

Abstract
Pseudouridine is the most prevalent RNA modification and has been found in both eukaryotes and prokaryotes. Currently, pseudouridine has been demonstrated in several kinds of RNAs, such as small nuclear RNA, rRNA, tRNA, mRNA, and small nucleolar RNA. Therefore, its significance to academic research and drug development is understandable. Through biochemical experiments, the pseudouridine site identification has produced good outcomes, but these lab exploratory methods and biochemical processes are expensive and time consuming. Therefore, it is important to introduce efficient methods for identification of pseudouridine sites. In this study, an intelligent method for pseudouridine sites using the deep-learning approach was developed. The proposed prediction model is called iPseU-CNN (identifying pseudouridine by convolutional neural networks). The existing methods used handcrafted features and machine-learning approaches to identify pseudouridine sites. However, the proposed predictor extracts the features of the pseudouridine sites automatically using a convolution neural network model. The iPseU-CNN model yields better outcomes than the current state-of-the-art models in all evaluation parameters. It is thus highly projected that the iPseU-CNN predictor will become a helpful tool for academic research on pseudouridine site prediction of RNA, as well as in drug discovery.
Keywords: convolution neural network, deep learning, iPseU-CNN, pseudouridine sites, RNA
Graphical Abstract
Introduction
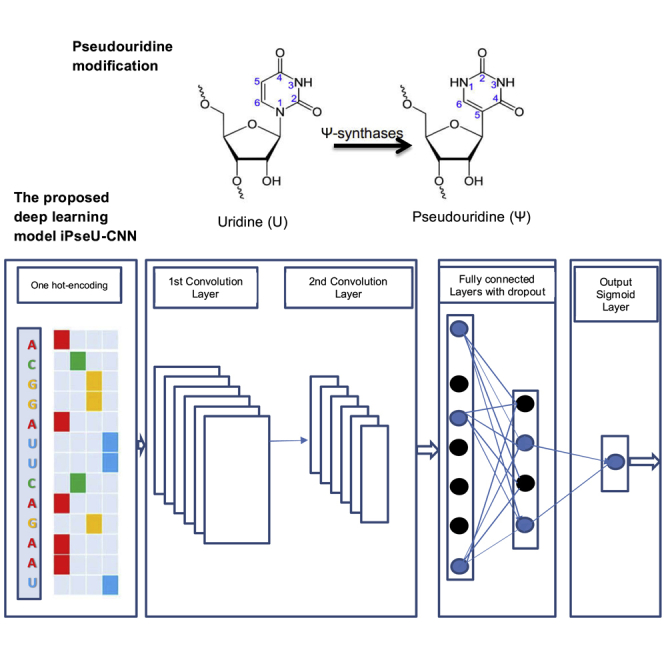
Pseudouridine (Ψ) is a common RNA modification that has been found in both eukaryotes and prokaryotes.1 Currently, Ψ has been demonstrated in various categories of RNAs.2 The Ψ synthase enzyme catalyzes Ψ, the isomer of uridine, by removing uridine residue base from its sugar followed by the isomer of uridine, rotating it 180° along the N3–C6 axis, and ultimately, again linking the base’s 5-carbon to the 1′-carbon of the sugar, as shown in Figure 1.3 Currently, Ψ modification is considered to be an important process in the molecular mechanism, including stabilization of the tRNA structure,3, 4 and is important for gene regulation machinery, i.e., in the spliceosome. The presence of Ψ modifications in regions involved with RNA-protein or RNA-RNA interaction enhances the reaction and assembly of the spliceosome that is responsible for producing a functional mRNA, i.e., in AU/AC intron splicing.5 Furthermore, incorporation of Ψ into mRNA may inhibit the RNA-elicited innate immune response and enhance the translation efficiency of that mRNA.6 Although many researchers have unveiled the role of Ψ modification in most RNA systems, its biological functions and action mechanisms have yet to be identified. Therefore, it is important to highlight the Ψ modification sites in the transcriptome that govern the related biological principle.
Figure 1.

Illustration of the Pseudouridine Modification
Although some lab exploratory techniques have been introduced to identify Ψ sites, they are costly and labor intensive.7, 8, 9 Because of the increasing availability of genomics and proteomics samples produced in the post-genomics era, it is necessary to develop robust, fast, low-cost computational models to predict Ψ sites on the RNA sequence. In previous works, several machine-learning-based computational methods or statistical-learning techniques have been introduced to identify Ψ sites.10, 11, 12 Li et al.13 introduced a computational method, PPUS, for the identification of Ψ-synthase (PUS)-specific Ψ sites in Saccharomyces cerevisiae and Homo sapiens. The method used the support vector machine (SVM) for classification and nucleotides around Ψ as the features. Similarly, the identifying RNA Ψ (iRNA-PseU) method was introduced by Chen et al.,14 for the identification of Ψ sites in Mus musculus, S. cerevisiae, and H. sapiens. This method combines the occurrence frequency density distributions of the nucleotides and their chemical properties into pseudo K-tuple nucleotide composition (PseKNC). Most recently, the Ψ identification (PseUI) model was developed by He et al.15 for identification of Ψ sites from RNA samples in M. musculus, S. cerevisiae, and H. sapiens. This model used five types of feature-extraction technique, including dinucleotide composition (DC), nucleotide composition (NC), position-specific dinucleotide propensity (PSDP), position-specific nucleotide propensity (PSNP), and pseudodinucleotide composition (PseDNC). Then, a sequential forward-feature-selection strategy was used to select a relevant feature combination and a support vector machine as a classifier.16, 17
More recently, PseKNC has been effectively and widely used in the predation of several RNA/DNA regulatory elements, such as the nucleosome-positioning sequence,18, 19 RNA modification sites,20, 21, 22 DNA recombination spots,23, 24 translation initiation site,25 promoter,26 and origin of replication.27, 28 Although the above studies have illustrated that PseKNC is one of the most often used feature-extraction techniques to formulate RNA/DNA sequences, all of them used type-I PseKNC, which mixes various physicochemical properties. Because various properties may play various roles, the type-II PseKNC could handle these variances and improve the description of sequences. Recently, type II PseKNC was used in various DNA element identification and achieved good results.29, 30 On the other hand, the main focus of our work was use of a deep-learning technique for automatically extracting the important features directly from the sequence itself for classification.
The performance of the above predictors and methods can be further improved by proposing other robust machine-learning or deep-learning methods. The existing methods use hand-designed input features based on domain knowledge. However, the proposed system can automatically learn the features from RNA sequences by using a deep-learning technique. Deep learning has produced better outcomes in natural language processing,31 information retrieval,32 speech recognition,33 and image recognition.34, 35, 36 Recently, a large number of genomics methods and techniques have been introduced based on deep-learning mechanisms—for example, CNNclust,37 BiRen,38 iDeepS,39 RNA branch point prediction,40 alternative splicing site prediction,41 and iRNA-PseKNC(2methyl).42
We introduce an efficient computational architecture for prediction of Ψ sites, using machine-learning and deep-learning approaches. In machine learning, two simple feature-extraction techniques were used as baselines—n-gram and multivariate mutual information (MMI)—and SVM was used as the classifier. In deep learning, we used a convolution neural network (CNN) model. As shown in the result and discussion sections, the deep-learning method produced better outcomes than the machine-learning ones. The proposed prediction iPseU-CNN (identifying Ψ by convolutional neural networks) model is based on a CNN. It is an efficient and simple architecture for Ψ site prediction and is evaluated on three various training benchmark datasets and two independent testing benchmark datasets. The proposed model achieves a more efficient outcome than the current state-of-the-art methods published recently in the literature. To the best of our knowledge, the proposed iPseU-CNN prediction model is the first model, automatically capture important features from RNA sequences using CNN for identification of Ψ sites.
Results and Discussion
In recent studies, four statistical parameters, Matthews’s correlation coefficient (MCC), sensitivity (Sen), specificity (Sp), and accuracy (Acc), have been used to define the effectiveness and performance of the computational methods.43, 44, 45, 46, 47 These parameters are expressed as:
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
In this work, we implemented two simple machine-learning baselines. These methods are based on using n-gram and MMI for feature extraction and SVM as a classifier. The n-gram and MMI feature-extraction techniques are simple and are used widely in many applications. Table 1 shows the success rate of n-gram, MMI, and the proposed iPseU-CNN. It can be seen that the n-gram-based method outperformed the MMI-based one in the H. sapiens (H)_990, S. cerevisiae (S)_628, and M. musculus (M)_944 datasets. However, the CNN-based method markedly outperformed both machine-learning-based techniques. More specifically, iPseU-CNN improved accuracy by 6.68%, sensitivity by 13.49%, and MCC by 0.14 in the H_990 dataset. On the other hand, iPseU-CNN improved the performance of the S_628 dataset by 5.42%, 9.63%, and 0.12 in terms of accuracy, specificity, and MCC, respectively. Furthermore, iPseU-CNN improved the performance of the M_944 dataset by 9.1%, 9.75%, 8.73%, and 0.19 in terms of accuracy, sensitivity, specificity, and MCC, respectively. Thus, it is clear that the proposed iPseU-CNN predictor outperforms the baseline machine-learning methods.
Table 1.
The Success Rates of iPseU-CNN and the Baseline Methods with the Training Datasets
| Training Dataset | Methods | Accuracy (%) | Sensitivity (%) | Specificity (%) | MCC |
|---|---|---|---|---|---|
| H_990 | n-gram | 60.00 | 51.51 | 68.48 | 0.20 |
| MMI | 58.78 | 47.47 | 70.10 | 0.18 | |
| CNN | 66.68 | 65.00 | 68.78 | 0.34 | |
| S_628 | n-gram | 62.73 | 64.64 | 60.82 | 0.25 |
| MMI | 60.19 | 67.51 | 52.86 | 0.20 | |
| CNN | 68.15 | 66.36 | 70.45 | 0.37 | |
| M_944 | n-gram | 62.71 | 65.04 | 60.38 | 0.25 |
| MMI | 58.26 | 63.13 | 53.38 | 0.16 | |
| CNN | 71.81 | 74.79 | 69.11 | 0.44 |
The prediction outcomes of the iPseU-CNN model were measured on two independent datasets, i.e., S_200 and H_200, and are illustrated in Table 2. We showed experimentally that the success rate of our iPseU-CNN model based on deep learning was better than that of the machine-learning baseline methods. More specifically, iPseU-CNN method improved the accuracy, sensitivity, and MCC on H_200 dataset by 2%, 19.72%, and 0.05, respectively. On the other hand, the success rates of the S_200 dataset were improved by 3%, 6.82%, and 0.06 in terms of accuracy, specificity, and MCC, respectively.
Table 2.
The Success Rates of iPseU-CNN and the Baseline Methods with Two Independent Testing Datasets
| Testing Dataset | Methods | Accuracy (%) | Sensitivity (%) | Specificity (%) | MCC |
|---|---|---|---|---|---|
| H_200 | n-gram | 67.00 | 57.00 | 78.00 | 0.35 |
| MMI | 63.50 | 58.00 | 69.00 | 0.27 | |
| CNN | 69.00 | 77.72 | 60.81 | 0.40 | |
| S_200 | n-gram | 70.50 | 70.00 | 71.00 | 0.41 |
| MMI | 69.50 | 72.00 | 67.00 | 0.39 | |
| CNN | 73.50 | 68.76 | 77.82 | 0.47 |
It is clear that that the CNN-based approach outperforms the machine-learning-based approaches with a big margin in the different evaluation metrics as shown in Tables 1 and 2 and Figure 2.
Figure 2.

The Success Rates of the iPseU-CNN and Baseline Methods
Finally, the prediction performance comparison of the iPseU-CNN model with the existing methods, such as iRNA-PseU14 and PseUI,15 is shown in Table 3. iRNA-PseU14 combines the occurrence frequency density distributions of the nucleotides and their chemical properties into PseKNC for feature extraction to identify Ψ sites. PseUI15 uses five feature-extraction techniques to identify Ψ sites.
Table 3.
The Success Rates of iPseU-CNN and State-of-the-Art Methods with the Training Datasets
| Training Dataset | Models | Accuracy (%) | Sensitivity (%) | Specificity (%) | MCC |
|---|---|---|---|---|---|
| H_990 | iPseU-CNN | 66.68 | 65.00 | 68.78 | 0.34 |
| PseUI | 64.24 | 64.85 | 63.64 | 0.28 | |
| iRNA-PseU | 60.40 | 61.01 | 59.80 | 0.21 | |
| S_628 | iPseU-CNN | 68.15 | 66.36 | 70.45 | 0.37 |
| PseUI | 65.13 | 62.74 | 67.52 | 0.30 | |
| iRNA-PseU | 64.49 | 64.65 | 64.33 | 0.29 | |
| M_944 | iPseU-CNN | 71.81 | 74.79 | 69.11 | 0.44 |
| PseUI | 70.44 | 79.87 | 70.34 | 0.41 | |
| iRNA-PseU | 69.07 | 73.31 | 64.83 | 0.38 |
The results in Table 3 show that the iPseU-CNN model improved all evaluation metrics for the H_990 dataset by 2.44%, 0.15%, 5.14%, and 0.06 in terms of accuracy, sensitivity, specificity, and MCC, respectively.
In addition, iPseU-CNN improved all evaluation metrics for the S_628 dataset by 1.71%, 3.02%, 2.93%, and 0.07 in terms of specificity, sensitivity, accuracy, and MCC, respectively, and it improved accuracy and MCC for the M_944 dataset by 1.37% and 0.03, respectively.
Furthermore, the performance of iPseU-CNN on independent datasets has been compared with those of iRNA-Pse and PseUI, as given in Table 4. It can be observed that the iPseU-CNN model improved all evaluation metrics for the S_200 dataset by 5.82%, 3.76%, 5%, and 0.1 in terms of specificity, sensitivity, accuracy, and MCC, respectively, and it improved accuracy, sensitivity and MCC for the H_200 dataset by 3.5%, 14.72%, and 0.09, respectively.
Table 4.
The Success Rates of the iPseU-CNN and State-of-the-Art Methods with Two Independent Testing Datasets
| Testing Dataset | Models | Accuracy (%) | Sensitivity (%) | Specificity (%) | MCC |
|---|---|---|---|---|---|
| H_200 | iPseU-CNN | 69.00 | 77.72 | 60.81 | 0.40 |
| PseUI | 65.50 | 63.00 | 68.00 | 0.31 | |
| RNA-PseU | 61.50 | 58.00 | 65.00 | 0.23 | |
| S_200 | iPseU-CNN | 73.50 | 68.76 | 77.82 | 0.47 |
| PseUI | 68.50 | 65.00 | 72.00 | 0.37 | |
| iRNA-PseU | 60.00 | 63.00 | 57.00 | 0.20 |
It is clear that the CNN-based approach outperforms the current predictors in different evaluation metrics, as displayed in Tables 3 and 4 and Figure 3.
Figure 3.

The Success Rates of the iPseU-CNN and State-of-the-Art Methods
Recently, the main direction of bioinformatics applications is in preparing databases48, 49 and establishing efficient web servers.22, 50 Therefore, our future work is to improve the performance and build a user-friendly web server for our developed tools.
To conclude, we developed a deep-learning mechanism to identify Ψ sites from RNA samples—namely, iPseU-CNN. Machine-learning and deep-learning mechanisms were used; however, the performance of the deep-learning approach outperformed the machine-learning ones. We applied n-gram and MMI to extract the features in the machine-learning approach and SVM for classification. The deep-learning approach used a CNN model. The iPseU-CNN model automatically learned the features from RNA sequences compared with previous works that employ handcrafted features for classification. The proposed iPseU-CNN prediction model is the first model to full automatically capture important feature from RNA sequences using CNNs for identification of Ψ sites. The success rate indicates that the proposed prediction model is more stable and accurate than the current methods in terms of evaluation parameters. It is highly expected that the iPseU-CNN prediction model may be helpful in drug-related applications and academia.
Materials and Methods
We introduce the proposed model and benchmark datasets used for training and testing.
The Proposed Model
We introduce an efficient computational architecture for prediction of Ψ sites using machine-learning and deep-learning approaches.
In machine-learning approaches, we used two different feature spaces, MMI and n-gram,51, 52 to extract the numerical features from RNA samples and SVM as an operation engine. Second, a deep-learning approach uses CNNs to identify Ψ sites from RNA/DNA samples directly. The CNN model automatically captures the key features from the input samples during training.
Machine-Learning Approach
We selected simple feature-extraction methods to work as baselines for comparison with the proposed deep-learning method.
n-gram
In this feature-extraction technique, n-gram is expressed as (vi, ci), where vi represents the feature and ci represents the total number of this feature in the protein or DNA/RNA sample.53 For instance, in the case of 3-g, v represents the three-nucleotide combination set and c represents the total number of combination occurrences inside the complete sequence. In this work, we constructed a feature vector containing from 1-g to 3-g. The n-gram can be mathematically expressed as:
| (Equation 5) |
where S represents the combination list of nucleotides, S1, S2, and S3, with the 41, 42, and 43 features, respectively, and generates an 84-dimensional vector.
MMI
In prior work,54, 55, 56, 57 MMI has been widely adopted in protein samples to extract features. In the same manner, the nucleotide samples in RNA/DNA can be represented using the MMI feature-extraction technique. In this method, the RNA/DNA samples are represented by 2-tuple and 3-tuple as follows:
| (Equation 6) |
There is no relationship with the order of the nucleotides for the MMI in a tuple. The K2 has 10 elements and K3 has 20 elements.
The 2-tuple mutual information (MI) for the nucleotide pair in K2 can be defined as below:
| (Equation 7) |
The 3-tuple MI for the nucleotide pair in K3 can be defined as below:
| (Equation 8) |
where is a fraction of each nucleotide in the sequence and and are the occurrence frequency of 2-tuple and 3-tuple, respectively.
SVM
SVM is a learning tool for regression, classification, and pattern recognition. It has achieved more efficient results than other machine-learning methods or techniques.47, 58, 59, 60 In the current study, the LIBSVM package was used for implementing the SVM model, in which the radial basis function (RBF) was used as the kernel function. The kernel of RBF includes two parameters, g and c, that are set to 5.5 and 0.0035, respectively. The concrete values of these parameters are determined through the optimization procedure called a grid-search algorithm on the benchmark dataset.61, 62, 63, 64, 65
Deep-Learning Approach
We used a CNN to predict Ψ sites from RNA/DNA samples, and during training, it automatically searched the key features in the input samples. The CNN model took a single RNA sequence as an input (n = 21 for the M_944 and H_900 datasets and n = 31 for the S_628 dataset) and produced a real value. The input is represented by a one-hot vector with four channels A, C, G, and U. Its length depends on the value of n. For more details, A is denoted by (1 0 0 0), C is denoted by (0 1 0 0), G is denoted by (0 0 1 0), and U is denoted by (0 0 0 1). Figure 4 illustrates the architecture of the proposed CNN model.
Figure 4.

Illustration of the Architecture of the iPseU-CNN Model
A one-step process in deep learning is represented by a layer that could be a convolution layer, a pooling layer, a normalization layer, a ReLU layer, a dropout layer, a loss layer, or a fully connected layer. The grid-search method was used for selecting the best-performing hyper-parameters. The tuned parameters are the number of filters, number of convolution layers, size of the filters, the strides, and the dropout probability. For the proposed CNN model, the list of tuned hyper-parameters is shown in Table 5.
Table 5.
The Ranges of the Tuned Hyper-Parameters
| Hyper-Parameter | Range |
|---|---|
| Convolution layers | [1,2] |
| Filters | [5,7,9] |
| Filter size | [3,5,7] |
| Stride | [1,2] |
| Dropout | [0.25, 0.50] |
The best parameters were selected based on validation loss. The sigmoid function outputs normalized class probabilities for a given input. The convolution layer is mathematically represented and computed as
| (Equation 9) |
where R represents the input of the RNA sample, f denotes the index of the filter, and j denotes the index of the output position. Each filter Wf is an S × N weight matrix of size S channels of N. The rectified linear function (ReLU) is expressed as:
| (Equation 10) |
The output layer is transformed to [0, 1] by a sigmoid function that is used for Ψ sites predictions.
| (Equation 11) |
In this study, the Keras framework was used to implement the iPseU-CNN model.66 The Adam optimizer with a learning rate of 0.001 was used, epochs were set to 50, and the batch size was set to 10.
Benchmark Datasets
In this study, three different benchmark datasets—M_944, S_628, and H_990—were used for training, where M, S, and H denoted M. musculus, S. cerevisiae, and H. sapiens, respectively, and each dataset contained 944, 628, and 990 samples, respectively. These three benchmark datasets of pseudouridylation sites were taken from the additional materials of Chen et al.14, who also introduced two various independent testing datasets for S. cerevisiae and H. sapiens denoted S_200 and H_200, respectively. The H_990, M_944, and S_628 datasets consisted of 495, 472, and 314 positive subsets of RNA samples, and every RNA sample had a uridine at the center position that could be pseudouridylated. Similarly, H_990, M_944, and S_628 datasets contained 495, 472, and 314 negative subsets of RNA samples, and each RNA sample had a uridine at the center position, but it could not be pseudouridylated. The RNA sample of these three datasets can be mathematically formulated as:
| (Equation 12) |
where represents the RNA sample, the center U denotes uridine, denotes the upstream and denotes the downstream of the central uridine for all ξ-th elements.
In H_990 and M_944 datasets, the length of each RNA sample was 21 nt, whereas in the S_628 dataset, the length of each RNA samples was 31 nt. Specifically, the value of ξ was 15 and the length of the RNA sample was 1 + 2 × 15 for the S_628 dataset. On the other hand, the value of ξ is 10 and the length of the RNA samples was 1 + 2 × 10 for the M_944 and H_900 datasets.
Cross-Validation
The error rate used in the machine- and deep-learning methods to evaluate the performance of the operation engine. In this regard, the dataset was divided into different mutually exclusive folds. In this work, we used a k-fold cross-validation test where a particular dataset can be divided into k-fold for cross-validation.61, 62, 67, 68 In this type of validation test, for the testing purpose, 1-fold was reserved, whereas for training a particular model, the remaining k − 1 folds were used. This is a k-time recursive process where every fold is tested once.62, 69 We applied a 5-fold cross-validation test to measure the four performance parameters.
Author Contributions
Conceptualization, M.T. and H.T.; Methodology, M.T. and H.T.; Investigation, M.T., H.T., and K.T.C.; Writing – Original Draft, M.T. and H.T.; Writing – Review & Editing, M.T., H.T., and K.T.C.; Visualization, M.T., and H.T.; Supervision, K.T.C.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the Brain Research Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (NRF-2017M3C7A1044815).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.03.010.
Contributor Information
Hilal Tayara, Email: hilaltayara@jbnu.ac.kr.
Kil To Chong, Email: kitchong@jbnu.ac.kr.
Supplemental Information
References
- 1.Hudson G.A., Bloomingdale R.J., Znosko B.M. Thermodynamic contribution and nearest-neighbor parameters of pseudouridine-adenosine base pairs in oligoribonucleotides. RNA. 2013;19:1474–1482. doi: 10.1261/rna.039610.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ge J., Yu Y.-T. RNA pseudouridylation: new insights into an old modification. Trends Biochem. Sci. 2013;38:210–218. doi: 10.1016/j.tibs.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charette M., Gray M.W. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000;49:341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 4.Davis D.R., Veltri C.A., Nielsen L. An RNA model system for investigation of pseudouridine stabilization of the codon-anticodon interaction in tRNALys, tRNAHis and tRNATyr. J. Biomol. Struct. Dyn. 1998;15:1121–1132. doi: 10.1080/07391102.1998.10509006. [DOI] [PubMed] [Google Scholar]
- 5.Basak A., Query C.C. A pseudouridine residue in the spliceosome core is part of the filamentous growth program in yeast. Cell Rep. 2014;8:966–973. doi: 10.1016/j.celrep.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karijolich J., Yu Y.-T. The new era of RNA modification. RNA. 2015;21:659–660. doi: 10.1261/rna.049650.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlile T.M., Rojas-Duran M.F., Zinshteyn B., Shin H., Bartoli K.M., Gilbert W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lovejoy A.F., Riordan D.P., Brown P.O. Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE. 2014;9:e110799. doi: 10.1371/journal.pone.0110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz S., Bernstein D.A., Mumbach M.R., Jovanovic M., Herbst R.H., León-Ricardo B.X., Engreitz J.M., Guttman M., Satija R., Lander E.S. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W., Feng P., Tang H., Ding H., Lin H. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics. 2016;107:255–258. doi: 10.1016/j.ygeno.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Sun W.-J., Li J.-H., Liu S., Wu J., Zhou H., Qu L.-H., Yang J.H. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016;44(D1):D259–D265. doi: 10.1093/nar/gkv1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Züst R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J., Szretter K.J., Baker S.C., Barchet W., Diamond M.S. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y.-H., Zhang G., Cui Q. PPUS: a web server to predict PUS-specific pseudouridine sites. Bioinformatics. 2015;31:3362–3364. doi: 10.1093/bioinformatics/btv366. [DOI] [PubMed] [Google Scholar]
- 14.Chen W., Tang H., Ye J., Lin H., Chou K.-C. iRNA-PseU: Identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids. 2016;5:e332. doi: 10.1038/mtna.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He J., Fang T., Zhang Z., Huang B., Zhu X., Xiong Y. PseUI: Pseudouridine sites identification based on RNA sequence information. BMC Bioinformatics. 2018;19:306. doi: 10.1186/s12859-018-2321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ververidis D., Kotropoulos C. Proceedings of the 13th European Signal Processing Conference. 2005. Sequential forward feature selection with low computational cost; pp. 1–4. [Google Scholar]
- 17.Wang L., Shen C., Hartley R. Proceedings of the International Conference on Digital Image Computing Techniques and Applications (DICTA) 2011. On the optimality of sequential forward feature selection using class separability measure; pp. 203–208. [Google Scholar]
- 18.Chen W., Feng P., Ding H., Lin H., Chou K.-C. Using deformation energy to analyze nucleosome positioning in genomes. Genomics. 2016;107:69–75. doi: 10.1016/j.ygeno.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Guo S.-H., Deng E.-Z., Xu L.-Q., Ding H., Lin H., Chen W., Chou K.C. iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics. 2014;30:1522–1529. doi: 10.1093/bioinformatics/btu083. [DOI] [PubMed] [Google Scholar]
- 20.Chen W., Feng P., Yang H., Ding H., Lin H., Chou K.-C. iRNA-3typeA: identifying three types of modification at RNA’s adenosine sites. Mol. Ther. Nucleic Acids. 2018;11:468–474. doi: 10.1016/j.omtn.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng P., Yang H., Ding H., Lin H., Chen W., Chou K.-C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics. 2019;111:96–102. doi: 10.1016/j.ygeno.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Yang H., Lv H., Ding H., Chen W., Lin H. iRNA-2OM: a sequence-based predictor for identifying 2′-O-methylation sites in homo sapiens. J. Comput. Biol. 2018;25:1266–1277. doi: 10.1089/cmb.2018.0004. [DOI] [PubMed] [Google Scholar]
- 23.Chen W., Feng P.-M., Lin H., Chou K.-C. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gks1450. e68–e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H., Qiu W.-R., Liu G., Guo F.-B., Chen W., Chou K.-C., Lin H. iRSpot-Pse6NC: Identifying recombination spots in Saccharomyces cerevisiae by incorporating hexamer composition into general PseKNC. Int. J. Biol. Sci. 2018;14:883–891. doi: 10.7150/ijbs.24616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W., Feng P.-M., Deng E.-Z., Lin H., Chou K.-C. iTIS-PseTNC: a sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem. 2014;462:76–83. doi: 10.1016/j.ab.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 26.Lin H., Deng E.-Z., Ding H., Chen W., Chou K.-C. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014;42:12961–12972. doi: 10.1093/nar/gku1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li F., Li C., Wang M., Webb G.I., Zhang Y., Whisstock J.C., Song J. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics. 2015;31:1411–1419. doi: 10.1093/bioinformatics/btu852. [DOI] [PubMed] [Google Scholar]
- 28.Li W.-C., Deng E.-Z., Ding H., Chen W., Lin H. iORI-PseKNC: a predictor for identifying origin of replication with pseudo k-tuple nucleotide composition. Chemometrics and Intelligent Laboratory Systems. 2015;141:100–106. [Google Scholar]
- 29.Dao F.-Y., Lv H., Wang F., Feng C.-Q., Ding H., Chen W., Lin H. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics. 2018:bty943. doi: 10.1093/bioinformatics/bty943. [DOI] [PubMed] [Google Scholar]
- 30.Feng C.-Q., Zhang Z.-Y., Zhu X.-J., Lin Y., Chen W., Tang H., Lin H. iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty827. Published online September 21, 2018. [DOI] [PubMed] [Google Scholar]
- 31.Collobert R., Weston J., Bottou L., Karlen M., Kavukcuoglu K., Kuksa P. Natural language processing (almost) from scratch. J. Mach. Learn. Res. 2011;12:2493–2537. [Google Scholar]
- 32.Qu W., Wang D., Feng S., Zhang Y., Yu G. A novel cross-modal hashing algorithm based on multimodal deep learning. Sci. China Inf. Sci. 2017;60:092104. [Google Scholar]
- 33.Hinton G., Deng L., Yu D., Dahl G.E., Mohamed A.-r., Jaitly N., Senior A., Vanhoucke V., Nguyen P., Sainath T.N., Kingsbury B. Deep neural networks for acoustic modeling in speech recognition: The shared views of four research groups. IEEE Signal Process. Mag. 2012;29:82–97. [Google Scholar]
- 34.Krizhevsky A., Sutskever I., Hinton G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012;25:1097–1105. [Google Scholar]
- 35.Tayara H., Chong K.T. Object Detection in Very High-Resolution Aerial Images Using One-Stage Densely Connected Feature Pyramid Network. Sensors (Basel) 2018;18:E3341. doi: 10.3390/s18103341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tayara H., Soo K.G., Chong K.T. Vehicle Detection and Counting in High-Resolution Aerial Images Using Convolutional Regression Neural Network. IEEE Access. 2018;6:2220–2230. [Google Scholar]
- 37.Aoki G., Sakakibara Y. Convolutional neural networks for classification of alignments of non-coding RNA sequences. Bioinformatics. 2018;34:i237–i244. doi: 10.1093/bioinformatics/bty228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang B., Liu F., Ren C., Ouyang Z., Xie Z., Bo X., Shu W. BiRen: predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics. 2017;33:1930–1936. doi: 10.1093/bioinformatics/btx105. [DOI] [PubMed] [Google Scholar]
- 39.Pan X., Rijnbeek P., Yan J., Shen H.-B. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics. 2018;19:511. doi: 10.1186/s12864-018-4889-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nazari I., Tayara H., Chong K.T. Branch Point Selection in RNA Splicing Using Deep Learning. IEEE Access. 2018;7:1800–1807. [Google Scholar]
- 41.Oubounyt M., Louadi Z., Tayara H., Chong K.T. Deep Learning Models Based on Distributed Feature Representations for Alternative Splicing Prediction. IEEE Access. 2018;6:58826–58834. [Google Scholar]
- 42.Tahir M., Tayara H., Chong K.T. iRNA-PseKNC(2methyl): Identify RNA 2′-O-methylation sites by convolution neural network and Chou’s pseudo components. J. Theor. Biol. 2019;465:1–6. doi: 10.1016/j.jtbi.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 43.Chen W., Ding H., Zhou X., Lin H., Chou K.-C. iRNA(m6A)-PseDNC: Identifying N6-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018;561-562:59–65. doi: 10.1016/j.ab.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Cheng X., Lin W.-Z., Xiao X., Chou K.-C. pLocbal-mAnimal: predict subcellular localization of animal proteins by balancing training dataset and PseAAC. Bioinformatics. 2019;35:398–406. doi: 10.1093/bioinformatics/bty628. [DOI] [PubMed] [Google Scholar]
- 45.Liu B., Li K., Huang D.-S., Chou K.-C. iEnhancer-EL: identifying enhancers and their strength with ensemble learning approach. Bioinformatics. 2018;34:3835–3842. doi: 10.1093/bioinformatics/bty458. [DOI] [PubMed] [Google Scholar]
- 46.Qiu W.-R., Sun B.-Q., Xiao X., Xu Z.-C., Jia J.-H., Chou K.-C. iKcr-PseEns: Identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics. 2018;110:239–246. doi: 10.1016/j.ygeno.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Tahir M., Hayat M. iNuc-STNC: a sequence-based predictor for identification of nucleosome positioning in genomes by extending the concept of SAAC and Chou’s PseAAC. Mol. Biosyst. 2016;12:2587–2593. doi: 10.1039/c6mb00221h. [DOI] [PubMed] [Google Scholar]
- 48.Liang Z.-Y., Lai H.-Y., Yang H., Zhang C.-J., Yang H., Wei H.-H., Chen X.X., Zhao Y.W., Su Z.D., Li W.C. Pro54DB: a database for experimentally verified sigma-54 promoters. Bioinformatics. 2017;33:467–469. doi: 10.1093/bioinformatics/btw630. [DOI] [PubMed] [Google Scholar]
- 49.Zhang T., Tan P., Wang L., Jin N., Li Y., Zhang L., Yang H., Hu Z., Zhang L., Hu C. RNALocate: a resource for RNA subcellular localizations. Nucleic Acids Res. 2017;45(Suppl D1):D135–D138. doi: 10.1093/nar/gkw728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen W., Lv H., Nie F., Lin H. i6mA-Pred: Identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics. 2019 doi: 10.1093/bioinformatics/btz015. Published online January 8, 2019. [DOI] [PubMed] [Google Scholar]
- 51.Ding Y., Tang J., Guo F. Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinformatics. 2016;17:398. doi: 10.1186/s12859-016-1253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan G., Jiang L., Tang J., Guo F. A novel computational method for detecting DNA methylation sites with DNA sequence information and physicochemical properties. Int. J. Mol. Sci. 2018;19:511. doi: 10.3390/ijms19020511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nanni L. Hyperplanes for predicting protein–protein interactions. Neurocomputing. 2005;69:257–263. [Google Scholar]
- 54.Cao J., Xiong L. Protein sequence classification with improved extreme learning machine algorithms. BioMed Res. Int. 2014;2014:103054. doi: 10.1155/2014/103054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caragea C., Silvescu A., Mitra P. Protein sequence classification using feature hashing. Proteome Sci. 2012;10(Suppl 1):S14. doi: 10.1186/1477-5956-10-S1-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cerf N.J., Adami C. Information theory of quantum entanglement and measurement. Physica D. 1998;120:62–81. [Google Scholar]
- 57.Nanni L., Lumini A. An ensemble of K-local hyperplanes for predicting protein-protein interactions. Bioinformatics. 2006;22:1207–1210. doi: 10.1093/bioinformatics/btl055. [DOI] [PubMed] [Google Scholar]
- 58.Chen W., Yang H., Feng P., Ding H., Lin H. iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics. 2017;33:3518–3523. doi: 10.1093/bioinformatics/btx479. [DOI] [PubMed] [Google Scholar]
- 59.Tahir M., Hayat M. Machine learning based identification of protein-protein interactions using derived features of physiochemical properties and evolutionary profiles. Artif. Intell. Med. 2017;78:61–71. doi: 10.1016/j.artmed.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 60.Tahir M., Hayat M., Kabir M. Sequence based predictor for discrimination of enhancer and their types by applying general form of Chou’s trinucleotide composition. Comput. Methods Programs Biomed. 2017;146:69–75. doi: 10.1016/j.cmpb.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 61.Hayat M., Iqbal N. Discriminating protein structure classes by incorporating pseudo average chemical shift to Chou’s general PseAAC and support vector machine. Comput. Methods Programs Biomed. 2014;116:184–192. doi: 10.1016/j.cmpb.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 62.Hayat M., Khan A. Predicting membrane protein types by fusing composite protein sequence features into pseudo amino acid composition. J. Theor. Biol. 2011;271:10–17. doi: 10.1016/j.jtbi.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 63.Hayat M., Tahir M. PSOFuzzySVM-TMH: identification of transmembrane helix segments using ensemble feature space by incorporated fuzzy support vector machine. Mol. Biosyst. 2015;11:2255–2262. doi: 10.1039/c5mb00196j. [DOI] [PubMed] [Google Scholar]
- 64.Kabir M., Hayat M. iRSpot-GAEnsC: identifing recombination spots via ensemble classifier and extending the concept of Chou’s PseAAC to formulate DNA samples. Mol. Genet. Genomics. 2016;291:285–296. doi: 10.1007/s00438-015-1108-5. [DOI] [PubMed] [Google Scholar]
- 65.Tahir M., Hayat M., Khan S.A. iNuc-ext-PseTNC: an efficient ensemble model for identification of nucleosome positioning by extending the concept of Chou’s PseAAC to pseudo-tri-nucleotide composition. Mol. Genet. Genomics. 2018;294:199–210. doi: 10.1007/s00438-018-1498-2. [DOI] [PubMed] [Google Scholar]
- 66.Keras Keras: Deep learning library for theano and tensorflow. 2015. https://keras.io
- 67.Feng P., Ding H., Yang H., Chen W., Lin H., Chou K.-C. iRNA-PseColl: identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Mol. Ther. Nucleic Acids. 2017;7:155–163. doi: 10.1016/j.omtn.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hayat M., Khan A. Discriminating outer membrane proteins with Fuzzy K-nearest Neighbor algorithms based on the general form of Chou’s PseAAC. Protein Pept. Lett. 2012;19:411–421. doi: 10.2174/092986612799789387. [DOI] [PubMed] [Google Scholar]
- 69.Tahir M., Hayat M., Khan S.A. A Two-Layer Computational Model for Discrimination of Enhancer and Their Types Using Hybrid Features Pace of Pseudo K-Tuple Nucleotide Composition. Arab. J. Sci. Eng. 2018;43:6719–6727. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.