

Summary
Aim
Antiepileptic drugs that modulate GABA have the potential to aggravate or improve the symptoms of absence epilepsy. PF‐06372865 is a positive allosteric modulator (PAM) of α2/3/5 subunit‐containing GABAA receptors with minimal activity at α1‐containing receptors, which are believed to mediate many of the adverse events associated with benzodiazepines. The aim of this study was to assess the antiepileptic effect of PF‐06372865 in a preclinical model of absence seizures.
Methods
Genetic absence epilepsy rats from Strasbourg (GAERS) was implanted with four cortical electrodes over the frontoparietal cortex, and the number and cumulated duration of spike‐and‐wave discharges (SWDs) were recorded for 10‐90 minutes following administration of vehicle, PF‐06372865, and positive controls diazepam and valproate.
Results
PF‐06372865 (0.3, 1, 2, 10 mg kg−1) dose‐dependently reduced the expression of SWDs, including full suppression at the highest doses by 30 minutes after administration.
Conclusions
PF‐06372865 demonstrated robust efficacy in suppressing SWDs in the GAERS model of absence epilepsy. To our knowledge, this is the first demonstration of antiepileptic activity of an α2/3/5‐subtype‐selective GABAA PAM in a model of absence epilepsy. Further study of the antiepileptic properties of PF‐06372865 is warranted in patients with absence seizures.
Keywords: absence, epilepsy, GABA, GAERS, PF‐06372865 (total ≥5, ≤8)
1. INTRODUCTION
Absence epilepsy is a particular form of epileptic syndrome in which patients show generalized nonconvulsive seizures characterized by a brief unresponsiveness to environmental stimuli and cessation of activity. Clinically, typical absence seizures are associated with bilateral, synchronous, and regular spike‐and‐wave discharges (SWDs).1 This form of epilepsy presents a specific pharmacology different from that observed in other types of epilepsies. Therefore, careful evaluation of new antiepileptic drugs (AEDs) in clinical development is advisable to assess whether there may be an aggravation of SWD in idiopathic generalized epilepsies,2, 3 and particularly in absence epilepsy, which may prohibit their use in these patients.
The genetic absence epilepsy rat from Strasbourg (GAERS) is a selectively inbred strain of Wistar rats exhibiting spontaneous SWDs. Cortical electroencephalography (EEG) recording in the GAERS model is characterized by ~7‐11 Hz field oscillations (SWDs) lasting 17‐25 seconds, with a recurrence of approximately 70 SWDs per hour. SWDs start and end abruptly and are associated with a behavioral arrest lasting the time of the discharge, analogous to the impaired consciousness associated with SWDs in humans.4 Given these rats present with behavioral, electrophysiological, and pharmacological features of absence seizures,5 it is a favorable model to enable translation of novel AEDs with varied mechanisms of action to the clinic. Accordingly, both clinically and in the GAERS model, SWDs are suppressed by some AEDs (eg, valproate,6, 7 ethosuximide,7, 8 levetiracetam,9, 10 and diazepam11), whereas other AEDs can exacerbate SWDs, particularly carbamazepine,12, 13 and phenytoin.13, 14
Various classes of GABAergic drugs have also been shown to both suppress and aggravate absence epilepsy. For example, both the GABA reuptake inhibitor tiagabine and vigabatrin, an irreversible inhibitor of GABA transaminase, the enzyme responsible for the metabolism of GABA, nonselectively potentiate GABA by virtue of increasing the availability of GABA for receptor binding on the surfaces of postsynaptic cells. Both drugs have been reported to aggravate absence epilepsy.15, 16, 17, 18 One of the AEDs most implicated to aggravate seizures, including absence seizures, is carbamazepine, and this is via the indirect activation of GABAA receptors.19 Nonetheless, benzodiazepines (BZDs), nonselective positive allosteric modulators (PAMs) of GABAA receptors, are highly efficacious in epilepsy. The majority of GABAA receptors present in the CNS contain two α, two β, and a single γ subunit, and those that contain an α1, α2, α3, or α5 subunit in conjunction with a γ2 subunit20 are sensitive to BZDs. Unfortunately, prolonged BZD use has been dose‐limited by adverse effects believed to be attributed to potentiation of α1 subunit‐containing GABAA receptors, even at low receptor occupancy.21 As such, there has been a concerted effort to develop drugs capable of maintaining the antiepileptic efficacy offered by BZD‐site activation but avoiding the undesired characteristics of GABAA α1 modulation.
PF‐06372865 is a novel small molecule high‐affinity ligand for the BZD site of the GABAA receptor, with functional selectivity in vitro and in vivo for receptors containing α2/3/5 subunits compared with those containing the α1 subunit.22 The compound has demonstrated antiseizure activity in animal models, including amygdala kindling in rat and pentylenetetrazol in mice,23 and clinically in a proof‐of‐principle study in patients with photosensitive epilepsy.24 Additionally, PF‐06372865 has been shown to be safe and well tolerated in both healthy volunteers and patients in the clinic.22, 24, 25, 26 In this study, we examine whether subtype‐selective potentiation of GABAA receptors is efficacious in reducing the occurrence of SWDs in the GAERS model using PF‐06372865.
2. MATERIALS AND METHODS
2.1. Animals
Adult male GAERS were obtained from Dr Antoine Depaulis (INSERM, Grenoble Institute of Neurosciences, Grenoble, France) and allowed to acclimate for at least 1 week before experiments. Prior to surgery, animals were socially housed in cages with wood litter and ad libitum access to food and water. Cages were maintained under artificial lighting between 7:30 am to 7:30 pm in a room with controlled ambient temperature (22 ± 2°C) and relative humidity. Following initial recordings as described below, animals exhibiting prototypical cortical SWDs at 3 months of age were included in the study.5
All experiments were approved by the ethical committee of the High Technology Animal Platform, University Grenoble Alpes, and performed in accordance with the European Committee Council directive of September 22, 2010 (2010/63/EU). All efforts were made to minimize animal suffering and reduce the number of animals used.
2.2. Compounds and administration
PF‐06372865 was provided by Pfizer, Inc (Groton, CT, USA). Diazepam was purchased from Roche (Paris, France), valproate was obtained from Sigma‐Aldrich (Paris, France), and both were used as the positive controls. PF‐06372865 (0.3, 1, 3, and 10 mg kg−1) and diazepam (3 mg kg−1) were suspended in a solution containing 0.1% methylcellulose and 1% HPMC acetate succinate in 20 mmol L−1 Tris buffer (pH 7.4) immediately before administration to the animals (dose volume 5 mL kg−1). Animals were randomized to treatment order using a Latin‐square crossover protocol, and blinding to treatment conditions were maintained throughout data acquisition and analysis. In a first arm, vehicle, PF‐06372865 (1, 3, and 10 mg kg−1) and valproate (500 mg kg−1) were tested. In a second arm, in the same animals, 3 weeks after completion of the first arm, PF‐06372865 at 0.3 mg kg−1 and diazepam at 3 mg kg−1 were tested.
2.3. Surgery
Stereotaxic implantation of electrodes was performed under general anesthesia (isoflurane; 2%‐2.5% in oxygen). Once animals were nonreactive to stimuli, rats were placed in the stereotaxic frame, and body temperature was maintained at 37°C throughout the surgery. An ophthalmic gel was placed on the eyes to prevent drying of the cornea. The depth of anesthesia was adjusted throughout the surgery to maintain the breathing rate and cardiac rhythm. The surgical site was first sterilized and prepped with betadine. Following the incision, the skull was cleaned, and four (4) ~1 mm holes were drilled bilaterally over frontal (AP: +2 mm, ML: ±3 mm) and parietal (AP: −7 mm, ML: ±3 mm) to accommodate skull screws for EEG recordings. A female connector was then attached to the screws and fixed on the skull to enable chronic EEG recordings. At the end of the surgery, animals received an intraperitoneal injection of buprenorphine (0.01 mg kg−1). Animals were then housed individually and allowed to recover for at least 1 week prior to experiments.
2.4. EEG recording
On the day of the recording session, GAERS animals (N = 10) were placed in a recording chamber and connected to a digital acquisition system, SystemPlus Evolution (Micromed, Macon, France, 512 Hz sampling rate, low pass filter: 150 Hz, high pass filter: 0.008 Hz) to record EEG activity. A 1 hour habituation period was allowed before starting the EEG recording session. Recordings were performed on freely moving animals for 20 minutes preadministration (baseline period) and between 10 and 90 minutes postadministration using SystemPlus Evolution (Micromed). Because the drug administration process disturbs the occurrence of SWDs, the first 10 minutes following administration were not considered for analysis. Given that SWDs commonly occur during quiet wakefulness, animals were maintained in this state by the experimenters by introducing objects in the recording chambers when necessary.
2.5. Data analysis
EEG recordings were analyzed offline and quantified blindly manually by an expert to identify SWDs. Briefly, SWDs were hand‐scored by an expert during the 20 minutes baseline period (immediately before compound administration) and for a period of 80 minutes between 10 and 90 minutes after compound administration. For each animal and administration, data were computed for number and cumulative duration of SWDs in 20 minutes epochs.
2.6. Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Statistical analyses were performed using Prism (GraphPad version 7.02, GraphPad Software, La Jolla, CA, USA) using a two‐way ANOVA for repeated measures, followed by paired comparisons vs baseline periods, vs vehicle, and vs diazepam using the Bonferroni’s t test. The significance level was set at P < 0.05.
2.7. Plasma sample collection and bioanalysis
To confirm expected plasma exposures were achieved in this model, 12 GAERS rats from a second satellite batch of animals were randomly distributed into four groups of three animals. All groups were treated with PF‐06372865 at 10 mg kg−1: administrations were performed 24, 8, 3, or 1 hours before blood collection. To collect blood, the animals were deeply anesthetized with isoflurane (2%‐3% in oxygen), and a terminal blood sample was collected by intracardiac puncture into K2/EDTA tubes and immediately stored on ice. Within 10 minutes of collection, blood samples were centrifuged at 4°C and 3000 g for 10 minutes, and the plasma was collected and divided into two plastic tubes. All plasma samples were stored at −80°C until shipment to Pfizer for confirmation of exposure.
Plasma samples were analyzed by protein precipitation with volumes of internal standard containing acetonitrile (5:1 ratio with sample), followed by mixing and centrifugation to pellet protein. Supernatant was then mixed (1:1) with water prior to analysis by LC‐MS/MS monitoring a multiple reaction monitoring transition for PF‐06372865:441.4 > 348.2. Limits of quantification of 0.5 ng mL−1 were achieved.
3. RESULTS
At 10 mg kg−1, the mean plasma exposure at 1 hour postadministration was 1840 ng mL−1 (SD ± 602 ng mL−1), which was within 2‐fold of that previously reported.22 All the GAERS enrolled in this study showed prototypical cortical SWDs at 3 months of age.5 On average, the animals displayed 74.5 ± 8 SWDs per 60 minutes (Figure 1, “Base 0‐20 minutes”). From the 10 GAERS enrolled at the beginning of the study, two were removed from the study. One lost its recording system and did not complete the crossover, and the second animal did not respond to the positive control compound valproate.
Figure 1.
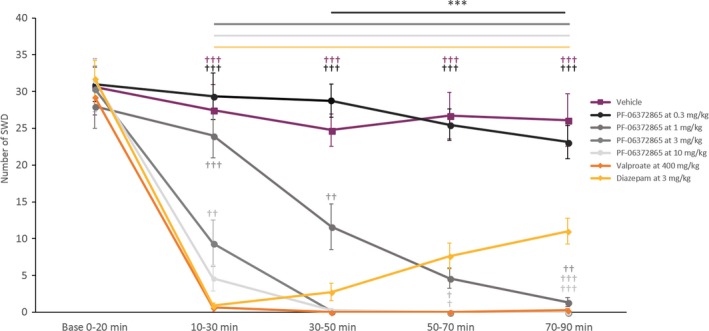
Number of SWD (mean ± SEM, n = 8) during baseline and postadministration periods, in all pharmacological conditions. ***P < 0.001; as compared to the vehicle; ††,†††,†††† P < 0.01, 0.001, and 0.0001, respectively, as compared to diazepam
3.1. PF‐06372865 significantly reduced SWDs in the GAERS model of absence epilepsy
Following treatment with PF‐06372865, we observed a dose‐dependent decrease in the incidence of SWDs (Figure 1), demonstrating significant differences between the treatments and time points (F‐values: Time: F 4,28 = 111.2, P < 0.0001; Treatment: F 6,42 = 47.8, P < 0.0001; Interaction Time × Treatment: F 24,168 = 16.6, P < 0.0001).
3.2. Effect of PF‐06372865 on the number of SWDs compared to the baseline period
Compared to the predose baseline period, PF‐06372865 (0.3 mg kg−1) showed a significant decrease at 70‐90 minutes postdose (P < 0.0001). For the intermediate dose of PF‐06372865 (1 mg kg−1), a significant reduction was observed between 30 and 90 minutes postadministration (P < 0.001 for all remaining time points). The two higher doses of PF‐06372865 (3 and 10 mg kg−1) showed a significant reduction in the number of SWDs at all time points after dosing (P < 0.001 for all time points).
The positive controls diazepam and valproate showed a significant reduction between 10 and 90 minutes postadministration (P < 0.001 for all the time points).
3.3. Effect of PF‐06372865 on the number of SWDs compared to vehicle
The lowest dose of PF‐06372865 (0.3 mg kg−1) did not significantly reduce SWDs compared to vehicle at any time period tested, and the intermediate dose of 1 mg kg−1 showed a significant reduction in the number of SWDs between 30 and 90 minutes postadministration (P < 0.0001). Interestingly, the two highest doses of PF‐06372865 tested (3 and 10 mg kg−1) significantly reduced the incidence of SWDs at all time periods after dosing, with full suppression observed between 30 and 90 minutes postrecording (P < 0.0001), which is consistent with expected pharmacokinetic parameters previously reported.22
With diazepam, the maximal effect was observed as early as 10‐30 minutes (P < 0.0001); however, this effect gradually decreased over the remaining 30‐90 minutes. For the other positive control, valproate, a complete reduction in the number of SWD was observed at the 30‐50 minutes time period and lasting until the end of the recording (P < 0.0001). This is concordant with expected pharmacokinetics of these drugs based on in‐house data.
3.4. Effect of PF‐06372865 on the occurrence of SWDs compared to diazepam and valproate
The kinetics of SWD suppression between PF‐06372865 and the reference compound diazepam was strikingly different. The maximum suppression of SWDs by diazepam was observed between 10 and 30 minutes post‐treatment, followed by a gradual decrease in efficacy from the 30‐ to 50‐minutes time period to the end of the recording (between 30 and 90 minutes postadministration for the two highest dose). Conversely, the efficacy profile of PF‐06372865 demonstrated complete suppression of SWDs from 30‐50 minutes onward (Figure 1). In comparison with the second positive control (valproate at 500 mg kg−1), the kinetics of effect is also different. Valproate showed an almost complete reduction in SWD between 10 and 30 minutes postadministration, this effect being complete for the rest of the recording. A significant difference was seen between valproate at 500 mg kg−1 and PF‐06372865 at 3 mg kg−1 for the 10‐30 minutes time period (P = 0.0028). Comparable results were obtained for the cumulated duration of SWDs (Figure 2).
Figure 2.
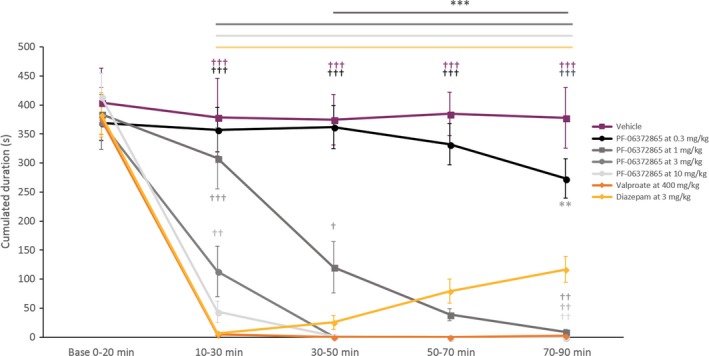
Cumulated duration of SWD (mean ± SEM, n = 8) during baseline and postadministration periods, in all pharmacological conditions. ***P < 0.001; as compared to the vehicle; ††,††† P < 0.01, 0.001, respectively, as compared to diazepam
4. DISCUSSION
To our knowledge, this is the first ever preclinical study of an α2/3/5‐subtype‐selective GABAA PAM in a model of absence epilepsy. Outstanding levels of efficacy were observed with PF‐06372865, enabling a clear conclusion to be reached with a small number of animals. The resulting data are testament to the effectiveness of the study design and conduct in generating a good basis in which the efficacy of PF‐06372865 could be assessed.
In general, nonselective benzodiazepines are efficacious in epilepsy models at <20% receptor occupancy (RO); however, this occupancy comes with adverse effects driven by binding to the GABAA α1‐containing receptors that limit their use as a long‐term treatment option.21, 27 Previous reports have estimated the RO of PF‐06372865 in rodents,22 enabling a better understanding of the required modulation to drive efficacy in the GAERS model. Here, we show that at the lowest dose of PF‐06372865 (0.3 mg kg−1), which is expected to achieve total receptor occupancy at GABAA receptors in brain of approximately 40%, there was no effect on the expression of SWDs within the timeframe that was examined compared to vehicle. Delayed efficacy was noted with the 1 mg kg−1 dose (~50% RO), while the two highest doses (3 and 10 mg kg−1, resulting in ~65% and ~80% RO, respectively) resulted in a significant reduction in SWDs at the early time points that persisted throughout the experiment. To examine the effect of PF‐06372865 over time was not feasible within this study, however, there is further indication in the GAERS model, as with other pharmacodynamic data with this compound, that efficacy is closely related to receptor occupancy.22, 25 Moreover, the analysis of the baseline recordings over the crossover design did not show any long‐lasting effect of PF‐06372865. The difference in RO required to achieve efficacy is one aspect of how the pharmacology of PF‐06372865 is significantly different from that of BZDs, such as diazepam, which are nonselective full PAMs of the GABAA BZD‐binding site. PF‐06372865 was designed to lack α1 activity to minimize sedation and be a functionally selective α2/3/5 PAM, which should retain some presumably α2‐driven anticonvulsant activity based on animal studies.28 However, PF‐06372865 is only a partial PAM at α2/3/5‐containing receptors.22 Partial GABAA PAMs need to occupy a greater proportion of the receptors to produce the same behavioral effect as a full PAM in anxiety animal models,21 and there is some evidence for efficacy in epilepsy models.23, 28 In the first clinical study of an α2/3/5‐subtype‐selective GABAA PAM in patients with clinically diagnosed epilepsy, PF‐06372865 demonstrated robust efficacy. In the photosensitivity model proof‐of‐principle study, both 17.5 mg and 52.5 mg single doses of PF‐06372865 (doses expected to achieve >60% receptor occupancy) resulted in full abolition of the photosensitivity response in six out of seven patients.24 The current preclinical data provide supporting evidence that PF‐06372865 may have robust functional efficacy in absence epilepsy at RO similar to those used in the prior clinical study.
While there can be little argument to the important role that GABAA receptors play in convulsant pathways, at the outset of this study, there was some uncertainty as to whether a suppression of SWDs would be observed with PF‐06372865. This was due to the lack of data with subtype‐selective GABAA PAMs in absence seizures both preclinically and clinically, the potentially important contribution of the α1 subunit to an absence epilepsy phenotype (as shown by the GABRA1 gene association29), and the varied picture of both aggravation and suppression of SWD with other GABAergic AEDs. While it has been demonstrated that increased GABAA receptor‐mediated tonic inhibition in the thalamus plays a role in experimental genetic and pharmacological models of absence epilepsy,30 this was not an expected effect of PF‐06372865 because tonic inhibition is thought to arise predominantly from activity of GABA at extrasynaptic GABAA receptors containing α4 and α6 subunits, to which PF‐06372865 (and BZDs) does not bind.22
Some inconsistencies regarding relative contributions of the GABAA α subunits to the anticonvulsant activity of BZDs have been reported. For instance, the anticonvulsant efficacy of diazepam has been ascribed primarily to the α1 subunit in molecular studies in mice with α subunits rendered insensitive to BZDs (H101R mutants31). However, comprehensive work using both subtype‐selective GABAA PAMs together with transgenic mice with point mutations altering the BZD‐binding site at other GABAA subtypes (α2‐H101R, α5‐H105R) demonstrated that no single GABAA receptor subtype is solely responsible for the anticonvulsant effects of GABA.28 Based on that work, which was carried out in nonabsence epilepsy models, it was reported that the α2 subunit played a greater role than the α1‐containing receptors, concluding that efficacy at more than a single GABAA receptor subtype can be achieved and that the α1‐ and α2‐containing receptors act synergistically, at least in animal models. Based on the current data, it is reasonable to conclude that the α1 subunit is less important in conferring antiabsence activity of PF‐06372865 than α2/3/5. However, further work to elucidate the precise role of the GABAA subunits in absence epilepsy preclinically would be informative.
Clinically, only five genes (GABRA1, GABRA3, GABRB3, GABRG2, and GABRD) encoding the α1‐, α3‐, β3‐, γ2‐, and δ‐subunits of the GABAA receptor have been directly associated with epilepsy.29, 32 Both the GABRA1 and GABRA3 rare loss‐of‐function variants increase the risk for a varying combination of epilepsies (including absence), indicating a potential role of the α3 subunit in absence epilepsy. This finding perhaps supports the hypothesis that a drug with the specific pharmacology of PF‐06372865 has therapeutic potential in this condition and merits further exploration.
Based on prior evidence of the predictive validity of the GAERS model,33 although acknowledging that translation of preclinical models to the clinic can be difficult, our data herein provide optimism that PF‐06372865 could be efficacious in patients with absence epilepsy.
CONFLICT OF INTEREST
RG and DLB are or were employees of Pfizer at the time of this research and may own stock in the company.
Duveau V, Buhl DL, Evrard A, et al. Pronounced antiepileptic activity of the subtype‐selective GABAA‐positive allosteric modulator PF‐06372865 in the GAERS absence epilepsy model. CNS Neurosci Ther. 2019;25:255–260. 10.1111/cns.13046
Funding information
This work was funded by Pfizer Inc.
REFERENCES
- 1. Loiseau P, Duché B, Pédespan JM. Absence epilepsies. Epilepsia. 1995;36(12):1182‐1186. [DOI] [PubMed] [Google Scholar]
- 2. Manning J‐PA, Richards DA, Bowery NG. Pharmacology of absence epilepsy. Trends Pharmacol Sci. 2003;24:542‐549. [DOI] [PubMed] [Google Scholar]
- 3. Genton P. When antiepileptic drugs aggravate epilepsy. Brain Dev. 2000;22(2):75‐80. [DOI] [PubMed] [Google Scholar]
- 4. Gibbs FA, Davis H, Lennox WG. The electroencephalogram in epilepsy and in conditions of impaired consciousness. Arch Neurol Psychiatr. 1935;34:1133‐1148. [Google Scholar]
- 5. Depaulis A, David O, Charpier S. The genetic absence epilepsy rat from Strasbourg as a model to decipher the neuronal and network mechanisms of generalized idiopathic epilepsies. J Neurosci Methods. 2016;260:159‐174. [DOI] [PubMed] [Google Scholar]
- 6. Dedeurwaerdere S, van Raay L, Morris MJ, Reed RC, Hogan RE, O'Brien TJ. Fluctuating and constant valproate administration gives equivalent seizure control in rats with genetic and acquired epilepsy. Seizure. 2011;20(1):72‐79. [DOI] [PubMed] [Google Scholar]
- 7. Glauser T, Ben‐Menachem E, Bourgeois B, et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54(3):551‐563. [DOI] [PubMed] [Google Scholar]
- 8. Nehlig A, Vergnes M, Marescaux C, Boyet S. Cerebral energy metabolism in rats with genetic absence epilepsy is not correlated with the pharmacological increase or suppression of spike‐wave discharges. Brain Res. 1993;618(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 9. Gower AJ, Hirsch E, Boehrer A, Noyer M, Marescaux C. Effects of levetiracetam, a novel antiepileptic drug, on convulsant activity in two genetic rat models of epilepsy. Epilepsy Res. 1995;22(3):207‐213. [DOI] [PubMed] [Google Scholar]
- 10. Vrielynck P. Current and emerging treatments for absence seizures in young patients. Neuropsychiatr Dis Treat. 2013;9:963‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michelletti G, Vergnes M, Marescaux C, et al. Antiepileptic drug evaluation in a new animal model: spontaneous petit mal epilepsy in the rat. Drug Res. 1985;483‐485. [PubMed] [Google Scholar]
- 12. Wallengren C, Li S, Morris MJ, Jupp B, O'Brien TJ. Aggravation of absence seizures by carbamazepine in a genetic rat model does not induce neuronal c‐Fos activation. Clin Neuropharmacol. 2005;28(2):60‐65. [DOI] [PubMed] [Google Scholar]
- 13. Somerville ER. Some treatments cause seizure aggravation in idiopathic epilepsies (especially absence epilepsy). Epilepsia. 2009;50(Suppl 8):31‐36. [DOI] [PubMed] [Google Scholar]
- 14. Gurbanova AA, Aker R, Berkman K, Onat FY, van Rijn CM, van Luijtelaar G. Effect of systemic and intracortical administration of phenytoin in two genetic models of absence epilepsy. Br J Pharmacol. 2006;148(8):1076‐1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coenen A, Blezer E, Van Luijtelaar E. Effects of the GABA‐uptake inhibitor tiagabine on electroencephalogram, spike‐wave discharge and behaviour of rats. Epilepsy Res. 1995;21:89‐94. [DOI] [PubMed] [Google Scholar]
- 16. Knake S, Hamer H, Schomburg U, Oertel W, Rosenow F. Tiagabine‐induced absence status in idiopathic generalized epilepsy. Seizure. 1999;8:314‐317. [DOI] [PubMed] [Google Scholar]
- 17. Vergnes M, Marescaux C, Micheletti G, Depaulis A, Rumbach L, Warter JM. Enhancement of spike and wave discharges by GABAmimetic drugs in rats with spontaneous petit‐mal‐like epilepsy. Neurosci Lett. 1984;44(1):91‐94. [DOI] [PubMed] [Google Scholar]
- 18. Panayiotopoulos CP, Agathonikou A, Sharoqui IA, Parker AP. Vigabatrin aggravates absences and absence status. Neurology. 1997;49(5):1467. [DOI] [PubMed] [Google Scholar]
- 19. Liu L, Zheng T, Morris MJ, et al. The mechanism of carbamazepine aggravation of absence seizures. JPET. 2006;319:790‐798. [DOI] [PubMed] [Google Scholar]
- 20. McKernan RM, Whiting PJ. Which GABAA‐receptor subtypes really occur in the brain? Trends Neurosci. 1996;19(4):139‐143. [DOI] [PubMed] [Google Scholar]
- 21. Atack JR. GABAA receptor alpha2/alpha3 subtype‐selective modulators as potential nonsedating anxiolytics. Curr Top Behav Neurosci. 2010;2:331‐360. [DOI] [PubMed] [Google Scholar]
- 22. Nickolls SA, Gurrell R, van Amerongen G, et al. Pharmacology in translation; the preclinical and early clinical profile of the novel α2/3 functionally selective GABAA receptor positive allosteric modulator PF‐06372865. Br J Pharmacol. 2018;175:708‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buhl DL, DaSilva JK, Tyszkiewicz C, et al. PF‐06372865, An α2/3/5‐Subtype Selective Partial Positive Allosteric Modulator, is a Potent Anticonvulsant in Animal Models of Epilepsy. Washington, DC: American Epilepsy Society abstract; 2017. [Google Scholar]
- 24. Gurrell R, , Gorman D , Whitlock M, French J. PF-06232865, an α2/3/5-subtype selective GABAA partial positive allosteric modulator, has promising efficacy in the photosensitivity model. American Epilepsy Society abstract, Washington, DC, 2017. [Google Scholar]
- 25. Gurrell R, Dua P, Feng G, et al. A randomised, placebo‐controlled clinical trial with the α2/3/5 subunit selective GABAA positive allosteric modulator PF‐06372865 in patients with chronic low back pain. Pain. 2018;159(9):1742‐1751. [DOI] [PubMed] [Google Scholar]
- 26. Simen A, Whitlock M, Ruolun Q, et al. An 8‐Week, randomized, phase 2, double‐blind, sequential parallel‐group comparison study of two dose levels of PF‐06372865 compared to placebo as an adjunctive treatment in outpatients with inadequate response to standard of care for generalized anxiety disorder. J Clin Psychopharm. 2018. [DOI] [PubMed] [Google Scholar]
- 27. Riss J, Cloyd J, Gates J, Collins S. Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurol Scand. 2008;118:69‐86. [DOI] [PubMed] [Google Scholar]
- 28. Fradley RL, Guscott MR, Bull S, et al. Differential contribution of GABAA receptor subtypes to the anticonvulsant efficacy of benzodiazepine site ligands. Psychopharm. 2007;21(4):384‐391. [DOI] [PubMed] [Google Scholar]
- 29. Macdonald RL, Kang J‐Q, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010;588(11):1861‐1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cope DW, Di Giovanni G, Fyson SJ, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rudolph U, Crestani F, Benke D, et al. Benzodiazepine actions mediated by specific gamma‐aminobutyric acid (A) receptor subtypes. Nature. 1999;401(6755):796‐800. [DOI] [PubMed] [Google Scholar]
- 32. Niturad CE, Lev D, Kalscheuer VM, et al. Rare GABRA3 variants are associated with epileptic seizures, encephalopathy and dysmorphic features. Brain. 2017;140(11):2879‐2894. [DOI] [PubMed] [Google Scholar]
- 33. van Luijtelaar E, Drinkenburg W, van Rijn CM, Coenen A. Rat models of genetic absence epilepsy: what do EEG spike‐wave discharges tell us about drug effects? Methods Find Exp Clin Pharmacol. 2002;24:65‐70. [PubMed] [Google Scholar]