

Highlights
-
•
In myocardial ischemia, the integrity of the cardiac sarcolemma is severely stressed in the critical earliest moments upon reperfusion. Bolstering sarcolemma integrity improves myocyte survival.
-
•
This review focuses on cardiac sarcolemma stability and its role as a therapeutic target in ischemia-reperfusion injury.
-
•
Synthetic block copolymers have been shown to interface with the muscle membrane to confer membrane stabilization during stress.
-
•
Integrated multidisciplinary research teams, spanning cardiology, physiology, chemistry, and chemical engineering are essential to guide future mechanistic and translational studies of novel chemical-based membrane stabilizers for preserving viable heart muscle during ischemia-reperfusion injury in human patients.
Key Words: copolymer, heart, ischemia, reperfusion
Summary
The phospholipid bilayer membrane that surrounds each cell in the body represents the first and last line of defense for preserving overall cell viability. In several forms of cardiac and skeletal muscle disease, deficits in the integrity of the muscle membrane play a central role in disease pathogenesis. In Duchenne muscular dystrophy, an inherited and uniformly fatal disease of progressive muscle deterioration, muscle membrane instability is the primary cause of disease, including significant heart disease, for which there is no cure or highly effective treatment. Further, in multiple clinical forms of myocardial ischemia-reperfusion injury, the cardiac sarcolemma is damaged and this plays a key role in disease etiology. In this review, cardiac muscle membrane stability is addressed, with a focus on synthetic block copolymers as a unique chemical-based approach to stabilize damaged muscle membranes. Recent advances using clinically relevant small and large animal models of heart disease are discussed. In addition, mechanistic insights into the copolymer-muscle membrane interface, featuring atomistic, molecular, and physiological structure-function approaches are highlighted. Collectively, muscle membrane instability contributes significantly to morbidity and mortality in prominent acquired and inherited heart diseases. In this context, chemical-based muscle membrane stabilizers provide a novel therapeutic approach for a myriad of heart diseases wherein the integrity of the cardiac muscle membrane is at risk.
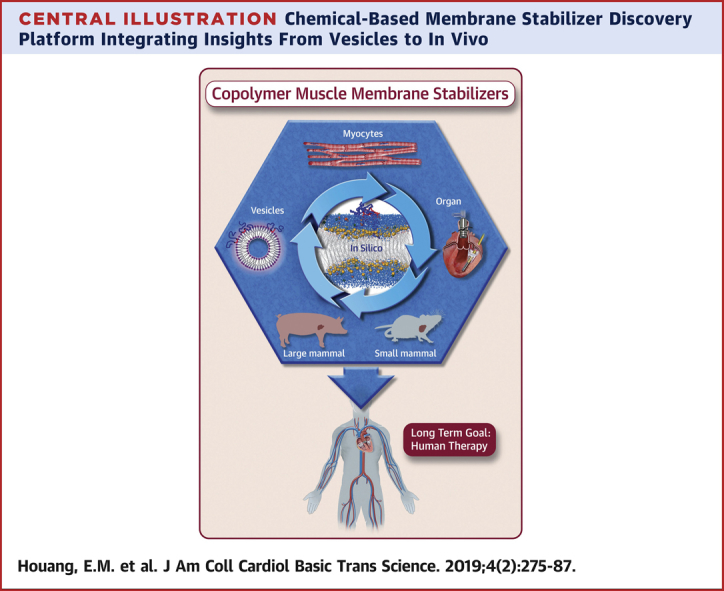
Central Illustration
Overview: Cardiac Muscle Membrane Integrity and Heart Performance
Cardiac muscle membrane instability is a hallmark of myocardial ischemia-reperfusion (I/R) injury 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29. It is well-documented that in the very earliest moments of reperfusion cardiac myocytes swell markedly due to significant myoplasmic osmolyte accumulation during the ischemic period 21, 25, 28, 30, 31. Myocyte swelling, along with myocyte contracture, exerts a severe stress on the cardiac membrane (sarcolemma), leading to membrane tears, pores, blebs, and instability 19, 20, 25, 26, 27, 28. Loss in the primary barrier function of the cardiac membrane leads to catastrophic outcomes in terms of overall myocyte viability and function. It follows that preserving cardiac sarcolemma integrity in I/R holds promise for enhancing cardiac muscle viability thus improving overall heart function during reperfusion (Figure 1).
Figure 1.

Health Relevance
Clinically relevant pathways to cardiac reperfusion injury with focus on cardiac membrane instability and the novel use of synthetic membrane stabilizers. CPR = cardiopulmonary resuscitation; MI = myocardial infarction.
Myocardial I/R causes millions of deaths per year 2, 10, 15, 17, 19, 32, 33, 34, 35, 36, 37, 38, 39 underscoring the urgency for detailed understanding. There are multiple pathological pathways to cardiac reperfusion injury; nonetheless, in the face of this complexity, a key unifying component of I/R injury centers on cardiac muscle membrane instability 19, 20, 21, 23, 24, 25, 26, 29, 31, 37, 40. In cardiac I/R, sarcolemmal rupture and necrotic cell death are well documented in the critical minutes of reperfusion after an ischemic event 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19. Whereas the pathways leading to cardiac I/R injury are multifold and complex 18, 41, 42, we and others have established a sound premise that a clear approach for a clinically meaningful treatment would be one that would directly enhance cardiac membrane stability in I/R (43). Given that cardiac sarcolemma damage is a well-established hallmark of I/R injury, it is surprising this has received relatively little direct investigation as a possible target for I/R treatment development (44).
In this review, we detail the design and implementation of chemical-based synthetic muscle membrane stabilizers for the diseased heart (Central Illustration). Muscle membrane stabilizers target and interface with, but do not transit across, the cell outer membrane to confer sarcolemma stabilization (Figure 2) 45, 46, 47. We recently showed in several genetic models of cardiac muscle membrane instability that synthetic block copolymer-based membrane stabilizers prevent progressive cardiomyopathy 48, 49. We focus here on the mechanism by which block copolymer muscle membrane stabilizers interface with the sarcolemma to preserve cardiac myocyte viability. A structure-function approach is enabled by leveraging insights across disciplinary lines, extending from chemistry to physiology and cardiology. Collectively, we advance synthetic membrane stabilizers as a unique first-in-class cell extrinsic strategy for directly protecting cardiac muscle membranes during I/R in vivo 48, 49, 50, 51, 52, 53. We propose that membrane stabilizers, in the form of synthetic copolymers, provide a unique tool to investigate the mechanism of cardiac I/R injury and provide a template toward developing novel therapies designed to stabilize the muscle membrane in acquired heart disease. Thus, this review is centered on novel means and mechanisms to preserve viable myocardium during reperfusion.
Central Illustration.

Chemical-Based Membrane Stabilizer Discovery Platform Integrating Insights From Vesicles to In Vivo
Overarching goal: stabilizing the cardiac muscle membrane in disease using synthetic chemistry.
Figure 2.

Schematic Representation of Block Copolymer P188 Interaction With the Cardiac Sarcolemma
Blue spheres = polyethylene oxide block; red spheres = polypropylene oxide block.
The Cardiac Membrane as a Direct Therapeutic Target in Myocardial I/R
Cardiac I/R injury exacts a tremendous toll on morbidity and mortality in humans 54, 55, 56, 57, 58, 59, and recent clinical cardioprotective trials have been disappointing (60). The hypothesis advanced here is that during the critical earliest stages of I/R the cardiac myocyte's endogenous membrane stabilization/repair machinery is overwhelmed and that synthetic copolymers serve to rapidly stabilize the membrane as a bridging mechanism. In this way, copolymers permit the cell intrinsic membrane repair pathways adequate time to fully repair the damaged membrane and preserve cardiac myocyte viability in vivo. Block copolymers offer a direct approach to preserve membrane integrity in I/R, thereby representing a novel therapeutic development opportunity with the potential for high clinical impact.
Heart muscle has multiple cell intrinsic/endogenous mechanisms to stabilize/repair the fragile/damaged sarcolemma during stress 61, 62, 63, 64. These include sarcolemma phospholipid fluid mosaic re-structuring that can rapidly repair very small disruptions/injury to the membrane. For more significant damage, which would occur during cardiac I/R, key endogenous mechanisms are activated to repair damage to preserve muscle cell integrity and viability. Published data supports endogenous dysferlin-mediated muscle membrane repair 65, 66. Dysferlin repairs muscle membranes in a Ca2+-dependent manner whereby dysferlin regulates fusion of repair vesicles with the sarcolemma to accomplish membrane repair. Mitsugumin 53 has also been identified as a crucial component in membrane repair (67). Recently, a new cell intrinsic membrane stabilization pathway has been elucidated: thrombospondin 4(Thbs4)–mediated striated muscle membrane stabilization 68, 69, 70. The Thbs4 mechanism of cell intrinsic membrane stabilization (69) is restricted to cardiac and skeletal muscle and its expression is induced with injury or disease in the heart, including ischemic injury 69, 70, 71, 72. Thbs4 is therefore distinct from other Thbs gene family members that are secreted glycoproteins. Within striated muscles, Thbs4 directly enhances intracellular vesicular trafficking and chaperones the critical dystrophin-glycoprotein complex and integrin attachment to the muscle membrane, leading to increased membrane stability of heart muscle 69, 70, 71, 72, 73. Data show that genetic ablation of the Thbs4 pathway in otherwise healthy animals directly causes sarcolemma weakness (69). Further, overexpression of Thbs4 markedly enhances vesicular trafficking of dystrophin-glycoprotein complex and integrin complexes to augment muscle membrane integrity to enable efficient membrane repair.
On the physiological timescale, the myocyte's intrinsic membrane repair and stabilization pathways, including dysferlin, mitsugumin 53, Thbs4, and others, can take up to several minutes or longer to repair sarcolemma damage 61, 74, 75, 76, 77. In I/R, this delay in repair can be catastrophic, leading to the demise of the cell. To address this, we and others have pursued cell-extrinsic synthetic chemistries as a rapid means to protect damaged striated muscle membranes (Figures 1, 2, and 3). We were the first to show that the first-in-class synthetic membrane stabilizer, triblock copolymer poloxamer 188 (P188), protects the heart (Figure 2) (48). The poloxamer family of block copolymers has been in wide use for drug delivery and other biomedical applications (78). P188 has been safely used in animals and humans, including advanced clinical trials for limiting vaso-occlusive crisis in sickle cell anemia 79, 80, 81. As reviewed in detail below, synthetic membrane stabilizers are amphiphilic long-chain macromolecular block copolymers that interact with and protect the muscle sarcolemma during stress (Central Illustration, Figure 2). The goal of this review is to illuminate the role of membrane integrity in I/R by disseminating recent insights into copolymer-membrane interactions. We further provide an overview of the potential for translation of this therapeutic approach into clinically relevant beneficial outcomes in the setting of myocardial I/R in vivo.
Figure 3.

Timing and Location of Membrane Stabilizer Delivery are Essential Elements to Clinical Efficacy in STEMI/Percutaneous Coronary Intervention
Myocardial I/R Injury Clinical Features
Coronary heart disease is the most common form of human heart disease in the United States, accounting for 20% of all deaths (82). Coronary heart disease most often presents as a myocardial infarction (MI), during which cardiac tissue becomes ischemic due to coronary arterial blockage, and this blockage must be rapidly reversed to help preserve functional heart muscle 4, 30, 33, 34, 36, 37, 83, 84, 85, 86, 87, 88. In the United States, >250,000 patients per year have ST-segment elevation MI (STEMI) treated with primary percutaneous coronary intervention (PCI) that involves intracoronary placement of a stent to open up the blocked vessel. Urgent restoration of blood flow, where “time is muscle” is the cardiologist's main imperative. Paradoxically, restoration of blood flow causes a second wave of cardiac muscle damage termed myocardial I/R injury 10, 12, 38, 89.
Along with MI, other clinically relevant scenarios involving myocardial ischemia (or low flow) and reperfusion include cardiopulmonary resuscitation (CPR) and heart organ procurement and transplantation (Figure 1) 90, 91. In the case of CPR, discordant electrical activity of the heart leads to marked reductions in heart pump performance requiring external compressions of the chest cavity (CPR) to provide low-flow perfusion. Upon reacquisition of sinus rhythm, flow can be restored; however, as in MI, the reintroduction of oxygenated blood results in reperfusion injury. Further, heart organ transplantation necessitates organ procurement and a prolonged ischemic period that can last hours before transplantation and reperfusion 59, 92. Each of these common clinical scenarios presents unique conditions for the development of I/R injury, and each provides unique opportunities to assess the efficacy of membrane stabilization to improve myocardial viability and function.
There are 2 basic clinical presentations that result from sudden onset of profound myocardial ischemia. Whereas they differ in the distribution of the ischemia and resulting reperfusion, the underlying mechanisms of cellular injury are similar. The first, MI, occurs in approximately 600,000 people per year. Of these, STEMI accounts for 30% of cases (82). STEMI causes mortality in 6% of cases, with up to 20% of patients going on to develop overt heart failure requiring lifelong therapy (93). STEMI is caused by complete occlusion of a coronary arterial, severely limiting or eliminating blood flow to a region of the heart. This is most commonly caused by sudden formation of thrombus at the site of an atherosclerotic plaque. Advances in treatment of STEMI over the last few decades have focused on reperfusing the muscle by removing the obstruction. This began initially with thrombolytic therapy to dissolve the obstructing thrombus. Presently, frontline clinical treatment focuses on the use of PCI in the cardiac catheterization laboratory, where balloons and/or stents are deployed to resolve the occlusion 94, 95. The use of PCI has markedly increased the likelihood of reperfusion, increasing from 40% to 55% with thrombolytics to >90% with PCI (96).
Block Copolymers: First-in-Class Synthetic Muscle Membrane Stabilizers
The scientific premise guiding this review is that maintenance of cardiac membrane integrity is severely challenged from the very earliest moments of reperfusion. Thus, the stability of the muscle membrane represents a potentially highly efficacious target for therapeutic development. In this light, we and others have advanced synthetic chemistries featuring block copolymers as a unique class of molecules with membrane stabilization functionalities (Central Illustration). Synthetic block copolymers include a range of soft materials with wide ranging industrial and biological applications 45, 81, 97. The most well-known synthetic block copolymers in biomedicine are triblock copolymers known as poloxamers (or pluronics), which are composed of a hydrophobic polypropylene oxide (PPO) block flanked by hydrophilic polyethylene oxide (PEO) chains (Central Illustration, Figures 2 and 3). In principle, an immeasurable number of block copolymers of distinct physicochemical properties could be obtained by varying the lengths of the PEO and PPO blocks as well as their architectural assembly. These various combinations give rise to amphiphilic molecules with complex membrane interface behavior that have been found to directly interact with cellular membranes as lysis agents and membrane stabilizers, directly dependent on their unique chemical composition 48, 97, 98, 99, 100, 101.
Block copolymers can be designed to vary markedly in size, composition, and architecture (Figures 2 and 4) (102). By far, the most well-studied block copolymer in biomedical research has been the triblock P188 (or PEO75-PPO30-PEO75, PPO/PEO = 0.20 and MW = 8.4 kDa) (Figures 2 and 3, Table 1). P188 was first approved by the Food and Drug Administration as an anti-viscosity agent added to blood and has been previously tested in clinical trials for sickle cell anemia and MI 81, 103, 104, 105, 106, 107, 108, 109, 110. The pharmacokinetic profile of P188 in healthy males has been obtained in a cohort of volunteers, and it has been determined that elimination occurs primarily through renal clearance 111, 112. The formulation of P188 purified from small molecular weight impurities is well tolerated (113). P188's safety profile was further validated for long-term use in human patient sickle cell anemia trials, showing an overall excellent safety profile in humans 81, 103, 104.
Figure 4.

Copolymer-Muscle Membrane Interface
(A) Diblock copolymer structures with unique single chemical end groups where blue represents polyethylene oxide units and red represents polypropylene oxide units. (B) Conceptualization of anchor and chain working model, wherein the relatively more hydrophobic tert-butyl end group anchors the polypropylene oxide block more deeply in the phospholipid bilayer (right).
Table 1.
Chemical Features of Various Synthetic Block Copolymers
| Architecture | Polymer | PEO∗ | PPO∗ | End Group† | Mass‡ | PEO%§ |
|---|---|---|---|---|---|---|
| Triblock copolymer/P188‖ | PEO75PPO30PEO75 | 150 | 30 | — | 8,400 | 80 |
| Triblock copolymer/P338‖ | PEO140PPO44PEO140 | 280 | 44 | — | 14,600 | 84 |
| Triblock copolymer/P331‖ | PEO7PPO54PEO7 | 14 | 54 | — | 3,700 | 26 |
| Diblock copolymers | PEO75PPO15−H | 75 | 15 | −H | 4,200 | 80 |
| PEO75PPO15−C4 | 75 | 15 | −C(CH3)3 | 4,430 | 77 | |
| Control homopolymer | PEO198 | 198 | 0 | — | 8,700¶ | 100 |
EO = ethylene oxide; NMR = nuclear magnetic resonance; PEO = polyethylene oxide; PO = propylene oxide; PPO = polypropylene oxide.
Total number of EO or PO monomer units.
Chemical end group.
Molecular weight in g/mol by 1H NMR end-group analysis.
PEO weight percent to total molecular weight.
Manufacturer BASF, Florham Park, New Jersey.
Number average molecular weight.
P188 has shown membrane stabilization capability in a diverse range of pathophysiological settings. These include irradiation and burn injury 114, 115, blunt impact–induced cartilage damage and joint degeneration (116), amyotrophic lateral sclerosis (117), neuronal cell death (118), traumatic brain injury–induced blood-brain barrier damage, brain edema (119), and apoptosis-induced neuronal cell death after oxygen-glucose deprivation (120). Membrane stabilization by P188 has also been well established in Duchenne muscular dystrophy models 48, 49, 53, 121, 122 where purified clinical grade P188 is currently under a Phase II, single-site, open-label clinical trial for Duchenne muscular dystrophy patients. Furthermore, in vivo delivery of P188 was shown to protect neuronal cells during spinal cord compression and acute intracranial hemorrhage (123). P188 has also been used as an additive enhancing blood oxygenation, cardiopulmonary bypass, in heart-lung bypass, and as a rheological agent to lessen blood viscosity and platelet aggregation 124, 125, 126, 127, 128, 129.
Of direct relevance to STEMI and I/R injury, a previous study showed that intravenous injection of P188 significantly improved blood flow to ischemic brain areas induced by surgical occlusion of the middle cerebral arterial in a rabbit model of focal cerebral ischemia (130). Because P188 was reported to improve blood flow without reducing blood viscosity and without hemodilution, it has been hypothesized that P188 improves blood flow by reducing adhesive interactions between blood cells and vessel walls and between fibrin and fibrinogen in the microcirculation (130). Moreover, P188 was found in one study to diminish I/R-induced brain injury and improve long-term functional recovery after focal cerebral ischemia in mice (131). There is evidence that P188 can provide long-term protective effects on cerebral I/R injury both in vivo and in vitro and that neuroprotection offered by P188 involves several mechanisms, including preserving blood-brain barrier impermeability, intervening in the nuclear factor kappa light chain enhancer signaling pathway by inhibiting MMP-9, and reducing cell death. P188 was also found to rescue cultured hippocampal neurons after excitotoxicity and oxidative stress (132), both central mechanisms of hypoxia injury-induced neurodegeneration. Another study showed that P188 decreased the activation of autophagy in neuronal cells under oxygen-glucose deprivation in vitro and that intravenous injection of P188 reduced infarct volume and neurological/motor deficits in a mouse model of cerebral ischemia in vivo (133).
A recent investigation further showed that cardiac I/R injury, which is significantly amplified in animal models of sarcolemmal loss-of-function, was abrogated by application of copolymer-based membrane stabilizers (42). P188 treatment of rat cardiac myocytes exposed to simulated ischemia/reperfusion in vitro prevented membrane leakage of intracellular lactate dehydrogenase. Furthermore, P188 mediated membrane stabilization was shown to prevent I/R-functional deficits in isolated hearts ex vivo (42). Taken together, these studies provide strong evidence that the neuronal and cardiac cell membranes can be targeted for membrane stabilization to improve viability and function in I/R.
Copolymer-Based Membrane Stabilizers in Myocardial I/R Injury
In the setting of reperfusion injury after MI, membrane stabilizers were first tested in small and large animal studies in the 1990s. The first study, published in 1991, used a dog model of left anterior descending arterial occlusion with infusion of P188 for the 15 min before reperfusion and the first 45 min of reperfusion (134). P188 reduced infarct size by 50% in that model. Subsequent studies went on to replicate the reduced infarct size using the same canine model (110). At that time, these positive effects were attributed to the hemorheological and antithrombotic effects of P188, as it was shown to reduce red blood cell aggregation (81), facilitate clot lysis by recombinant tissue plasminogen activator (135), and reduce leukocyte chemotaxis (110). It was later discovered that P188 also stabilizes the sarcolemma (132).
In the clinical setting of I/R, the first human trial enrolled patients from 11 centers between March 1992 and August 1993 (107). All patients had ongoing chest pain of >30 min duration and STEMI on electrocardiogram. Study participants were only included if they were candidates for thrombolytic therapy, as that was the state-of-the-art reperfusion therapy available at that time. The initial phase of the trial randomized the first 45 patients to either control or low-dose P188 (150 mg/kg/h for 1 h then 15 mg/kg/h for 47 h). The second phase randomized the following 69 patients to placebo or high-dose P188 (300 mg/kg/h for 1 h then 30 mg/kg/h for 47 h). P188 showed improvement in infarct size (38% reduction), increased median myocardial salvage (9% absolute increase), and improved left ventricular (LV) function (6% absolute increase in LV ejection fraction [EF]). The P188 group also had a 12% absolute reduction in re-infarcts.
The CORE (Collaborative Organization for RheothRx Evaluation) trial was a phase II open label clinical trial, including 2,948 patients with STEMI receiving thrombolytic treatment, with the trial beginning in 1994 (136). The trial randomized patients to placebo (n = 963) or 1 of multiple dosing regimens with P188. In this large trial, no significant clinical benefit of P188 was observed with similar rates of mortality, reinfarction, and cardiogenic shock between groups. It was also noted in a subset of elderly patients with pre-existing renal dysfunction that acute renal dysfunction was more frequent in the P188 group. In this context, subsequent studies have examined this in detail showing that the commercially available (nonpurified) P188 resulted in vacuolization of the proximal tubule epithelium without signs of irreversible injury or necrosis (113). Purification of P188, which removes low molecular weight contaminants, fully prevented the renal effects of P188 in humans and animal models.
In retrospect, and in the full light of contemporary clinical management of STEMI, it can now be readily discerned that multiple important limitations were inherent in the CORE trial design. First, CORE trial patients were treated with thrombolytics which left at least one-half of the patients without any reperfusion. It follows that the goal of treating reperfusion injury was not possible in the patients that did not achieve reperfusion. As discussed above, PCI is now the standard of care in STEMI, resulting in significantly higher rates of reperfusion. Second, in the CORE trial, P188 was administered intravenously through the PVC approximately 30 min after thrombolytic therapy was administered. This delay is not explicitly described in the publication; however, randomization was recommended within 15 min of thrombolytic infusion, and P188 infusion was initiated within 15 min of randomization. Given the absolute requirement for rapid delivery of therapies targeting reperfusion injury and the rapid decay in benefit over the minutes following reperfusion (137), this 30-min delay is a key parameter in understanding the significantly reduced efficacy of membrane stabilization therapy in the CORE trial. In addition, the trial's PVC delivery dilutes the effect and further minimizes the possibility of detecting a therapeutic benefit.
The methodological limitations of the CORE trial were recently investigated in a porcine model of STEMI (137). Anterior STEMI was established in adult pigs via 45 min of endovascular occlusion of the mid left anterior descending coronary arterial. The pigs were randomized to 1 of 4 groups including vehicle control, immediate intracoronary P188 delivery, delayed peripheral P188, and intracoronary polyethylene glycol (PEG, or PEO)–8,000 infusion serving as a mass equivalent hydrophilic control for P188. After 45 min of ischemia, the endovascular balloon was deflated and removed and the appropriate treatment was infused through the coronary guide catheter in all groups other than the delayed peripheral P188 group where 30 min was allowed to elapse and P188 was then infused via PVC similar to the CORE trial. The pigs were maintained for 4 h at which time the hearts were removed for evaluation of mitochondrial function or staining for quantification of the infarct size. The study revealed significantly improved mitochondrial structure, respiration, and calcium retention in the setting of the immediate intracoronary delivery of P188. Infarct size and serum troponin I levels were reduced by 66% and 75%, respectively, with immediate intracoronary P188 delivery. Importantly, animals receiving the delayed peripheral P188, similar to the CORE trial protocol, resulted in no benefit. In addition, immediate intracoronary PEG infusion also provided no benefit, suggesting that the mechanism was not due to the hemorheological effects of P188. Taken together, these results are evidence that the lack of benefit observed in the CORE trial could be ascribed to a suboptimal treatment protocol rather than a lack of a P188 therapeutic effect. Evidence has accumulated that timing (immediate) and location (intracoronary) are essential parameters of copolymer-based membrane stabilization treatment in STEMI-I/R. It follows that further study in STEMI patients receiving PCI therapy with immediate intracoronary P188 infusion are warranted and should be investigated through future clinical studies (Figure 3).
To date, there have been no studies conducted on patients suffering cardiac arrest with P188 intervention. Recently, P188 was studied as part of a bundle of care strategy in a porcine model of prolonged cardiac arrest (138). Here, pigs had ventricular fibrillation induced electrically followed by 17 min of untreated downtime. Animals were then randomized to control CPR or bundle therapy including ischemic post-conditioning with stutter CPR, inhaled sevoflurane, and P188. Pigs receiving the bundle of therapy showed improved hemodynamics during CPR, improved rates of return of spontaneous circulation, reduced need for epinephrine and defibrillation after return of spontaneous circulation, reduced troponin and measures of end-organ dysfunction, increased LVEF, and improved neurologic function and freedom from major adverse events. It is difficult to delineate the effect of each component of the bundle in terms of contribution to therapeutic outcome. In this context, it is appropriate that further study be initiated to determine the potential for P188 to prevent cardiac and neurological injury in the setting of cardiac arrest.
Overall, the use of block copolymer–based membrane stabilizers, and the available evidence for their use, is starkly contrasted between the STEMI population and the cardiac arrest population. Membrane stabilizers have a 2-decade history of benefit in controlled animal models of STEMI and inconsistent benefits in human studies of STEMI where duration of ischemia and reperfusion are less well-controlled. Current technologies that increase reperfusion success and provide a definite time of reperfusion, such as PCI, can be expected to improve the consistency of therapeutic benefit in humans. In addition, the opportunity to infuse P188 directly into the coronary artery provides the opportunity for immediate site of action delivery, while minimizing the potential effects on off-target organs such as the kidneys. In contrast to STEMI, evidence is presently lacking for evaluating the efficacy of membrane stabilizers in the setting of cardiac arrest. This population has a broad systemic injury and would then require systemic infusion. In addition, patients often receive bystander or first-responder CPR, which initiates inefficient reperfusion at least 5 to 10 min before initiation of an intravenous catheter, and may limit the benefits of P188 infusion. Alternatively, systemic injury provides a potential opportunity to reduce injury to other organs, including potentially the brain. The dysregulation of the blood-brain barrier and the importance of brain injury in the outcomes after cardiac arrest provide an important target for beneficial effects of membrane stabilizers.
The Copolymer-Membrane Interface: Toward the Mechanism of Muscle Membrane Stabilization
It has been established that block copolymer molecular design impacts molecular interaction with phospholipid membranes and this, in turn, significantly affects muscle membrane stabilization efficacy 50, 51, 52, 53, 139. From a structure-function perspective, there is a significant opportunity to gain mechanistic insights with the long-range goal to improve and optimize membrane stabilization properties of block copolymers (Table 1). To date, only a very limited subset of the vast possible chemical landscape of the copolymer superfamily has been investigated. There is growing evidence showing that membrane stabilization efficacy is highly dependent on the composition, size, and architecture of the block copolymer. For example, P188 was shown to be superior to a 70% PEO, 13-kDa PEO-PPO-PEO triblock copolymer, but inferior to 60% vinylpyrrolidone–40% vinyl acetate random copolymer VA64 in resealing cortical and hippocampal cells after controlled cortical impact (140). Conversely, the same 70% PEO, 13-kDa hydrophilic PEO-PPO-PEO triblock copolymer was superior to P188 in maintaining cell viability after transient photoacoustic permeabilization for molecular delivery (141). On the other hand, increased molecular weight, while maintaining 80% PEO content, improved dystrophic myofiber protection in culture, and also impacted its pharmacodynamics in dystrophic skeletal muscles in vivo 50, 51.
Physicochemical models of cellular membranes have been developed to provide new mechanistic insights into copolymer-membrane interactions. Copolymer interactions with lipid membranes were first described via Langmuir trough experiments in which compression and expansion of simple model lipid monolayers were used to assess the surface pressures at which copolymer insertion occurred (98). P188 was found to only insert at surface pressure below that of an intact cell membrane and then was squeezed out above that threshold (97). This experiment directly correlates copolymer insertion with membrane lipid packing density. Additionally, the surface pressure of insertion depends on copolymer hydrophobicity. Copolymers with comparatively higher PPO/PEO ratios show higher squeeze-out pressures. Furthermore, hydrophobic copolymers insert at faster rates and can increase membrane permeability 142, 143. These findings indicate an important relationship between the PPO/PEO ratio and molecular weight in determining copolymer-membrane interactions. These experimental studies are supported by recent in silico studies using molecular dynamics to simulate interactions of copolymers with a lipid bilayer under lateral mechanical stress (52). P188 insertion into stretched lipid bilayers significantly increases the lateral pressure at which membrane rupture occurs. In contrast, highly hydrophobic copolymers (e.g., PEO7-PPO54-PEO7) decrease the lateral pressure required for membrane rupture. This is consistent with experimental findings showing that above a specific hydrophobicity threshold copolymer insertion leads to increased susceptibility to mechanical stress (52).
Copolymer-bilayer interactions have also been investigated using pulsed-field gradient nuclear magnetic resonance spectroscopy to measure the diffusion of block polymers in the presence and absence of phospholipid vesicles (144). This technique further confirmed that increased copolymer molecular weight and increased relative hydrophobicity caused increased binding and liposome coverage relative to smaller, more hydrophilic copolymers. This differential diffusivity enables quantification of the extent of copolymer interaction with the phospholipid membrane. Recent studies have shown higher interaction with increased copolymer molecular weight and hydrophobicity. For example, surface plasmon resonance (SPR) experiments have been used to measure binding kinetics for copolymers with supported phospholipid bilayers (139). These SPR experiments further quantify the extensive duration of P188-bilayer interaction and revealed the ability of the hydrophilic PEO block itself to engage phospholipid membranes at high concentrations. These findings support the hypothesis that the hydrophilic block of poloxamers is not merely a passive bystander to the hydrophobic block’s interaction with the core of the phospholipid bilayer, but rather also contributes to membrane interaction. These results motivate further design and engineering of the copolymer hydrophilic block(s) for improved membrane interactions.
Block copolymers can be synthesized with distinct chemical blocks in a variety of sizes, compositions, and architectures (Figure 4, Table 1) (102). Recent papers have addressed the impact of block copolymer architecture on membrane stabilization 50, 51, 52, 53, 139, 144. In particular, a PEO75-PPO16 diblock, essentially half of P188 (Figure 4), was shown to provide protection to cultured myoblasts in response to hypotonic shock and isotonic recovery (51) Moreover, it was shown that addition of a single hydrophobic tert-butoxy moiety to the PPO end of the diblock enhances membrane stabilization efficacy both in vitro and in vivo in a muscular dystrophy mouse model (50). Molecular dynamics studies further showed that the tert-butoxy moiety acts like an anchor, keeping the PPO end of the diblock deep within the acyl chains of the bilayer, whereas absence of this moiety led to the PPO group preferring to interact with interfacial water (50). From these studies, an “anchor and chain” model of copolymer-membrane interface has emerged 50, 51. Stronger interaction with the bilayer without actual percolation across the membrane could be envisioned to be beneficial to pharmacokinetics and pharmacodynamics properties in vivo. The benefit of such a hydrophobic anchor was further substantiated by the lack of efficacy of diblocks with shortened hydrophobic PPO blocks. The ability to modulate performance with focused investigation of copolymer composition and architecture leads to substantial optimism for mechanistic insight and enhanced activity with broader systematic changes in copolymer design. Notably, such systematic evaluation is facilitated by the use of the diblock architecture, which is more synthetically efficient than the triblock architecture and affords the opportunity to manipulate end-group chemistry independent of the hydrophobic block characteristics.
Studies using dynamic light scattering and isothermal titration calorimetry suggest that copolymers that adsorb but do not insert into the intact bilayer affect the hydration shell at the bilayer-solvent interface (143). P188 and its mass equivalent PEO homopolymer PEG8000 were shown to prevent the diffusion of a free radical lipid peroxidation initiator into the lipid bilayer 100, 145. In contrast, more hydrophobic copolymers such as P335 (PEO38-PPO54-PEO38), P333 (PEO20-PPO54-PEO20), and P181 (PEO2-PPO30-PEO2) did not prevent free radical–induced lipid peroxidation. This result was confirmed by Cheng et al. (143) using dynamic light scattering, isothermal titration calorimetry, and small molecule–directed lipid peroxidation of liposomes. The PPO/PEO ratio was shown to be a key feature in effectively protecting intact liposomes from peroxidation. Copolymers that adsorb at the membrane surface without penetration into the bilayer core, such as P188 and PEG8000, presumably affect the initial hydration shells of the bilayer via interactions with water molecules at the interface. Presence of a “water shield” of hydrated copolymers would physically suppress the diffusion of the free radical lipid peroxidation initiator into the lipid bilayer, thereby preventing the initiation of lipid peroxidation. More hydrophobic poloxamers, for example P335 (PEO38-PPO54-PEO38), P333 (PEO20-PPO54-PEO20), and P181 (PEO2-PPO30-PEO2) have significant heat of partitioning indicative of insertion into the liposomal membrane (143) and do not prevent lipid peroxidation (146).
More detailed structure-function analysis of copolymer block chemistry, length, and structural characteristics will be important. Future experiments are needed to expand our understanding of copolymer-membrane interactions. In addition to the use of physicochemical models to quantify the kinetics and extent of membrane interaction for different polymers and membrane compositions, it will be important to develop more extensive biophysical methods to determine the localization of polymers within the membrane and within organs/tissues to help determine engagement with target tissues and the pharmacokinetic profile of copolymer variants. The vast unexplored copolymer chemical landscape and novel structure-function insights to follow will help guide copolymer design for advancing copolymers in applications for I/R injury and STEMI.
Summary and Future Opportunities
Block copolymers represent a class of organic materials with unique biological properties, including direct interfacing with biological membranes. In the context of a myriad of acquired and inherited heart diseases that involve, directly or indirectly, destabilization of the cardiac muscle membrane, block copolymers provide a unique potential therapeutic strategy to preserve myocyte and heart organ function.
By combining diverse expertise in chemistry, chemical engineering, muscle physiology, and cardiology, the design and implementation of synthetic block copolymers as novel membrane stabilizing agents has come to the forefront as a potential therapy for clinical disorders involving loss in muscle membrane integrity. Structure-function studies, guided by strategic molecular design, provide insights into the fundamental basis of the copolymer-membrane interface. An anchor-and-chain working model has been proposed to account, mechanistically, for the copolymer-membrane interface that leads to membrane stabilization during stress, including I/R. Exciting opportunities lie ahead to decipher atomic to organ physiology levels of discovery in this research space.
There is a significant opportunity to advance mechanistic insights and enhanced performance into how copolymers function as membrane stabilizers. To date, structure-function and efficacy studies have been achieved with a comparatively limited evaluation, in terms of the vast chemical space of copolymers. The ability to strategically modulate copolymer-membrane function with detailed investigations of copolymer size, composition, and architecture leads to substantial optimism for mechanistic insights—and ultimately enhanced activity—with expansive and systematic changes in copolymer design. Notably, such systematic evaluation is facilitated by the use of the diblock architecture (Figure 4), which is more synthetically efficient than triblocks, and affords the opportunity to manipulate end-group chemistry independent of the hydrophobic block characteristics. Moreover, well-designed cellular and molecular assays enable efficient evaluation of copolymer designs and mechanisms.
The localization of P188 both within the membrane hydrophobic core and along the hydrophilic head groups is consistent with the anchor-and-chain mechanism, while hinting at more extensive interaction of the PEO component with the hydrophilic head groups than initially considered; notably, this is consistent with the aforementioned SPR results that indicate significant PEO-membrane interaction. Moreover, the discovery that P188 is distributed throughout the 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer alters the perception of the depth of interaction. More extensive studies with copolymer variants and altered membrane composition will further elucidate copolymer localization and the mechanism of interaction. These insights, in turn, will lead to deeper understanding of the mechanism of membrane stabilization.
Future directions in clinical models are essential to extend basic science discoveries toward clinical impact. Along with reperfusion studies in the context of STEMI, studies are warranted in CPR and organ transplantation. Having demonstrated the essential features of copolymer-based membrane stabilization, shown to be critically dependent upon time of delivery and precise location of delivery (Figure 3), there is now a significant opportunity to conduct STEMI patient trials with these key parameters clearly aligned in clinical protocols during PCI-based therapeutic trials.
Acknowledgments
This work was supported by grants from the NIH, MDA and AHA. The authors thank Cynthia DeKay for the schematics presented in Figures 2 and 3.
Footnotes
Drs. Metzger and Yannopoulos have received grants from the National Institutes of Health. Dr. Metzger is on the scientific advisory board of and holds zero value equity shares in Phrixus Pharmaceuticals Inc., a company developing novel therapeutics for heart failure and DMD, and this is actively managed by the UMN Office of Institutional Compliance. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and U.S. Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
References
- 1.Hausenloy D.J., Yellon D.M. Preconditioning and postconditioning: united at reperfusion. Pharmacol Ther. 2007;116:173–191. doi: 10.1016/j.pharmthera.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Hausenloy D.J., Yellon D.M. The therapeutic potential of ischemic conditioning: an update. Nat Rev Cardiol. 2011;8:619–629. doi: 10.1038/nrcardio.2011.85. [DOI] [PubMed] [Google Scholar]
- 3.Hausenloy D.J., Yellon D.M. “Conditional Conditioning” in cardiac bypass surgery. Basic Res Cardiol. 2012;107:1–6. doi: 10.1007/s00395-012-0258-4. [DOI] [PubMed] [Google Scholar]
- 4.Hausenloy D.J., Boston-Griffiths E., Yellon D.M. Cardioprotection during cardiac surgery. Cardiovasc Res. 2012;94:253–265. doi: 10.1093/cvr/cvs131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yellon D.M., Downey J.M. Spotlight on preconditioning. Cardiovasc Res. 2002;55:425–428. doi: 10.1016/s0008-6363(02)00488-1. [DOI] [PubMed] [Google Scholar]
- 6.Yellon D.M., Downey J.M. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 7.Yellon D.M., Hausenloy D.J. Realizing the clinical potential of ischemic preconditioning and postconditioning. Nat Clin Pract Cardiovasc Med. 2005;2:568–575. doi: 10.1038/ncpcardio0346. [DOI] [PubMed] [Google Scholar]
- 8.Yellon D.M., Opie L.H. Postconditioning for protection of the infarcting heart. Lancet. 2006;367:456–458. doi: 10.1016/S0140-6736(06)68157-9. [DOI] [PubMed] [Google Scholar]
- 9.Yellon D.M., Hausenloy D.J. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 10.Piper H.M., Meuter K., Schafer C. Cellular mechanisms of ischemia-reperfusion injury. Ann Thorac Surg. 2003;75:S644–S648. doi: 10.1016/s0003-4975(02)04686-6. [DOI] [PubMed] [Google Scholar]
- 11.Piper H.M., Balser C., Ladilov Y.V. The role of Na+/H+ exchange in ischemia-reperfusion. Basic Res Cardiol. 1996;91:191–202. doi: 10.1007/BF00788905. [DOI] [PubMed] [Google Scholar]
- 12.Piper H.M., Garcia-Dorado D. Prime causes of rapid cardiomyocyte death during reperfusion. Ann Thorac Surg. 1999;68:1913–1919. doi: 10.1016/s0003-4975(99)01025-5. [DOI] [PubMed] [Google Scholar]
- 13.Piper H.M., Garcia-Dorado D., Ovize M. A fresh look at reperfusion injury. Cardiovasc Res. 1998;38:291–300. doi: 10.1016/s0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 14.Piper H.M., Siegmund B., Ladilov Y.V., Schluter K.D. Myocardial protection during reperfusion. Thorac Cardiovasc Surg. 1996;44:15–19. doi: 10.1055/s-2007-1011976. [DOI] [PubMed] [Google Scholar]
- 15.Piper H.M., Abdallah Y., Schafer C. The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res. 2004;61:365–371. doi: 10.1016/j.cardiores.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Piper H.M., Garcia-Dorado D. Cardiac protection takes off. Cardiovasc Res. 2009;83:163–164. doi: 10.1093/cvr/cvp186. [DOI] [PubMed] [Google Scholar]
- 17.Piper H.M., Garcia-Dorado D. Reducing the impact of myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2012;94 doi: 10.1093/cvr/cvs133. 167–7. [DOI] [PubMed] [Google Scholar]
- 18.Kung G., Konstantinidis K., Kitsis R.N. Programmed necrosis, not apoptosis, in the heart. Circ Res. 2011;108:1017–1036. doi: 10.1161/CIRCRESAHA.110.225730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jennings R.B. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013;113:428–438. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- 20.Jennings R.B., Steenbergen C., Jr., Reimer K.A. Myocardial ischemia and reperfusion. Monogr Pathol. 1995;37:47–80. [PubMed] [Google Scholar]
- 21.Jennings R.B., Reimer K.A. The cell biology of acute myocardial ischemia. Annu Rev Med. 1991;42:225–246. doi: 10.1146/annurev.me.42.020191.001301. [DOI] [PubMed] [Google Scholar]
- 22.Jennings R.B., Murry C.E., Steenbergen C., Jr., Reimer K.A. Development of cell injury in sustained acute ischemia. Circulation. 1990;82:II2–II12. [PubMed] [Google Scholar]
- 23.Jennings R.B., Murry C., Reimer K.A. Myocardial effects of brief periods of ischemia followed by reperfusion. Adv Cardiol. 1990;37:7–31. doi: 10.1159/000418813. [DOI] [PubMed] [Google Scholar]
- 24.Jennings R.B., Reimer K.A. Pathobiology of acute myocardial ischemia. Hosp Pract (Off Ed) 1989;24:89–101. doi: 10.1080/21548331.1989.11703644. 105. [DOI] [PubMed] [Google Scholar]
- 25.Sage M.D., Jennings R.B. Myocyte swelling and plasmalemmal integrity during early experimental myocardial ischemia in vivo. Scanning Microsc. 1988;2:477–484. [PubMed] [Google Scholar]
- 26.Steenbergen C., Hill M.L., Jennings R.B. Cytoskeletal damage during myocardial ischemia: changes in vinculin immunofluorescence staining during total in vitro ischemia in canine heart. Circ Res. 1987;60:478–486. doi: 10.1161/01.res.60.4.478. [DOI] [PubMed] [Google Scholar]
- 27.Jennings R.B., Reimer K.A., Steenbergen C. Myocardial ischemia revisited. The osmolar load, membrane damage, and reperfusion. J Mol Cell Cardiol. 1986;18:769–780. doi: 10.1016/s0022-2828(86)80952-x. [DOI] [PubMed] [Google Scholar]
- 28.Steenbergen C., Hill M.L., Jennings R.B. Volume regulation and plasma membrane injury in aerobic, anaerobic, and ischemic myocardium in vitro. Effects of osmotic cell swelling on plasma membrane integrity. Circ Res. 1985;57:864–875. doi: 10.1161/01.res.57.6.864. [DOI] [PubMed] [Google Scholar]
- 29.Reimer K.A., Jennings R.B., Tatum A.H. Pathobiology of acute myocardial ischemia: metabolic, functional and ultrastructural studies. Am J Cardiol. 1983;52:72A–81A. doi: 10.1016/0002-9149(83)90180-7. [DOI] [PubMed] [Google Scholar]
- 30.Mirzaei M., Truswell A.S., Taylor R., Leeder S.R. Coronary heart disease epidemics: not all the same. Heart. 2009;95:740–746. doi: 10.1136/hrt.2008.154856. [DOI] [PubMed] [Google Scholar]
- 31.Ganote C.E., Seabra-Gomes R., Nayler W.G., Jennings R.B. Irreversible myocardial injury in anoxic perfused rat hearts. Am J Pathol. 1975;80:419–450. [PMC free article] [PubMed] [Google Scholar]
- 32.Wijns W., Naber C.K. Reperfusion delay in patients with high-risk ST-segment elevation myocardial infarction: every minute counts, much more than suspected. Eur Heart J. 2018;39:1075–1077. doi: 10.1093/eurheartj/ehy069. [DOI] [PubMed] [Google Scholar]
- 33.Yannopoulos D., Bartos J.A., Raveendran G. Coronary arterial disease in patients with out-of-hospital refractory ventricular fibrillation cardiac arrest. J Am Coll Cardiol. 2017;70:1109–1117. doi: 10.1016/j.jacc.2017.06.059. [DOI] [PubMed] [Google Scholar]
- 34.Yannopoulos D., Bartos J.A., Martin C. Minnesota Resuscitation Consortium's Advanced Perfusion and Reperfusion Cardiac Life Support Strategy for Out-of-Hospital Refractory Ventricular Fibrillation. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003732. pii:e003732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank A., Bonney M., Bonney S., Weitzel L., Koeppen M., Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16:123–132. doi: 10.1177/1089253211436350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Dorado D., Barba I., Inserte J. Twenty-five years of preconditioning: are we ready for ischaemia? From coronary occlusion to systems biology and back. Cardiovasc Res. 2011;91:378–381. doi: 10.1093/cvr/cvr140. [DOI] [PubMed] [Google Scholar]
- 37.Schwartz L.L., Kloner R.A., Arai A.E. New horizons in cardioprotection: recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation. 2011;124:1172–1179. doi: 10.1161/CIRCULATIONAHA.111.032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Dorado D., Ruiz-Meana M., Piper H.M. Lethal reperfusion injury in acute myocardial infarction: facts and unresolved issues. Cardiovasc Res. 2009;83:165–168. doi: 10.1093/cvr/cvp185. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Dorado D. Myocardial reperfusion injury: a new view. Cardiovasc Res. 2004;61:363–364. doi: 10.1016/j.cardiores.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 40.Jennings R.B., Reimer K.A., Hill M.L., Mayer S.E. Total ischemia in dog hearts, in vitro. 1. Comparison of high energy phosphate production, utilization, and depletion, and of adenine nucleotide catabolism in total ischemia in vitro vs. severe ischemia in vivo. Circ Res. 1981;49:892–900. doi: 10.1161/01.res.49.4.892. [DOI] [PubMed] [Google Scholar]
- 41.Whelan R.S., Kaplinskiy V., Kitsis R.N. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martindale J.J., Metzger J.M. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J Mol Cell Cardiol. 2014;67:26–37. doi: 10.1016/j.yjmcc.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong S.W., Yao Y., Hong Y. Preventive effects of poloxamer 188 on muscle cell damage mechanics under oxidative stress. Ann Biomed Eng. 2017;45:1083–1092. doi: 10.1007/s10439-016-1733-0. [DOI] [PubMed] [Google Scholar]
- 44.See Hoe L.E., May L.T., Headrick J.P., Peart J.N. Sarcolemmal dependence of cardiac protection and stress-resistance: roles in aged or diseased hearts. Br J Pharmacol. 2016;173:2966–2991. doi: 10.1111/bph.13552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Padanilam J.T., Bischof J.C., Lee R.C. Effectiveness of poloxamer 188 in arresting calcein leakage from thermally damaged isolated skeletal muscle cells. Ann N Y Acad Sci. 1994;720:111–123. doi: 10.1111/j.1749-6632.1994.tb30439.x. [DOI] [PubMed] [Google Scholar]
- 46.Lee R.C., River L.P., Pan F.S., Ji L., Wollmann R.L. Surfactant-induced sealing of electropermeabilized skeletal muscle membranes in vivo. Proc Natl Acad Sci U S A. 1992;89:4524–4528. doi: 10.1073/pnas.89.10.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee R.C., Canaday D.J., Hammer S.M. Transient and stable ionic permeabilization of isolated skeletal muscle cells after electrical shock. J Burn Care Rehabil. 1993;14:528–540. doi: 10.1097/00004630-199309000-00007. [DOI] [PubMed] [Google Scholar]
- 48.Yasuda S., Townsend D., Michele D.E., Favre E.G., Day S.M., Metzger J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. doi: 10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- 49.Townsend D., Turner I., Yasuda S. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120:1140–1150. doi: 10.1172/JCI41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Houang E.M., Haman K.J., Kim M. Chemical end group modified diblock copolymers elucidate anchor and chain mechanism of membrane stabilization. Mol Pharm. 2017;14:2333–2339. doi: 10.1021/acs.molpharmaceut.7b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim M., Haman K.J., Houang E.M. PEO-PPO diblock copolymers protect myoblasts from hypo-osmotic stress in vitro dependent on copolymer size, composition, and architecture. Biomacromolecules. 2017;18:2090–2101. doi: 10.1021/acs.biomac.7b00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Houang E.M., Bates F.S., Sham Y.Y., Metzger J.M. All-atom molecular dynamics-based analysis of membrane-stabilizing copolymer interactions with lipid bilayers probed under constant surface tensions. J Phys Chem B. 2017;121:10657–10664. doi: 10.1021/acs.jpcb.7b08938. [DOI] [PubMed] [Google Scholar]
- 53.Houang E.M., Haman K.J., Filareto A. Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle. Mol Ther Methods Clin Dev. 2015;2:15042. doi: 10.1038/mtm.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benjamin E.J., Blaha M.J., Chiuve S.E. Heart disease and stroke statistics–2017 Update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murray C.J., Vos T., Lozano R. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- 56.Lozano R., Naghavi M., Foreman K. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helis E., Augustincic L., Steiner S., Chen L., Turton P., Fodor J.G. Time trends in cardiovascular and all-cause mortality in the “old” and “new” European Union countries. Eur J Cardiovasc Prev Rehabil. 2011;18:347–359. doi: 10.1177/1741826710389361. [DOI] [PubMed] [Google Scholar]
- 58.Mathers C.D., Boerma T., Ma F.D. Global and regional causes of death. Br Med Bull. 2009;92:7–32. doi: 10.1093/bmb/ldp028. [DOI] [PubMed] [Google Scholar]
- 59.Lopez A.D., Murray C.C. The global burden of disease, 1990–2020. Nat Med. 1998;4:1241–1243. doi: 10.1038/3218. [DOI] [PubMed] [Google Scholar]
- 60.Fordyce C.B., Gersh B.J., Stone G.W., Granger C.B. Novel therapeutics in myocardial infarction: targeting microvascular dysfunction and reperfusion injury. Trends Pharmacol Sci. 2015;36:605–616. doi: 10.1016/j.tips.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 61.McNeil P.L., Steinhardt R.A. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- 62.Miyake K., McNeil P.L. Mechanical injury and repair of cells. Crit Care Med. 2003;31:S496–S501. doi: 10.1097/01.CCM.0000081432.72812.16. [DOI] [PubMed] [Google Scholar]
- 63.McNeil P.L., Vogel S.S., Miyake K., Terasaki M. Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci. 2000;113(Pt 11):1891–1902. doi: 10.1242/jcs.113.11.1891. [DOI] [PubMed] [Google Scholar]
- 64.McNeil P.L., Steinhardt R.A. Loss, restoration, and maintenance of plasma membrane integrity. J Cell Biol. 1997;137:1–4. doi: 10.1083/jcb.137.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doherty K.R., McNally E.M. Repairing the tears: dysferlin in muscle membrane repair. Trends Mol Med. 2003;9:327–330. doi: 10.1016/s1471-4914(03)00136-9. [DOI] [PubMed] [Google Scholar]
- 66.Bansal D., Campbell K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 67.Cai C., Weisleder N., Ko J.K. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J Biol Chem. 2009;284:15894–15902. doi: 10.1074/jbc.M109.009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stenina-Adognravi O., Plow E.F. Thrombospondin-4 in tissue remodeling. Matrix Biol. 2019 doi: 10.1016/j.matbio.2017.11.006. 75-76:300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vanhoutte D., Schips T.G., Kwong J.Q. Thrombospondin expression in myofibers stabilizes muscle membranes. Elife. 2016;5 doi: 10.7554/eLife.17589. pii:e17589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lynch J.M., Maillet M., Vanhoutte D. A thrombospondin-dependent pathway for a protective ER stress response. Cell. 2012;149:1257–1268. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brody M.J., Schips T.G., Vanhoutte D. Dissection of thrombospondin-4 domains involved in intracellular adaptive endoplasmic reticulum stress-responsive signaling. Mol Cell Biol. 2016;36:2–12. doi: 10.1128/MCB.00607-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frolova E.G., Sopko N., Blech L. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012;26:2363–2373. doi: 10.1096/fj.11-190728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cingolani O.H., Kirk J.A., Seo K. Thrombospondin-4 is required for stretch-mediated contractility augmentation in cardiac muscle. Circ Res. 2011;109:1410–1414. doi: 10.1161/CIRCRESAHA.111.256743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McNeil P.L., Kirchhausen T. An emergency response team for membrane repair. Nat Rev Mol Cell Biol. 2005;6:499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
- 75.Bansal D., Miyake K., Vogel S.S. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 76.Blazek A.D., Paleo B.J., Weisleder N. Plasma membrane repair: a central process for maintaining cellular homeostasis. Physiology (Bethesda ) 2015;30:438–448. doi: 10.1152/physiol.00019.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cai C., Masumiya H., Weisleder N. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kabanov A.V., Batrakova E.V., Alakhov V.Y. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82:189–212. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 79.Guler N., Abro S., Emanuele M., Iqbal O., Hoppensteadt D., Fareed J. The protective effect of poloxamer-188 on platelet functions. Clin Appl Thromb Hemost. 2017;23:987–991. doi: 10.1177/1076029616669785. [DOI] [PubMed] [Google Scholar]
- 80.Emanuele R.M. FLOCOR: a new anti-adhesive, rheologic agent. Expert Opin Investig Drugs. 1998;7:1193–1200. doi: 10.1517/13543784.7.7.1193. [DOI] [PubMed] [Google Scholar]
- 81.Adams-Graves P., Kedar A., Koshy M. RheothRx (poloxamer 188) injection for the acute painful episode of sickle cell disease: a pilot study. Blood. 1997;90:2041–2046. [PubMed] [Google Scholar]
- 82.Go A.S., Mozaffarian D., Roger V.L. Heart disease and stroke statistics—2014 update: a report from the american heart association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hausenloy D.J., Yellon D.M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gershlick A.H., Banning A.P., Myat A., Verheugt F.W., Gersh B.J. Reperfusion therapy for STEMI: is there still a role for thrombolysis in the era of primary percutaneous coronary intervention? Lancet. 2013;382:624–632. doi: 10.1016/S0140-6736(13)61454-3. [DOI] [PubMed] [Google Scholar]
- 85.O'Gara P.T., Kushner F.G., Ascheim D.D. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the American College of Emergency Physicians and Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol. 2013;62:485–510. [Google Scholar]
- 86.Braunwald E. The rise of cardiovascular medicine. Eur Heart J. 2012;33:838–845. doi: 10.1093/eurheartj/ehr452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McMurray J.J. Clinical practice. Systolic heart failure. N Engl J Med. 2010;362:228–238. doi: 10.1056/NEJMcp0909392. [DOI] [PubMed] [Google Scholar]
- 88.Aronow W.S. Heart disease and aging. Med Clin North Am. 2006;90:849–862. doi: 10.1016/j.mcna.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 89.Kass R.S., Lindegger N., Hagen B., Lederer W.J. Another calcium paradox in heart failure. J Mol Cell Cardiol. 2008;45:28–31. doi: 10.1016/j.yjmcc.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 90.Yannopoulos D., Halperin H.R. During CPR, push hard and fast and please do not stop! Resuscitation. 2011;82:1475–1476. doi: 10.1016/j.resuscitation.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 91.Yannopoulos D., Aufderheide T. Acute management of sudden cardiac death in adults based upon the new CPR guidelines. Europace. 2007;9:2–9. doi: 10.1093/europace/eul126. [DOI] [PubMed] [Google Scholar]
- 92.Rickenbacher P., Pfisterer M., Burkard T. Why and how do elderly patients with heart failure die? Insights from the TIME-CHF study. Eur J Heart Fail. 2012;14:1218–1229. doi: 10.1093/eurjhf/hfs113. [DOI] [PubMed] [Google Scholar]
- 93.Kontos M.C., Rennyson S.L., Chen A.Y., Alexander K.P., Peterson E.D., Roe M.T. The association of myocardial infarction process of care measures and in-hospital mortality: a report from the NCDR(R) Am Heart J. 2014;168:766–775. doi: 10.1016/j.ahj.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 94.Doost H.A., Moloi S., Chandrasekhar J., Farshid A. Mortality pattern and cause of death in a long-term follow-up of patients with STEMI treated with primary PCI. Open Heart. 2016;3:e000405. doi: 10.1136/openhrt-2016-000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frohlich G.M., Meier P., White S.K., Yellon D.M., Hausenloy D.J. Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J. 2013;34:1714–1722. doi: 10.1093/eurheartj/eht090. [DOI] [PubMed] [Google Scholar]
- 96.Armstrong P.W., Gershlick A.H., Van de Werf F. Fibrinolysis or primary PCI in myocardial infarction. N Engl J Med. 2013;369:280–281. doi: 10.1056/NEJMc1305999. [DOI] [PubMed] [Google Scholar]
- 97.Maskarinec S.A., Hannig J., Lee R.C., Lee K.Y. Direct observation of poloxamer 188 insertion into lipid monolayers. Biophys J. 2002;82:1453–1459. doi: 10.1016/S0006-3495(02)75499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frey S.L., Zhang D., Carignano M.A., Szleifer I., Lee K.Y. Effects of block copolymer's architecture on its association with lipid membranes: experiments and simulations. J Chem Phys. 2007;127:114904. doi: 10.1063/1.2768947. [DOI] [PubMed] [Google Scholar]
- 99.Wu G., Lee K.Y. Interaction of poloxamers with liposomes: an isothermal titration calorimetry study. J Phys Chem B. 2009;113:15522–15531. doi: 10.1021/jp906331m. [DOI] [PubMed] [Google Scholar]
- 100.Wu G., Majewski J., Ege C., Kjaer K., Weygand M.J., Lee K.Y. Interaction between lipid monolayers and poloxamer 188: an x-ray reflectivity and diffraction study. Biophys J. 2005;89:3159–3173. doi: 10.1529/biophysj.104.052290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang J.Y., Chin J., Marks J.D., Lee K.Y. Effects of PEO-PPO-PEO triblock copolymers on phospholipid membrane integrity under osmotic stress. Langmuir. 2010;26:12953–12961. doi: 10.1021/la101841a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bates F.S., Hillmyer M.A., Lodge T.P., Bates C.M., Delaney K.T., Fredrickson G.H. Multiblock polymers: panacea or Pandora's box? Science. 2012;336:434–440. doi: 10.1126/science.1215368. [DOI] [PubMed] [Google Scholar]
- 103.Orringer E.P., Casella JF, Ataga K.I. Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease: a randomized controlled trial. JAMA. 2001;286:2099–2106. doi: 10.1001/jama.286.17.2099. [DOI] [PubMed] [Google Scholar]
- 104.Ballas S.K., Files B., Luchtman-Jones L. Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin. 2004;28:85–102. doi: 10.1081/hem-120035919. [DOI] [PubMed] [Google Scholar]
- 105.Toth K., Bogar L., Juricskay I. The effect of RheothRx injection on the hemorheological parameters in patients with acute myocardial infarction. Clin Hemorheol Microcirc. 1997;17:117–125. [PubMed] [Google Scholar]
- 106.Chareonthaitawee P., Gibbons R.J., Roberts R.S., Christian T.F., Burns R., Yusuf S. The impact of time to thrombolytic treatment on outcome in patients with acute myocardial infarction. For the CORE investigators (Collaborative Organisation for RheothRx Evaluation) Heart. 2000;84:142–148. doi: 10.1136/heart.84.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schaer G.L., Spaccavento L.J., Browne K.F. Beneficial effects of RheothRx injection in patients receiving thrombolytic therapy for acute myocardial infarction. Results of a randomized, double-blind, placebo-controlled trial. Circulation. 1996;94:298–307. doi: 10.1161/01.cir.94.3.298. [DOI] [PubMed] [Google Scholar]
- 108.Kelly R.F., Hursey T.L., Patel R.B., Parrillo J.E., Schaer G.L. Effect of poloxamer 188 on collateral blood flow, myocardial infarct size, and left ventricular function in a canine model of prolonged (3-hour) coronary occlusion and reperfusion. J Thromb Thrombolysis. 1998;5:239–247. doi: 10.1023/A:1008848026759. [DOI] [PubMed] [Google Scholar]
- 109.O'Keefe J.H., Grines C.L., DeWood M.A. Poloxamer-188 as an adjunct to primary percutaneous transluminal coronary angioplasty for acute myocardial infarction. Am J Cardiol. 1996;78:747–750. doi: 10.1016/s0002-9149(96)00414-6. [DOI] [PubMed] [Google Scholar]
- 110.Schaer G.L., Hursey T.L., Abrahams S.L. Reduction in reperfusion-induced myocardial necrosis in dogs by RheothRx injection (poloxamer 188 N.F.), a hemorheological agent that alters neutrophil function. Circulation. 1994;90:2964–2975. doi: 10.1161/01.cir.90.6.2964. [DOI] [PubMed] [Google Scholar]
- 111.Grindel J.M., Jaworski T., Piraner O., Emanuele R.M., Balasubramanian M. Distribution, metabolism, and excretion of a novel surface-active agent, purified poloxamer 188, in rats, dogs, and humans. J Pharm Sci. 2002;91:1936–1947. doi: 10.1002/jps.10190. [DOI] [PubMed] [Google Scholar]
- 112.Grindel J.M., Jaworski T., Emanuele R.M., Culbreth P. Pharmacokinetics of a novel surface-active agent, purified poloxamer 188, in rat, rabbit, dog and man. Biopharm Drug Dispos. 2002;23:87–103. doi: 10.1002/bdd.297. [DOI] [PubMed] [Google Scholar]
- 113.Emanuele M., Balasubramaniam B. Differential effects of commercial-grade and purified poloxamer 188 on renal function. Drugs R D. 2014;14:73–83. doi: 10.1007/s40268-014-0041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Greenebaum B., Blossfield K., Hannig J. Poloxamer 188 prevents acute necrosis of adult skeletal muscle cells following high-dose irradiation. Burns. 2004;30:539–547. doi: 10.1016/j.burns.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 115.Collins J.M., Despa F., Lee R.C. Structural and functional recovery of electropermeabilized skeletal muscle in-vivo after treatment with surfactant poloxamer 188. Biochim Biophys Acta. 2007;1768:1238–1246. doi: 10.1016/j.bbamem.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Coatney G.A., Abraham A.C., Fischenich K.M., Button K.D., Haut R.C., Haut Donahue T.L. Efficacy of P188 on lapine meniscus preservation following blunt trauma. J Mech Behav Biomed Mater. 2015;47:57–64. doi: 10.1016/j.jmbbm.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Riehm J.J., Wang L., Ghadge G. Poloxamer 188 decreases membrane toxicity of mutant SOD1 and ameliorates pathology observed in SOD1 mouse model for ALS. Neurobiol Dis. 2018;115:115–126. doi: 10.1016/j.nbd.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Serbest G., Horwitz J., Jost M., Barbee K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006;20:308–310. doi: 10.1096/fj.05-4024fje. [DOI] [PubMed] [Google Scholar]
- 119.Bao H., Yang X., Zhuang Y. The effects of poloxamer 188 on the autophagy induced by traumatic brain injury. Neurosci Lett. 2016;634:7–12. doi: 10.1016/j.neulet.2016.09.052. [DOI] [PubMed] [Google Scholar]
- 120.Shelat P.B., Plant L.D., Wang J.C., Lee E., Marks J.D. The membrane-active tri-block copolymer pluronic F-68 profoundly rescues rat hippocampal neurons from oxygen-glucose deprivation-induced death through early inhibition of apoptosis. J Neurosci. 2013;33:12287–12299. doi: 10.1523/JNEUROSCI.5731-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Townsend D., Yasuda S., Metzger J. Cardiomyopathy of Duchenne muscular dystrophy: pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert Rev Cardiovasc Ther. 2007;5:99–109. doi: 10.1586/14779072.5.1.99. [DOI] [PubMed] [Google Scholar]
- 122.Markham B.E., Kernodle S., Nemzek J., Wilkinson J.E., Sigler R. Chronic dosing with membrane sealant poloxamer 188 NF improves respiratory dysfunction in dystrophic Mdx and Mdx/utrophin-/- Mice. PLoS One. 2015;10:e0134832. doi: 10.1371/journal.pone.0134832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Borgens R.B., Bohnert D., Duerstock B., Spomar D., Lee R.C. Subcutaneous tri-block copolymer produces recovery from spinal cord injury. J Neurosci Res. 2004;76:141–154. doi: 10.1002/jnr.20053. [DOI] [PubMed] [Google Scholar]
- 124.Adams J.E., Owens G., Mann G., Headrick J.R., Munoz A., Scott H.W., Jr. Experimental evaluation of pluronic F68 (a non-ionic detergent) as a method of diminishing systemic fat emboli resulting from prolonged cardiopulmonary bypass. Surg Forum. 1960;10:585–589. [PubMed] [Google Scholar]
- 125.Miyauchi Y., Inoue T., Paton B.C. Adjunctive use of a surface-active agent in extracorporeal circulation. Circulation. 1966;33:I71–I77. doi: 10.1161/01.cir.33.4s1.i-71. [DOI] [PubMed] [Google Scholar]
- 126.Hymes A.C., Safavian M.H., Gunther T. The influence of an industrial surfactant Pluronic F-68, in the treatment of hemorrhagic shock. J Surg Res. 1971;11:191–197. doi: 10.1016/0022-4804(71)90060-6. [DOI] [PubMed] [Google Scholar]
- 127.Hymes A.C., Robb H.J., Margulis R.R. Influence of an industrial surfactant (pluronic F-68) on human amniotic fluid embolism. Am J Obstete Gynecol. 1970;107:1217–1222. doi: 10.1016/s0002-9378(15)30372-0. [DOI] [PubMed] [Google Scholar]
- 128.Hymes A.C., Beck K. A comparison of pluronic F-68, low molecular weight dextran, mannitol, and saline as priming agents in the heart-lung apparatus. II. The in vitro influence on oxygen consumption of certain fluids used as priming agents in a heart-lung bypass apparatus. J Thorac Cardiovasc Surg. 1968;56:23–27. [PubMed] [Google Scholar]
- 129.Hymes A.C., Safavian M.H., Arbulu A., Baute P. A comparison of pluronic F-68, low molecular weight dextran, mannitol, and saline as priming agents in the heart-lung apparatus. I. Pluronic F-68: first use as a plasma substitute. J Thorac Cardiovasc Surg. 1968;56:16–22. [PubMed] [Google Scholar]
- 130.Colbassani H.J., Barrow D.L., Sweeney K.M., Bakay R.A., Check I.J., Hunter R.L. Modification of acute focal ischemia in rabbits by poloxamer 188. Stroke. 1989;20:1241–1246. doi: 10.1161/01.str.20.9.1241. [DOI] [PubMed] [Google Scholar]
- 131.Gu J.H., Ge J.B., Li M., Xu H.D., Wu F., Qin Z.H. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS One. 2013;8:e61641. doi: 10.1371/journal.pone.0061641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Marks J.D., Pan C.Y., Bushell T., Cromie W., Lee R.C. Amphiphilic, tri-block copolymers provide potent membrane-targeted neuroprotection. FASEB J. 2001;15:1107–1109. doi: 10.1096/fj.00-0547fje. [DOI] [PubMed] [Google Scholar]
- 133.Wang T., Chen X., Wang Z. Poloxamer-188 can attenuate blood-brain barrier damage to exert neuroprotective effect in mice intracerebral hemorrhage model. J Mol Neurosci. 2015;55:240–250. doi: 10.1007/s12031-014-0313-8. [DOI] [PubMed] [Google Scholar]
- 134.Justicz A.G., Farnsworth W.V., Soberman M.S. Reduction of myocardial infarct size by poloxamer 188 and mannitol in a canine model. Am Heart J. 1991;122:671–680. doi: 10.1016/0002-8703(91)90510-o. [DOI] [PubMed] [Google Scholar]
- 135.Carr M.E., Jr., Powers P.L., Jones M.R. Effects of poloxamer 188 on the assembly, structure and dissolution of fibrin clots. Thromb Haemost. 1991;66:565–568. [PubMed] [Google Scholar]
- 136.Effects of RheothRx on mortality, morbidity, left ventricular function, and infarct size in patients with acute myocardial infarction. Collaborative Organization for RheothRx Evaluation (CORE) Circulation. 1997;96:192–201. [PubMed] [Google Scholar]
- 137.Bartos J.A., Matsuura T.R., Tsangaris A. Intracoronary poloxamer 188 prevents reperfusion injury in a porcine model of ST-segment elevation myocardial infarction. J Am Coll Cardiol Basic Trans Science. 2016;1:224–234. doi: 10.1016/j.jacbts.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bartos J.A., Matsuura T.R., Sarraf M. Bundled postconditioning therapies improve hemodynamics and neurologic recovery after 17 min of untreated cardiac arrest. Resuscitation. 2015;87:7–13. doi: 10.1016/j.resuscitation.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim M., Vala M., Ertsgaard C.T. Surface plasmon resonance study of the binding of PEO-PPO-PEO triblock copolymer and PEO homopolymer to supported lipid bilayers. Langmuir. 2018;34:6703–6712. doi: 10.1021/acs.langmuir.8b00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mbye L.H., Keles E., Tao L. Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2012;32:515–524. doi: 10.1038/jcbfm.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sengupta A., Dwivedi N., Kelly S.C., Tucci L., Thadhani N.N., Prausnitz M.R. Poloxamer surfactant preserves cell viability during photoacoustic delivery of molecules into cells. Biotechnol Bioeng. 2015;112:405–415. doi: 10.1002/bit.25363. [DOI] [PubMed] [Google Scholar]
- 142.Cheng C.Y., Wang J.Y., Kausik R., Lee K.Y., Han S. An ultrasensitive tool exploiting hydration dynamics to decipher weak lipid membrane-polymer interactions. J Magn Reson. 2012;215:115–119. doi: 10.1016/j.jmr.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 143.Cheng C.Y., Wang J.Y., Kausik R., Lee K.Y., Han S. Nature of interactions between PEO-PPO-PEO triblock copolymers and lipid membranes: (II) role of hydration dynamics revealed by dynamic nuclear polarization. Biomacromolecules. 2012;13:2624–2633. doi: 10.1021/bm300848c. [DOI] [PubMed] [Google Scholar]
- 144.Zhang W., Haman K.J., Metzger J.M., Hackel B.J., Bates F.S., Lodge T.P. Quantifying binding of ethylene oxide-propylene oxide block copolymers with lipid bilayers. Langmuir. 2017;33:12624–12634. doi: 10.1021/acs.langmuir.7b02279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu G., Majewski J., Ege C., Kjaer K., Weygand M.J., Lee K.Y. Lipid corralling and poloxamer squeeze-out in membranes. Phys Rev Lett. 2004;93:028101. doi: 10.1103/PhysRevLett.93.028101. [DOI] [PubMed] [Google Scholar]
- 146.Wang J.Y., Marks J., Lee K.Y. Nature of interactions between PEO-PPO-PEO triblock copolymers and lipid membranes: (I) effect of polymer hydrophobicity on its ability to protect liposomes from peroxidation. Biomacromolecules. 2012;13:2616–2623. doi: 10.1021/bm300847x. [DOI] [PMC free article] [PubMed] [Google Scholar]