of animals developmentally exposed to MeHg [157], thus underpins the theory of “developmental origin of health and disease” [158]. Perinatal exposure to MeHg (0.5 ppm, 14 days) could induce a long-lasting depressive behavior in adult male mice, which might be associated with a decrease in the expression of brain-derived neurotrophic factor (BDNF) and a decrease in histone H3 acetylation [150]. Furthermore, it has been reported that exposure to MeHg (1~ 100 nM, 24 h) was able to promote epigenetic transgenerational inheritance associated with epimutation in zebrafish [157]. Though it is still unclear whether these are direct effects or indirect effects by modulation of thiol-containing proteins by MeHg, these studies pave the way to the understanding of epigenetic modifications induced by MeHg and its relation to neurodevelopmental disorders.
4.3. PTMs induced by oxidative stress
Redox protein and lipid modifications are essential for normal physiology, as long as ROS are produced under a fine control by specific pathways. Mounting evidence has corroborated that pathological dysfunction arises not because of a non-specific oxidation in biomolecules, but rather as a consequence of damage in the specific signaling domain control which could lead to bigger impacts in the system [159]. In other words, the loss of control of non-physiological redox modifications and the toxic gain of function of oxidized proteins and lipids are the core of oxidative stress consequences [160]. Indeed, in order to maintain the redox state of the cell, redox-active molecules promote reversible oxidative PTMs to promote signal transduction and modulation of the cellular function [161, 162]. As would be expected, oxidative post-translational modification of proteins has been associated with several CNS pathologies [163–165].
It is well documented that mercury has a strong affinity for thiol and selenol groups, and by binding to these groups mercury can impair protein function [166, 167]. The classical decrease in the antioxidant enzyme activities is explained by the inhibition of enzymes such as glutathione reductase (GR), glutathione peroxidases (GPxs), and superoxide dismutases (SODs) [168]. However, recent studies have demonstrated that in addition to a direct effect on proteins and other biomolecules (more details will be discussed in part 4.4), MeHg can modulate several signaling pathways by PTMs. An interesting study on the activity of CYP1A1, a member of the cytochrome P450 enzymes family, showed that MeHg affects the activity of this enzyme through PTMs. More specifically, the researchers treated human hepatoma HepG2 cells with a halogenated aromatic hydrocarbon (2,3,7,8-tetrachlorodibenzo-p-dioxin, named TCDD) that is able to induce the activity of CYP1A1. They found that MeHg (1.25–5 μM, 24 h) was able to inhibit TCDD-mediated induction of CYP1A1 activity by a post-translational mechanism [169]. A study using Neuro-2a cells, a mouse neuroblastoma cell line, showed that in addition to reducing cell viability and increasing oxidative stress damage, MeHg (3 μM, 0.5 h) was also able to increase the phosphorylation of ERK1/2 and p38MAPK. The authors suggest that the mitochondria-dependent apoptotic pathway might be regulated by signals of ROS-mediated ERK1/2 and p38MAPK activation [109]. Furthermore, Petroni and colleagues have demonstrated that neuroblastoma cells (SH-SY5Y) incubated with low concentrations of MeHg (50 nM, 6 h) showed an increase in tau phosphorylation, possibly through an oxidative stress-dependent mechanism [170]. Treatment with antioxidants blocked the MeHg-induced ROS generation and significantly reduced the effects of MeHg on tau phosphorylation. Moreover, a calpain inhibitor, MDL-28170, attenuated MeHg-induced tau phosphorylation and cytotoxic effects. These findings are consistent with findings by Sakaue et al. who noted that the toxic effects of MeHg in CGC were dependent on calpain activation (30 nM MeHg, 48 h) [171].
In addition, omics studies can provide greater systematic landscape on the toxic effects of MeHg and its affected molecular pathways. A more recent metabolome research using fathead minnow as a model demonstrated that metabolites involved in lipid metabolism and neurotransmission could be significantly altered in fathead minnow larvae exposed to maternally-transferred dietary MeHg (0.72 ppm) [172]. The number of proteomic studies related to MeHg poisoning has been progressively mounting. Li and collaborators [173] used proteomic and high-throughput mRNA sequencing (RNA-seq) to evaluate the cellular responses of human neuroblastoma SK-N-SH cells to MeHg. Interestingly, the authors observed that MeHg alters RNA splicing via splicesome, where a total of 658 aberrant RNA alternative splicing events were observed post MeHg treatment. Another study using RNA-seq analysis in C. elegans (larval 4 stage) exposed to MeHg (10 μM) revealed several changes in the gene transcription. It was found that 541 genes were upregulated and 261 genes down-regulated, encoding proteins related to oxidative stress responses and ER stress pathways [174]. Additionally, a sophisticated proteome study using marmoset to identify and analyze the differential expression induced by MeHg (1.5 ppm, 14 days) showed that from a total of 1045 and 1062 proteins identified in the frontal lobe and occipital lobe, 62 and 89 proteins were altered after MeHg exposure, respectively. Among them, the authors highlight that apolipoprotein E (ApoE) and GPx1 could be possible key proteins targeted by MeHg in occipital lobe [175]. In addition, another study using an aquatic organism (beluga), demonstrated that animals exposed to MeHg (diet containing 0.8 ppm MeHg for 70 days) presented significant changes in the proteome. Some of the identified proteins were involved in metabolism, protein folding, cell division, and signal transduction [61]. Similarly, MeHg effects have been also observed in systems other than CNS. For instance, rats treated with MeHg (oral daily dose of 0.04 mg/kg for 60 days) showed a significant modification in the proteomic profile in the salivary glands, especially in proteins related to structural components of cytoskeleton, metabolic pathways, and redox homeostasis [176].
Several groups have extensively investigated the role of PTMs as modulators of metabolic pathways, and its effects on mitochondrial proteins (Fig. 2) [177]. In fact, PTM enzymes such as deacetylase, desuccinylase and demalonylase and ADP-ribosylase, as well as kinase or phosphatase activity are localized within mitochondria. Furthermore, many mitochondrial PTMs have been described in signal transduction pathways, modulating mitochondrial energy generation, apoptosis, autophagy, metabolism, and tissue response to ischemic injury [178]. As mentioned above, MeHg-induced neurotoxicity is widely associated with excessive generation of ROS in mitochondrial respiration chain, as well as inhibition of antioxidant enzymes [6]. Consequently, the disrupted cellular redox homeostasis leads to oxidative stress, which alters cellular function [179]. In this context, increased oxidative stress seems to alter the quantity of mitochondria per cell as well as the copy number and integrity of mtDNA [74, 180]. In fact, it has been demonstrated that mitochondrial redox status correlates with mitochondrial morphology and is also associated with processing of important mitochondrial PTMs in proteins that regulate mitochondrial dynamics, such as optic atrophy 1 (OPA1) proteolysis, ubiquitin proteasome mediated degradation of A-kinase anchor protein 121 (AKAP121), and dynamin-relate protein 1 (DRP1) phosphorylation [181]. MeHg (500 nM, 12 h) was shown to reduce significantly both mtDNA content and mtDNA transcript levels in cultured rat CGNs. These effects could be a result from a ROS-induced mutation in the origins of replication and transcription in MeHg-treated CGNs [182]. Likewise, a study performed to evaluate the effects of MeHg (2.5 μM, 24 h) on myotube formation has demonstrated that along with the reduction in Myogenic factor (MyoG) expression (a transcription factor involved in myocyte differentiation) there was a significant decrease in many other factors involved in mitochondrial biogenesis and mtDNA transcription and translation (mitochondrial Transcription factor A, mitochondrial single-stranded DNA-binding protein and citrate synthase), which may implicate a role for mitochondria in mediating MeHg-induced impairment in myocyte differentiation and myotube formation [183]. Furthermore, an in vitro study using human neural progenitor cells (hNPCs) demonstrated that low-dose MeHg (10 nM, 24 h) was able to upregulate mitochondrial genes transcription followed by higher levels of mtDNA mutations and ROS production [184]. Despite the potential relevance of mitochondrial PTMs in MeHg toxicity, very little was investigated about this phenomenon thus far. Nonetheless, the close proximity of the mtDNA to the main source of ROS generation make mtDNA highly susceptible to oxidative damage, added to this the absence of a histone protection, and relatively less efficient DNA repair mechanisms [185]. It is noteworthy that there is an important communication between the mitochondria and the nucleus. When, mitochondrial function is controlled by the nucleus, a mechanism referred to as ‘anterograde regulation’ promotes biogenesis and regulation of the mitochondrial activity according to the cellular demand. Also, the nucleus can be controlled by the mitochondria through a ‘retrograde response’, thus modifying cellular function by reprogramming its metabolism. This mitonuclear communication is an important network which maintains cellular homeostasis in response to stress [186].
Fig. 2. ROS and MeHg-induced damages of protein synthesis and functions.
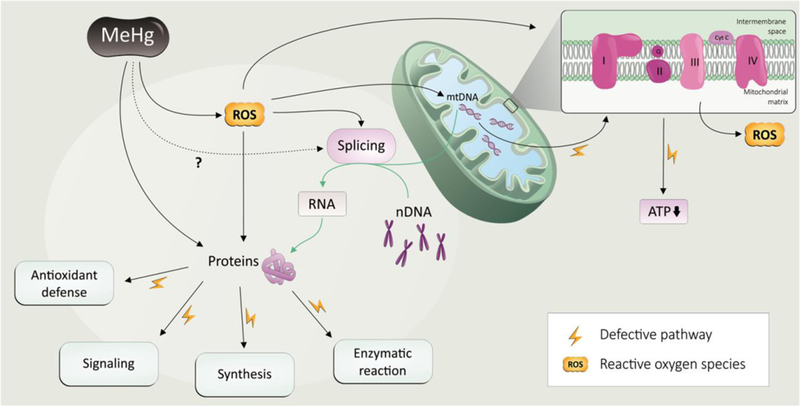
Reactive oxygen species (ROS) are known to mediate MeHg-induced neurotoxicity. MeHg induce oxidative stress via overproduction of ROS and decrease in the synthesis of antioxidants directly. Further, ROS induced by MeHg are able to promote an imbalance in the number of copies of the mtDNA as well as in its quality control of important protein such as the complex I of the mitochondrial respiratory chain, which in turn favor the overproduction of ROS and decrease the ATP synthesis. Several proteins are affected by redox imbalance induced by MeHg: oxidative damage via reaction with thiol and selenol groups can cause impairment in the protein function. ROS induced by MeHg alters the proteome of the cell by compromising the splicing and PTM of the proteins involved in antioxidant defense, signaling, synthesis and enzymatic reactions in general. Plain black plain arrows means effect of MeHg exposure based on literature data; dotted line arrow means a likely but not documented event; green arrow means physiological events. Yellow bolt cartoon represents the pathways that are defective due to MeHg exposure.
MeHg can increase or decrease protein nitrosylation. As a form of PTMs, nitrosylation describes the covalent attachment of nitrogen monoxide (NO) group to the thiol side chain of cysteine. In an experiment to analyze the role of NMDA in MeHg-induced neurotoxicity in the developing cortical neurons, the author found that MeHg (10 ppm, 7 days) induced a marked accumulation of nitrotyrosine, a product of peroxynitrite and L-tyrosine. MK-801, a non-competitive antagonist of NMDA, suppressed accumulation of nitrotyrosine [187]. Another study using SH-SY5Y cells as experimental model showed that treatment with MeHg (1 μM, 24 h) caused 50% reduction of nitrosylation of the catalytic domains of protein disulfide isomerase (PDI) [188].
4.4. PTMs of antioxidant proteins
4.4.1. Nuclear factor erythroid 2-related factor 2 (Nrf2)
Nrf2 functions mainly as a transcription factor to initiate gene expression of phase II detoxifying enzymes to combat oxidative stress resultant from xenobiotics exposure. It has been known that, upon 0.25 μM MeHg exposure, the antioxidant transcription factor Nrf2 is activated in SH-SY5Y cells [189]. Numerous studies, using different models including cell lines [189], primary cultures [190, 191], Nrf2-deficient mice [192], drosophila [193] and C. elegans [194, 195] verified that Nrf2 plays a pivotal role in mitigating MeHg toxicity. Due to its importance in cellular responses to oxidative stress, Nrf2 activity is regulated by complex pathways upon MeHg exposure (Fig. 3). A recent study from our group, using primary cortical astrocytes as an experimental model, showed that exposure to 5 μM MeHg for 6 h induced decreased transcription as well as nuclear protein levels of the Src family kinase, Fyn, which is a negative regulator of Nrf2 nuclear translocation by phosphorylation of Nrf2 [196]. Based on the concomitant finding that the phosphorylation levels of Akt were increased by MeHg treatment, we suggested that the Akt/ GSK-3β/Fyn pathway is one of mechanisms of MeHg-induced Nrf2 activation. Another study using primary culture of neural progenitor cell showed that, at low level of MeHg (10 nM), GSK-3β was up-regulated 3h after exposure, and that GSK-3β inhibitors protected neural progenitor cell death against MeHg cytotoxicity [197]. Interestingly, it has been known that GSK-3β can directly phosphorylate Nrf2 to signal its nuclear exclusion [198]. Although there is no direct evidence yet, the possibility that GSK-3β is a negative regulator of MeHg-induced Nrf2 activation should be considered. In the same experiment, we also found that MeHg decreased the level of Kelch-like ECH-associated protein 1 (Keap1), a major negative regulator of Nrf2 [196]. Nrf2/Keap1 complex facilitates ubiquitination of Nrf2, and the disassociation of Nrf2 from Keap1 is believed to be a cause, at least in part for the increased levels of Nrf2. There are two sequences in Nrf2 that are the binding sites for Keap1, namely, the high-affinity ETGE and low-affinity DLG motifs. Both of the sequences are bound by different Keap1 sub-units, and are required for Cul3-Rbx1 to ubiquitylate Nrf2. The ubiquitylation of Nrf2 follows a cycle that the formation of Nrf2/ Keap1 complex enables Cul3-Rbx1 to ubiquitinate Nrf2, and consequently the released Keap1 can bind to a new Nrf2 molecule [199].
Fig. 3. Mechanism of MeHg-induced Nrf2 activation.
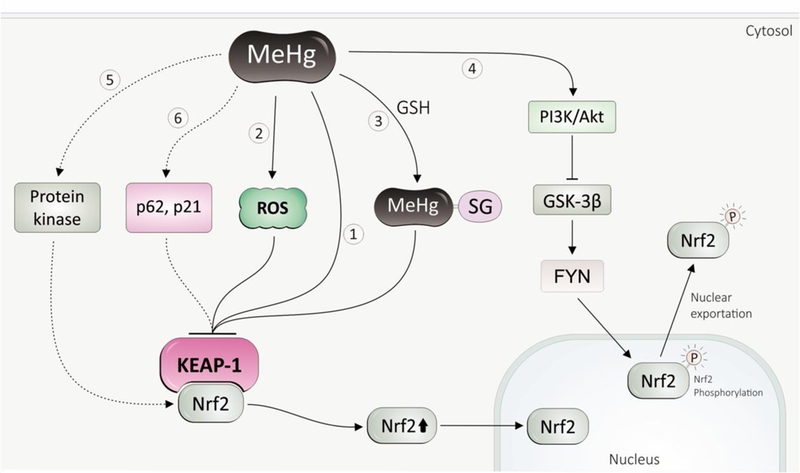
(1) MeHg directly binds with thiol groups in Keapl, inhibiting Keapl activity, thus compromise Nrf2 ubiquinition. (2) MeHg-induced ROS oxidizes Keapl, and leads to disassociation of Nrf2 from Keapl. (3) The MeHg-GSH adduct, a major form of MeHg excretion, can inhibit Keapl. (4) MeHg inhibits Nrf2 nuclear exportation by inhibiting GSK-3 via PI3K. (5) Activation of Nrf2 by MeHg can be possible via protein kinase pathways. (6) p21, p62 play protective role in MeHg toxicity, which might function through Nrf2 activation. Dashed line presents possible mechanisms.
It was originally suggested that the release of Nrf2 from Keap1 is a result of oxidative modifications of thiol groups of the two proteins [200], and it is possible that MeHg-induced activation of Nrf2 was mediated by increased levels of ROS. Due to the fact that Keap1 contains at least 25 cysteine residues and Nrf2 has 7 cysteines, later experiments revealed that a direct binding of MeHg with thiol groups in Keap1 is also involved in MeHg-dependent activation of Nrf2. By using matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF/MS) analysis, three cysteine residues (Cys151, Cys368, and Cys489) in Keap1 were identified as binding sites for MeHg. As Cys151 is one of essential residues for regulating Nrf2 function [201], the author concluded that binding of MeHg with Cys151 in Keap1 potentially causes a structural alteration, releasing Nrf2 (see review [202]). The same group also found that the MeHg-SG adduct, formed by the reaction of MeHg with GSH, can modify Keap1, activating Nrf2 [203]. The above conclusion supported a notion that it is not oxidization of Keap1 but conformational changes in Keap1 that result in release of Nrf2. Since oxidative stress plays a major role in MeHg toxicity, we cannot preclude the possibility that increased oxidative species upon MeHg exposure leads to protein oxidation, thus causes alterations of binding activities of Keap1 or Nrf2.
As far as we know, Nrf2 activity is largely controlled by Keap1, and it is believed that proteins that can compete with Keap1 to bind Nrf2 are potential positive regulators of Nrf2. Chemically, many biomolecules, including proteins and low-molecular weight molecules, are potential targets of MeHg due to its electrophilic property. Some of these proteins or molecules are modified by MeHg, either directly or indirectly, and may participate in the molecular networks that modulate MeHg toxicity. p21, also known as Waf1 or Cip1, is a cyclin-dependent kinase inhibitor, and experiments with primary embryonic cells (2 μM MeHg exposure for 24 h) and adult mice (10 ppm MeHg exposure for 4 weeks) showed that both mRNA and protein levels of p21 are markedly increased following MeHg exposure [204]. Later experiment also showed that p21 mRNA was up-regulated in neural stem cells exposed to low level of MeHg (2.5 or 5.0 nM for 48 h) [205]. It has been shown that p21 can bind with the DLG and ETGE motifs in Nrf2 via its KRR motif, competing with Keap1 to compromise Nrf2 ubiquitination [206]. Moreover, p62 is believed to be a competitor of Keap1 for binding to Nrf2 [207], and experiments in MEFs cells (1 μM MeHg exposure for 4 h), astrocytes (1 μM MeHg exposure for 6 h), and SH-SY5Y (1 μM MeHg exposure for 24 h) showed that p62 was up-regulated and plays a protective role in MeHg toxicity [208–210]. Therefore, whether these molecules function via Nrf2 activation pathway warrants further investigations.
The activation of Nrf2 by MeHg is far more complex than originally thought. An experiment with SH-SY5Y cells showed that carbon monoxide (CO) and its producing enzyme heme oxygenase-2 (HO-2) protects cells against MeHg cytotoxicity (1 μM, 24 h) by Nrf2 activation, and a possible mechanism accounting for this observation is CO activation of Nrf2 via MAPK or ERK pathway [211]. As noted earlier, different signaling pathways and kinases are involved in toxic consequences upon MeHg exposure, and these kinases, such as PKC, PI3K, MAPKs, are also involved in modulation of Nrf2 activity [212]. The functional importance of Nrf2 in antioxidant response, especially in MeHg toxicity, warrants further investigations on its role and regulation.
4.4.2. Glutathione system
The glutathione system includes reduced glutathione (GSH) and oxidized forms of glutathione (GSSG), the enzymes that are required for GSH synthesis and enzymes such as glutathione s-transferases (GSTs) and GPxs that use GSH in the antioxidant defense activities. GSH is a tripeptide that plays an essential role in antioxidant defenses. Due to its high concentration in cells, GSH may act as the first target for ROS generated by MeHg [213, 214]. MeHg exposure causes GSH depletion and a decreased ratio of GSH/GSSG, a consequence of the pro-oxidative events caused by MeHg. Notably, the abundance of GSH in the brain tissue is in the low millimolar range [215], whereas a nanomolar range of MeHg can induce neurotoxicity. Therefore, these events are not consequence of the simple interaction between MeHg and GSH, but rather resultant from the increased levels of oxidants that are secondary to inhibition of antioxidant selenoenzymes, such as GPx and TrxR after MeHg exposure [6, 216]. MeHg- GSH adduct is the major form of detoxification. There is a large body of epidemiology studies corroborating that polymorphisms in GSH-related genes was associated with body burdens of MeHg, and a recent epidemiology study showed that polymorphism in glutamate cysteine ligase (GCL) has a significant association with hair Hg content [217]. Experimental evidence has shown that GCL transcription levels in the placenta of C57Bl/6 mice were up-regulated after exposure to 1 ppm MeHg for 32 days [218].
Selenium (Se) is an essential nutrient required by selenoproteins. GPxs represent a family of peroxide-detoxifying enzymes; most of them are selenoproteins whose catalytic activity depends on the reducing power of a selenol group located at the active site. MeHg preferentially targets selenol-containing molecules than thiol-containing molecules (see review [14]), and reduced activity of GPxs is an important pro-oxidative event in MeHg neurotoxicity. Experimental data showed that GPx activity was significantly inhibited in mice after 21-day exposure of 40 ppm MeHg. Incubation of isolated mitochondrial with exogenous GPx totally blocked MeHg-induced lipid peroxidation. Moreover, the inhibitory effect of 1 μM MeHg exposure for 24 h on GPx activity was observed in SH-SY5Y cells [219]. Although the inhibitory effects of MeHg on GPxs’ activity was believed to be the chemical modification of the selenol group in the protein by MeHg, experiments on selenium supplementation did not find a full recovery of enzyme activity [66, 220]. The reason behind this might be that MeHg can sequester selenium into nonbioavailable forms, and the enzyme activity can’t be fully reversed due to the sustained structural modification. Of particular importance, the protective effect of synthetic organic selenium compounds against MeHg toxicity has received much attention. Study with slices of cerebral cortex of rats revealed that 1 μM MeHg exposure for 30 min induced hyperphosphorylation of high molecular weight neurofilament subunit (NF-H). Both ebselen (5 μM) and diphenyl diselenide (15 μM), which mimic GPx activity, presented a protective effect against MeHg-induced hyperphosphorylation of NF-H [221]. Experiments with primary cortical astrocytes showed that pretreatment with ebselen (10 μM) partially reversed the 1 μM MeHg-induced astrocytic inhibition of glutamine uptake and reversed the 1 μM MeHg-induced changes in mitochondrial membrane potential. Meanwhile, ebselen pretreatment inhibited MeHg-induced phosphorylation of ERK and blocked MeHg-induced activation of caspase-3 [123].
4.4.3. Thioredoxin system
The thioredoxin system comprises thioredoxin (Trx), thioredoxin reductase (TrxR) and NADPH [222]. The Trx system is the major system responsible for maintaining the redox state of cells and this function involves thiol reduction mediated by selenol groups in TrxRs. TrxR is an early target of MeHg. Using human neuroblastoma cells as an in vitro model, TrxR activity was significantly decreased by MeHg, with 1 μM this metal causing 50% inhibition in TrxR activity [223]. MeHg can bind to selenocysteine residues present in the catalytic site of TrxR, in turn, causing enzyme inhibition that can compromise the redox state of cells. MeHg caused a significant inhibition of TrxR in a tissue-dependent manner. In vitro assay with purified enzyme from brain, liver and kidney of mice showed that 0.05 μM MeHg caused a significant inhibition of TrxR in brain, but the net effect was lower than that in liver or kidney [224]. In vivo experiment also showed that exposure to 5 ppm MeHg for 24 h caused a significant inhibitory effect on TrxR activity in liver and kidney, but not in brain [224]. Study in zebra-seabreams showed that both TrxR and Trx were affected by exposure to 0.5 ~ 2 ppb MeHg for 28 days. The thioredoxin system in the brain was also affected. In contrast to TrxR, Trx was only affected in the later stages of exposure [225]. In SH-SY5Y cells, the oxidation of Trx1 can be induced at dosage of 5 μM MeHg. Depletion of GSH is directly linked to increased oxidation of Trx1. Meanwhile, the phosphorylation of apoptosis signaling kinase 1 (ASK-1) was increased [167]. Different species of mercurial exert varied inhibitory effect on Trx. A previous in vitro study showed that Hg2+ can make a disulfide bridge between adjacent thiols and promotes dimerization of Trx molecules, whereas MeHg can only bind to one thiol at a time, making Hg2+ a stronger inhibitor of Trx [226].
Although MeHg presents significant pro-oxidative properties (mainly due to its electrophilic profile) and has been reported to cause several PTMs, a clear mechanistic relationship between both phenomena is lacking in the literature. Moreover, it is not clear if the effects of MeHg on antioxidant systems represent a relevant cause of PTMs. Although there is an evident need for further research on this topic, recent data begin to emerge in the literature. In this regard, a recent in vitro study with cultured SH-SY5Y cells has shown that, due to its affinity for nucleophilic groups, MeHg can cause S-mercuration (which represents a PTM), and that this phenomenon was responsible for the modulation of signaling pathways involved in cell survival and apoptosis [227]. Thus, MeHg-induced PTMs can be a direct consequence of its electrophilic/oxidant effects toward nucleophilic groups of biomolecules. On the other hand, it is important to mention that the well-reported inhibitory effects of MeHg on GPx [219] may lead to increased levels of hydroperoxides [29, 228]. Because hydrogen peroxide (H2O2) presents a central role in regulating several signaling pathways through the reaction with cysteine regulatory switches [229], one may posit that the increased hydroperoxide levels resulting from MeHg-mediated GPx inhibition may contribute to oxidative PTMs that modulate myriad of signaling pathways. In this scenario, it is noteworthy that cells present diverse thiol protein sensors, with different reactivity of their sulfhydryl groups towards H2O2, being activated by different concentrations and times of exposure to H2O2 [230]. Thus, it is provocative to hypothesize that several of the PTMs induced by MeHg can represent a secondary (indirect) consequence of its effects toward antioxidant systems. However, this is a newly opened research field that deserves significant efforts in order to elucidate the relationship between the effects of MeHg on antioxidant systems and its ability to cause PTM. Taking into account that (i) the pro-oxidative effects of MeHg can also lead (even indirectly) to increased levels of NO and GSSG [231–233], and that (ii) S-nitrosylation [234] and glutathionylation [235] also represent pivotal signaling PTMs modulating both physiological and pathological events, a potential role of S-nitrosylation and glutathionylation in MeHg-induced toxicity cannot be ruled out, representing a promising new research field.
5. Conclusions and perspectives
MeHg exposure continues to remain an environmental health burden due to its toxic effects, especially on the brain. Population whose diet consists largely of fish are at the greatest risk for exposure. The developing fetus and children are susceptible to its neurotoxic effects. Studies on the molecular mechanism of MeHg-neurotoxicity are pivotal to our understanding of its toxic effects and the development of preventive measures. MeHg is a soft electrophile that has extremely high affinity for nucleophilic thiol (-SH) and selenol (-SeH) groups, thus a large pool of cellular molecules could be its candidate targets. PTMs, such phosphorylation, ubiquitination, and acytelation, are essential mechanisms in the biosynthesis of proteins and play pivotal role in the regulation of cellular environment. MeHg modulates the activities of MAPKs (ERK1/2, JNK, p38MAPK), PKA and PKC, which are important in modulating pathways related to its neurotoxicity, such as cellular redox status, cell differentiation, and cell death. Mitochondrial damage and increased ROS generation caused by MeHg are important steps in the process of PTMs of proteins that mark MeHg neurotoxicity. Nfr2 is important in combating oxidative stress, and many pathways have been proved to be involved in MeHg-induced activation of Nrf2. Accumulated data shows that antioxidant proteins (such as GPxs and TrxR) are likely to be modulated by MeHg via a PTMs mechanism. Although researches on MeHg-neurotoxicity have illustrated many important aspects of its mechanism, such as oxidative stress, calcium dyshomeostasis, and disrupted glutamine recycling, etc, we still lack knowledge on the molecular events triggered by MeHg. For example, cells contain more than 19,000 -SH containing proteins and 25 selenoproteins, however, we do not know with which of these proteins MeHg directly interacts. The PTMs modification can be a result of direct inhibition of the proteins by MeHg or can be indirect either inhibition or activation via modulation of up-stream thiol- or selenol-containing proteins. Since PTMs is a conserved and essential process for maintaining cellular homeostasis and function, and many newly defined cellular machineries are carried out by proteins that are modified by multiple PTMs mechanisms, current investigations should offer new insight on the relation between PTMs and MeHg neurotoxicity.
Highlights.
PTMs are an important molecular mechanism involved in the neurotoxic effects of MeHg.
MeHg can modulate PTMs in a developmentally dependent and cell-type specific manner.
Selective target of PTMs that mediated by MeHg’s pro-oxidative events represents a promising new research field.
Acknowledgments
This work was supported by the National Institutes of Health to MA [grant numbers NIEHS R01ES07331, NIEHS R01ES10563 and NIEHS R01ES020852]. The authors also would like to thank Ms Priscila B. Rosa for the help with the graphic illustrations.
Abreviations:
- Hg
Mercury
- EtHg
ethylmercury
- MeHg
methylmercury
- FMD
Fetal Minamata disease
- CGC
cerebellar granule cells
- ROS
reactive oxygen species
- PTMs
post translational modifications
- OXPHOS
oxidative phosphorylation
- ETC
electron transport chain
- AIF
apoptosis inducing factor
- NMDA
N-methyl D-aspartate
- AD
Alzheimer’s disease
- HATs
histone acetylases
- HDACs
histone deacetylases
- MAPKs
mitogen-activated protein kinases
- PKA
protein kinase A
- PKC
protein kinase C
- ERK1/2
extracellular signal-regulated kinases
- JNKs
c-Jun N-terminal kinases
- LTP
long term potentiation
- cAMP
5’-cyclic-adenosine monophosphate
- ACs
adenylyl cyclases
- DUBs
deubiquitinases
- UCH-L1
ubiquitin carboxy-terminal hydrolase L1
- UBE2L3
ubiquitin conjugating enzyme E2L3
- BDNF
brain-derived neurotrophic factor
- GR
glutathione reductase
- GPxs
glutathione peroxidases
- SODs
superoxide dismutases
- ER
endoplasmic reticulum
- ApoE
apolipoprotein E
- CGNs
cerebellar granule neurons
- hNPCs
human neural progenitor cells
- MALDI-TOF/MS
matrix-assisted laser desorption and ionization time-of-flight mass spectrometry
- GSH
glutathione
- GSTs
glutathione s-transferases
- GCL
glutamate cysteine ligase
- NF-H
high molecular weight neurofilament subunit
- TrxR
thioredoxin reductase
- ASK-1
apoptosis signaling kinase 1
- Nrf2
Nuclear factor erythroid 2-related factor 2
- Keap1
Kelch-like ECH-associated protein 1
- PDI
protein disulfide isomerase
- NO
nitrogen monoxide
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.Clarkson TW, The three modern faces of mercury. Environ Health Perspect, 2002. 110 Suppl 1: p. 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eto K, Pathology of Minamata disease. Toxicol Pathol, 1997. 25(6): p. 614–23. [DOI] [PubMed] [Google Scholar]
- 3.Snyder RD, Congenital mercury poisoning. N Engl J Med, 1971. 284(18): p. 1014–6. [DOI] [PubMed] [Google Scholar]
- 4.Hamada R,OM, Minamata disease and other Mercury syndromes, in Toxicology of metals, LW C, Editor. 1996, CRC Press: Boca Raton (FL). p. 337–51. [Google Scholar]
- 5.Clarkson TW, Magos L, and Myers GJ, The toxicology of mercury - Current exposures and clinical manifestations. New England Journal of Medicine, 2003. 349(18): p. 1731–1737. [DOI] [PubMed] [Google Scholar]
- 6.Farina M, Aschner M, and Rocha JB, Oxidative stress in MeHg-induced neurotoxicity. Toxicol Appl Pharmacol, 2011. 256(3): p. 405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kershaw TG, Clarkson TW, and Dhahir PH, The relationship between blood levels and dose of methylmercury in man. Arch Environ Health, 1980. 35(1): p. 28–36. [DOI] [PubMed] [Google Scholar]
- 8.Zareba G, et al. , Thimerosal distribution and metabolism in neonatal mice: comparison with methyl mercury. J Appl Toxicol, 2007. 27(5): p. 511–8. [DOI] [PubMed] [Google Scholar]
- 9.Aschner M and Clarkson TW, Uptake of methylmercury in the rat brain: effects of amino acids. Brain Res, 1988. 462(1): p. 31–9. [DOI] [PubMed] [Google Scholar]
- 10.Syversen T and Kaur P, The toxicology of mercury and its compounds. J Trace Elem Med Biol, 2012. 26(4): p. 215–26. [DOI] [PubMed] [Google Scholar]
- 11.Costa LG, et al. , Developmental neuropathology of environmental agents. Annu Rev Pharmacol Toxicol, 2004. 44: p. 87–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansson C, et al. , Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox Res, 2007. 11(3–4): p. 241–60. [DOI] [PubMed] [Google Scholar]
- 13.Grandjean P and Herz KT, Methylmercury and brain development: imprecision and underestimation of developmental neurotoxicity in humans. Mt Sinai J Med, 2011. 78(1): p. 107–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farina M and Aschner M, Methylmercury-Induced Neurotoxicity: Focus on Pro-oxidative Events and Related Consequences. Adv Neurobiol, 2017. 18: p. 267–286. [DOI] [PubMed] [Google Scholar]
- 15.Ceccatelli S, Dare E, and Moors M, Methylmercury-induced neurotoxicity and apoptosis. Chem Biol Interact, 2010. 188(2): p. 301–8. [DOI] [PubMed] [Google Scholar]
- 16.Limke TL, Bearss JJ, and Atchison WD, Acute exposure to methylmercury causes Ca2+ dysregulation and neuronal death in rat cerebellar granule cells through an M3 muscarinic receptor-linked pathway. Toxicol Sci, 2004. 80(1): p. 60–8. [DOI] [PubMed] [Google Scholar]
- 17.Falluel-Morel A, et al. , Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J Neurochem, 2007. 103(5): p. 1968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi BH, The effects of methylmercury on the developing brain. Prog Neurobiol, 1989. 32(6): p. 447–70. [DOI] [PubMed] [Google Scholar]
- 19.Pinheiro MC, et al. , Mercury pollution and childhood in Amazon riverside villages. Environ Int, 2007. 33(1): p. 56–61. [DOI] [PubMed] [Google Scholar]
- 20.Aschner M, et al. , Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz J Med Biol Res, 2007. 40(3): p. 285–91. [DOI] [PubMed] [Google Scholar]
- 21.Charleston JS, et al. , Increases in the number of reactive glia in the visual cortex of Macaca fascicularis following subclinical long-term methyl mercury exposure. Toxicol Appl Pharmacol, 1994. 129(2): p. 196–206. [DOI] [PubMed] [Google Scholar]
- 22.Charleston JS, et al. , Changes in the number of astrocytes and microglia in the thalamus of the monkey Macaca fascicularis following long-term subclinical methylmercury exposure. Neurotoxicology, 1996. 17(1): p. 127–38. [PubMed] [Google Scholar]
- 23.Aschner M, et al. , Methylmercury-induced alterations in excitatory amino acid transport in rat primary astrocyte cultures. Brain Res, 1993. 602(2): p. 181–6. [DOI] [PubMed] [Google Scholar]
- 24.Aschner M and Costa LG, The role of glia in neurotoxicity. 2nd ed 2005, Boca Raton, FL: CRC Press; 456p.,12 p. of plates. [Google Scholar]
- 25.Shanker G and Aschner M, Methylmercury-induced reactive oxygen species formation in neonatal cerebral astrocytic cultures is attenuated by antioxidants. Brain Res Mol Brain Res, 2003. 110(1): p. 85–91. [DOI] [PubMed] [Google Scholar]
- 26.Shanker G and Aschner M, Identification and characterization of uptake systems for cystine and cysteine in cultured astrocytes and neurons: evidence for methylmercury-targeted disruption of astrocyte transport. J Neurosci Res, 2001. 66(5): p. 998–1002. [DOI] [PubMed] [Google Scholar]
- 27.Juarez BI, et al. , Methylmercury increases glutamate extracellular levels in frontal cortex of awake rats. Neurotoxicol Teratol, 2002. 24(6): p. 767–71. [DOI] [PubMed] [Google Scholar]
- 28.Park ST, et al. , Methylmercury-induced neurotoxicity in cerebral neuron culture is blocked by antioxidants and NMDA receptor antagonists. Neurotoxicology, 1996. 17(1): p. 37–45. [PubMed] [Google Scholar]
- 29.Manfroi CB, et al. , Maternal milk as methylmercury source for suckling mice: Neurotoxic effects involved with the cerebellar glutamatergic system. Toxicological Sciences, 2004. 81(1): p. 172–178. [DOI] [PubMed] [Google Scholar]
- 30.Allen JW, Mutkus LA, and Aschner M, Methylmercury-mediated inhibition of 3H-D-aspartate transport in cultured astrocytes is reversed by the antioxidant catalase. Brain Res, 2001. 902(1): p. 92–100. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds IJ and Hastings TG, Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci, 1995. 15(5 Pt 1): p. 3318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orrenius S, Nicotera P, and Zhivotovsky B, Cell Death Mechanisms and Their Implications in Toxicology. Toxicological Sciences, 2011. 119(1): p. 3–19. [DOI] [PubMed] [Google Scholar]
- 33.Marty MS and Atchison WD, Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicology and Applied Pharmacology, 1997. 147(2): p. 319–330. [DOI] [PubMed] [Google Scholar]
- 34.Marty MS and Atchison WD, Elevations of intracellular Ca2+ as a probable contributor to decreased viability in cerebellar granule cells following acute exposure to methylmercury. Toxicology and Applied Pharmacology, 1998. 150(1): p. 98–105. [DOI] [PubMed] [Google Scholar]
- 35.Sirois JE and Atchison WD, Methylmercury affects multiple subtypes of calcium channels in rat cerebellar granule cells. Toxicology and Applied Pharmacology, 2000. 167(1): p. 1–11. [DOI] [PubMed] [Google Scholar]
- 36.Sakamoto M, Ikegami N, and Nakano A, Protective effects of Ca2+ channel blockers against methyl mercury toxicity. Pharmacology & Toxicology, 1996. 78(3): p. 193–199. [DOI] [PubMed] [Google Scholar]
- 37.Yuan Y and Atchison WD, Multiple Sources of Ca2+ Contribute to Methylmercury-Induced Increased Frequency of Spontaneous Inhibitory Synaptic Responses in Cerebellar Slices of Rat. Toxicol Sci, 2016. 150(1): p. 117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bradford AB, Mancini JD, and Atchison WD, Methylmercury-Dependent Increases in Fluo4 Fluorescence in Neonatal Rat Cerebellar Slices Depend on Granule Cell Migrational Stage and GABAA Receptor Modulation. J Pharmacol Exp Ther, 2016. 356(1): p. 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan Y and Atchison WD, Action of methylmercury on GABA(A) receptor-mediated inhibitory synaptic transmission is primarily responsible for its early stimulatory effects on hippocampal CA1 excitatory synaptic transmission. J Pharmacol Exp Ther, 1997. 282(1): p. 64–73. [PubMed] [Google Scholar]
- 40.Yuan Y and Atchison WD, Methylmercury differentially affects GABA(A) receptor-mediated spontaneous IPSCs in Purkinje and granule cells of rat cerebellar slices. J Physiol, 2003. 550(Pt 1): p. 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atchison WD, Is chemical neurotransmission altered specifically during methylmercury-induced cerebellar dysfunction? Trends in Pharmacological Sciences,2005. 26(11): p. 549–557. [DOI] [PubMed] [Google Scholar]
- 42.Faro LRF, et al. , Effects of methyl mercury on the in vivo release of dopamine and its acidic metabolites DOPAC and HVA from striatum of rats. Ecotoxicology and Environmental Safety, 1997. 38(2): p. 95–98. [DOI] [PubMed] [Google Scholar]
- 43.Faro LR, et al. , Protective effects of glutathione and cysteine on the methylmercury-induced striatal dopamine release in vivo. Life Sci, 2005. 77(4): p. 444–51. [DOI] [PubMed] [Google Scholar]
- 44.Faro LRF, et al. , Protection of methylmercury effects on the in vivo dopamine release by NMDA receptor antagonists and nitric oxide synthase inhibitors. Neuropharmacology, 2002. 42(5): p. 612–618. [DOI] [PubMed] [Google Scholar]
- 45.Dare E, et al. , Effects of prenatal exposure to methylmercury on dopamine-mediated locomotor activity and dopamine D2 receptor binding. Naunyn Schmiedebergs Arch Pharmacol, 2003. 367(5): p. 500–8. [DOI] [PubMed] [Google Scholar]
- 46.Tiernan CT, et al. , The role of de novo catecholamine synthesis in mediating methylmercury-induced vesicular dopamine release from rat pheochromocytoma (PC12) cells. Toxicol Sci, 2013. 133(1): p. 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tiernan CT, et al. , Methylmercury impairs canonical dopamine metabolism in rat undifferentiated pheochromocytoma (PC12) cells by indirect inhibition of aldehyde dehydrogenase. Toxicol Sci, 2015. 144(2): p. 347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao YT and Chan HM, Effects of methylmercury on dopamine release in MN9D neuronal cells. Toxicology Mechanisms and Methods, 2015. 25(8): p. 637–644. [DOI] [PubMed] [Google Scholar]
- 49.Shao YT, et al. , Methylmercury can induce Parkinson’s-like neurotoxicity similar to 1-methyl-4-phenylpyridinium: a genomic and proteomic analysis on MN9D dopaminergic neuron cells. Journal of Toxicological Sciences, 2015. 40(6): p. 817–828. [DOI] [PubMed] [Google Scholar]
- 50.Dave V, et al. , Astrocytes as Mediators of Methylmercury Neurotoxicity - Effects on D-Aspartate and Serotonin Uptake. Developmental Neuroscience, 1994. 16(3–4): p. 222–231. [DOI] [PubMed] [Google Scholar]
- 51.Beyrouty P, et al. , Effects of prenatal methylmercury exposure on brain monoamine oxidase activity and neurobehaviour of rats. Neurotoxicology and Teratology, 2006. 28(2): p. 251–259. [DOI] [PubMed] [Google Scholar]
- 52.Puty B, et al. , Ascorbic Acid Protects Against Anxiogenic-Like Effect Induced by Methylmercury in Zebrafish: Action on the Serotonergic System. Zebrafish, 2014. 11(4): p. 365–370. [DOI] [PubMed] [Google Scholar]
- 53.Koopman WJ, et al. , OXPHOS mutations and neurodegeneration. EMBO J, 2013. 32(1): p. 9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nunnari J and Suomalainen A, Mitochondria: in sickness and in health. Cell, 2012. 148(6): p. 1145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jezek P and Hlavata L, Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol, 2005. 37(12): p. 2478–503. [DOI] [PubMed] [Google Scholar]
- 56.Bondy SC and McKee M, Disruption of the potential across the synaptosomal plasma membrane and mitochondria by neurotoxic agents. Toxicol Lett, 1991. 58(1): p. 13–21. [DOI] [PubMed] [Google Scholar]
- 57.Fowler BA and Woods JS, Ultrastructural and biochemical changes in renal mitochondria during chronic oral methyl mercury exposure: the relationship to renal function. Exp Mol Pathol, 1977. 27(3): p. 403–12. [DOI] [PubMed] [Google Scholar]
- 58.Verity MA, Brown WJ, and Cheung M, Organic mercurial encephalopathy: in vivo and in vitro effects of methyl mercury on synaptosomal respiration. J Neurochem, 1975. 25(6): p. 759–66. [DOI] [PubMed] [Google Scholar]
- 59.Murphy MP, How mitochondria produce reactive oxygen species. Biochem J, 2009. 417(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Srinivas Bharath MM, Post-Translational Oxidative Modifications of Mitochondrial Complex I (NADH: Ubiquinone Oxidoreductase): Implications for Pathogenesis and Therapeutics in Human Diseases. J Alzheimers Dis, 2017. 60(s1): p. S69–S86. [DOI] [PubMed] [Google Scholar]
- 61.Yee S and Choi BH, Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology, 1996. 17(1): p. 17–26. [PubMed] [Google Scholar]
- 62.Sone N, Larsstuvold MK, and Kagawa Y, Effect of methyl mercury on phosphorylation, transport, and oxidation in mammalian mitochondria. J Biochem, 1977. 82(3): p. 859–68. [DOI] [PubMed] [Google Scholar]
- 63.Herculano AM, et al. , Methylmercury intoxication activates nitric oxide synthase in chick retinal cell culture. Braz J Med Biol Res, 2006. 39(3): p. 415–8. [DOI] [PubMed] [Google Scholar]
- 64.Mori N, Yasutake A, and Hirayama K, Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Arch Toxicol, 2007. 81(11): p. 769–76. [DOI] [PubMed] [Google Scholar]
- 65.Glaser V, et al. , Diphenyl diselenide administration enhances cortical mitochondrial number and activity by increasing hemeoxygenase type 1 content in a methylmercury-induced neurotoxicity mouse model. Mol Cell Biochem, 2014. 390(1–2): p. 1–8. [DOI] [PubMed] [Google Scholar]
- 66.Glaser V, et al. , Effects of inorganic selenium administration in methylmercury-induced neurotoxicity in mouse cerebral cortex. Int J Dev Neurosci, 2010. 28(7): p. 631–7. [DOI] [PubMed] [Google Scholar]
- 67.Caito SW and Aschner M, NAD+ Supplementation Attenuates Methylmercury Dopaminergic and Mitochondrial Toxicity in Caenorhabditis Elegans. Toxicol Sci, 2016. 151(1): p. 139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patel M, Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol Sci, 2016. 37(9): p. 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravagnan L, Roumier T, and Kroemer G, Mitochondria, the killer organelles and their weapons. J Cell Physiol, 2002. 192(2): p. 131–7. [DOI] [PubMed] [Google Scholar]
- 70.Ward MW, Kogel D, and Prehn JH, Neuronal apoptosis: BH3-only proteins the real killers? J Bioenerg Biomembr, 2004. 36(4): p. 295–8. [DOI] [PubMed] [Google Scholar]
- 71.Limke TL and Atchison WD, Acute exposure to methylmercury opens the mitochondrial permeability transition pore in rat cerebellar granule cells. Toxicol Appl Pharmacol, 2002. 178(1): p. 52–61. [DOI] [PubMed] [Google Scholar]
- 72.Shenker BJ, et al. , Mercury-induced apoptosis in human lymphocytes: caspase activation is linked to redox status. Antioxid Redox Signal, 2002. 4(3): p. 379–89. [DOI] [PubMed] [Google Scholar]
- 73.Tamm C, et al. , High susceptibility of neural stem cells to methylmercury toxicity: effects on cell survival and neuronal differentiation. J Neurochem, 2006. 97(1): p. 69–78. [DOI] [PubMed] [Google Scholar]
- 74.Mattson MP and Liu D, Energetics and oxidative stress in synaptic plasticity and neurodegenerative disorders. Neuromolecular Med, 2002. 2(2): p. 215–31. [DOI] [PubMed] [Google Scholar]
- 75.Polunas M, et al. , Role of oxidative stress and the mitochondrial permeability transition in methylmercury cytotoxicity. Neurotoxicology, 2011. 32(5): p. 526–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang YC, Peterson SE, and Loring JF, Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Research, 2014. 24(2): p. 143–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jensen ON, Interpreting the protein language using proteomics. Nature Reviews Molecular Cell Biology, 2006. 7(6): p. 391–403. [DOI] [PubMed] [Google Scholar]
- 78.Walsh CT, Garneau-Tsodikova S, and Gatto GJ, Protein posttranslational modifications: The chemistry of proteome diversifications. Angewandte Chemie-International Edition, 2005. 44(45): p. 7342–7372. [DOI] [PubMed] [Google Scholar]
- 79.Oliveira AP and Sauer U, The importance of post-translational modifications in regulating Saccharomyces cerevisiae metabolism. Fems Yeast Research, 2012. 12(2): p. 104–117. [DOI] [PubMed] [Google Scholar]
- 80.Oliver Bonham-Carter JP, Lotfollah Najjar, Dhundy Bastola, Modeling the effects of microgravity on oxidation in mitochondria: a protein damage assessment across a diverse set of life forms. Comput Biol Med, 2014(53): p. 179–89. [DOI] [PubMed] [Google Scholar]
- 81.Peng LR, et al. , Ubiquitinated Sirtuin 1 (SIRT1) Function Is Modulated during DNA Damage-induced Cell Death and Survival. Journal of Biological Chemistry, 2015. 290(14): p. 8904–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peuget S, et al. , Oxidative stress-induced p53 activity is enhanced by a redox-sensitive TP53INP1 SUMOylation. Cell Death and Differentiation, 2014. 21(7): p. 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Minguez P, et al. , PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic Acids Research, 2013. 41(D1): p. D306–D311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cohen P, The origins of protein phosphorylation. Nature Cell Biology, 2002. 4(5): p. E127–E130. [DOI] [PubMed] [Google Scholar]
- 85.Didonna A and Benetti F, Post-translational modifications in neurodegeneration. Aims Biophysics, 2016. 3(1): p. 27–49. [Google Scholar]
- 86.Humphrey SJ, James DE, and Mann M, Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends in Endocrinology and Metabolism, 2015. 26(12): p. 676–687. [DOI] [PubMed] [Google Scholar]
- 87.Yang XJ and Seto E, Lysine acetylation: Codified crosstalk with other posttranslational modifications. Molecular Cell, 2008. 31(4): p. 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peleg S, et al. , Altered Histone Acetylation Is Associated with Age-Dependent Memory Impairment in Mice. Science, 2010. 328(5979): p. 753–756. [DOI] [PubMed] [Google Scholar]
- 89.Saha RN and Pahan K, HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death and Differentiation, 2006. 13(4): p. 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haberland M, Montgomery RL, and Olson EN, The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature Reviews Genetics, 2009. 10(1): p. 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hua ZH and Vierstra RD, The Cullin-RING Ubiquitin-Protein Ligases. Annual Review of Plant Biology, Vol 62, 2011. 62: p. 299–334. [DOI] [PubMed] [Google Scholar]
- 92.Heride C, Urbe S, and Clague MJ, Ubiquitin code assembly and disassembly. Current Biology, 2014. 24(6): p. R215–R220. [DOI] [PubMed] [Google Scholar]
- 93.Iwai K and Tokunaga F, Linear polyubiquitination: a new regulator of NF-kappaB activation. EMBO Rep, 2009. 10(7): p. 706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu GQ and Jaffrey SR, Proteomic identification of protein ubiquitination events. Biotechnology and Genetic Engineering Reviews, Vol 29, Issue1, 2013. 29(1): p. 73–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang Q, et al. , Adaptive Posttranslational Control in Cellular Stress Response Pathways and Its Relationship to Toxicity Testing and Safety Assessment. Toxicological Sciences, 2015. 147(2): p. 302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Soti C and Csermely P, Protein stress and stress proteins: implications in aging and disease. Journal of Biosciences, 2007. 32(3): p. 511–515. [DOI] [PubMed] [Google Scholar]
- 97.Wani R and Murray BW, Analysis of Cysteine Redox Post-Translational Modifications in Cell Biology and Drug Pharmacology. Methods Mol Biol, 2017. 1558: p. 191–212. [DOI] [PubMed] [Google Scholar]
- 98.Chung HS, et al. , Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res, 2013. 112(2): p. 382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yagame H, et al. , Differential effects of methylmercury on the phosphorylation of protein species in the brain of acutely intoxicated rats. Toxicology, 1994. 92(1–3): p. 101–13. [DOI] [PubMed] [Google Scholar]
- 100.Sarafian T and Verity MA, Altered patterns of protein phosphorylation and synthesis caused by methyl mercury in cerebellar granule cell culture. J Neurochem, 1990. 55(3): p. 922–9. [DOI] [PubMed] [Google Scholar]
- 101.Sarafian T and Verity MA, Methyl mercury stimulates protein 32P phospholabeling in cerebellar granule cell culture. J Neurochem, 1990. 55(3): p. 913–21. [DOI] [PubMed] [Google Scholar]
- 102.Yoshida E, et al. , Methylmercury promotes prostacyclin release from cultured human brain microvascular endothelial cells via induction of cyclooxygenase-2 through activation of the EGFR-p38 MAPK pathway by inhibiting protein tyrosine phosphatase 1B activity. Toxicology, 2017. 392: p. 40–46. [DOI] [PubMed] [Google Scholar]
- 103.Heimfarth L, et al. , Delayed neurochemical effects of prenatal exposure to MeHg in the cerebellum of developing rats. Toxicol Lett, 2018. 284: p. 161–169. [DOI] [PubMed] [Google Scholar]
- 104.Fujimura M and Usuki F, Methylmercury causes neuronal cell death through the suppression of the TrkA pathway: in vitro and in vivo effects of TrkA pathway activators. Toxicol Appl Pharmacol, 2015. 282(3): p. 259–66. [DOI] [PubMed] [Google Scholar]
- 105.Parran DK, Barone S Jr., and Mundy WR, Methylmercury inhibits TrkA signaling through the ERK1/2 cascade after NGF stimulation of PC12 cells. Brain Res Dev Brain Res, 2004. 149(1): p. 53–61. [DOI] [PubMed] [Google Scholar]
- 106.Heimfarth L, et al. , Developmental neurotoxicity of the hippocampus following in utero exposure to methylmercury: impairment in cell signaling. Arch Toxicol, 2018. 92(1): p. 513–527. [DOI] [PubMed] [Google Scholar]
- 107.Yin Z, et al. , Methylmercury-induced alterations in astrocyte functions are attenuated by ebselen. Neurotoxicology, 2011. 32(3): p. 291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sakaue M, et al. , Acceleration of methylmercury-induced cell death of rat cerebellar neurons by brain-derived neurotrophic factor in vitro. Brain Res, 2009. 1273: p. 155–62. [DOI] [PubMed] [Google Scholar]
- 109.Lu TH, et al. , Involvement of oxidative stress-mediated ERK1/2 and p38 activation regulated mitochondria-dependent apoptotic signals in methylmercury-induced neuronal cell injury. Toxicol Lett, 2011. 204(1): p. 71–80. [DOI] [PubMed] [Google Scholar]
- 110.Fujimura M and Usuki F, Site-specific neural hyperactivity via the activation of MAPK and PKA/CREB pathways triggers neuronal degeneration in methylmercury-intoxicated mice. Toxicol Lett, 2017. 271: p. 66–73. [DOI] [PubMed] [Google Scholar]
- 111.Pierozan P, et al. , Neurotoxicity of Methylmercury in Isolated Astrocytes and Neurons: the Cytoskeleton as a Main Target. Mol Neurobiol, 2017. 54(8): p. 5752–5767. [DOI] [PubMed] [Google Scholar]
- 112.Fujimura M, et al. , Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain. Neurotoxicology, 2009. 30(6): p. 1000–7. [DOI] [PubMed] [Google Scholar]
- 113.Guida N, et al. , p38/Sp1/Sp4/HDAC4/BDNF Axis Is a Novel Molecular Pathway of the Neurotoxic Effect of the Methylmercury. Front Neurosci, 2017. 11: p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Posser T, et al. , Human neuroblastoma cells transfected with tyrosine hydroxylase gain increased resistance to methylmercury-induced cell death. Toxicol In Vitro, 2010. 24(6): p. 1498–503. [DOI] [PubMed] [Google Scholar]
- 115.Noguchi Y, et al. , Astrocytes protect neurons against methylmercury via ATP/P2Y(1) receptor-mediated pathways in astrocytes. PLoS One, 2013. 8(2): p. e57898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shinozaki Y, et al. , Microglia trigger astrocyte-mediated neuroprotection via purinergic gliotransmission. Sci Rep, 2014. 4: p. 4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saijoh K, et al. , Effects of methylmercury on protein kinase A and protein kinase C in the mouse brain. Environ Res, 1993. 63(2): p. 264–73. [DOI] [PubMed] [Google Scholar]
- 118.Haykal-Coates N, et al. , Effects of gestational methylmercury exposure on immunoreactivity of specific isoforms of PKC and enzyme activity during post-natal development of the rat brain. Brain Res Dev Brain Res, 1998. 109(1): p. 33–49. [DOI] [PubMed] [Google Scholar]
- 119.Cargnello M and Roux PP, Activation and function of the MAPKs and their substrates, the MAPK-activatedprotein kinases. Microbiol Mol Biol Rev, 2011. 75(1): p. 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim EK and Choi EJ, Compromised MAPK signaling in human diseases: an update. Arch Toxicol, 2015. 89(6): p. 867–82. [DOI] [PubMed] [Google Scholar]
- 121.Parran DK, Barone S, and Mundy WR, Methylmercury inhibits TrkA signaling through the ERK1/2 cascade after NGF stimulation of PC12 cells. Developmental Brain Research, 2004. 149(1): p. 53–61. [DOI] [PubMed] [Google Scholar]
- 122.Heimfarth L, et al. , Developmental neurotoxicity of the hippocampus following in utero exposure to methylmercury: impairment in cell signaling. Archives of Toxicology, 2018. 92(1): p. 513–527. [DOI] [PubMed] [Google Scholar]
- 123.Yin ZB, et al. , Methylmercury-induced alterations in astrocyte functions are attenuated by ebselen. Neurotoxicology, 2011. 32(3): p. 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sakaue M, et al. , Acceleration of methylmercury-induced cell death of rat cerebellar neurons by brain-derived neurotrophic factor in vitro. Brain Research, 2009. 1273: p. 155–162. [DOI] [PubMed] [Google Scholar]
- 125.Lu TH, et al. , Involvement of oxidative stress-mediated ERK1/2 and p38 activation regulated mitochondria-dependent apoptotic signals in methylmercury-induced neuronal cell injury. Toxicology Letters, 2011. 204(1): p. 71–80. [DOI] [PubMed] [Google Scholar]
- 126.Coffey ET, Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci, 2014. 15(5): p. 285–99. [DOI] [PubMed] [Google Scholar]
- 127.Kumar A, et al. , JNK pathway signaling: a novel and smarter therapeutic targets for various biological diseases. Future Med Chem, 2015. 7(15): p. 2065–86. [DOI] [PubMed] [Google Scholar]
- 128.Cordero-Herrera I, et al. , Molecular mechanisms involved in the protective effect of selenocystine against methylmercury-induced cell death in human HepG2 cells. Food and Chemical Toxicology, 2013. 59: p. 554–563. [DOI] [PubMed] [Google Scholar]
- 129.Takanezawa Y, et al. , Atg5-dependent autophagy plays a protective role against methylmercury-induced cytotoxicity. Toxicology Letters, 2016. 262: p. 135–141. [DOI] [PubMed] [Google Scholar]
- 130.Costa AP, et al. , Differential Activation of Mitogen-Activated Protein Kinases, ERK 1/2, p38(MAPK) and JNK p54/p46 During Postnatal Development of Rat Hippocampus. Neurochem Res, 2016. 41(5): p. 1160–9. [DOI] [PubMed] [Google Scholar]
- 131.Lee JK and Kim NJ, Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules, 2017. 22(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guida N, et al. , p38/Sp1/Sp4/HDAC4/BDNF Axis Is a Novel Molecular Pathway of the Neurotoxic Effect of the Methylmercury. Frontiers in Neuroscience, 2017. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Battaini F and Pascale A, Protein kinase C signal transduction regulation in physiological and pathological aging. Ann N Y Acad Sci, 2005. 1057: p. 177–92. [DOI] [PubMed] [Google Scholar]
- 134.Zhang Y, et al. , Protein kinase M zeta and the maintenance of long-term memory. Neurochem Int, 2016. 99: p. 215–220. [DOI] [PubMed] [Google Scholar]
- 135.Rajanna B, et al. , Modulation of protein kinase C by heavy metals. Toxicol Lett, 1995. 81(2–3): p. 197–203. [DOI] [PubMed] [Google Scholar]
- 136.Li L, et al. , The effects of Chinese medicines on cAMP/PKA signaling in central nervous system dysfunction. Brain Res Bull, 2017. 132: p. 109–117. [DOI] [PubMed] [Google Scholar]
- 137.Kelly MP, Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal, 2018. 42: p. 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bishop P, Rocca D, and Henley JM, Ubiquitin C-terminal hydrolase L1 (UCH-L1): structure, distribution and roles in brain function and dysfunction. Biochem J, 2016. 473(16): p. 2453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hwang GW, et al. , Deletion of the ubiquitin-conjugating enzyme Ubc2 confers resistance to methylmercury in budding yeast by promoting Whi2 degradation. J Toxicol Sci, 2013. 38(2): p. 301–3. [DOI] [PubMed] [Google Scholar]
- 140.Furuchi T, Hwang GW, and Naganuma A, Overexpression of the ubiquitin-conjugating enzyme Cdc34 confers resistance to methylmercury in Saccharomyces cerevisiae. Mol Pharmacol, 2002. 61(4): p. 738–41. [DOI] [PubMed] [Google Scholar]
- 141.Hwang GW, Sasaki D, and Naganuma A, Overexpression of Rad23 confers resistance to methylmercury in saccharomyces cerevisiae via inhibition of the degradation of ubiquitinatedproteins. Mol Pharmacol, 2005. 68(4): p. 1074–8. [DOI] [PubMed] [Google Scholar]
- 142.Hwang GW, Ishida Y, and Naganuma A, Identification of F-box proteins that are involved in resistance to methylmercury in Saccharomyces cerevisiae. Febs Letters, 2006. 580(30): p. 6813–6818. [DOI] [PubMed] [Google Scholar]
- 143.Hwang GW, et al. , Overexpression of the novel F-box protein Ymr258c confers resistance to methylmercury in Saccharomyces cerevisiae. Journal of Toxicological Sciences, 2009. 34(4): p. 413–416. [DOI] [PubMed] [Google Scholar]
- 144.Toyama T, et al. , S-Mercuration of ubiquitin carboxyl-terminal hydrolase L1 through Cys152 by methylmercury causes inhibition of its catalytic activity and reduction of monoubiquitin levels in SH-SY5Y cells. J Toxicol Sci, 2015. 40(6): p. 887–93. [DOI] [PubMed] [Google Scholar]
- 145.Shao Y, et al. , Methylmercury can induce Parkinson’s-like neurotoxicity similar to 1-methyl-4- phenylpyridinium: a genomic and proteomic analysis on MN9D dopaminergic neuron cells. J Toxicol Sci, 2015. 40(6): p. 817–28. [DOI] [PubMed] [Google Scholar]
- 146.Landrigan PJ, et al. , Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect, 2005. 113(9): p. 1230–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Petersen MS, et al. , Impact of dietary exposure to food contaminants on the risk of Parkinson’s disease. Neurotoxicology, 2008. 29(4): p. 584–90. [DOI] [PubMed] [Google Scholar]
- 148.Ardley HC and Robinson PA, The role of ubiquitin-protein ligases in neurodegenerative disease. Neurodegener Dis, 2004. 1(2–3): p. 71–87. [DOI] [PubMed] [Google Scholar]
- 149.Onishchenko N, et al. , Developmental exposure to methylmercury alters learning and induces depression-like behavior in male mice. Toxicol Sci, 2007. 97(2): p. 428–37. [DOI] [PubMed] [Google Scholar]
- 150.Onishchenko N, et al. , Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J Neurochem, 2008. 106(3): p. 1378–87. [DOI] [PubMed] [Google Scholar]
- 151.Karpova NN, et al. , TrkB overexpression in mice buffers against memory deficits and depression-like behavior but not all anxiety- and stress-related symptoms induced by developmental exposure to methylmercury. Front Behav Neurosci, 2014. 8: p. 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hasin DS, et al. , Epidemiology of Adult DSM-5 Major Depressive Disorder and Its Specifiers in the United States. JAMA Psychiatry, 2018. 75(4): p. 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wagner GC, et al. , Behavioral and neurochemical sensitization to amphetamine following early postnatal administration of methylmercury (MeHg). Neurotoxicology, 2007. 28(1): p. 59–66. [DOI] [PubMed] [Google Scholar]
- 154.Lee Y, et al. , Efficacy of antidepressants on measures of workplace functioning in major depressive disorder: A systematic review. Journal of Affective Disorders, 2018. 227: p. 406–415. [DOI] [PubMed] [Google Scholar]
- 155.Basu N, Goodrich JM, and Head J, Ecogenetics of Mercury: From Genetic Polymorphisms and Epigenetics to Risk Assessment and Decision-Making. Environmental Toxicology and Chemistry, 2014. 33(6): p. 1248–1258. [DOI] [PubMed] [Google Scholar]
- 156.Cardenas A, et al. , Persistent DNA methylation changes associated with prenatal mercury exposure and cognitive performance during childhood. Scientific Reports, 2017. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Carvan MJ 3rd, et al. , Mercury-induced epigenetic transgenerational inheritance of abnormal neurobehavior is correlated with sperm epimutations in zebrafish. PLoS One, 2017. 12(5): p. e0176155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wadhwa PD, et al. , Developmental Origins of Health and Disease: Brief History of the Approach and Current Focus on Epigenetic Mechanisms. Seminars in Reproductive Medicine, 2009. 27(5): p. 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dodson M, Darley-Usmar V, and Zhang J, Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med, 2013. 63: p. 207–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wende AR, et al. , Redox biology and the interface between bioenergetics, autophagy and circadian control of metabolism. Free Radic Biol Med, 2016. 100: p. 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kramer PA, et al. , The Measurement of Reversible Redox Dependent Post-translational Modifications and Their Regulation of Mitochondrial and Skeletal Muscle Function. Front Physiol, 2015. 6: p. 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Levonen AL, et al. , Redox regulation of antioxidants, autophagy, and the response to stress: implications for electrophile therapeutics. Free Radic Biol Med, 2014. 71: p. 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Halliwell B, Free radicals and antioxidants: updating a personal view. Nutr Rev, 2012. 70(5): p. 257–65. [DOI] [PubMed] [Google Scholar]
- 164.Kalyanaraman B, Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol, 2013. 1: p. 244–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sies H, Oxidative stress: a concept in redox biology and medicine. Redox Biol, 2015. 4: p. 180–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stohs SJ and Bagchi D, Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med, 1995. 18(2): p. 321–36. [DOI] [PubMed] [Google Scholar]
- 167.Branco V, et al. , Impaired cross-talk between the thioredoxin and glutathione systems is related to ASK-1 mediated apoptosis in neuronal cells exposed to mercury. Redox Biology, 2017. 13: p. 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Berntssen MH, Aatland A, and Handy RD, Chronic dietary mercury exposure causes oxidative stress, brain lesions, and altered behaviour in Atlantic salmon (Salmo salar) parr. Aquat Toxicol, 2003. 65(1): p. 55–72. [DOI] [PubMed] [Google Scholar]
- 169.Amara IE, Anwar-Mohamed A, and El-Kadi AO, Posttranslational mechanisms modulating the expression of the cytochrome P450 1A1 gene by methylmercury in HepG2 cells: a role of heme oxygenase-1. Toxicol Lett, 2013. 219(3): p. 239–47. [DOI] [PubMed] [Google Scholar]
- 170.Petroni D, et al. , Low-dose methylmercury-induced oxidative stress, cytotoxicity, and tau-hyperphosphorylation in human neuroblastoma (SH-SY5Y) cells. Environ Toxicol, 2012. 27(9): p. 549–55. [DOI] [PubMed] [Google Scholar]
- 171.Sakaue M, Okazaki M, and Hara S, Very low levels of methylmercury induce cell death of cultured rat cerebellar neurons via calpain activation. Toxicology, 2005. 213(1–2): p. 97–106. [DOI] [PubMed] [Google Scholar]
- 172.Bridges KN, et al. , Alterations to the Intestinal Microbiome and Metabolome of Pimephales promelas and Mus musculus Following Exposure to Dietary Methylmercury. Environmental Science & Technology, 2018. 52(15): p. 8774–8784. [DOI] [PubMed] [Google Scholar]
- 173.Li Y, et al. , Methylmercury exposure alters RNA splicing in human neuroblastoma SK-N-SH cells: Implications from proteomic and post-transcriptional responses. Environ Pollut, 2018. 238: p. 213–221. [DOI] [PubMed] [Google Scholar]
- 174.Rudgalvyte M, et al. , Methylmercury exposure increases lipocalin related (lpr) and decreases activated in blocked unfolded protein response (abu) genes and specific miRNAs in Caenorhabditis elegans. Toxicol Lett, 2013. 222(2): p. 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shao Y, et al. , Proteome profiling reveals regional protein alteration in cerebrum of common marmoset (Callithrix jacchus) exposed to methylmercury. Toxicology, 2016. 347–349: p. 29–39. [DOI] [PubMed] [Google Scholar]
- 176.Bittencourt LO, et al. , Oxidative Biochemistry Disbalance and Changes on Proteomic Profile in Salivary Glands of Rats Induced by Chronic Exposure to Methylmercury. Oxid Med Cell Longev, 2017. 2017: p. 5653291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kim SC, et al. , Substrate and functional diversity of lysine acetylation revealed by a proteomicssurvey. Mol Cell, 2006. 23(4): p. 607–18. [DOI] [PubMed] [Google Scholar]
- 178.Stram AR and Payne RM, Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol Life Sci, 2016. 73(21): p. 4063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Garg TK and Chang JY, Methylmercury causes oxidative stress and cytotoxicity in microglia: attenuation by 15-deoxy-delta 12, 14-prostaglandin J2. J Neuroimmunol, 2006. 171(1–2): p. 17–28. [DOI] [PubMed] [Google Scholar]
- 180.Yang JL, et al. , Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst), 2008. 7(7): p. 1110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tsushima K, et al. , Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ Res, 2018. 122(1): p. 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Qu M, et al. , Protective effects of lycopene against methylmercury-induced neurotoxicity in cultured rat cerebellar granule neurons. Brain Res, 2013. 1540: p. 92–102. [DOI] [PubMed] [Google Scholar]
- 183.Prince LM and Rand MD, Methylmercury exposure causes a persistent inhibition of myogenin expression and C2C12 myoblast differentiation. Toxicology, 2018. 393: p. 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang X, et al. , Low-Dose Methylmercury-Induced Apoptosis and Mitochondrial DNA Mutation in Human Embryonic Neural Progenitor Cells. Oxid Med Cell Longev, 2016. 2016: p. 5137042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yakes FM and Van Houten B, Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A, 1997. 94(2): p. 514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Quiros PM, Mottis A, and Auwerx J, Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol, 2016. 17(4): p. 213–26. [DOI] [PubMed] [Google Scholar]
- 187.Miyamoto K, et al. , Involvement of enhanced sensitivity of N-methyl-D-aspartate receptors in vulnerability of developing cortical neurons to methylmercury neurotoxicity. Brain Research, 2001. 901(1–2): p. 252–258. [DOI] [PubMed] [Google Scholar]
- 188.Makino K, et al. , Correlation Between Attenuation of Protein Disulfide Isomerase Activity Through S-Mercuration and Neurotoxicity Induced by Methylmercury. Neurotoxicity Research, 2015. 27(2): p. 99–105. [DOI] [PubMed] [Google Scholar]
- 189.Toyama T, et al. , Cytoprotective role of Nrf2/Keap1 system in methylmercury toxicity. Biochem Biophys Res Commun, 2007. 363(3): p. 645–50. [DOI] [PubMed] [Google Scholar]
- 190.Ni M, et al. , Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia, 2011. 59(5): p. 810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ni M, et al. , Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicol Sci, 2010. 116(2): p. 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Toyama T, et al. , Isothiocyanates reduce mercury accumulation via an Nrf2-dependent mechanism during exposure of mice to methylmercury. Environ Health Perspect, 2011. 119(8): p. 1117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rand MD, Dao JC, and Clason TA, Methylmercury disruption of embryonic neural development in Drosophila. Neurotoxicology, 2009. 30(5): p. 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Martinez-Finley EJ, et al. , The Role of skn-1 in methylmercury-induced latent dopaminergic neurodegeneration. Neurochem Res, 2013. 38(12): p. 2650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Vanduyn N, et al. , SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci, 2010. 118(2): p. 613–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Culbreth M, Zhang Z, and Aschner M, Methylmercury augments Nrf2 activity by downregulation of the Src family kinase Fyn. Neurotoxicology, 2017. 62: p. 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Fujimura M and Usuki F, Low concentrations of methylmercury inhibit neural progenitor cell proliferation associated with up-regulation of glycogen synthase kinase 3beta and subsequent degradation of cyclin E in rats. Toxicol Appl Pharmacol, 2015. 288(1): p. 19–25. [DOI] [PubMed] [Google Scholar]
- 198.Salazar M, et al. , Glycogen synthase kinase-3 beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. Journal of Biological Chemistry, 2006. 281(21): p. 14841–14851. [DOI] [PubMed] [Google Scholar]
- 199.Hayes JD and Dinkova-Kostova AT, The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci, 2014. 39(4): p. 199–218. [DOI] [PubMed] [Google Scholar]
- 200.He X and Ma Q, NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol Pharmacol, 2009. 76(6): p. 1265–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhang DD and Hannink M, Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol, 2003. 23(22): p. 8137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kumagai Y, et al. , The role of the Keap1/Nrf2 pathway in the cellular response to methylmercury. Oxid Med Cell Longev, 2013. 2013: p. 848279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yoshida E, Abiko Y, and Kumagai Y, Glutathione adduct of methylmercury activates the Keap1-Nrf2 pathway in SH-SY5Y cells. Chem Res Toxicol, 2014. 27(10): p. 1780–6. [DOI] [PubMed] [Google Scholar]
- 204.Ou YC, et al. , Induction of the cell cycle regulatory gene p21 (Waf1, Cip1) following methylmercury exposure in vitro and in vivo. Toxicol Appl Pharmacol, 1999. 157(3): p. 203–12. [DOI] [PubMed] [Google Scholar]
- 205.Bose R, et al. , Inherited effects of low-dose exposure to methylmercury in neural stem cells. Toxicol Sci, 2012. 130(2): p. 383–90. [DOI] [PubMed] [Google Scholar]
- 206.Chen WM, et al. , Direct Interaction between Nrf2 and p21(Cip1/WAF1) Upregulates the Nrf2-Mediated Antioxidant Response. Molecular Cell, 2009. 34(6): p. 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Komatsu M, et al. , The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keapl. Nat Cell Biol, 2010. 12(3): p. 213–23. [DOI] [PubMed] [Google Scholar]
- 208.Takanezawa Y, et al. , Sequestosome1/p62 protects mouse embryonic fibroblasts against low-dose methylercury-induced cytotoxicity and is involved in clearance of ubiquitinatedproteins. Sci Rep, 2017. 7(1): p. 16735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yuntao F, et al. , Role of autophagy in methylmercury-induced neurotoxicity in rat primary astrocytes. Arch Toxicol, 2016. 90(2): p. 333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Takanezawa Y, et al. , Atg5-dependent autophagy plays a protective role against methylmercury-induced cytotoxicity. Toxicol Lett, 2016. 262: p. 135–141. [DOI] [PubMed] [Google Scholar]
- 211.Toyama T, et al. , Carbon monoxide derived from heme oxygenase-2 mediates reduction of methylmercury toxicity in SH-SY5Y cells. Toxicol Appl Pharmacol, 2010. 249(1): p. 86–90. [DOI] [PubMed] [Google Scholar]
- 212.Pi JB, et al. , Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radical Biology and Medicine, 2007. 42(12): p. 1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Franco JL, et al. , Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: protective effects of quercetin. Chem Res Toxicol, 2007. 20(12): p. 1919–26. [DOI] [PubMed] [Google Scholar]
- 214.Shanker G, et al. , Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Brain Res Mol Brain Res, 2005. 137(1–2): p. 11–22. [DOI] [PubMed] [Google Scholar]
- 215.Janaky R, et al. , Glutathione and signal transduction in the mammalian CNS. J Neurochem, 1999. 73(3): p. 889–902. [DOI] [PubMed] [Google Scholar]
- 216.Farina M, Aschner M, and Rocha JBT, Special Issue: Environmental Chemicals and Neurotoxicity Oxidative stress in MeHg-induced neurotoxicity. Toxicology and applied pharmacology, 2011. 256(3): p. 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wahlberg K, et al. , Maternal polymorphisms in glutathione-related genes are associated with maternal mercury concentrations and early child neurodevelopment in a population with a fish-rich diet. Environ Int, 2018. 115: p. 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Diaz D, et al. , Effect of methylmercury on glutamate-cysteine ligase expression in the placenta and yolk sac during mouse development. Reprod Toxicol, 2004. 19(1): p. 117–29. [DOI] [PubMed] [Google Scholar]
- 219.Franco JL, et al. , Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radical Biology and Medicine, 2009. 47(4): p. 449–457. [DOI] [PubMed] [Google Scholar]
- 220.Branco V, et al. , Mercury and selenium interaction in vivo: effects on thioredoxin reductase and glutathione peroxidase. Free Radic Biol Med, 2012. 52(4): p. 781–93. [DOI] [PubMed] [Google Scholar]
- 221.Funchal C, et al. , Diphenyl ditelluride- and methylmercury-induced hyperphosphorilation of the high molecular weight neurofilament subunit is prevented by organoselenium compounds in cerebral cortex of young rats. Toxicology, 2006. 222(1–2): p. 143–53. [DOI] [PubMed] [Google Scholar]
- 222.Holmgren A, Thioredoxin and glutaredoxin systems. J Biol Chem, 1989. 264(24): p. 13963–6. [PubMed] [Google Scholar]
- 223.Meinerz DF, et al. , Diphenyl diselenide protects against methylmercury-induced inhibition of thioredoxin reductase and glutathione peroxidase in human neuroblastoma cells: a comparison with ebselen. J Appl Toxicol, 2017. 37(9): p. 1073–1081. [DOI] [PubMed] [Google Scholar]
- 224.Wagner C, et al. , In vivo and in vitro inhibition of mice thioredoxin reductase by methylmercury. Biometals, 2010. 23(6): p. 1171–7. [DOI] [PubMed] [Google Scholar]
- 225.Branco V, et al. , Inhibition of the thioredoxin system in the brain and liver of zebra-seabreams exposed to waterborne methylmercury. Toxicol Appl Pharmacol, 2011. 251(2): p. 95–103. [DOI] [PubMed] [Google Scholar]
- 226.Carvalho CM, et al. , Inhibition of the human thioredoxin system. A molecular mechanism of mercury toxicity. J Biol Chem, 2008. 283(18): p. 11913–23. [DOI] [PubMed] [Google Scholar]
- 227.Unoki T, et al. , Methylmercury, an environmental electrophile capable of activation and disruption of the Akt/CREB/Bcl-2 signal transduction pathway in SH-SY5Y cells. Scientific Reports, 2016. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Farina M, et al. , Probucol Increases Glutathione Peroxidase-1 Activity and Displays Long-Lasting Protection against Methylmercury Toxicity in Cerebellar Granule Cells. Toxicological Sciences, 2009. 112(2): p. 416–426. [DOI] [PubMed] [Google Scholar]
- 229.Riquier S, et al. , Peroxiredoxin post-translational modifications by redox messengers. Redox Biology, 2014. 2: p. 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Marinho HS, et al. , Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biology, 2014. 2: p. 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yamashita T, et al. , Role of nitric oxide in the cerebellar degeneration during methylmercury intoxication. Biochimica Et Biophysica Acta-General Subjects, 1997. 1334(2–3): p. 303–311. [DOI] [PubMed] [Google Scholar]
- 232.Herculano AM, et al. , Methylmercury intoxication activates nitric oxide synthase in chick retinal cell culture. Brazilian Journal of Medical and Biological Research, 2006. 39(3): p. 415–418. [DOI] [PubMed] [Google Scholar]
- 233.Robitaille S, Mailloux RJ, and Chan HM, Methylmercury alters glutathione homeostasis by inhibiting glutaredoxin 1 and enhancing glutathione biosynthesis in cultured human astrocytoma cells. Toxicology Letters, 2016. 256: p. 1–10. [DOI] [PubMed] [Google Scholar]
- 234.Evangelista AM, Kohr MJ, and Murphy E, S-Nitrosylation: Specificity, Occupancy, and Interaction with Other Post-Translational Modifications. Antioxidants & Redox Signaling, 2013. 19(11): p. 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ghezzi P, Protein glutathionylation in health and disease. Biochimica Et Biophysica Acta-General Subjects, 2013. 1830(5): p. 3165–3172. [DOI] [PubMed] [Google Scholar]