

Summary
Background
Treatments immediately after spinal cord injury (SCI) are anticipated to decrease neuronal death, disruption of neuronal connections, demyelination, and inflammation, and to improve repair and functional recovery. Currently, little can be done to modify the acute phase, which extends to the first 48 hours post‐injury. Efforts to intervene have focused on the subsequent phases — secondary (days to weeks) and chronic (months to years) — to both promote healing, prevent further damage, and support patients suffering from SCI.
Methods
We used a contusion model of SCI in female mice, and delivered a small molecule reagent during the early phase of injury. Histological and behavioral outcomes were assessed and compared.
Results
We find that the reagent Pifithrin‐μ (PFT‐μ) acts early and directly on microglia in vitro, attenuating their activation. When administered during the acute phase of SCI, PFT‐μ resulted in reduced lesion size during the initial inflammatory phase, and reduced the numbers of pro‐inflammatory microglia and macrophages. Treatment with PFT‐μ during the early stage of injury maintained a stable anti‐inflammatory environment.
Conclusions
Our results indicate that a small molecule reagent PFT‐μ has sustained immunomodulatory effects following a single dose after injury.
Keywords: 2‐phenylethynesulfonamide, cell death, inflammation, mice, PES, PFT‐μ, phagocytosis, spinal cord
1. INTRODUCTION
The National Spinal Cord Injury Statistics Center estimates that ~17 000 new cases of spinal cord injury (SCI) occur in the United States each year.1 This permanent debilitating condition results in acute necrotic death of neurons and glia at the site of injury and disrupts the conduction of sensory and motor signals between the brain and the body.36 The initial injury triggers a series of biochemical and cellular events that mediate secondary apoptotic cell death, cause demyelination, and prompt an immune response.4, 37
Microglia, the resident immune cells of the CNS, play a central role in the inflammatory response that follows SCI. Subsequent gene induction causes microglia to quickly migrate to the site of damage to remove cell debris and release pro‐inflammatory cytokines which recruit additional microglia/macrophages to the injury site.5 Following injury, microglia adopt differential phenotypes,6 which variably contribute to pathology. The pro‐inflammatory “M1‐like” state is characterized by the production of TNF‐α, IL‐1β, and IL‐6,7, 8 and the anti‐inflammatory “M2‐like” state is characterized by the production of IL‐10, Arginase 1 (Arg 1), and transforming growth factor‐β (TGF‐ β).9, 10 In several injury models, the shift toward an M2‐like population has been deemed beneficial for cell regeneration and remyelination of damaged axons,11, 12 while the pro‐inflammatory phenotype is considered detrimental for tissue healing and repair.
Several anti‐inflammatory drugs have been used in animal studies to reduce the activation of pro‐inflammatory microglia and improve disease outcome. Our lab has shown that targeting microglial function during the secondary phase of SCI (ie, days to weeks after injury) improves the injury outcome by attenuating microglial activation and enhancing oligodendrocyte differentiation and remyelination.13, 14 Despite this success, there still remains no option to treat SCI during the early phase (ie, first 48 hours). In this work, we assess the anti‐inflammatory effects of 2‐phenylethynesulfonamide, a small molecule inhibitor also known as Pifithrin‐ μ (PFT‐μ), and propose its therapeutic use during the early phase of SCI.
Pifithrin‐ μ was isolated from a library of compounds known to modulate apoptosis15 and was shown to inhibit the association of p53 with mitochondria by reducing its affinity for apoptotic proteins BcL‐xL and Bcl‐2 without affecting the transcriptional activity of p53.15 Inhibiting the association of p53 with mitochondria using PFT‐μ reduced mitochondrial release of cytochrome c, caspase‐3 activation, lesion size, and neuronal damage in a model of perinatal hypoxic‐ischemic brain damage.16 In this model, PFT‐μ had a therapeutic window of 6 hours, indicating that it acted acutely (ie, during the first few hours post‐injury) to produce these beneficial effects. PFT‐μ has also been shown to inhibit HSP70 substrate binding and decrease autophagic flux. The protein LC3 is responsible for forming autophagosomes, and is mediated by HSP70 activity.17 Furthermore, it has been recognized that LC3‐associated phagocytosis is mediated by TLR receptor activation and recognition of cellular apoptosis.18
Here we show that PFT‐μ treatment modulated the inflammatory response of microglia directly in culture by reducing the levels of pro‐inflammatory cytokines, decreasing aberrant phagocytosis, and permitting stromal cell wound closure. We extended these experiments in vivo, in a contusion model of SCI, and show that treatment with PFT‐μ reduced the pro‐inflammatory responses that accompany the acute phase of injury. These results support further investigation of PFT‐μ as an anti‐inflammatory agent and suggest studies for its use in SCI.
2. METHODS
2.1. Contusion model of spinal cord injury
Two‐month female C57BL/6 (wild‐type) or MacGreen (Csf1R‐EGFP) mice on the C57BL/6 background were used for this study.19 Female animals were used because both in our experience and in the literature females survive SCI and recover locomotor function much better than male animals.20 The Csf1R‐EGFP animals21 express EGFP under the control of the microglia/macrophage Colony Stimulating Factor 1 Receptor promoter, allow for easy (EGFP) identification of microglia/macrophages within the complex injury environment, and offer an advantage to immunohistochemistry. Mice were anesthetized with isoflurane and a dorsal laminectomy was performed between thoracic levels 8‐10. An impactor tip 1.25 mm in diameter was applied from a height of 2.5 cm at a force of 50 kdynes using an Infinite Horizon Impactor (Precision Systems and Instrumentation).22 The overlying muscle and skin were sutured. Sham‐operated groups underwent laminectomy without contusion. Postoperatively, mice were injected with buprenorphine (0.03 mg/kg) to reduce pain and placed on a heating pad overnight. Bladder expression was performed twice daily or until the mice regained bladder control. Mice were injected with a 5% dextrose/saline solution to prevent extreme weight loss during the first week following surgery. Animals that dropped more than 10% of their body weight, or that showed signs of limb or tail autophagy, were removed from the analysis.
2.2. Drug administration
Pifithrin‐ μ (Sigma‐Aldrich, Steinheim, Germany) was administered intraperitoneally at a dose of 8 mg/kg body weight (dose based on previous studies16). The drug was diluted in 4% DMSO in phosphate buffered saline (PBS). Control mice received an injection of 4% DMSO in PBS. Mice were treated with PFT‐μ immediately after SCI.
2.3. Perfusion and tissue collection
Mice were deeply anesthetized with 2.5% avertin and transcardially perfused with PBS followed by 4% PFA. The spinal cords were collected, post‐fixed, and transferred to a 30% sucrose solution for cryoprotection. The spinal cords were frozen in OCT and cut into 20‐μm‐thick slices.
2.4. Immunofluorescence
Sections were blocked for one hour with blocking solution (3% BSA in PBS) and then incubated with primary antibodies overnight at 4°C. The primary antibodies used were: Collagen IV (Abcam, 1:500), GFAP (Cell Signaling, 1:1000), CD206 (Cell Signaling, 1:100), and CD86 (Cell Signaling, 1:100). Sections were washed and incubated with secondary antibodies for one hour. Slides were washed and mounted with DAPI fluoromount.
2.5. Terminal deoxynucleotidyl transferase dUTP nick end labeling staining
Transferase dUTP nick end labeling (TUNEL) staining was performed as per the manufacturers protocol (ROCHE #11767291910). Tissue was collected at 24 hours post‐injury and prepared as above. Tissue sections were washed in PBS, permeabilized in 0.1% Triton X‐100/0.1% sodium citrate, blocked using 3% normal goat serum in PBST, and were then incubated with TUNEL reaction mixture (5‐μL TUNEL‐Enzyme + 45 μL TUNEL‐Label mix) or TUNEL‐label mix alone for negative control at 37.5°C for 1 hour. Slides were mounted with DAPI Fluoromount‐G (Southern Biotech). FITC+ cells ± 800 μm from the injury epicenter were quantified.
2.6. Subcellular fractionation
Subcellular fractionation was performed as previously described.16, 23 In brief, ~10 mm of spinal cord tissue surrounding the injury was dissected from mice 24 hours after contusion SCI. The tissue was homogenized and suspended in ice cold buffer containing 70 mmol/L sucrose, 210 mmol/L mannitol, 5 mmol/L HEPES, 1 mmol/L EDTA, and protease inhibitors, and was adjusted to pH 7.35 with KOH. Fractionation by centrifugation was conducted, and cytoplasmic or mitochondrial supernatants were collected and the total protein concentration was quantified using the Pierce BCA protein assay kit (Thermo Scientific #23225). Supernatants were stored at −80°C until use.
2.7. Immunoblotting
Protein samples were separated via SDS‐PAGE and transferred to a PVDF membrane. Membranes were blocked with 3% non‐fat milk at room temperature for 1 hour and then incubated with primary antibodies overnight: moue anti‐p53 (CST #2524), goat anti‐HSP60 (sc‐1052), mouse anti‐Cytochrome C (BD #556433), Rabbit anti‐total Caspase 9 (CST #9504), mouse anti‐beta actin (Sigma #A5441). Membranes were washed in TBST and incubated with secondary antibodies conjugated with HRP. SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific #34087) was used to detect relative amounts of proteins of interest and developed on autoradiography film (Denville Scientific). Densitometry was performed using Fiji.24
2.8. Quantification of lesion volume
To analyze lesion volume, alternating sagittal spinal cord sections were stained with the astrocytic marker GFAP or Collagen IV. Lesion size was analyzed from single plane confocal images of five alternate sections that had been immunofluorescently stained as described above. ImageJ software was used to calculate the area occupied by the lesion, which was defined as the area bordered by GFAP+ signal. In the case of Collagen IV, the lesion site was defined as the area occupied by positive staining. Data from each time point and treatment group were compared using the two‐way ANOVA statistical method.
2.9. Quantification of microglial infiltration into the lesion site
Tissue sections from injured MacGreen mice were stained for Collagen IV to identify the injury epicenter. Within the delineated injury, the area of E‐GFP+ signal was quantified by a blind observer, and the relative fluorescence intensity was calculated using ImageJ. For co‐labeling experiments, microglia/macrophage cells positive for immunostaining and DAPI were quantified within the injury epicenter and the average cell number, per biological replicate, was plotted. The ratio of average CD206+ to CD86+ cells was then compared. Data from each time point and treatment group were compared using the one‐way ANOVA statistical method.
2.10. Imaging
Immunofluorescent‐stained sections were photographed at a digital resolution of 1024 × 1024 with either a Zeiss confocal microscope using LSM 510 Meta software, or a Leica SP8x confocal microscope using LASX software.
2.11. Primary neonatal cell cultures
Cerebral cortices from postnatal day 1 C57BL/6J mice were dissected, digested with trypsin (0.25% in HBSS) for 15 minutes at 37°C, and mechanically dissociated by trituration as described elsewhere.25 Mixed cortical cells were plated in DMEM medium, 10% FBS, 1% sodium pyruvate, and gentamycin on poly‐D‐lysine‐coated tissue culture plates. After 10 days, microglial cells were separated from the astrocytic monolayer by the addition of 12 mM lidocaine, and the isolated microglia were seeded on 24 well plates at a density of 15 000 cells/mL for experiments. Astrocytic monolayers were shaken to remove oligodendrocyte progenitors, and the remaining mixed cortical cells were seeded in 6 well plates and grown to confluency to be used for scratch wound assays as previously described.26
2.12. Enzyme‐linked immunosbsorbent assay
Twenty‐four hours after plating, microglia were treated with PFT‐μ at doses of 1, 3, and 5 μM. LPS was used as a positive control because it is a potent inducer of pro‐inflammatory markers in microglia.27 Approximately six hours after treatment, the media from the cells were collected, spun down, and used for Enzyme‐linked immunosbsorbent assay (ELISA). Levels of TNF‐α, and IL‐10 were determined using the eBiosciences quantitative sandwich enzyme immunoassay following the manufacturer's protocol. Briefly, 96 well plates were coated with diluted Capture antibody and incubated overnight at 4°C. The plates were blocked for one hour at room temperature followed by a two‐hour incubation with standard or sample. The wells were then incubated for 1 hour with working detector solution followed by incubation with substrate solution for 30 minutes. The reaction was stopped with 50 μL of 1N H2SO4. The absorbance of each well was read at a 450 nm light wavelength.
2.13. Scratch wound assay
Microglia conditioned medium (MGCM) was collected from the N9 microglial cell line after culturing the cells for 24 hours in DMEM, 10% FBS, 1% penicillin‐streptomycin, and kept at 5% CO2 and 37°C. The media collected were either the control media (MGCM) or media generated after different treatments of the cells, either with 200 ng/mL LPS (MGCM + LPS) or with both LPS and 5 μM PFT‐μ (MGCM + LPS + PFT‐μ). LPS treatment of N9 cells results in a robust pro‐inflammatory response, analogous to cultures of primary microglia,28 and the use of this cell line allowed for the collection of larger volumes of media to be used for experiments. Cells were preincubated for 2 hours with PFTμ prior to the addition of LPS. Conditioned media were collected, spun down to remove dead cells and debris, and stored at −80°C.
Reference lines were etched horizontally into the bottom of 6 well plates using a razor. Scratches were then made, perpendicularly, within the confluent mixed cortical cultures (MCC). Three horizontal etches and three vertical scratch wounds made a total of 9 intersecting regions of interest within each well. After scratch induction, MCCs were treated with either control media, 5 μM PFTμ, MGCM, MGCM + LPS, MGCM + LPS + PFTu, or MGCM + LPS with the addition of 5 μM PFTμ to the MCC. After 48 hours of incubation, cells were fixed in 4% PFA, blocked with 3% NGS/PBST, and immunostained with GFAP (Cell Signaling, 1:1000) and Texas Red‐X Phalloidin (Invitrogen #T7471). Aqueous DAPI was added to the wells for 10 minutes and the wells were then washed with PBS. Well plates were imaged using a Zeiss Axiovert 200 M inverted microscope. One‐way ANOVA was used to compare treatment groups. Nine images were taken per experiment, which was independently replicated three times.
2.14. Phagocytosis assay
Adult primary microglia from Macgreen mice were sorted from cortical tissue based on GFP fluorescence and cultured in DMEM, 10% FBS, 1% Anti‐Anti, 1% Sodium pyruvate, and 40 μg/mL Gentamycin and kept at 5% CO2 and 37°C. Cells were seeded on coverslips for 24 hours and preincubated for 2 hours with 5 μM PFTμ prior to the addition of 100 ng/mL LPS. Cells were treated for 24 hours and then incubated with a final concentration of 0.1 μg/mL red fluorescent beads for 2 hours (Latex beads, carboxylate‐modified polystyrene, fluorescent red. 0.5‐μm mean particle size. Sigma, L3280). Coverslips were mounted with DAPI, and 40× confocal images were taken for analysis. At least 50 cells per group were imaged in each experiment. The experiment was independently replicated three times.
2.15. Animal behavior — Rotarod test
Motor performance was assessed in intact and injured mice using a Rotarod (Med Associates, Inc.) as described elsewhere.14 Briefly, mice were placed on a moving rod that accelerated from 4‐40 rpm over the course of 5 minutes. Once the mouse fell off, both the speed of the rod and the latency to fall were automatically recorded by the apparatus. Mice were subjected to three consecutive trials and the average of the trials was used for subsequent analysis. Rotarod performance was recorded the day before injury and then again at 7, 14, 21, and 30 dpi. No significant weight differences were noted between vehicle‐ and PFTμ ‐treated animals.
2.16. Animal behavior — Footprint analysis
The footprint test was used to assess gait and posture in spinal cord injured animals. To obtain footprints, both the forelimbs and the hind limbs of the mice were coated with black non‐toxic ink. The animals were allowed to walk along a 50‐cm‐long, 10‐cm‐wide strip of filter paper bounded by 10‐cm‐high walls as described elsewhere.29, 30 Forelimb and hind limb prints were collected in sham mice and in SCI mice at 7 and 30 dpi. In the case of sham mice, the rear track width was measured (in centimeters) as the perpendicular distance from a given left hind limb print to a line connecting its proceeding right hind limb print. Five consecutive hind limb prints were measured and the average was calculated to obtain stride width. After injury, the mice produced hind limb foot tracks instead of hind limb prints due to paralysis. In this case, rear track width was measured as the distance between foot tracks on five representative points along the traveled distance. The average value was calculated for analysis.
2.17. Experimental design and statistical analysis
All statistics were performed using Statview (v4.0), Graphpad Prism 6 for Windows, GraphPad Software (http://www.graphpad.com) or R (https://www.r-project.org/). Image analysis was conducted using ImageJ (https://imagej.nih.gov/ij/) or Fiji (https://imagej.net/Fiji) software. All image analyses were conducted by an observer blinded to the treatment groups. Data were considered statistically significant when P < .05. For GFAP lesion immunohistochemistry experiments, adult female mice were used; at 7 dpi; Vehicle n = 9, PFT‐μ n = 6. At 30 dpi; Vehicle n = 5, PFT‐μ n = 9. Welch two‐sample t test, two‐sided was used to determine the significance between experimental and control groups. For collagen immunohistochemistry and quantification of E‐GFP+ signal within the SCI lesion, a second group of adult female mice were used: at 7 dpi; Vehicle n = 4, PFT‐μ n = 3. At 30 dpi; Vehicle n = 3, PFT‐μ n = 6. Welch two‐sample t test, two‐sided test was used to determine the significance between experimental and control groups. For immunoblotting, additional groups of adult female mice were used for each western blot: vehicle n = 3, PFT‐μ n = 5. 4 mice overlapped between experiments (1 vehicle, and 3 PFT‐μ). One PFT‐μ treated sample did not produce a useable β actin band, and was therefore removed from both the cytochrome C and caspase 9 results, rendering a total PFT‐μ n = 4. Two‐tailed Mann‐Whitney test was used to compare relative densities normalized to β actin between experimental and control groups. For TUNEL stains, an additional group of adult female mice were used: n = 5 per group, 1 section per group containing the injury epicenter was used. FITC+ cells were counted ± 800 μm from the injury epicenter. Two‐tailed Mann‐Whitney test was used to compare experimental and control groups. The same set of animals used for TUNEL staining was also used to immunohistochemically detect the mature oligodendrocyte marker, CC1. Two animals per group did not produce staining of high enough quality to quantify cell numbers, and were therefore eliminated from the results producing n = 3 per group. 1‐2 sections containing the injury epicenter were used for analysis. CC1+ cells were counted ±800 μm from the injury epicenter. Two‐tailed Mann‐Whitney test was used to compare experimental and control groups. For CD86/CD206 microglia/macrophage population analysis via immunohistochemistry, separate groups of adult female mice were used for 7 dpi and 30 dpi. (n = 3 per group). Six images were taken per tissue section, and 5 sections were imaged per biological replicate, making a total of 30 images analyzed per animal. One‐way ANOVA followed by Tukey's HSD post hoc was used to determine the significance between experimental and control groups.
For the ELISA experiment, three independent experiments were performed and one‐way ANOVA followed by Bonferroni's post hoc test was used to determine the significance between experimental and control groups. For the scratch wound assay, three reference lines and three individual scratches made a total of 9 regions of interest per condition. Two wells only rendered 8 images for analysis, the rest rendered 9 images. All images were used for analysis. One‐way ANOVA followed by Bonferroni's post hoc test was used to determine the significance between experimental and control groups. For the phagocytosis assay, 8 images were taken across each well. The number of beads per cell were quantified and subjected to bin analysis. A threshold was set at greater than 10 beads per cell, and the number of cells over this threshold was compared. One‐way ANOVA followed by Bonferroni's post hoc test was used to determine the significance between experimental and control groups.
Statistical information for each figure is listed in the Torres et al Supplement.
3. RESULTS
3.1. PFT‐μ polarized cultured microglia to the anti‐inflammatory M2 state
A previous report on PFT‐μ has shown that it exerts anti‐inflammatory effects on the cultured BV2 microglia cell line.31 We examined a potential direct effect of PFT‐μ on primary microglia, as they represent more reliably native cell activity. Microglia were treated with PFT‐μ at different concentrations, and LPS was used as a positive control for microglial activation.27 Approximately 6 hours after treatment, levels of cytokines were determined by ELISA in the culture media. LPS significantly increased the levels of secreted TNF‐α, whereas in the presence of PFT‐μ LPS‐induced TNF‐α levels were significantly reduced at all the doses examined (Figure 1A). There was a dose‐dependent increase in IL‐10 levels with PFT‐μ treatment which was significant over no treatment at the 3 and 5 μM doses (Figure 1B). Together, these data indicate that PFT‐μ polarizes cultured primary microglia to “M2‐like” state under pro‐inflammatory conditions. These data are in agreement with those reported using BV2 cells31 and a model of diabetic neuropathy.32
Figure 1.
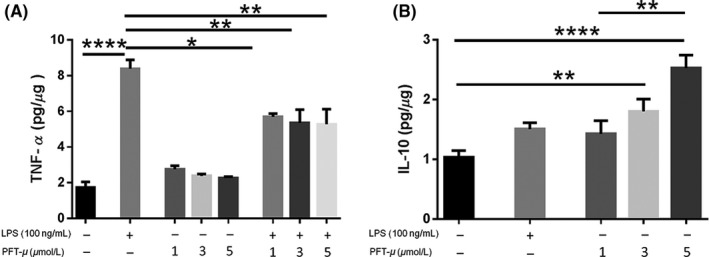
PFT‐μ reduced the production of TNF‐α and increased the release of IL‐10 in cultured primary microglia. A, B, TNF‐α and IL‐10 levels after treatment of cultured microglia with PFT‐μ and/or LPS were measured by ELISA. Data were normalized to total protein. C, The ratio of IL‐10 to TNF‐α is shown. One‐way ANOVA followed by Bonferroni's post hoc test was used to determine the significance between experimental and control groups. Data are shown as mean ± SEM of three independent experiments. *P < .05, **P < .01, ****P < .0001
3.2. PFT‐μ decreases phagocytosis under inflammatory conditions
Initial activation of microglia during injury results in increases in their phagocytic activity. We assessed the phagocytic behavior of primary microglia in the presence of PFT‐μ. As PFT‐μ has shown to decrease autophagic flux potentially through HSP70, LC3, and TLR receptor activation,17 we hypothesized that PFT‐μ may be exerting its effects by directly modulating the phagocytic pathway in microglia. Microglia were primed with LPS and treated with or without PFT‐μ in the presence of fluorescently labeled beads. Binning analysis from the assay revealed that a basal level of phagocytic activity was evident in primary microglia (Figure 2A,B). When we set a threshold above this basal level, microglia significantly increase their phagocytic activity under inflammatory conditions (LPS priming), visualized by an increase in the uptake of fluorescent beads per cell (Figure 2C). This increase was reversed by treatment with PFT‐μ, supporting a role for PFT‐μ in directly affecting microglial activation under inflammatory conditions. These results contrast the reports on BV2 cells, possibly due to the differences between the primary and the cultured cells.
Figure 2.
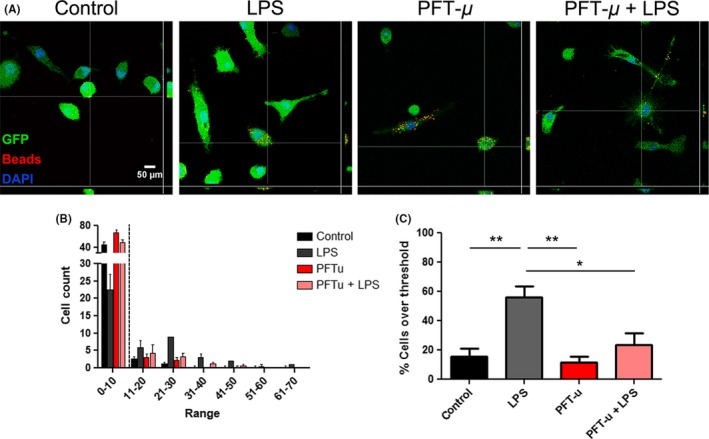
PFT‐μ modulates microglial phagocytic activity under inflammatory conditions. A, Representative confocal images of GFP + adult primary microglia exposed to latex beads for 2 hours following treatment. Orthogonal view of colocalization indicates cellular phagocytosis. Scale bar = 50 μm. B, Binning analysis was applied to the number of beads consumed per cell. A minimum of 40 cells were counted per replicate (n = 4). Dotted line indicates threshold at which cells were considered phagocytically active. C. Percent cells over threshold (greater than 10 beads per cell) were quantified. One‐way ANOVA followed by Bonferroni's post hoc test was used to determine the significance between experimental and control groups. Data are shown as mean ± SEM. *P < .05, **P < .01
3.3. PFT‐μ modulates cell migration
To further examine how PFT‐μ may modulate microglia activity, we evaluated the immunomodulatory function of PFT‐μ in a system relevant to SCI: we performed a scratch assay to investigate cell migration. A small scratch wound was made in a confluent monolayer of mixed cortical cultures (MCCs). The resulting monolayer of MCCs is devoid of microglia and oligodendrocytes and primarily consists of stromal cells, such as astrocytes and epithelial cells.25, 26 These cells play a key role in glial scar formation and maintenance following SCI, and are inhibitory to newly sprouting axons.14
Mixed cortical cultures were treated with conditioned medium collected from microglial cells that had been exposed to LPS with or without PFT‐μ. MCCs treated with microglia conditioned media in which microglia were primed with LPS (MGCM + LPS) displayed a significant reduction in wound closure, as evident by the unchanged scratch size and decrease in number of cells migrating into the wound (Figure 3B panel iv, C). The cells appeared to “wall‐off” the scratch, reminiscent of thick glial scar borders that form in the secondary phase of SCI. Interestingly, this effect was reversed when conditioned media from microglia cells concomitantly treated with MGCM + LPS + PFT‐μ was applied to MCCs, but not when PFT‐μ was administered separately to MCCs following MGCM+LPS addition (Figure 3B panel v and vi, C), indicating that the effects of PFT‐μ were linked to microglial cell polarization. Conditioned medium from untreated microglial cells (MGCM) served as an additional control, indicating that the effects observed in the MCC scratch assay were correlated with the activation state of the cells in response to inflammatory LPS stimulation and PFT‐μ. There was no difference in GFAP+ astrocyte numbers (Figure 3C), thus ruling out potential differences in MCC cellular differentiation and responses (One‐way ANOVA, P = .2032. No differences found with Tukey's Multiple Comparison). The data from this experiment suggest that the anti‐inflammatory effects of PFT‐μ may allow for a more permissive environment in which stromal cells can support and repair the injured CNS tissue. This result also supports the idea that PFT‐μ exerts this anti‐inflammatory effect by modulating microglia directly.
Figure 3.
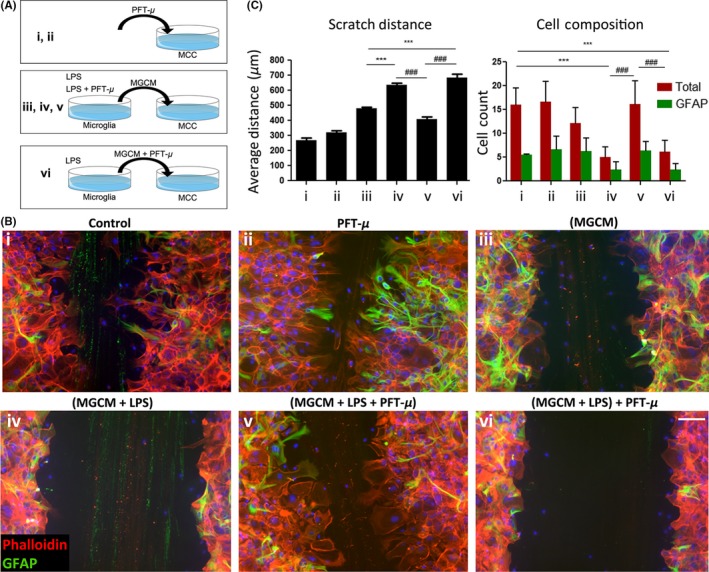
PFT‐μ modulates inflammatory response and cellular migration. A, Experimental scheme detailing treatment combinations. B, Representative images of scratch wound from phalloidin/GFAP‐stained MCCs, 48 hours following treatments. Eight images were taken across three individual scratches per well. DAPI counterstain in blue. C, Average scratch distance (left) and number of migrating cells (right). MGCM—microglia‐conditioned medium. One‐way ANOVA followed by Bonferroni's post hoc test was used to determine the significance between experimental and control groups. Data are shown as mean ± SEM. *P < .05, **P < .01, ***P < .001. * = comparisons made against (MGCM). # = comparisons made against (MGCM + LPS + PFT‐μ). ^ = comparisons made against control
3.4. PFT‐μ significantly reduced lesion volume after SCI
Our culture data reveal a robust anti‐inflammatory effect of PFT‐μ. To examine whether this role is also effective in vivo, we used PFT‐μ in a contusion model of SCI. We hypothesized that this anti‐inflammatory effect may be beneficial for inhibiting aberrant inflammation following the early SCI stages.
Environmental cues present at the SCI lesion site induce astrocytes to undergo hypertrophy, proliferation, and migration, forming a dense cellular network that surrounds the epicenter of the lesion.33 GFAP+ astrocytes demarcate the lesion site and separate the injured tissue from spared areas. Immunohistochemical analysis using GFAP+ astrocytes to mark lesion borders shows a significant reduction in lesion size in animals treated with PFT‐μ immediately following SCI (Figure 4A,B). By 30 dpi, the lesion volume of treated animals remained the same, while the lesions size of control animals decreased to that of levels seen with PFT‐μ treatment, suggesting that PFT‐μ may either expedite the natural wound closure process, or prevent excessive damage from occurring if administered during the early phase of injury.
Figure 4.
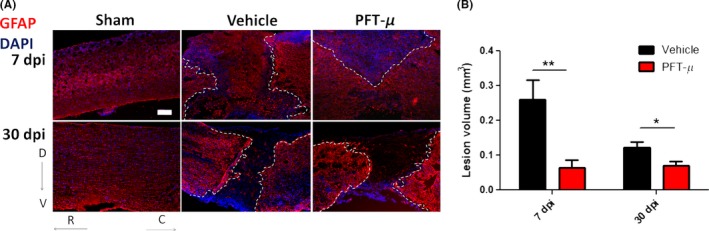
PFT‐μ significantly reduced SCI lesion volume: A, Tissue sections from injured wild‐type mice taken at 7 and 30 dpi were stained for the astrocytic marker GFAP. Injured mice had been injected ip with either vehicle or 8 mg/kg of PFT‐μ. Sham controls underwent laminectomy without contusion. Dotted lines outline the lesion border. Scale bar = 200 μm. B, Lesion volume was quantified by an observer blind to the treatment group using ImageJ. Welch two‐sample t test, two‐sided was used to determine the significance between experimental and control groups. At 7 dpi; Vehicle n = 9, PFT‐μ n = 6. At 30 dpi; Vehicle n = 5, PFT‐μ n = 9. Data are shown as mean ± SEM *P < .05, **P < .01
Collagen‐producing fibroblasts also participate in the scar formation process by closely interacting with astrocytes.34 Collagen1α+ fibroblasts begin accumulating on day 4 post‐SCI, become clearly visible by day 7, and persist up to day 56 post‐injury.35 A similar reduction in lesion volume was observed at 7 dpi in tissue sections immunofluorescently labeled with an anti‐Collagen IV antibody (Figure S1). In this case, the injury site was defined as the area occupied by Collagen IV‐positive staining. This result corroborates the results observed in the scratch assay, in which PFT‐μ treatment allowed for a more permissive environment and promoted wound closure.
3.5. PFT‐μ modulates microglia/macrophage cell populations following SCI
Under steady‐state conditions in the CNS, microglia use their processes to continuously scan the surrounding extracellular space and to communicate directly with glia, neurons, and blood vessels.38, 39 This surveying state allows them to respond quickly to damage or infection by transforming into an activated phenotype and turning on gene expression that ultimately leads to their proliferation and migration to the site of injury. To examine whether PFT‐μ alters the presence of microglia/macrophages at the lesion site, we used Csf1R‐EGFP/MacGreen mice,19 which express E‐GFP under the CSF1R promoter for easy identification of microglia/macrophages by E‐GFP fluorescence. Compared to vehicle treatment, the intensity of E‐GFP signal in the injury site was significantly decreased in PFT‐μ‐treated animals at 30 dpi, but not 7 dpi (Figure 5A,B). This result suggests that PFT‐μ has no effect on microglia/macrophage infiltration during the early phase of SCI, but rather may influence their inflammatory phenotype and responses.
Figure 5.
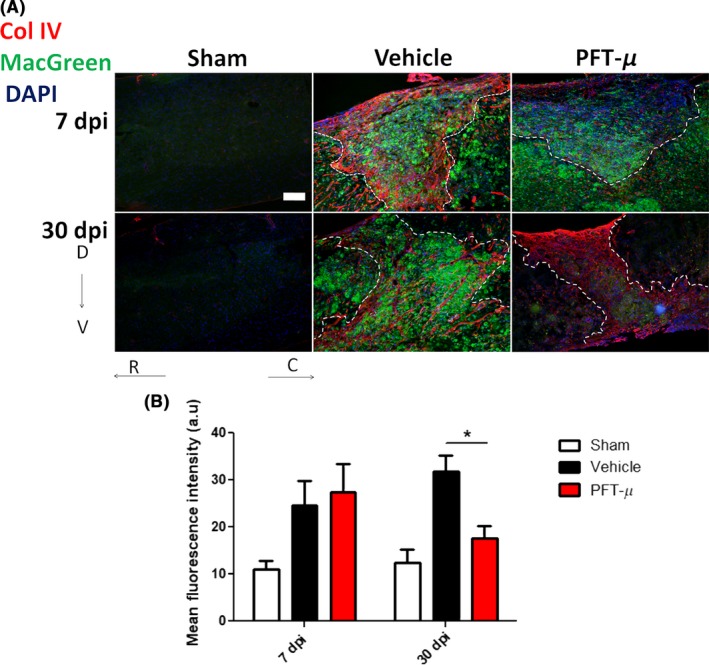
PFT‐μ significantly reduced the presence of microglia/macrophages in the SCI lesion site at 30 dpi: (A). Tissue sections from injured MacGreen mice were stained for Collagen IV to delineate the lesion border. Dotted lines outline the injury epicenter. Sham controls underwent laminectomy without contusion. Scale bar = 200 μm (B). The intensity of E‐GFP (microglia/macrophage) signal contained within the lesion site was quantified by an observer blind to the treatment group using ImageJ. At 7 dpi; Vehicle n = 7, PFT‐μ n = 6. At 30 dpi; Vehicle n = 4, PFT‐μ n = 8. Welch two‐sample t test, two‐sided was used to determine the significance between experimental and control groups. Data are shown as mean ± SEM. *P < .05
Several studies describe the predominant population of microglia at the injury site to be pro‐inflammatory, with the anti‐inflammatory response being relatively transient.11 To determine whether PFT‐μ affects microglial polarization in vivo, tissue sections from vehicle‐treated and PFT‐μ‐treated injured MacGreen mice were immunofluorescently labeled with either the pro‐inflammatory marker CD86 or the anti‐inflammatory marker CD206. Confocal images were taken from microglia/macrophages located within the lesion site. The percentage of cells positive for either CD86 or CD206 was calculated over the total number of E‐GFP+ cells. Cells positive for either CD86 or CD206, but not E‐GFP, were excluded from quantification. Following PFT‐μ treatment, a significant decrease in the percentage of E‐GFP+ CD86+ cells was evident at 7 dpi compared to the vehicle control, while there was no difference in CD206 expression (Figure 6). In agreement with the in vitro results, a decrease in the ratio of CD86+ to CD206+ cells was observed in the mice that received PFT‐μ treatment (Figure 6D), indicating a shift toward an “M2‐like” population. A similar analysis was performed for day 30 after SCI (Figure 7A‐D). This reduction in pro‐inflammatory CD86+ microglia/macrophage populations persisted up to 30 dpi (Figure 7A,B). At 30 dpi, there was a significant increase in EGFP+CD206+ cells (Figure 7C). The population ratios were again compared at 30 dpi, revealing a predominantly anti‐inflammatory environment, similar to that of sham injury at this time point (Figure 7D). This demonstrates lasting immunomodulatory effects following PFT‐μ treatment during the acute phase of SCI. Taken together, these data demonstrate that early administration of PFT‐μ following SCI reduces “M1‐like” microglia/macrophage (MG/MP) populations during the initial recovery phase of injury, and that this effect persists up to 30 days following injury. This also suggests that PFT‐μ can affect microglia/macrophage polarization in vivo.
Figure 6.
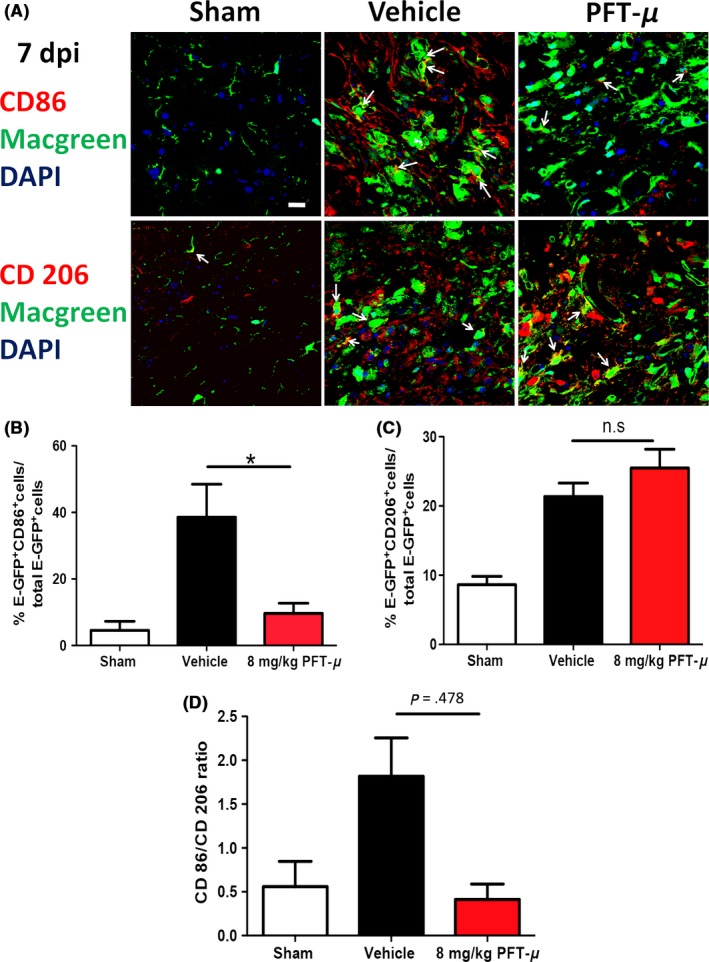
PFT‐μ reduced the expression of CD86 and increased the expression of CD206 at 7 dpi. A, Representative images from tissue sections from MacGreen mice at 7 dpi that were immunofluorescently labeled with CD86 (top) or CD206 (bottom). Images were taken of microglia/macrophages present within the injury site. Six images were taken per section and 5 sections were imaged per biological replicate. White arrows point to double positive cells. Scale = 20 μm. n = 3 per group. B, C, Quantification of the percentage of microglia/macrophages positive for CD86 and CD206. D, The ratio of CD86+ to CD206+ microglia/macrophages is shown. n = 3 per group. One‐way ANOVA followed by Tukey's HSD post hoc was used to determine the significance between experimental and control groups. Data are shown as mean ± SEM. *P < .05
Figure 7.
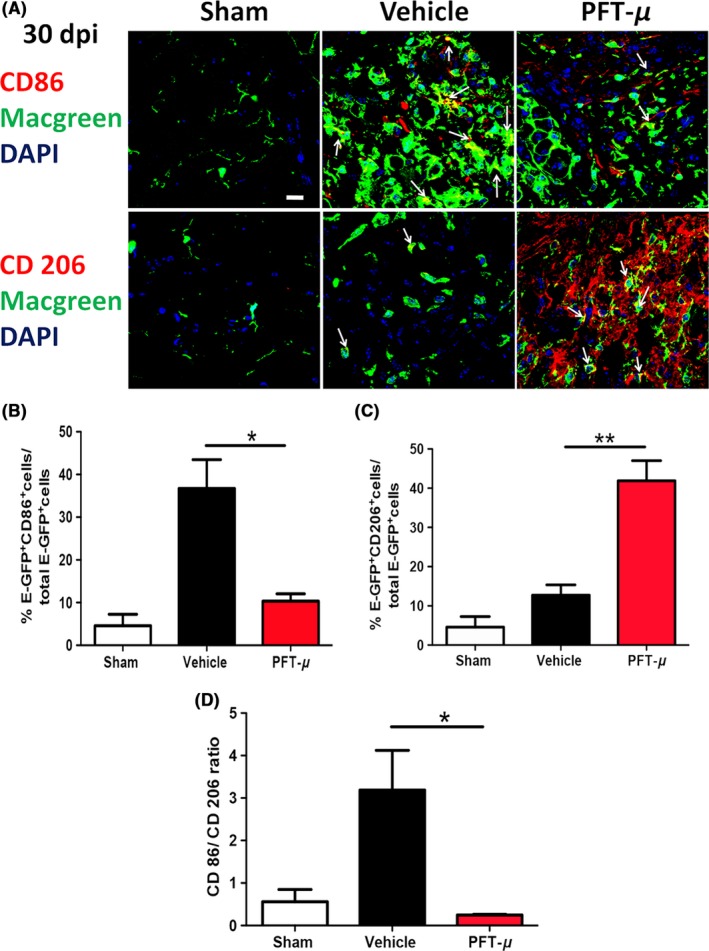
PFT‐μ maintained the anti‐inflammatory phenotype 30 days post‐SCI. A, Representative images from tissue sections from MacGreen mice at 30 dpi that were immunofluorescently labeled with CD86 (top) or CD206 (bottom). Images were taken of microglia/macrophages present within the injury site. Six images were taken per section and 5 sections were imaged per biological replicate. White arrows point to double positive cells. Scale = 20 μm. n = 3 per group. B, C, Quantification of the percentage of microglia/macrophages positive for CD86 and CD206. D, The ratio of CD86+ to CD206+ microglia/macrophages is shown. n = 3 per group. One‐way ANOVA followed by Tukey's HSD post hoc was used to determine the significance between experimental and control groups. Data are shown as mean ± SEM. *P < .05, **P < .01
3.6. PFT‐μ does not reduce p53‐mediated apoptosis following spinal contusion injury
Pifithrin‐ μ is reported to attenuate the translocation of p53 to mitochondria and inhibit cellular apoptosis.16 Translocation of p53 to the mitochondria induces mitochondrial cytochrome C (CyC) release, and activation of a caspase cascade, which leads to programmed cell death.40, 41 Peak activation of caspase levels was reported at 24 hours following SCI in rats.42 We investigated whether, in addition to the robust anti‐inflammatory effects of PFT‐μ in vitro and in vivo, it may also support an anti‐apoptotic role following SCI. We examined the release of CyC into the cytoplasm as well as the activation of caspase 9. 24 hours after SCI, PFT‐μ‐treated mice did not display a consistent decrease in CyC release (Figure 8A). CyC release induces the formation of cellular apoptosomes, which form complexes with pro‐caspase 9 activating it.41 This represents a “point of no return” and is a signature of apoptotic cell death. Caspase 9 activation 24 hours after SCI was not significantly affected with 8 mg/kg PFT‐μ (Figure 8B).
Figure 8.
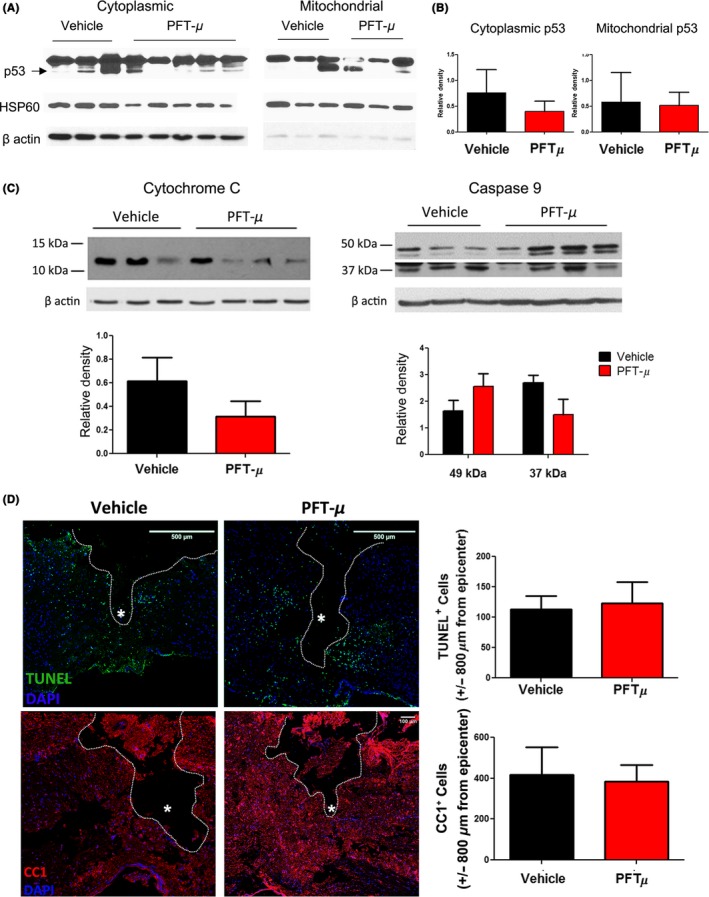
PFT‐μ does not affect cell death following SCI. A, Cytoplasmic cytochrome C release and, B, cytoplasmic caspase 9 levels in fractionated spinal cord tissue, 24 hours following injury. Protein levels normalized to β actin. Vehicle n = 3, PFT‐μ n = 4. C, Representative images from TUNEL stained and D, CC1‐stained sagittal tissue sections, 24 hours following SCI. Cells were quantified ± 800 μm from the injury epicenter. White asterisk marks epicenter. n = 5 per group for TUNEL and n = 3 per group for CC1. Data are shown as mean ± SEM
Transferase dUTP nick end labeling staining of the tissue was also performed 24 hours following SCI. We did not observe any changes in the number of cells undergoing cell death following PFT‐μ treatment during the early phase of injury (Figure 8C). Oligodendrocytes are critically susceptible to apoptosis following spinal cord trauma, and this death causes the demyelination of axons and subsequent neuronal cell death.43, 44 Preservation of these cell types is key to recovery following SCI. Immunostaining for the mature oligodendrocyte marker CC1 showed no difference in the number of spared oligodendrocytes following SCI at 24 hours with PFT‐μ treatment (Figure 8D). Taken together, PFT‐μ treatment does not reduce cell death 24 hours following SCI, but instead may be affecting SCI pathology by exerting immunomodulatory effects on microglia/macrophages directly, promoting a more anti‐inflammatory environment conducive to wound healing.
3.7. Early administration of PFT‐μ affects functional outcome following SCI
We next examined whether early administration of PFT‐μ would yield improved functional outcome following SCI. To test the effect of treatment on motor coordination and balance, injured mice were tested on the Rotarod at days 7, 14, 21, and 30 post‐SCI as previously described.14 Minimal improvement in Rotarod performance was observed with PFT‐μ at the time points examined (Figure 9A). However, a qualitative improvement in the gait of the injured mice was observed, and thus the animals were subjected to hind limb footprint analysis. Foot prints were collected in sham mice and in SCI mice at 7 and 30 dpi (Figure 9B). Rear track width has been described previously as a sensitive measure of balance and posture, and as a measure of functional outcome in wild‐type mice following SCI.45, 46 SCI mice treated with PFT‐μ exhibited a significant increase in rear track width over vehicle‐treated animals at 7 dpi, indicating moderate functional recovery (Figures 9B,C); however, this was not statistically significant at 30 dpi, despite an increasing trend toward improved functional outcome. These results indicate that PFT‐μ treatment during the early phases of injury resulted in small but significant improvements in motor coordination and posture following SCI.
Figure 9.

PFT‐μ significantly improved motor coordination and posture. A, Injured mice were tested in the Rotarod at days 7, 14, 21, and 30 post‐SCI. B, Footprint analysis was conducted on days 7 and 30 post‐SCI. Shown are representative images of footprints taken at 7 days post‐injury. Red arrows show the distance measured to obtain stride width. C, Quantification of stride width (measured in cm). One‐way ANOVA followed by Welch two‐sample t test, two‐sided was used to determine the significance between experimental and control groups. Data are shown as mean ± SEM. **P < .01. Vehicle n = 9 PFT‐μ n = 6
4. DISCUSSION
Here we show that the small molecule PFT‐μ affords anti‐inflammatory effects on primary microglia cells in vitro. Our data suggest that PFT‐μ directly modulates microglia to produce the anti‐inflammatory cytokine IL‐10, while also inhibiting inflammatory‐mediated phagocytosis. In addition to direct effects, PFT‐μ treatment of microglia also resulted in the production of soluble factors that enhanced cellular migration of CNS stromal cells and wound closure in vitro. PFT‐μ effects were tested in a model of SCI, where significant improvements in anatomical recovery during the initial phase of injury were observed when PFT‐μ was given during the acute window of pathology. The increase in IL‐10 production may explain the observed reduction in lesion volume after PFT‐μ treatment since this cytokine has been shown to attenuate astroglial reactivity which correlates with the formation of glial scar.45
Pro‐inflammatory microglia/macrophages are prevalent at the spinal cord lesion site for at least 28 days after injury, and drive debilitating pathogenesis.11 It has been shown that anti‐inflammatory M2 shifts are beneficial for remyelination and regeneration following CNS injury.12 As PFT‐μ treatment results in the formation of an anti‐inflammatory milieu in vitro, we propose that its effects in vivo on improved anatomical recovery following SCI are a result of microglia/macrophage polarization shifts, and not a result of inhibiting immune cell infiltration to the site of injury. Consistent with this idea is the observed downregulation of “M1‐like” microglia/macrophage populations throughout the chronic phase of injury, which rendered a persistent “M2‐like” population up to 30 days following SCI. However, we cannot at this point rule out any indirect effects PFT‐μ may be having on SCI pathology.
The mechanism of action of PFT‐μ has been associated with both the inhibition of mitochondria translocation of p53, and the inhibition of heat shock protein 70 (Hsp70).17, 31, 46, 47 In our experiments, PFT‐μ results in a shift of the polarization of microglia/macrophages from an “M1‐like” to an “M2‐like” phenotype, consistent with studies in peripheral models demonstrating that PFT‐μ reduces the expression of inflammatory markers, such as TNF‐α, IL‐6, and iNOS, through inhibition of Hsp70.50 These results also corroborate the findings that PFT‐μ has direct anti‐inflammatory effects on the BV2 microglia cell line.31, 49 Although PFT‐μ is canonically described as being neuroprotective by inhibiting p53‐mediated mitochondrial‐induced apoptosis, these studies were conducted in neuron‐specific injury models.16, 51, 52 We did not observe anti‐apoptotic effects of PFT‐μ 24 hours following SCI, where p53 activation and cell death are at their peak.42 Additionally, spared oligodendrocytes remained comparable between control and treated groups, suggesting an alternative mechanism for PFT‐μ in scenarios of trauma. It should be noted that PFT‐μ's anti‐apoptotic effect was observed in models of excitotoxic neuronal injury, which differ from the one presented here. As such, more work needs to be done to describe the potential mechanisms PFT‐μ has following SCI.
Despite the anti‐inflammatory effects seen when PFT‐μ was given during the early stages of SCI, only modest improvement was observed when measuring functional outcomes. This result indicates that treating the early phase alone may stimulate an anti‐inflammatory and permissive environment following SCI, but may not be sufficient in providing full recovery. Recently, neuroprotective effects of rapamycin following SCI have been demonstrated.53, 54, 55, 56 In these reports, rapamycin was efficient in reducing pain and blocking microglial activation and immune cell infiltration into the lesioned spinal cord. In most cases, rapamycin was delivered either through a microosmotic pump or via a regimen of injections. In addition to rapamycin, the anti‐IL‐6 murine receptor antibody MR16‐1 has demonstrated improved functional outcomes following SCI in mice, primarily associated with attenuation of pain, but also changes in cytokine levels, such as TNFα, interferon‐γ, and IL4.57, 58, 59 In our study, we demonstrate that a single dose of PFT‐μ is effective in modulating inflammatory MG/MP populations, which differs from the delivery strategies of the two reagents above. Limiting dosage could mitigate any potential side effects experienced by patients. Additionally, the anti‐inflammatory and neuroprotective effects provided by rapamycin and/or MR16‐1 may synergize with PFT‐μ, as each attacks a separate target and such combination therapy has been shown to be beneficial to patients.58, 59, 60 PFT‐μ has also shown to have favorable effects, beyond its anti‐inflammatory and HSP70 inhibition properties.48 Together, we suggest that PFT‐μ should be considered as an adjunct to other therapies for SCI, as well as in conjunction with rehabilitation therapy.
There are currently no treatments for the early phases of SCI pathology, and it is difficult to manage patients during secondary phases as persistent inflammation and damage have been triggered from the inciting injurious events. Further studies need to be conducted to identify mechanisms by which PFT‐μ may be promoting an anti‐inflammatory phenotype in microglia/macrophages. This includes studying cells at the transcriptomic level as well as characterizing the effects PFT‐μ has on bone marrow‐derived macrophages. Significant anti‐inflammatory effects of PFT‐μ on SCI pathology were seen following the early administration of the drug, potentially highlighting the importance of targeting the early phase of SCI, and suggesting that PFT‐μ may be an interesting candidate to be considered for future work. The rapid and sustained response following PFT‐μ treatment on SCI lesion repair and modulation of inflammatory cells suggests that this compound could aid in the regulation of the subsequent phases of SCI.
5. CONCLUSIONS
A small molecular weight reagent PFT‐μ modulates in culture directly the microglial inflammatory response to stimulation by reducing the secretion of pro‐inflammatory cytokines and decreasing phagocytosis. It also allows for wound closure. When used in vivo, in a contusion model of SCI, PFT‐μ treatment results in the reduction of pro‐inflammatory responses that accompany the acute phase of injury. Therefore, PFT‐μ could be further investigated as an anti‐inflammatory agent in the context of SCI.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
No human research. All animal work complied with Stony Brook University guidelines, and was approved by the Stony Brook IACUC committee.
CONSENT FOR PUBLICATION
All authors have seen drafts of this manuscript and consent for publication.
AVAILABILITY OF SUPPORTING DATA
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS' CONTRIBUTIONS
LFT designed and performed experiments, analyzed data, and drafted the manuscript; MDC, designed and performed experiments, analyzed data, and drafted the manuscript; CR performed experiments; NR performed experiments; MA designed and performed experiments; JKR, designed experiments, analyzed data, and drafted the manuscript; and SET designed experiments, analyzed data, and drafted the manuscript.
Supporting information
ACKNOWLEDGMENTS
We are grateful to the members of the Tsirka and Robinson labs for advice and suggestions.
Caponegro MD, Torres LF, Rastegar C, et al. Pifithrin‐μ modulates microglial activation and promotes histological recovery following spinal cord injury. CNS Neurosci Ther. 2019;25:200–214. 10.1111/cns.13000
Funding information
Research was supported by the Turner Dissertation fellowship (LT), T32GM007518 and SBMS funds (MDC), and SBU TRO Fusion awarded by the Stony Brook School of Medicine and the Office of the Vice President for Research (SET).
Caponegro and Torres equally contributed to this work
REFERENCES
- 1. Spinal Cord Injury (SCI) . Facts and figures at a glance J Spinal Cord Med. 2016;39:493‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sekhon LH, Fehlings MG. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine (Phila Pa 1976) 2001;26(24 Suppl):S2‐S12. [DOI] [PubMed] [Google Scholar]
- 3. Liu XZ, Xu XM, Hu R, et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395‐5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beattie MS, Li Q, Bresnahan JC. Cell death and plasticity after experimental spinal cord injury. Prog Brain Res. 2000;128:9‐21. [DOI] [PubMed] [Google Scholar]
- 5. Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140‐155. [DOI] [PubMed] [Google Scholar]
- 6. Crotti A, Ransohoff RM. Microglial physiology and pathophysiology: insights from genome‐wide transcriptional profiling. Immunity. 2016;44:505‐515. [DOI] [PubMed] [Google Scholar]
- 7. Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp (Warsz). 2012;60:251‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colton C, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174‐191. [DOI] [PubMed] [Google Scholar]
- 9. Tyor WR, Avgeropoulos N, Ohlandt G, Hogan EL. Treatment of spinal cord impact injury in the rat with transforming growth factor‐beta. J Neurol Sci. 2002;200:33‐41. [DOI] [PubMed] [Google Scholar]
- 10. Thompson CD, Zurko JC, Hanna BF, Hellenbrand DJ, Hanna A. The therapeutic role of interleukin‐10 after spinal cord injury. J Neurotrauma. 2013;30:1311‐1324. [DOI] [PubMed] [Google Scholar]
- 11. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435‐13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miron VE, Boyd A, Zhao JW, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Emmetsberger J, Tsirka SE. Microglial inhibitory factor (MIF/TKP) mitigates secondary damage following spinal cord injury. Neurobiol Dis. 2012;47:295‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bukhari N, Torres L, Robinson JK, Tsirka SE. Axonal regrowth after spinal cord injury via chondroitinase and the tissue plasminogen activator (tPA)/Plasmin system. J Neurosci. 2011;31:14931‐14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strom E, Sathe S, Komarov PG, et al. Small‐molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006;2:474‐479. [DOI] [PubMed] [Google Scholar]
- 16. Nijboer CH, Heijnen CJ, van der Kooij MA, et al. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann Neurol. 2011;70:255‐264. [DOI] [PubMed] [Google Scholar]
- 17. Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Romao S, Munz C. LC3‐associated phagocytosis. Autophagy. 2014;10:526‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sasmono R, Oceandy D, Pollard J, et al. A macrophage colony‐stimulating factor receptor‐green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155‐1163. [DOI] [PubMed] [Google Scholar]
- 20. Farooque M, Suo Z, Arnold PM, et al. Gender‐related differences in recovery of locomotor function after spinal cord injury in mice. Spinal Cord. 2006;44:182‐187. [DOI] [PubMed] [Google Scholar]
- 21. Sasmono RT, Ehrnsperger A, Cronau SL, et al. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony‐stimulating factor receptor (CSF‐1R) as well as many other macrophage‐specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF‐1. J Leukoc Biol. 2007;82:111‐123. [DOI] [PubMed] [Google Scholar]
- 22. Scheff SW, Rabchevsky AG, Fugaccia I, Main JA, Lumpp JE Jr. Experimental modeling of spinal cord injury: characterization of a force‐defined injury device. J Neurotrauma. 2003;20:179‐193. [DOI] [PubMed] [Google Scholar]
- 23. Nijboer CH, Groenendaal F, Kavelaars A, Hagberg HH, van Bel F, Heijnen CJ. Gender‐specific neuroprotection by 2‐iminobiotin after hypoxia‐ischemia in the neonatal rat via a nitric oxide independent pathway. J Cereb Blood Flow Metab. 2007;27:282‐292. [DOI] [PubMed] [Google Scholar]
- 24. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bronstein R, Torres L, Nissen JC, Tsirka SE. Culturing microglia from the neonatal and adult central nervous system. J Vis Exp. 2013;78:50647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Etienne‐Manneville S. In vitro assay of primary astrocyte migration as a tool to study Rho GTPase function in cell polarization. Methods Enzymol. 2006;406:565‐578. [DOI] [PubMed] [Google Scholar]
- 27. Nakamura Y, Si QS, Kataoka K. Lipopolysaccharide‐induced microglial activation in culture: temporal profiles of morphological change and release of cytokines and nitric oxide. Neurosci Res. 1999;35:95‐100. [DOI] [PubMed] [Google Scholar]
- 28. Stansley B, Post J, Hensley K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer's disease. J Neuroinflammation. 2012;9:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carter RJ, Lione LA, Humby T, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248‐3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beastrom N, Lu H, Macke A, et al. mdx((5)cv) mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx Mice. Am J Pathol. 2011;179:2464‐2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fleiss B, Chhor V, Rajudin N, et al. The anti‐inflammatory effects of the small molecule pifithrin‐micro on BV2 microglia. Dev Neurosci. 2015;37:363‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jheng HF, Tsai PJ, Chuang YL, et al. Albumin stimulates renal tubular inflammation through an HSP70‐TLR4 axis in mice with early diabetic nephropathy. Dis Model Mech. 2015;8:1311‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. White R, Jakeman L. Don't fence me in: harnessing the beneficial roles of astrocytes for spinal cord repair. Restor Neurol Neurosci 2008;26:197‐214. [PMC free article] [PubMed] [Google Scholar]
- 34. Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146‐156. [DOI] [PubMed] [Google Scholar]
- 35. Soderblom C, Luo X, Blumenthal E, et al. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci. 2013;33:13882‐13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314‐1318. [DOI] [PubMed] [Google Scholar]
- 37. Arcuri C, Mecca C, Bianchi R, Giambanco I, Donato R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front Mol Neurosci. 2017;10:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577‐590. [DOI] [PubMed] [Google Scholar]
- 39. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621‐632. [DOI] [PubMed] [Google Scholar]
- 40. Zhang J, Cui Z, Feng G, et al. RBM5 and p53 expression after rat spinal cord injury: implications for neuronal apoptosis. Int J Biochem Cell Biol. 2015;60:43‐52. [DOI] [PubMed] [Google Scholar]
- 41. Casha S, Yu WR, Fehlings MG. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103:203‐218. [DOI] [PubMed] [Google Scholar]
- 42. Casha S, Yu WR, Fehlings MG. FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury. Exp Neurol. 2005;196:390‐400. [DOI] [PubMed] [Google Scholar]
- 43. Beare JE, Morehouse JR, DeVries WH, et al. Gait analysis in normal and spinal contused mice using the TreadScan system. J Neurotrauma. 2009;26:2045‐2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hill RL, Zhang YP, Burke DA, et al. Anatomical and functional outcomes following a precise, graded, dorsal laceration spinal cord injury in C57BL/6 mice. J Neurotrauma. 2009;26:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balasingam V, Yong VW. Attenuation of astroglial reactivity by interleukin‐10. J Neurosci. 1996;16:2945‐2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang R, Wang J, Hu Y, et al. Pifithrin‐mu attenuates acute sickness response to Lipopolysaccharide in C57BL/6J mice. Pharmacology. 2016;97:245‐250. [DOI] [PubMed] [Google Scholar]
- 47. Huang C, Lu X, Wang J, Tong L, Jiang B, Zhang W. Inhibition of endogenous heat shock protein 70 attenuates inducible nitric oxide synthase induction via disruption of heat shock protein 70/Na(+)/H(+) exchanger 1‐Ca(2+) ‐calcium‐calmodulin‐dependent protein kinase II/transforming growth factor beta‐activated kinase 1‐nuclear factor‐kappaB signals in BV‐2 microglia. J Neurosci Res. 2015;93:1192‐1202. [DOI] [PubMed] [Google Scholar]
- 48. Huang C, Wang J, Chen Z, Wang Y, Zhang W. 2‐phenylethynesulfonamide prevents induction of pro‐inflammatory factors and attenuates LPS‐induced liver injury by targeting NHE1‐Hsp70 complex in mice. PLoS One. 2013;8:e67582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chiu GS, Maj MA, Rizvi S, et al. Pifithrin‐mu prevents Cisplatin‐induced Chemobrain by preserving neuronal mitochondrial function. Cancer Res. 2017;77:742‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krukowski K, Nijboer CH, Huo X, Kavelaars A, Heijnen CJ. Prevention of chemotherapy‐induced peripheral neuropathy by the small‐molecule inhibitor pifithrin‐mu. Pain. 2015;156:2184‐2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao K, Wang YS, Yuan YJ, et al. Neuroprotective effect of rapamycin on spinal cord injury via activation of the Wnt/beta‐catenin signaling pathway. Neural Regen Res. 2015;10:951‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song Y, Xue H, Liu TT, Liu JM, Chen D. Rapamycin plays a neuroprotective effect after spinal cord injury via anti‐inflammatory effects. J Biochem Mol Toxicol. 2015;29:29‐34. [DOI] [PubMed] [Google Scholar]
- 53. Wang X, Li X, Huang B, Ma S. Blocking mammalian target of rapamycin (mTOR) improves neuropathic pain evoked by spinal cord injury. Transl Neurosci. 2016;7:50‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen HC, Fong TH, Hsu PW, Chiu WT. Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti‐inflammation, and neuroprotection. J Surg Res. 2013;179:e203‐e210. [DOI] [PubMed] [Google Scholar]
- 55. Murakami T, Kanchiku T, Suzuki H, et al. Anti‐interleukin‐6 receptor antibody reduces neuropathic pain following spinal cord injury in mice. Exp Ther Med. 2013;6:1194‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mukaino M, Nakamura M, Yamada O, et al. Anti‐IL‐6‐receptor antibody promotes repair of spinal cord injury by inducing microglia‐dominant inflammation. Exp Neurol. 2010;224:403‐414. [DOI] [PubMed] [Google Scholar]
- 57. Guerrero AR, Uchida K, Nakajima H, et al. Blockade of interleukin‐6 signaling inhibits the classic pathway and promotes an alternative pathway of macrophage activation after spinal cord injury in mice. J Neuroinflammation. 2012;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaiser M, Kuhnl A, Reins J, et al. Antileukemic activity of the HSP70 inhibitor pifithrin‐mu in acute leukemia. Blood Cancer J. 2011;1:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Monma H, Harashima N, Inao T, Okano S, Tajima Y, Harada M. The HSP70 and autophagy inhibitor pifithrin‐mu enhances the antitumor effects of TRAIL on human pancreatic cancer. Mol Cancer Ther. 2013;12:341‐351. [DOI] [PubMed] [Google Scholar]
- 60. Sekihara K, Harashima N, Tongu M, et al. Pifithrin‐mu, an inhibitor of heat‐shock protein 70, can increase the antitumor effects of hyperthermia against human prostate cancer cells. PLoS One. 2013;8:e78772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.