

Summary
Objective
Intracranial aneurysm (IA) is a life threatening cerebrovascular disease characterized by phenotypic modulation of vascular smooth muscle cells (VSMCs) and loss of vessel cells. In addition to environmental factors, genetic factors have been proposed to be a critical factor in the onset and progression of IA. The present study investigates the effects of serine‐arginine protein kinase 1 (SRPK1) on VSMC proliferation and apoptosis both in vivo and in vitro, as well as its role in vascular remodeling in vivo through PI3 K/Akt signaling in IA.
Methods
Differentially expressed genes related to IA were initially identified using microarray analysis. Immunohistochemistry was conducted to determine SRPK1 expression in the vascular walls in IA and normal cerebral vascular walls. TUNEL staining were applied to observe cell apoptosis patterns of VSMCs. VSMC proliferation and apoptosis in vitro were detected by cell counting kit‐8 (CCK8) assay and flow cytometry. The expressions of SRPK1, PI3 K/Akt signaling pathway‐ and apoptosis‐related genes were evaluated by RT‐qPCR and Western blot analysis.
Results
Microarray data of GSE36791 and GSE54083 were analyzed to determine the selection of SRPK1 gene. The vascular walls in IA rat models produced high levels of SRPK1 expression and an activated PI3 K/Akt signaling pathway. VSMCs treated with siRNA‐SRPK1 exhibited enhanced cell proliferation, repressed cell apoptosis, and increased vascular remodeling, all of which suggest the inhibition of the PI3 K/AKT pathway. Notably, PI3 K/AKT pathway reversed the effect of SRPK1 silencing.
Conclusion
Our results show that siRNA‐mediated silencing of SRPK1 gene inhibits VSMC apoptosis, and increases VSMCs proliferation and vascular remodeling in IA via the PI3 K/Akt signaling pathway. Our findings provide a novel intervention target for the molecular treatment of IA.
Keywords: intracranial aneurysm, PI3K/Akt signaling pathway, SRPK1, vascular remodeling, vascular smooth muscle cells
1. INTRODUCTION
Intracranial aneurysms (IAs) are cerebrovascular disorders that are characterized by localized dilation or expansion of the blood vessels. IA results from the weakness of either the wall of cerebral vein or cerebral artery.1 The spontaneous rupturing of IAs contributes to a substantial amount of morbidity and mortality in patients with IA.2 Rupture of an IA is the most frequently occurring cause of subarachnoid hemorrhage, which is a lethal acute cerebrovascular disorder that commonly affects the working‐age population.3 A lifelong follow‐up study pointed out that females are more prone to an aneurysm rupture, and Tobacco smoking is also an important factor that contributes to a higher possibility of rupture in patients with an unruptured IA (UIA) compared to UIA size factor.4 Pathological conditions and inflammation that alter the physiological hemodynamics in the cerebral vasculature can activate vascular remodeling characterized by vascular smooth muscle cell (VSMC) migration and apoptosis.5 Notably, there is evidence that inhibiting inflammation in the aortic wall coupled with promoting VSMC proliferation has an essential component in the treatment an aneurysm rupture.6 Recent studies are acknowledged for their findings pertaining to the molecular pathogenesis of IAs that focus on VSMC activities.7, 8
Serine‐arginine protein‐specific kinases (SRPKs) are pivotal regulators in transducing growth signals from the cell surface to the nucleus in order to regulate mRNA splicing.9 The structure of SRPKs comprises a conserved kinase domain which is separated by a large, non‐conserved insert domain approximately 250 amino acids in size.10 The serine‐arginine protein kinase 1 (SRPK1) has been found to induce the phosphorylation of SR proteins which include: SRp30, SRp40, SRp75, and SRSF1, as well as the nuclear SRPK1. These phosphorylated SR proteins promote the accumulation of other SR proteins in cytoplasm, and alters the downstream splicing events in malignant tumor cells.11, 12 SRPK1 in particular functions as an oncoprotein in human cancer due to its overexpression effects. Its overexpression has been implicated in promoting anchorage‐independent cell development in vitro as well as tumor growth in nude mice.13 Furthermore, SRPK1 plays a heterogeneous role in different cancers, such as regulating angiogenesis in prostate and colorectal cancers.14, 15, 16 SRPK1 is a downstream Akt target for transducing growth signals to regulate splicing. Its aberrant expression has been found to induce constitutive Akt activation.17 Evidence has demonstrated that the activation of the PI3 K/Akt signaling pathway significantly enhances VSMC proliferation induced by apelin‐13.18 VSMC activities have been found to be implicated in aortic aneurysms. The development of abdominal aortic aneurysm was suppressed through inhibiting the activation of the VEGF/PI3 K/Akt signaling pathway mediated by microRNA‐195.19 The abnormal proliferation and migration of VSMCs are events that contribute to the progression of cardiovascular diseases, including post‐angioplastic vascular remodeling.20 This study was performed to validate the effects of siRNA‐mediated silencing of SRPK1 on the proliferation and apoptosis of VSMCs. In addition, we aim to investigate siRNA‐mediated silencing of SRPK1 on vascular remodeling, in a rat model of IA, and the involvement of the PI3 K/Akt signaling pathway.
2. MATERIALS AND METHODS
2.1. Ethnics statement
The Ethics Committee of the First Hospital of China Medical University approved the study protocols that were used in this investigation. All participants provided written informed consents. All animal experimentation protocols were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Significant efforts were made in order to minimize the number of animals used as well as their suffering.
2.2. Microarray analysis
Using "intracranial aneurysm" as keyword, expression profiles of GSE36791 and GSE54083 were selected from GEO database (https://www.ncbi.nlm.nih.gov/geo/). We found 18 normal tissue samples and 43 IA rupture samples in GSE36791. Gene expression analysis was conducted on IA rupture microarray data against normal samples as the control. GSE54083 consists of 10 normal tissue samples, 8 IA rupture samples, and 5 UIA samples. Gene expressions in IA rupture samples and UIA samples were analyzed against the control sample, respectively. The differential analysis was carried out with an R. software "limma" package. The threshold values were set as P < 0.05 and |logFC|>2. Thermogram analysis of differentially expressed genes was conducted by “pheatmap” package in R language. The Venn diagram construction website (https://bioinformatics.psb.ugent.be/webtools/Venn/) was employed to find the intersection of the 3 differentially expressed datasets.
2.3. Patient recruitment
From January 2014 to January 2017, IA specimens were obtained from 53 patients with IA who underwent surgical treatment at the First Hospital of China Medical University. All the IA specimens were obtained without causing further complications and threatening the patient's life and operation safety. No endovascular embolization was performed before the operation. The participants included 27 women and 26 men, and their ages ranged from 19 to 63 years with a mean age of 39.38 ± 9.13 years. Thirty‐five cases of normal cerebral cortex arterial blood vessels of brain trauma were used as the control and designed as the normal group. The normal group consisted of 21 males and 14 females, and their age ranged from 21 to 66 years with a mean age of 42.06 ± 10.17 years.
2.4. Immunohistochemistry analysis
The IA and normal cortical artery tissues obtained from patients in the 2 groups were dehydrated by gradient alcohol, embedded in paraffin, and cut into 4 μm thick slices. The specimens were dewaxed, hydrated, and prepared by cell seeding. All sections were pretreated with 0.01 mol/L citrate buffer under high temperature and high pressure for antigen retrieval. The sections were blocked with goat serum at room temperature for 20 minutes, followed by discarding the excess serum. After that, 50 µL of primary antibodies rabbit‐anti‐human SRPK1, PI3 K, Akt, and p‐Akt (Abcam, Cambridge, MA, UK) were added to completely cover the sections. The sections were incubated overnight at 4°C and at 37°C for another 45 minutes. The sections were then washed for 3 times in phosphate buffered saline (PBS), 5 minutes each time, added with 40 to 50 µL goat‐anti‐mouse polyclonal antibody (Abcam) and incubated for 1 hour at 37°C. Following a 3‐time PBS rinse, 5 minutes each time, the sections were stained with diaminobenzidine (DAB) and allowed to develop for 5 to 10 minutes. The sections were then washed with PBS for 10 minutes and counterstained with hematoxylin for 2 minutes. Next, the sections were dehydrated, cleared, sealed by neutral gum, and observed under a light microscope. A positive result was defined as detection of light yellow or brownish‐yellow uneven particles in the nucleus. Positive cells were averaged by randomly selecting 5 visual fields. The sections were graded and counted by a half‐quantitative method: 0 point indicates no positive target cells; 1 point, ≤25% positive target cells; 2 points, 26%‐50%; 3 points, 51%‐57%; 4 points > 75%. Color visualization was evaluated and graded by the presence of color according to the following scale: 0 point, no cellular staining; 1 points, light yellow; 2 points, yellow‐brownish; and 3 points, brown. The final score was determined by the sum of the 2 scores: A score of ≤1 was deemed as negative, while a score of >1 was defined as positive.
2.5. Vector construction
Based on the SRPK1 gene sequence in the GenBank database, a full‐length cDNA primer sequence was designed and synthesized (Sangon Biotech Co. Ltd., Shanghai, China). Forward primer: 5'‐ CCACGCTCTTCGCCATTC‐ 3', Reverse primer: 5'‐ GAGACCGGTAACCGCCAG‐ 3'. Bam HI and Xho I restriction sites were inserted into the forward and reverse primers, respectively. The purified SRPK1 gene was inserted into the pEGFP‐N1 plasmid using PCR amplification, and the SRPK1 over‐expression vector was constructed. The corresponding base sequence of interfering SRPK1 expression is 5'‐ GTGCAGCAGAAATTAATT‐ 3'. The base sequence of the target sequence was transformed into an oligonucleotide template capable of expressing shRNA using an online design program of Ambion (Austin, TX, USA) and synthesized by Sangon Biotech Co. Ltd. (Shanghai, China). The synthesized oligonucleotides were mixed together in molar ratio 1:1, heated at 95°C for 5 minutes and were left to anneal very slowly to the room temperature. The pSilencerTM 3.1‐H1 Neo plasmid was ligated with the oligonucleotides and inserted into the target sequence of SRPK1 to obtain the SRPK1 gene silencing vector.
2.6. Model establishment
Eighty Sprague‐Dawlay (SD) male rats (weight: 200 to 300 g; Shanghai SLAC Laboratory Animal Co. Ltd., Shanghai, China) were used in this study. IA rat models were established in 70 randomly selected rats by ligating the left common carotid arteries and posterior branches of both renal arteries.21 Ligation of the left common carotid artery and the posterior branches of both renal arteries was performed under anesthesia with an intraperitoneal injection of 3% pentobarbital sodium. The same surgical incisions were made in 10 other SD rats in the sham group. In the sham group, the right carotid bifurcation and bilateral renal arteries were exposed and no ligation was performed. Ten out of the 70 rats without any other treatment were set as the IA group, while the other 60 rats were randomly classified into the SRPK1 group (injected through the caudal vein with SRPK1 over‐expression vector), si‐SRPK1 group (injected through the caudal vein with siRNA against SRPK1 vector), IGF‐1 group (injected through the caudal vein with 100 ng/mL PI3 K/Akt pathway activator IGF‐1), LY294002 group (injected through the caudal vein with 10 μmol/L PI3 K/Akt inhibitor LY294002), si‐SRPK1 + IGF‐1 group (injected through the caudal vein with siRNA against SRPK1 vector and IGF‐1), and SRPK1 + LY294002 group (injected through the caudal vein with SRPK1 over‐expression vector and LY294002) (10 rats in each group, each for 200 μL). After 30 days, under general anesthesia, the rats were perfused with 4% paraformaldehyde in PBS from the ascending aorta via the right ventricle. The circle of Willis was removed and immersed in 4% formaldehyde phosphate buffer solution. After dehydration using gradient alcohol, the specimens were embedded in paraffin wax and cut into 4 μm thick sections.
2.7. Hematoxylin‐eosin (HE) staining
Normal arterial tissue and aneurysm tissue specimens from the sham group and the IA group were dewaxed, hydrated, stained with hematoxylin for 5 minutes, and washed under tap water for 1 minute. Specimens were then differentiated in 1% hydrochloric acid alcohol for 30 seconds, soaked in tap water for 15 minutes, stained with 0.5% eosin for 3 minutes, and then rinsed with distilled water. The specimens were then dehydrated, cleared, sealed with neutral gum. The pathological morphology of the Willis arterial circle was observed under an optical microscope.
2.8. Terminal deoxynucleotidyl transferase (TdT)‐mediated dUTPbiotin nick end labeling (TUNEL) staining
Normal arterial tissue and aneurysm tissue specimens from the sham group and the IA group were dewaxed, hydrated, and incubated with protease solution at 37°C for 30 minutes. The specimens were rinsed with PBS 3 times with 5 minutes each time. Specimens were then incubated with TUNEL working solution for 60 minutes at 37°C in the dark and rinsed in PBS thrice. They were then dried in the dark at room temperature, stained with DAB and sealed. The positive cells showing brownish‐yellow nuclei were observed under a fluorescence microscope. Five visual fields (200×) were randomly selected from each section and counted for both normal cells and positive cells. The apoptotic rate was represented by averaging the number of positive stained cells/all cell number × 100%.
2.9. Cell culture
The tissue culture method was applied to culture VSMCs obtained from rats. After anesthesia, the entire thoracic aorta was separated from the SD rats. The outer layer of the blood vessel was clamped, and a cut was made using ophthalmic scissors. The vascular endothelial layer was washed with Dulbecco's Modified Eagle Medium (DMEM) twice, cut into 1 mm × 1 mm tissue blocks, and cultured in a 50 cm2 culture flask containing fetal bovine serum (FBS) culture medium. After 3 to 4 hours of cell culture, the flask was turned up vertically to allow the tissue to completely submerge in the medium, and left to culture in a 50 mL/L CO2 atmosphere at 37°C. After 5‐7 days, cells were released from tissue. Cells were passaged once the confluence reached 80% to 90%. The vascular smooth muscle α‐actin of the fourth to sixth passages of cells were detected by immunohistochemistry. After growing across a slide, cells were fixed in 4% paraformaldehyde and treated with 0.3% TritonX‐100 for 15 minutes. After washing with PBS, primary rabbit‐anti‐rat VSMC α‐actin antibody was added and incubated overnight. On the next day, fluorescein isothiocyanate (FITC)‐labeled goat‐anti‐rabbit secondary antibody was added and left to incubate for 60 minutes, and observed with a fluorescence confocal microscope.
2.10. Cell grouping and transfection
VSMCs were transfected with siRNA against SRPK1 vector (si‐SRPK1 group), SRPK1 overexpression vector (SRPK1 group), PI3 K/Akt pathway activator IGF‐1 100 ng/mL (IGF‐1 group), PI3 K/Akt pathway inhibitor LY294002 10 uM (LY294002 group), co‐transfected with SRPK1 gene silencing vector and PI3 K/Akt pathway activator IGF‐1 100 ng/mL (si‐SRPK1 + IGF‐1group), co‐transfected with SRPK1 over‐expression vector and PI3 K/Akt inhibitor LY294002 10 uM (SRPK1 + LY294002 group), the empty plasmid (negative control group, NC), respectively. Cells that were without treatment (blank group) were selected as the control group. Transfection was carried out using a Lipofectamine 2000 liposome transfection kit (Invitrogen Life Technologies, Carlsbad, CA, USA). One day prior to transfection, the cells were detached with 0.25% trypsin and counted. They were then transferred to a 6‐well plate with a density of 5 × 105 cells per well and covered with Royal Park Memorial Institute (RPMI) 1640 complete medium. The above plasmids were diluted with 100 μL of serum‐free RPMI 1640 medium. Next, 40 µL of Lipofectamine 2000 was diluted with 1000 μL serum‐free RPMI 1640 medium. The diluted plasmids were mixed with diluted Lipofectamine 2000 for 10 minutes, and left to sit at room temperature for 20 minutes. The total mixture of 250 μL was added into the 6‐well plate and mixed gently.
2.11. Cell counting kit‐8 (CCK‐8) assay
VSMCs at the logarithmic growth phase were inoculated in a 96‐well plate. Cells were counted 48 hours after adhering to the plates. The original culture medium was replaced by a 100‐μL fresh culture medium supplemented with 10‐μL CCK‐8 reagent (Beyotime Biotechnology Co., Shanghai, China), and continued to culture for 2 more hours. The Optical density (OD) at 450 nm was measured using an automatic plate reader (Bio‐Rad, Inc, Hercules, CA, USA), which was used to help determine cell proliferation. Six duplicates were set up for each experimental condition.
2.12. Flow cytometry
After 48 hours of transfection, VSMCs with a density of 1 × 106 cells/mL were extracted and stained in AnnexinV‐FITC (KeyGEN Bio‐TECH Co. Ltd., Nanjing, Jiangsu, China) for 15 minutes at room temperature in the dark. Cells were then centrifuged at 1000 r/min for 5 minutes, and then resuspended with 0.5 mL pre‐cooled binding buffer. After adding 10 μL of Propidiom iodide (PI) the samples were analyzed by a flow cytometer (Bio‐Rad, Inc, Hercules, CA, USA). In a scatter plot, viable cells were displayed in the lower left quadrant (Q4); early apoptotic cells were displayed in the lower right quadrant (Q3); necrosis and late apoptotic cells were displayed in the right upper quadrant (Q2). The Apoptosis rate (%) was represented as the total number of early apoptotic cells (Q3) and late apoptotic cells (Q2).
2.13. Reverse transcription quantitative polymerase chain reaction (RT‐qPCR)
The VSMCs were collected from each group. Total RNA was extracted with Trizol Reagent. The quality and quantity of RNA were determined by OD260 and OD280 on a spectrophotometer. The cDNA template was synthesized by reverse transcription PCR. The ABI7500 quantitative PCR instrument (ABI, Austen, TX, USA) was used to carry out real‐time quantitative PCR. One PCR reaction system typically contained: 1.5 μL of 10 × PCR buffer, 0.3 μL of dNTPs (10 mmol/L), forward and reverse primer (10 pmol/μL) each for 0.25, 0.25 μL of Taq polymerase (5 μ/μL, Takara Holdings Inc, Kyoto, Japan) all of which were mixed together with 1 μL cDNA template and brought up to 15 μL using ddH2O. The PCR mixture was initially pre‐denatured at 95°C for 10 minutes. It was then denatured at 95°C for 15 seconds, annealed at 60°C for 1 minute, and extended at 72°C for 40 seconds. The amplification cycles were repeated 50 times. The primers used in the reaction are shown in Table 1. β‐actin was used as an internal reference, and the fold was calculated by 2‐ΔΔCt, All the reactions were performed in triplicate and mean values were calculated.
Table 1.
RT‐qPCR primer sequence
| Gene | Sequence (5'−3') |
|---|---|
| SRPK1 | Forward: TTCCTCAACTGTAGGTCAGTCATTC |
| Reverse: TGTTCTTGCTCTTGTTCATCTTCAC | |
| PI3 K | Forward: CATCACTTCCTCCTGCTCTAT |
| Reverse: CAGTTGTTGGCAATCTTCTTC | |
| Akt | Forward: CTCATTCCAGACCCACGAC |
| Reverse: ACAGCCCGAAGTCCGTTA | |
| β‐actin | Forward: CACTCTTCCAGCCTTCCTTCC |
| Reverse: AGGTCTTTGCGGATGTCCAC |
RT‐qPCR, reverse transcription quantitative polymerase chain reaction; SRPK1, SR protein‐specific kinases; PI3 K, phosphatidylinositol‐3 kinase.
2.14. Western blot analysis
The total protein was extracted from VSMCs during the logarithmic growth phase using a protein lysis reagent (Beyotime Biotechnology Co., Shanghai, China). The extracted protein was quantified by the Bradford method (Thermo Fisher Scientific Inc, Waltham, MA, USA). Subsequently, 50 μg of total protein was separated using a 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The protein was then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore Corp, Billerica, MA, USA), and blocked in 5% skim milk at 37°C for 1 hour. The membranes were then incubated with the following primary antibodies at 4°C, overnight: rabbit anti‐mouse SRPK1 antibody, rabbit anti‐mouse PI3 K antibody, rabbit anti‐mouse AKT antibody, rabbit anti‐mouse p‐AKT antibody, apoptosis‐related protein Bcl‐2, Bax, Caspase 3, β‐actin monoclonal antibody (1:1000 dilution; Cell Signaling Technology, Beverly, MA, USA). After washing with Phosphate‐Buffered Saline/Tween (PBST) 3 times, 5 minutes each, the membrane was incubated with the corresponding horseradish peroxidase (HRP)‐labeled secondary goat anti‐rabbit antibody (1:4000 dilution; Cell Signaling Technology, Beverly, MA, USA) at room temperature for 2 hours and then rinsed. Immunobloting was developed in chemiluminescence solution which was prepared by mixing the Luminol Reagent and Peroxide Solution (Millipore Corp, Billerica, MA, USA) at a 1:1 ratio. This experiment was repeated 3 times in order to obtain the mean value.
2.15. Statistical analysis
Statistical analyses were conducted using SPSS 21.0 software (IBM Corp. Armonk, NY, USA). Enumeration data were expressed as a percentage (%), and a chi‐square test was used to analyze data among the groups. Measurement data were expressed as a mean ± standard deviation. Data differences between 2 groups were compared by a t test. Data between multiple groups were compared by a one‐way analysis of variance. Values of P < 0.05 indicated that the data was statistically significant.
3. RESULTS
3.1. SRPK1 and the PI3 K/AKT signaling pathway are related to the pathogenesis of IA
Two datasets of IA‐related gene expression profiles, GSE36791 and GSE54083 were retrieved from the GEO database. Differential expression analysis of GSE36791 showed that 31 genes were expressed aberrantly in IA (Figure 1A). GSE54083 consists of IA rupture and UIA. The gene expression levels of IA rupture and UIA in GSE54083 were analyzed with the normal sample as control (Figure 1B‐C). We found that there were 1788 differentially expressed genes in IA rupture compared with the control group, of which 939 genes were up‐regulated and 849 genes were down‐regulated. On the other hand, there were 1456 differentially expressed genes in UIA, of which 908 genes were up‐regulated and 548 genes were down‐regulated. In order to narrow down the genes that were related to IA, the aforementioned 3 groups of data were analyzed using a Venn diagram (Figure 1C). The results showed that only 2 genes, SRPK1 and HP, were identified in the intersection among all the 3 groups of data. SRPK1 was found to be significantly elevated in IA tissue. It has been reported that the HP gene is relevant to IA,22, 23 but few previous studies have reported the correlation between SRPK1 and IA. However, in some other tumor diseases, it has been reported that SRPK1 affects progression of diseases through the PI3 K/AKT signaling pathway.24, 25, 26 In addition, it was also reported that the AKT signaling pathway was involved in the progression of aneurysms,27, 28 but have not yet been demonstrated in IA. In summary, our results suggested that SRPK1 may affect the progression of IA by regulating the PI3 K/AKT signaling pathway.
Figure 1.
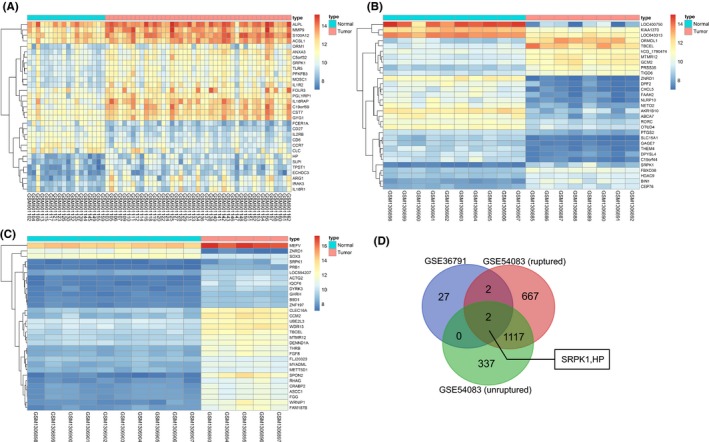
GEO data analysis shows that SRPK1 is associated with IA. Panel A, B, and C, heat map shows the differentially expressed genes in IA tissue. The heat map shows the analysis of IA expression microarray. The horizontal axis represents the samples, and the vertical axis represents the genes. The upper red and blue strip represents the sample type; whereby blue represents the normal sample, and the red represents the IA sample. The left dendrogram represents the clustering results among genes; each square indicates the expression of a gene in a sample. The upper right histogram color indicates the level of expression, whereby the red suggests a high expression, and the blue suggests a low expression value. Panel D, Venn diagram illustrates the overlap of differential expressed genes between IA tissue and normal tissue. The blue color represents the differentially expressed genes in the GSE36791 expression profile analysis, the red represents the differentially expressed genes of the IA rupture, and the green indicates the UIA in the GSE54083 expression profile analysis. The blue arrow shows the SRPK1 and HP1 genes in the intersection among the 3 data groups. The digital in each shape reflect the number of genes. IA, intracranial aneurysm; SRPK1, SR protein‐specific kinases
3.2. Highly expressed SRPK1 and activated PI3 K/AKT signaling pathway found in the aneurysm vascular wall of IA
The diameters of IAs generally vary from 0.7 to 3 cm, and most tumors grossly appear a brown or purplish red. Thrombus was observed in the aneurysm portion of the vascular structures. The thickness of the aneurysm wall was not in uniform; they appeared thick at the base, and thin superiorly. Immunohistochemistry analysis was used to detect IA and normal cerebral cortical artery tissues in the normal and IA groups. Results showed that SRPK1 was mainly expressed in the tunica media and tunica adventitia of the vascular wall, a little of which was detected in the tunica intima. The expression of SRPK1 was found to be higher in the IA group (66.04%) than that in the normal group (31.43%) (P < 0.05), suggesting that SRPK1 was up‐regulated in IA (Figure 2A‐B). In addition, we found that PI3 K, Akt and p‐Akt were mainly expressed in the tunica intima of vascular wall. The expressions of PI3 K, Akt, and p‐Akt in the IA group (75.47%, 83.02%, 73.58%) were significantly higher than that in the normal group (22.86%, 25.71%, 14.29%) (P < 0.05).
Figure 2.
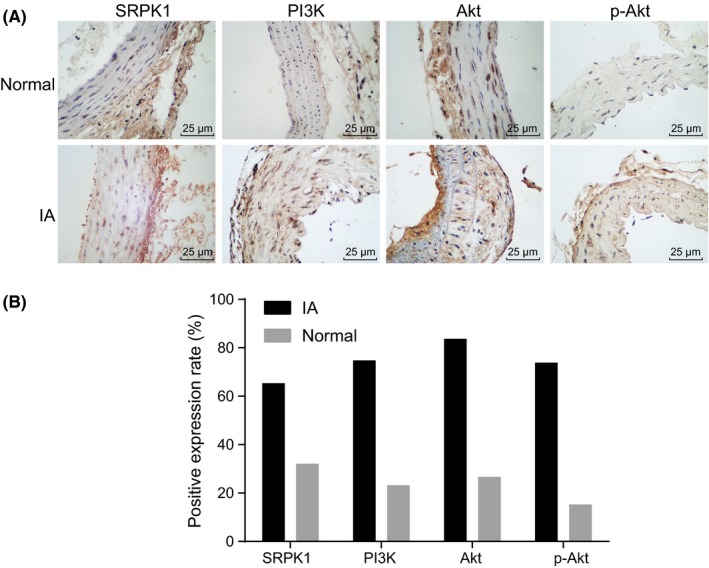
Immunohistochemical staining analysis for the expression of SRPK1 and the PI3 K/Akt signaling pathway‐related factors (PI3 K, AKT, and p‐AKT) in the aneurysm wall of IA and normal vasculature. Panel A, immunohistochemical staining of SRPK1, PI3 K, Akt, and p‐Akt in the normal group and the IA group (400×). SRPK1 was mainly localized in the tunica media and tunica adventitia of the vascular wall, and in very little amounts in the tunica intima. PI3 K, Akt, and p‐Akt were mainly located in the tunica intima of vascular wall.; Panel B, quantitative analysis indicates expressions of SRPK1, PI3 K, Akt, and p‐Akt were up‐regulated in IA; *P < 0.05 vs the normal group; IA, intracranial aneurysm; SRPK1, SR protein‐specific kinases; PI3 K, phosphatidylinositol‐3 kinase
3.3. Successful establishment of IA rat models
Our study simulated the high hemodynamic state of the whole body by ligating the posterior branch of bilateral renal artery while simultaneously ligating the left common carotid artery. This allowed us to simulate and induce a continuous impact of high hemodynamic changes on the vessel wall at the basilar artery ring region. As a result, we observed degenerative changes in the vessel wall structure of the right anterior cerebral artery/olfactory artery bifurcation which subsequently producing an aneurysm. At the end of the experiment, we observed that the rats in the sham group moved freely, and had normal physiological reactions. On the other hand, rats in the IA group became gradually thinner, had a slower reaction to stimulus, had the right eye ptosis, and developed lower limb paralysis. HE staining showed that rats in the sham group had VSMCs that were arranged in tight manner at the right anterior cerebral artery and olfactory artery bifurcation in cerebral artery, whereas the internal elastic membrane of rats in the IA group was fragmented and disappeared at the root of the aneurysm. VSMCs at the root of the aneurysm were different in their size, were arranged in a disorganized manner, and were necrotic (Figure 3). These results suggested that a stable rat model of IA was successfully established.
Figure 3.
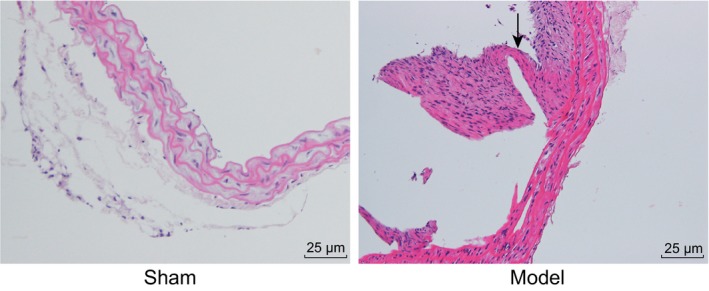
HE staining (200×) verifies the successful establishment of rat models of IA. Degenerative changes were observed in the vessel wall structure of the right anterior cerebral artery/olfactory artery bifurcation and eventually an aneurysm was formed. As indicated by the arrow, internal elastic lamina is injured, and the vascular wall is expanded, and the wall of the middle cerebral artery is enlarged. IA, intracranial aneurysm; HE, hematoxylin eosin
3.4. SRPK1 gene silencing promoted vascular remodeling in IA
HE staining was conducted to measure the intracranial arterial wall of rats in each group. Results showed (Figure 4) that cells in the tunica intima, VSMC layer and tunica adventitia of the sham group were intact and neatly arranged. In the IA group, the tunica intima of IA was destroyed; there was VSMCs degeneration and necrosis and thinning of the arterial wall. In the SRPK1, IGF‐1, and si‐SRPK1 + IGF‐1 groups, we observed the disappearance of the IA tunica intima, a destroyed elastic fiber, and the thinning of the arterial wall compared to the IA group. The integrity of tunica intima and VSMCs structure of the si‐SRPK1, LY294002, and SRPK1 + LY294002 groups appeared stronger than that of the IA group. These observations suggested that the gene silencing of SRPK1 promoted vascular remodeling in rats with IA.
Figure 4.
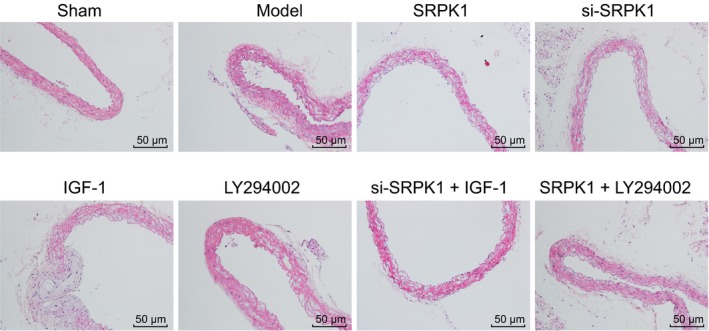
HE staining (200×) shows the effect of SRPK1 gene silencing mediated by siRNA on the histopathological changes of intracranial arterial wall. The gene silencing of SRPK1 promoted vascular remodeling in rats with IA. IA, intracranial aneurysm; HE, hematoxylin eosin; SRPK1, SR protein‐specific kinases
3.5. SRPK1 gene silencing inhibited rat VSMCs apoptosis
VSMC apoptosis occurred during the course of IA formation. TUNEL staining (Figure 5A‐B) accurately reflected the cell apoptosis conditions, by which apoptotic cells were identified by their yellow or brownish‐yellow appearance in color. Compared with the sham group, the other groups had an increased apoptosis rate of VSMCs in the intracranial arteries (P < 0.05). Compared with the IA group, the SRPK1 group, the IGF‐1 group, and the si‐SRPK1 + IGF‐1 group had a higher apoptosis rate of VSMCs. On the other hand, the si‐SRPK1 group, the LY294002 group, and the SRPK1 + LY294002 group had a significantly lower apoptosis rate of VSMCs in the intracranial artery (P < 0.05). The si‐SRPK1 group had significantly lower cell apoptosis rate, while the IGF‐1 group had significantly higher apoptosis rate compared with the si‐SRPK1 + IGF‐1 group. Compared with the SRPK1 + LY294002 group, the LY294002 group had significantly lower apoptosis rate, while the SRPK1 group had a markedly apoptosis rate. These results demonstrated that SRPK1 gene silencing inhibited the apoptosis of VSMCs of IA, which could be reversed by activating the PI3 K/Akt signaling pathway.
Figure 5.
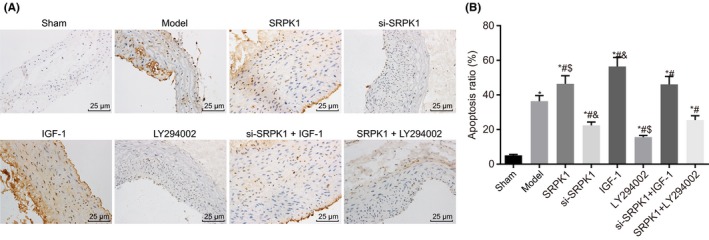
TUNEL staining (400×) shows that SPRK1 gene silencing reduces VSMC apoptosis in rats with IA. Panel A and B, TUNEL staining demonstrates apoptotic VSMCs in the intracranial artery. The si‐SRPK1 group had significantly reduced apoptosis rate, while the IGF‐1 group had significantly increased apoptosis rate. *P < 0.05 vs the sham group; #P < 0.05 vs the IA group; &P < 0.05 vs the si‐SRPK1 + IGF‐1 group; $P < 0.05 vs the SRPK1 + LY294002 group. VSMCs, vascular smooth muscle cells; TUNEL, Terminal deoxynucleotidyl transferase (TdT)‐mediated dUTPbiotin nick end labeling; IA, intracranial aneurysm; SRPK1, SR protein‐specific kinases
3.6. SRPK1 gene silencing promoted the proliferation of VSMCs
Under a light microscope, the morphology of normal VSMCs was spindle shaped. Under a fluorescence microscope, normal VSMCs were identified by their red fluorescence color and a positive rate >95% (Figure 6A). CCK‐8 assay (Figure 6B) was used to measure VSMC viability. The OD values of the SRPK1, IGF‐1, and si‐SRPK1 + IGF‐1 groups were significantly lower compared with the blank group and were significantly higher in the si‐SRPK1 group, LY294002 group, and SRPK1 + LY294002 groups. The si‐SRPK1 group had a significantly higher OD value of VSMCs, while the IGF‐1 group had significantly lower OD value compared with the si‐SRPK1 + IGF‐1 group. The OD value of the LY294002 group was significantly higher in the SRPK1 + LY294002 group, compared to the SRPK1 group. The results demonstrated that the silencing of SRPK1 gene increased VSMC proliferation. This process could also be reversed by the activation of PI3 K/Akt signaling pathway.
Figure 6.
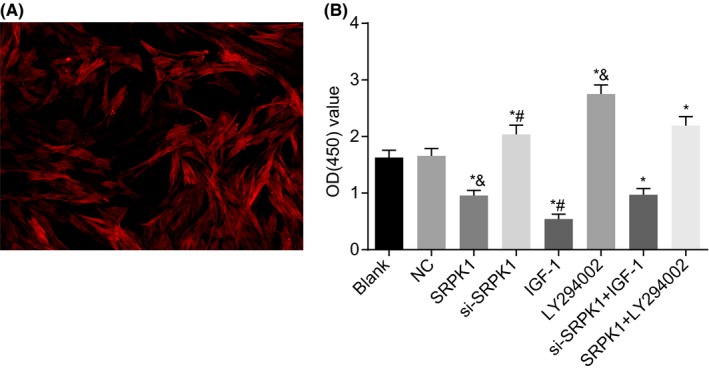
Microscopic examination and CCK‐8 assay show that SRPK1 gene silencing promotes VSMCs proliferation in rats with IA. Panel A, VSMCs were observed under a light microscope and a fluorescence confocal microscope (400×). VSMCs were identified by their red fluorescence appearance and a positive rate >95%; Panel B, cell viability detected by CCK‐8 assay was increased in the si‐SPRK1 group. *P < 0.05 vs the sham group; #P < 0.05 vs the si‐SRPK1 + IGF‐1 group; $P < 0.05 vs the SRPK1 + LY294002 group. VSMCs, vascular smooth muscle cells; CCK8, Cell counting kit‐8; IA, intracranial aneurysm; SRPK1, SR protein‐specific kinases
3.7. SRPK1 gene silencing inhibited the apoptosis of VSMCs
Apoptosis of VSMCs was assessed by flow cytometry (Figure 7A‐B). We found that the SRPK1 group, the IGF‐1 group and the si‐SRPK1 + IGF‐1 group had a significantly higher apoptosis rate, whereas the si‐SRPK1, LY294002, and SRPK1 + LY294002 groups had a reduced apoptosis rate compared with the blank group. The apoptosis rate of the si‐SRPK1 group was significantly lower, and the apoptosis rate of the IGF‐1 group was significantly higher than that of the si‐SRPK1 + IGF‐1 group. These results suggest that activated PI3 K/AKT signaling reverses the inhibitory effects of SRPK1. Compared with the SRPK1 + LY294002 group, the LY294002 group had a significantly lower apoptosis rate, whereas the apoptotic rate in the SRPK1 group was significantly higher. It suggests that over‐expressed SRPK1 is antagonized by PI3 K/AKT signaling deactivation. Compared with the blank group, the SRPK1, IGF‐1, and si‐SRPK1 + IGF‐1 groups had a significant reduction in the expression of apoptosis‐related protein Bcl‐2, and increased expression of Bax and Caspase 3 (Figure 7C‐D). The si‐SRPK1 group, the LY294002 group, and the SRPK1 + LY294002 group all exhibited significantly increased expression levels of Bcl‐2, and decreased expression of Bax and Caspase 3. Compared with the si‐SRPK1 + IGF‐1 group, the si‐SRPK1 group had higher Bcl‐2 expression, and reduced expression of Bax and Caspase 3. In contrast, the IGF‐1 group had a lower Bcl‐2 expression, and increased Bax and Caspase 3 expression. The LY294002 group had significantly higher Bcl‐2 expression and lower Bax and Caspase 3 expression compared to the SRPK1 + LY294002 group. Additionally, the SRPK1 group had a significantly lower expression of Bcl‐2 but a significantly increased expression of Bax and Caspase 3 than that in the SRPK1 + LY294002 group. Our results are consistent findings of the apoptotic cells produced by flow cytometry. In conclusion, the results further supports that the silencing of SRPK1 gene inhibited the apoptosis of VSMCs in vitro. This inhibitory effect however, could also be reversed by the activation of the PI3 K/Akt signaling pathway.
Figure 7.
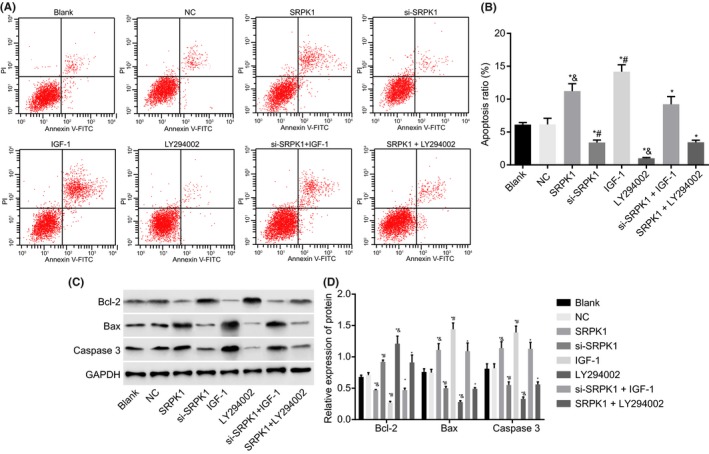
Flow cytometry and Western blot analysis show that SPRK1 gene silencing suppresses VSMC apoptosis in rats with IA. Panel A and B, Flow cytometry was used to detect the apoptosis rate, and showed that there was a significant decrease in cell apoptosis belonging in VSMCs belonging to the si‐SPRK1 group; Panel C and D, protein bands and levels of apoptosis‐related factors (Bcl‐2, Bax, Caspase3) in VSMCs were detected by Western blot. *P < 0.05 vs the NC group; #P < 0.05 vs the si‐SRPK1 + IGF‐1 group; $P < 0.05 vs the SRPK1 + LY294002 group. VSMCs, vascular smooth muscle cells; IA, intracranial aneurysm; NC, negative control; SRPK1, SR protein‐specific kinases; PI3 K, phosphatidylinositol‐3 kinase
3.8. SRPK1 gene silencing promoted VSMCs proliferation by inhibiting the PI3 K/Akt signaling pathway
Genes involved in the PI3 K/Akt signaling pathway in VSMCs were assessed on an mRNA and protein level by RT‐qPCR detection (Figure 8A) and Western blot analysis (Figure 8B‐C). Compared with the blank group, the SRPK1 and SRPK1 + LY294002 group had a significantly mRNA and protein expression of SRPK1 (P < 0.05), whereas the other groups had no significant changes in the mRNA and protein levels (P > 0.05). Compared with the blank group, the SRPK1, IGF‐1 and si‐SRPK1 + IGF‐1 groups had elevated mRNA expressions of PI3 K and Akt, and elevated protein levels of PI3, Akt, p‐Akt (P < 0.05). In contrast, the si‐SRPK1, LY294002, and SRPK1 + LY294002 groups had decreased mRNA expressions of PI3 K and Akt, as well as decreased protein levels of PI3, Akt, p‐Akt (P < 0.05). Compared with the si‐SRPK1 + IGF‐1 group, the si‐SRPK1 group had significantly lower mRNA expressions of Akt and PI3 K, protein levels of PI3 K, Akt, and p‐Akt, while the IGF‐1 group had significantly higher mRNA expressions of Akt and PI3 K, protein levels of PI3 K, Akt, and p‐Akt. Compared with the SRPK1 + LY294002 group, the LY294002 group had lower mRNA expressions of Akt and PI3 K, as well as lowered protein levels of PI3 K, Akt, and p‐Akt. The SRPK1 group produced the opposite trend by exhibiting a significantly higher level of mRNA expressions of Akt and PI3 K, as well as higher protein levels of PI3 K, Akt, and p‐Akt (P < 0.05). These findings illustrate that SRPK1 gene silencing promoted VSMCs proliferation through the inactivation of the PI3 K/Akt signaling pathway.
Figure 8.
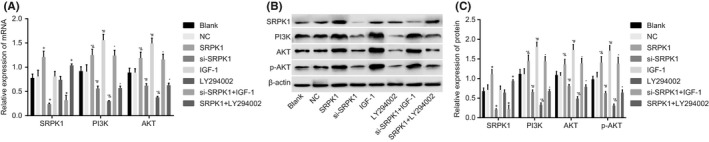
RT‐qPCR and Western blot analysis show that SRPK1 gene silencing promotes VSMCs proliferation by repressing the activation of the PI3 K/Akt signaling pathway in rats with IA. Panel A, The RT‐qPCR detection of mRNA expressions of SPRK1, PI3 K, and Akt, which were mostly reduced in the si‐SPRK1 group; Panel B and C, protein bands and levels of SPRK1, PI3 K, Akt, p‐Akt, and β‐actin were detected by Western blot, which were decreased mostly in the si‐SPRK1 group. *P < 0.05 vs the NC group; #P < 0.05 vs the si‐SRPK1 + IGF‐1 group; $P < 0.05 vs the SRPK1 + LY294002 group. VSMCs, vascular smooth muscle cells; IA, intracranial aneurysm; RT‐qPCR, reverse transcription quantitative polymerase chain reaction; NC, negative control; SRPK1, SR protein‐specific kinases; PI3 K, phosphatidylinositol‐3 kinase
4. DISCUSSION
IA represents the most severe case among all the cerebrovascular diseases, with a mortality rate of nearly 40% despite modern treatment approaches.1 Our present study investigated the effects of SPRK1 silencing on the regulation of proliferation, apoptosis, and vascular remodeling in IA, as well as the possible involvement of the PI3 K/Akt signaling pathway. Our findings provided evidence that si‐SPRK1 was able to inhibit the PI3 K/Akt signaling pathway, thereby increasing cell proliferation and vascular remodeling, as well as decreasing the apoptosis rate of VSMCs in rats with IA (Figure 9).
Figure 9.
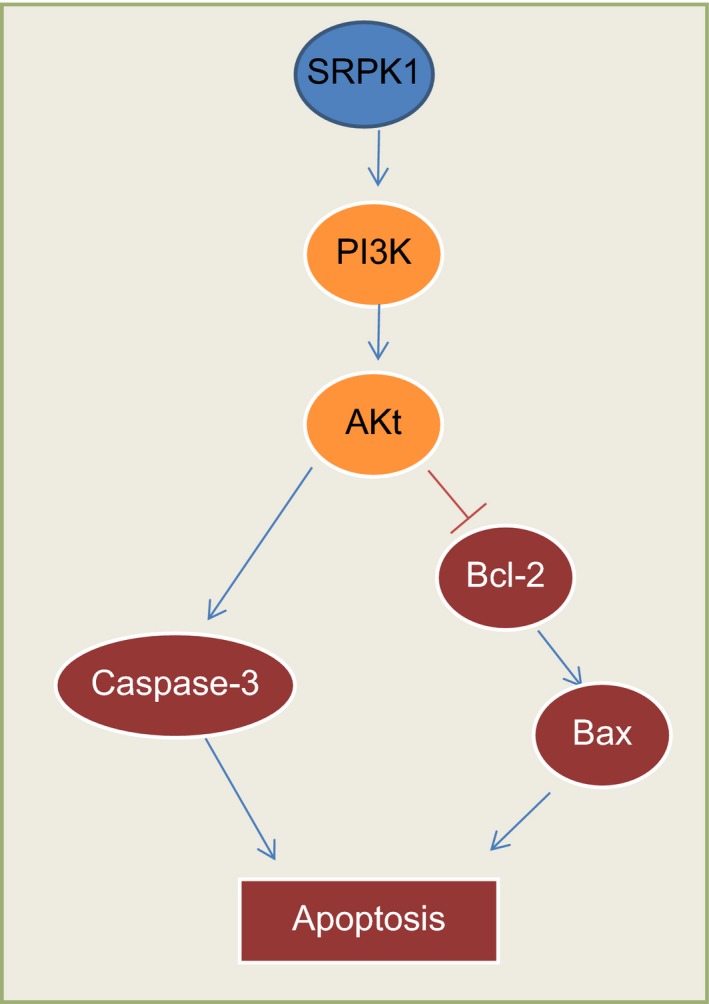
Map of molecular mechanisms involved in SRPK1 regulation in the apoptosis of intracranial aneurysm smooth muscle cells. SRPK1 is upregulated with a dysregulated PI3 K‐AKT signaling pathway in intracranial aneurysm. After SRPK1 gene silencing, the apoptosis of intracranial aneurysm smooth muscle cells was inhibited via the suppression of the PI3 K/AKT signaling pathway. SRPK1, SR protein‐specific kinases; PI3 K, phosphatidylinositol‐3 kinase
Our results show that SRPK1 is expressed at higher levels in the aneurysm wall of IA compared to those in the normal cerebral vessel wall. SRPK1 has been found to be expressed in both the cytoplasm and nucleus. SRPK1 can also translocate between these compartments under various conditions such as cell cycle and stress.29 SRPK1 is known to be expressed in the mammalian CNS and has been implicated in modulating the splicing of neuronal proteins.30 Additionally, SRPK1 has been proposed as a new molecular target that can be used to help treat patients with early stage gliomas. This is due to SRPK1’s ability to regulate cell growth, metastasis, chemosensitivity, and glioma angiogenesis.31 Erdö et al identified new pathways involved in the development of post‐ischemic brain injury, including the splicing factor SRPK1.32 Despite its many findings, the effects of SRPK1 on the apoptosis pathways in VSMCs still remain unclear. Microscopic examination and CCK8 results in our experiment demonstrated that SRPK1 gene silencing help promote VSMCs proliferation in rats with IA. Furthermore, TUNEL staining, flow cytometric detection, and Western blot analysis all revealed that SPRK1 gene silencing leads to attenuated VSMC apoptosis. The regulatory mechanism of this finding was found to be linked with the inactivation of the PI3 K/Akt signaling pathway.
The PI3 K/Akt signaling pathway has been widely documented due to its multifunctional role that includes the insulin signaling pathway in the brain, which promotes cell survival by inactivating the pro‐apoptotic pathways.33 Akt regulates the suppression of pro‐apoptotic genes: BAD and BIM, and the degradation of the tumor inhibitor protein p53 mediates programmed cell apoptosis all of which leads to increased cell survival.34 The Akt kinase system is recognized to be involved in the mediation of cell proliferation, migration, and apoptosis in mammalian cells.17 Activated Akt has been found to induce SRPK1 phosphorylation in vitro, which could also be inhibited by the Akt‐specific suppressor MK2206.9 Moreover, Zhou et al reported that activated Akt may lead to SRPK1 autophosphorylation, which causes SRPK1 to translocate into the nucleus and enhances the phosphorylation of downstream splicing. These findings suggest that SRPK1 may be located downstream of Akt,26 which corroborates with our results that the increased VMSC apoptosis rate after treatment of si‐SRPK1 was significantly lower and VMSC apoptosis after treatment of IGF‐1 was higher than that of the treatment of si‐SRPK1 + IGF‐1. According to Curcio et al, suppressed Akt leads to decreased VSMC proliferation, and an increase in the vascular apoptosis rate, which was observed in aged animals.35 Liu et al demonstrated that apelin‐13 induced rat VSMC proliferation through the PI3 K/Akt signaling pathway by confirming the effect of PI3 K inhibitor LY294002 and Akt inhibitor 1701‐1 on cell proliferation induced by apelin‐13.18 Additionally, Akt also mediates other genes of the PI3 K pathway such as Bcl‐2‐related proteins, glycogen synthase kinase‐3β (GSK3β) or MDM2.36 Akt also phosphorylates BAD, the proapoptotic Bcl‐2 family member, thereby decreasing BAD pro‐apoptotic functions.37
Apoptosis of VSMC is critical for normal vascular remodeling, in addition to the development of multiple pathological conditions, including aneurysms.5 Ruptured aneurysms induce oxidative stress, leukocyte migration, and vascular remodeling.38 Recently, it has been reported that the tissue transglutaminases have the potential to produce interactions of extracellular matrix fibrillar components with attachment sites on VSMCs.39 SRPK1 gene silencing promotes vascular remodeling in rats with IA whereby its actions can be reversed by the activation of the PI3 K/Akt signaling pathway. Tumors in SRPK1‐knockdown mice grew slower compared with those that expressed SRPK1 due to the suppression of angiogenesis. This finding was verified by a reduction in the micro‐vessel density in SRPK1‐knockdown tumors compared with the controls.29 It has been suggested by Giannakouros et al that SRPKs, and potential members of the Akt signaling pathway may play a key role during the start of mitosis by initially regulating the detachment of peripheral heterochromatin from the innermost nuclear membrane, and later by the deletion of HP1, consequently resulting in chromosome condensation.40 The PI3 K/Akt signaling pathway is also implicated in the proliferation and migration of VSMCs thus modulating vascular remodeling.41 Specifically, blockade of PI3 k/Akt signaling inhibited the VSMC proliferation both in vitro and in vivo.42
In summary, our study demonstrated that siRNA targeting of SRPK1 enhanced VSMC proliferation, vascular remodeling and inhibited VSMC apoptosis in rats with IA via inhibiting the activation of PI3 K/Akt signaling pathway. With further and extensive research, SRPK1 may serve as a potential novel therapeutic target to help enhance the vascular remodeling capacity in IAs. The findings of our current investigation should be supported by additional experiments with larger sample sizes to help develop clinical significance.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We would like to express our gratitude and acknowledge the reviewers for their helpful comments on this paper.
Li X‐G, Wang Y‐B. SRPK1 gene silencing promotes vascular smooth muscle cell proliferation and vascular remodeling via inhibition of the PI3K/Akt signaling pathway in a rat model of intracranial aneurysms. CNS Neurosci Ther. 2019;25:233–244. 10.1111/cns.13043
REFERENCES
- 1. Zhang G, Tu Y, Feng W, et al. Association of interleukin‐6‐572G/C gene polymorphisms in the Cantonese population with intracranial aneurysms. J Neurol Sci. 2011;306:94‐97. [DOI] [PubMed] [Google Scholar]
- 2. Liu J, Kuwabara A, Kamio Y, et al. Human mesenchymal stem cell‐derived microvesicles prevent the rupture of intracranial aneurysm in part by suppression of mast cell activation via a PGE2‐dependent mechanism. Stem Cells. 2016;34:2943‐2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Korja M, Kaprio J. Controversies in epidemiology of intracranial aneurysms and SAH. Nat Rev Neurol. 2016;12:50‐55. [DOI] [PubMed] [Google Scholar]
- 4. Korja M, Lehto H, Juvela S. Lifelong rupture risk of intracranial aneurysms depends on risk factors: a prospective Finnish cohort study. Stroke. 2014;45:1958‐1963. [DOI] [PubMed] [Google Scholar]
- 5. Penn DL, Witte SR, Komotar RJ, et al. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J Clin Neurosci. 2014;21:28‐32. [DOI] [PubMed] [Google Scholar]
- 6. Kovacevic M, Jonjic N, Stalekar H, et al. Apoptotic cell death and rupture of abdominal aortic aneurysm. Med Hypotheses. 2010;74:908‐910. [DOI] [PubMed] [Google Scholar]
- 7. Sun L, Zhao M, Zhang J, et al. MiR‐29b downregulation induces phenotypic modulation of vascular smooth muscle cells: implication for intracranial aneurysm formation and progression to rupture. Cell Physiol Biochem. 2017;41:510‐518. [DOI] [PubMed] [Google Scholar]
- 8. Guo D, Wang YW, Ma J, et al. Study on the role of Cathepsin B and JNK signaling pathway in the development of cerebral aneurysm. Asian Pac J Trop Med. 2016;9:499‐502. [DOI] [PubMed] [Google Scholar]
- 9. Zhou Z, Qiu J, Liu W, et al. The Akt‐SRPK‐SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell. 2012;47:422‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghosh G, Adams JA. Phosphorylation mechanism and structure of serine‐arginine protein kinases. FEBS J. 2011;278:587‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin JC, Lin CY, Tarn WY, et al. Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4‐regulated splicing events. RNA. 2014;20:1621‐1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aubol BE, Plocinik RM, Hagopian JC, et al. Partitioning RS domain phosphorylation in an SR protein through the CLK and SRPK protein kinases. J Mol Biol. 2013;425:2894‐2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein‐specific kinases. Chromosoma. 2013;122:191‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bullock N, Oltean S. The many faces of SRPK1. J Pathol. 2017;241:437‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mavrou A, Brakspear K, Hamdollah‐Zadeh M, et al. Serine‐arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene. 2015;34:4311‐4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goncalves V, Henriques AF, Pereira JF, et al. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor‐related Rac1b in colorectal cells. RNA. 2014;20:474‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang P, Zhou Z, Hu A, et al. Both decreased and increased SRPK1 levels promote cancer by interfering with PHLPP‐mediated dephosphorylation of Akt. Mol Cell. 2014;54:378‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu C, Su T, Li F, et al. PI3K/Akt signaling transduction pathway is involved in rat vascular smooth muscle cell proliferation induced by apelin‐13. Acta Biochim Biophys Sin (Shanghai). 2010;42:396‐402. [DOI] [PubMed] [Google Scholar]
- 19. Ma X, Yao H, Yang Y, et al. miR‐195 suppresses abdominal aortic aneurysm through the TNF‐alpha/NF‐kappaB and VEGF/PI3K/Akt pathway. Int J Mol Med. 2018;41:2350‐2358. [DOI] [PubMed] [Google Scholar]
- 20. Wang C, Wen J, Zhou Y, et al. Apelin induces vascular smooth muscle cells migration via a PI3K/Akt/FoxO3a/MMP‐2 pathway. Int J Biochem Cell Biol. 2015;69:173‐182. [DOI] [PubMed] [Google Scholar]
- 21. Kim C, Kikuchi H, Hashimoto N, et al. Involvement of internal elastic lamina in development of induced cerebral aneurysms in rats. Stroke. 1988;19:507‐511. [DOI] [PubMed] [Google Scholar]
- 22. Ruzevick J, Jackson C, Pradilla G, et al. Aneurysm formation in proinflammatory, transgenic haptoglobin 2–2 mice. Neurosurgery 2013;72:70–76; discussion 76. [DOI] [PubMed] [Google Scholar]
- 23. Kadirvel R, Ding YH, Dai D, et al. Differential expression of genes in elastase‐induced saccular aneurysms with high and low aspect ratios. Neurosurgery. 2010;66:578–584; discussion 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Q, Liu X, Liu Z, et al. MicroRNA‐1296 inhibits metastasis and epithelial‐mesenchymal transition of hepatocellular carcinoma by targeting SRPK1‐mediated PI3K/AKT pathway. Mol Cancer. 2017;16:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang Y, Wu Q, Tian T, et al. The influence of SRPK1 on glioma apoptosis, metastasis, and angiogenesis through the PI3K/Akt signaling pathway under normoxia. Tumour Biol. 2015;36:6083‐6093. [DOI] [PubMed] [Google Scholar]
- 26. Zhou B, Li Y, Deng Q, et al. SRPK1 contributes to malignancy of hepatocellular carcinoma through a possible mechanism involving PI3K/Akt. Mol Cell Biochem. 2013;379:191‐199. [DOI] [PubMed] [Google Scholar]
- 27. Hou WZ, Chen XL, Wu W, et al. MicroRNA‐370‐3p inhibits human vascular smooth muscle cell proliferation via targeting KDR/AKT signaling pathway in cerebral aneurysm. Eur Rev Med Pharmacol Sci. 2017;21:1080‐1087. [PubMed] [Google Scholar]
- 28. Laaksamo E, Ramachandran M, Frosen J et al. Intracellular signaling pathways and size, shape, and rupture history of human intracranial aneurysms. Neurosurgery. 2012;70:1565–1572; discussion 1572–1563. [DOI] [PubMed] [Google Scholar]
- 29. Oltean S, Gammons M, Hulse R, et al. SRPK1 inhibition in vivo: modulation of VEGF splicing and potential treatment for multiple diseases. Biochem Soc Trans. 2012;40:831‐835. [DOI] [PubMed] [Google Scholar]
- 30. Mytilinaios DG, Tsamis KI, Nikolakaki E, et al. Distribution of SRPK1 in human brain. J Chem Neuroanat. 2012;43:20‐27. [DOI] [PubMed] [Google Scholar]
- 31. Wu Q, Chang Y, Zhang L, et al. SRPK1 dissimilarly impacts on the growth, metastasis, chemosensitivity and angiogenesis of glioma in normoxic and hypoxic conditions. J Cancer. 2013;4:727‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Erdo F, Trapp T, Mies G, et al. Immunohistochemical analysis of protein expression after middle cerebral artery occlusion in mice. Acta Neuropathol. 2004;107:127‐136. [DOI] [PubMed] [Google Scholar]
- 33. Zhang H, Gao Y, Dai Z, et al. IGF‐1 reduces BACE‐1 expression in PC12 cells via activation of PI3‐K/Akt and MAPK/ERK1/2 signaling pathways. Neurochem Res. 2011;36:49‐57. [DOI] [PubMed] [Google Scholar]
- 34. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010;1:530‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Curcio A, Torella D, Indolfi C. Mechanisms of smooth muscle cell proliferation and endothelial regeneration after vascular injury and stenting: approach to therapy. Circ J. 2011;75:1287‐1296. [DOI] [PubMed] [Google Scholar]
- 36. Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13:234‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jang SW, Liu X, Fu H, et al. Interaction of Akt‐phosphorylated SRPK2 with 14‐3‐3 mediates cell cycle and cell death in neurons. J Biol Chem. 2009;284:24512‐24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chalouhi N, Hoh BL, Hasan D. Review of cerebral aneurysm formation, growth, and rupture. Stroke. 2013;44:3613‐3622. [DOI] [PubMed] [Google Scholar]
- 39. Schiffrin EL. Vascular remodeling in hypertension: mechanisms and treatment. Hypertension. 2012;59:367‐374. [DOI] [PubMed] [Google Scholar]
- 40. Giannakouros T, Nikolakaki E, Mylonis I, et al. Serine‐arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J. 2011;278:570‐586. [DOI] [PubMed] [Google Scholar]
- 41. Wang S, Wang Y, Jiang J, et al. 15‐HETE protects rat pulmonary arterial smooth muscle cells from apoptosis via the PI3K/Akt pathway. Prostaglandins Other Lipid Mediat. 2010;91:51‐60. [DOI] [PubMed] [Google Scholar]
- 42. Wang AB, Li HL, Zhang R, et al. A20 attenuates vascular smooth muscle cell proliferation and migration through blocking PI3k/Akt singling in vitro and in vivo. J Biomed Sci. 2007;14:357‐371. [DOI] [PubMed] [Google Scholar]