

Abstract
Introduction
Our previous study has confirmed that a novel curcumin derivate nicotinate‐curcumin (NC) can facilitate autophagic flux in THP‐1 cells induced by oxidized low‐density lipoprotein.
Aims
Given that autophagy plays critical roles in neurodegenerative diseases, the present study was carried out to investigate whether NC can improve cognitive function of rats with diabetes mellitus (DM) via restoring autophagic flux in CA1 hippocampus.
Results
Our results showed that NC treatment improved cognitive deficit and attenuated neuronal loss as well as cellular ultrastructure impairment in the CA1 region of DM rats induced by streptozotocin. Moreover, NC lowered the expressions of the apoptosis‐related proteins Bcl‐2, Bax, Cyt‐c, and cleaved Caspase‐3. Notably, NC treatment reversed autophagic flux impairment as evidenced by the deceases in LC3‐II and p62 protein levels, and autophagosome accumulation in the hippocampal CA1 region of DM rats. However, these protective effects of NC were abolished by cotreatment with 3‐methyladenine (an autophagy inhibitor) and chloroquine (an autophagic flux inhibitor), respectively. Furthermore, NC treatment decreased the expressions of phosphorylated mammalian target of rapamycin (mTOR) and p70 ribosomal protein S6 kinase (p70S6k) proteins in the CA1 region of DM rats.
Conclusions
These results indicate that NC ameliorates DM‐induced cognitive function impairment via restoring autophagic flux might by inhibiting mTOR/p70S6k activation in the CA1 region, and NC may be a promising agent for diabetic cognitive dysfunction prevention and treatment.
Keywords: autophagy, cognitive function, diabetes mellitus, hippocampus, mammalian target of rapamycin complex, nicotinate‐curcumin
1. INTRODUCTION
Compelling evidence has been indicated that diabetes mellitus (DM) is involved in the pathogenesis of its complications, such as cardiovascular diseases, kidney diseases, and peripheral nerve diseases.1, 2, 3 Furthermore, studies have revealed the close relationship between DM and cognitive deficits.4, 5 In addition, animal models with DM exhibit damage to the structure and function of the brain, particularly in the hippocampus, a brain area responsible for learning and memory.6, 7 Therefore, understanding the mechanisms by which DM results in cognitive function impairment will help to explore pharmacological treatments for this disease.
Autophagy is critical for sustaining neuronal homeostasis via selective clearance of damaged and aged organelles or misfolded proteins.8, 9, 10 Autophagy is mainly composed of the following three processes: the formation of autophagosome (AP), the fusion of AP with lysosome to form autophagolysosome (ALP), and the degradation of intra‐autophagosomal cargoes by lysosomal hydrolases.11 The above dynamic process is known as autophagic flux, which is strictly regulated by several signaling pathways. Among of those signaling, mammalian target of rapamycin (mTOR)/p70SK is a critical pathway that negatively modulates autophagy.12 It has been recognized that autophagy exhibits a dual function in neurodegenerative diseases. Recently, several studies have implied that autophagy impairment may contribute to diabetic encephalopathy, and targeting the autophagic signaling to restore autophagic flux may be neuroprotective in this setting.13, 14, 15
Curcumin, a major bioactive component of turmeric, can prevent cognitive impairment via its various pharmacological activities including regulation of autophagy, anti‐oxidative stress, and anti‐inflammation.16, 17, 18 However, the poor aqueous solubility and relatively low bioavailability of curcumin limits its utilization. To overcome those limitations, a series of curcumin derivatives have been developed.19, 20 Nicotinate‐Curcumin (NC), a novel curcumin derivative derived from nicotinate and curcumin, which has superior water solubility and bioavailability .21, 22 This compound exhibits potent anti‐inflammation and anti‐atherosclerosis properties. Recently, we found that NC can rescue the impaired autophagic flux likely via inhibiting hyperactivation of PI3k‐Akt‐mTOR signaling induced by Ox‐LDL in THP‐1 cells .22 However, it is unclear whether NC can restore hippocampus autophagic flux and prevent cognitive deficits in animals with DM.
In present work, we hypothesized that NC can rescue the impaired autophagic flux in the hippocampal CA1 region of DM rats induced by STZ (streptozotocin, STZ), and such a restoration promotes the elimination of damaged mitochondria that release Cyt‐c (Cytochrome, Cyt‐c) and proapoptotic proteins, thus preventing hippocampal neuron loss and cognitive deficits. For this purpose, we first explored whether NC can attenuate the impairment of spatial cognitive function and damage to hippocampal neurons in DM rats. Then, we investigated whether the above protective effects of NC are via restoring hippocampal autophagic flux and the underlying mechanisms. Our results reveal that NC improves learning and memory deficits by facilitating hippocampal autophagic flux of DM rats maybe through inhibiting hyperactivation of mTOR/p70S6K pathway.
2. MATERIALS AND METHODS
2.1. Materials
Nicotinate‐curcumin (purity >98%) was synthesized in our laboratory. 3‐Methyladenine (3‐MA), chloroquine diphosphate (CQ), and STZ were purchased from Sigma Aldrich (St. Louis, MO, USA). Antibodies against mTOR, p‐mTOR (Ser2448), p‐p70S6K, p‐p70S6K (Thr389) LC3I/II, p62, Cyt‐c, Bax, and Bcl‐2 were provided by Cell Signaling Biotechnology (Danvers, MA, USA). Rabbit antibody against actin was purchased from Proteintech Group, Inc (Chicago, IL, USA). Rapamycin (RAP) was obtained from Gene Operation (St. Louis, MO, USA).
2.2. Animals
A total of 85 male adult SD rats (16 weeks, 280‐300 g) were obtained from the Hunan SJA Laboratory animal Co. Ltd. (Changsha, china). The rats were under standard housing conditions (room temperature 25 ± 1°C and humidity 60%‐65%) with a 12‐hour light/12‐hour dark cycle (lights on at 7:00 am, lights off at 7:00 pm) and accessed to food and water freely. All animals used in this study were approved by the Animal Experimentation Ethics Committee of the University of South China (Permit Number: XYXK2015‐0001) and strictly conformed to the guidelines established by the “China Council on Animal Care’’.
2.3. Model building and animal grouping
Solid evidences indicate that animal type 2 DM models induced by an intraperitoneal injection of streptozotocin (STZ) exhibit obvious cognitive deficit. Therefore, in the present study, type 2 DM rat model established by STZ was used. All rats were fed with a high‐fat diet for 4 weeks prior to the experiment. After overnight fasting, 75 rats were randomly selected to inject with STZ (at a single dose of 50 mg/kg body weight) prepared in citrate buffer to induce diabetes. The remainder 10 rats served as normal control group which were injected with equal volume of vehicle. In the present study, fasting blood glucose levels before the experiment, at 72 hours post STZ injection and at the end of the experiment were measured by enzymatic GOD‐PAP (glucose oxidase peroxidase) diagnostic kits. The rats were considered to be diabetic if their fasting blood glucose levels exceeded 16.7 mmol/L .23 The rest 70 successful rat models were further randomly divided into the following five groups with 14 animals in each: (a) DM group (DM); (b) DM +NC group, the rats treated with NC (100 mg/kg/body weight); (c3) DM + RAP (RAP) group (1 mg/kg body weight); (d) DM + NC (100 mg/kg body weight) + CQ (40 mg/kg body weight); (e) DM + NC (100 mg/kg body weight) + 3‐MA (1.5 mg/kg body weight). The rats in both DM group and normal control group were injected with equal volume of vehicle (phosphate buffer saline). Vehicle or drugs were injected by intraperitoneal (IP) once daily for 35 consecutive days.
2.4. Novel object recognition test
The novel object recognition test (NOR) was performed to measure the ability of animals to identify a novel object in a familiar environment as previously described.24 In the NOR test, each rat is subjected to three tasks over 3 consecutive days. On day 1, the habituation phase, each rat was acclimated to the experimental area (50 × 50 × 40 cm) for 10 minutes once daily in the absent of any behavioral stimulus. On day 2, the training phase, two identical objects were placed into the area, and each animal was permitted to explore the objects for 5 minutes. On day 3, the retention phase, one of the old objects was replaced with a novel one, and each rat explored the novel object for 5 minutes. Exploration was referred to touching the novel object with the nose and/or pointing the nose to it at a distance <1 cm. To eliminate the odor cues, the field and the objects were fully decontaminated with 70% alcohol, dried, and ventilated between two tests. The time spent exploring each object was recorded using a video‐assisted tracking system. The cognitive function of rats was presented as the discrimination index (DI) [= (novel object exploration time − familiar object exploration time)/total exploration time × 100%].
2.5. Morris water maze test
After NC treatment for 35 days, spatial learning and memory ability of rats were determined by Morris water maze test (MWM) test as previously described.25 The experimental apparatus is composed of a circular water pool (180 cm in diameter, 60 cm in height) and filled with tepid water (25 ± 1°C) to a depth of 30 cm. The water in tank was colored with milk powder. The pool was equally divided into four quadrants, labeled 1‐2‐3‐4. A colorless escape platform was placed in one of the four quadrants (the target quadrant) and submerged 2.0 cm below the water surface during acquisition trails. On the day 1, each rat was trained to reach the visible platform. Locating the hidden platform, trial was performed to measure spatial learning function of rats for 4 consecutive days. In those tests, each rat was received three trials with an interval of 10 minutes between two trials, and duration for each trial is about 120 seconds unless the rats located the hidden platform. The rats were mildly guided to the hidden platform and permitted to remain there for 20 seconds for their failure to escape within 120 seconds. On the day 6, the hidden platform was cleared away from the pool and each rat was allowed to find the target quadrant within 120 seconds. The swimming time and routes of rats to find the target quadrant were recorded by a video camera linked to a computer tracking system. The escape latency and crossing the platform location times were analyzed by using the MT‐200 Morris image motion system (Chengdu Technology and Market Corp, Chengdu, China).
2.6. Hematoxylin and eosin staining
The rats (n = 3) were anaesthetized by pentobarbital (IP injection, 100 mg/kg body weight) and then transcardially perfused with ice‐cold phosphate buffer solution (PBS), followed by fixation with cold 4% paraformaldehyde for 30 minutes. Whole brain tissues were removed quickly and first fixed in 4% paraformaldehyde solution for 24 hours and then stored in a 4% paraformaldehyde solution containing 30% sucrose. The dehydrated brain specimens were embedded in paraffins, and then consecutive coronal paraffin sections (4 μm) of the whole hippocampus were performed according to the rat stereotaxic atlas. The sections were stained with hematoxylin and eosin staining (H&E). Cell morphology changes in the hippocampal CA1 region of rats were evaluated under a light microscope (200 magnification), and the cell number of this area was calculated by the following formula: cell counts in per mm hippocampal CA1 area = normal CA1 cell counts/CA1 length. Cells with morphological features of pyknosis or nuclear fragmentation will be excluded
2.7. Ultrastructural morphological observation using transmission electron microscope
After deeply anaesthetized, rats (n = 3) were transcardially prefixed with 4% paraformaldehyde and 2.5% glutaraldehyde in a cold (4°C) PBS. The hippocampal CA1 regions were quickly isolated and cut into 1mm3 size samples. The samples were immediately fixed with 2.5% glutaraldehyde in a cold neutral PBS for 6 hours. Then, the samples were washed with cold PBS and postfixed with 1% OSO4 in PBS for 1 hour. After embedding with Epon resin for 48 hours, the samples were stained with lead citrate and uranyl acetate. Consecutive ultrathin sections of 50 nm were cut with an ultramicrotome (Ultracut 7, Leica Microsystems, Wetzlar, Germany). For each sample, a minimum of 10 sections were examined with a transmission electron microscope (TEM, JEM‐1200EX; JEOL, Tokyo, Japan). Assessment of mitochondrial morphology and autophagy ultrastructure was done as previously described.22
2.8. Western blot analysis
Total proteins extracted from the hippocampus of rats (n = 4) as described in our previous study. The protein concentrations were subsequently quantified by using a BCA Protein Assay Kit (Beyotime, Shanghai, China). The extracted proteins were boiled in a buffer solution for 10 minutes. Equal amounts of the boiled proteins (20 μg per lane) from each sample were separated by a 12% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and then wet electro‐transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Darmstadt, Germany). After blocking with 5% bovine serum albumin at room temperature for 1 hour, the membranes were first incubated with primary antibody against actin (1:2000), LC3 (1:1000), p62 (1:1000), mTOR (1:1000), p‐mTOR (1:1000), p70S6K (1:1000), p‐p70S6K (1:1000), Bcl‐2 (1:1000), Bax (1:1000), and Caspase‐3 (1:1000) for 2 hours at room temperature. Next, the membranes were rinsed three times with TBST and subsequently incubated with horseradish peroxide conjugated secondary antibodies (1:3000) for 1 hour at room temperature. Finally, immunoreactivity was developed with ECL fluorescent detection reagent, and the immunoblots were analyzed using the NIH Image J software. Blots against β‐actin served as loading control.
2.9. Statistical analysis
The results were analyzed using the GraphPad Prism 7 statistical software. Data were expressed as mean ± SEM Student's t‐test (comparisons between two groups) or one‐way analysis of variance (comparisons among multiple groups) was used to perform statistical analysis. To clarify drug × group interaction, groups among DM + NC, DM + NC + CQ, and DM + NC + 3‐MA were compared using a two‐way ANOVA. Statistical significance was set at P < 0.05.
3. RESULTS
3.1. NC ameliorates the cognitive function of DM rats in novel object recognition test
We first confirmed whether cognitive function impaired in DM rats and whether NC treatment could reverse this impairment. The novel object recognition test was used to evaluate the cognitive function of rats. As shown in Figure 1A, the DI of DM rats was much lower than that of control ones. Compared with rats in DM group, the DI of rats significantly elevated in DM + RAP and DM + NC group, respectively, indicating that both RAP and NC reversed cognitive function impairment of DM rats induced by STZ. However, the DI of rats in DM + NC + CQ group and DM + NC + 3‐MA group remarkably decreased as compared with that in DM + NC group, respectively, implying that blocking autophagy‐lysosome pathway and autophagy abolished NC protective roles against cognitive function impairment of DM rats. Furthermore, our results demonstrated that there was no significant difference in the total exploration time among 6 groups (Figure 1B), indicating that the changes of DI of rats were not due to deference in olfactory discrimination. These data suggested that NC reversed cognitive function impairment of DM rats induced by STZ.
Figure 1.
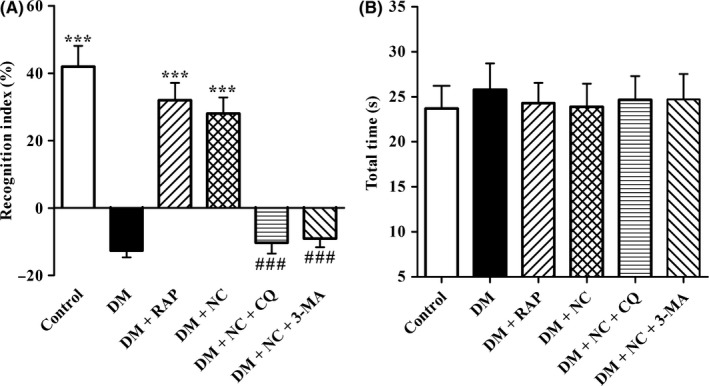
Effect of NC on the cognitive function of STZ‐induced diabetic rats in novel object recognition (NOR) test. Diabetic rats were treated with RAP (1 mg/kg/day, IP), NC (100 mg/kg/day, IP), NC (100 mg/kg/day, IP) + CQ (40 mg/kg/day, IP), NC (100 mg/kg/day, IP) + 3‐MA (1.5 mg/kg/day, IP) for 35 consecutive days. NOR test was performed to measure cognitive function of the rats. The discrimination index (A) and total exploration time (B) were analyzed. The data are expressed as mean ± SEM (n = 10‐14 per group). ***P < 0.001, compared with the DM group; ### P < 0.001 compared with the DM + NC group
3.2. NC improves spatial learning and memory function of DM rats in Morris water test
Next, we further investigated whether NC could ameliorate impairment in spatial learning and memory ability of DM rats by utilizing MWT assay. As shown in Figure 2A, the escape latency of each group to find the hidden platform decreased over 5 days training session, while the mean escape latency remarkably prolonged in DM group as compared with that in normal control group, indicating that DM resulted in damage to learning ability. However, treatment with RAP, an inducer of autophagy via inhibiting activation of mTOR, significantly shortened the escape latency of DM rats, suggesting that activation of autophagy ameliorated learning ability impairment in DM rats. Compared with rats in DM group, the escape latency of rats in NC treatment group decreased distinctly, indicating improvement in learning performance. However, the protective effects of NC could be canceled by cotreatment with CQ (an inhibitor of autophagy‐lysosome) or 3‐MA (an inhibitor of autophagy), implying that NC ameliorates impairment in learning ability of DM rats via facilitating autophagic flux.
Figure 2.
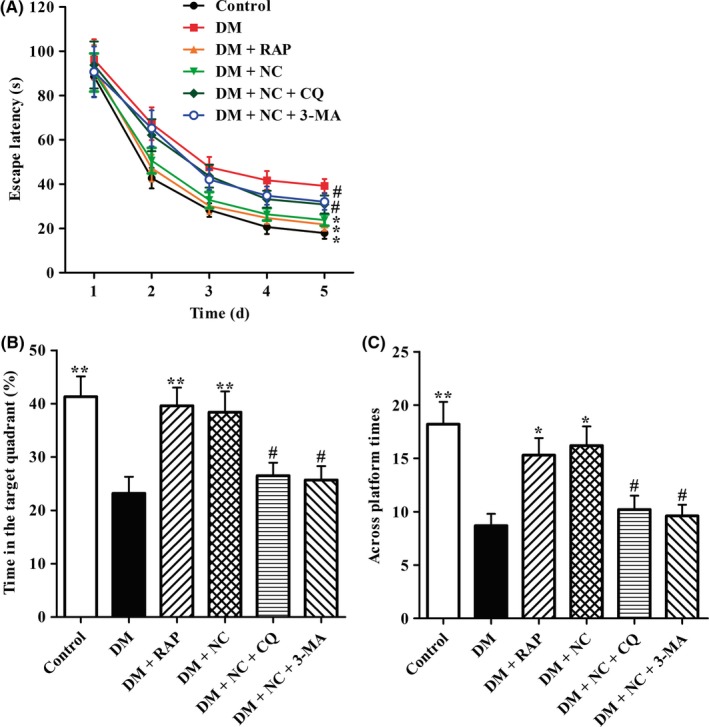
Effect of NC on the cognitive function of STZ‐induced diabetic rats in MWM test. Diabetic rats were treated with RAP (1 mg/kg/day, IP), NC (100 mg/kg/day, IP), NC (100 mg/kg/day, IP) + CQ (40 mg/kg/day, IP), NC (100 mg/kg/day, IP) + 3‐MA (1.5 mg/kg/day, IP) for 35 consecutive days. MWM test was used to evaluate the cognitive function of rats. The latency to find platform in acquisition phase was analyzed (A). The percentage of time spent in the target quadrant (B) and the times across invisible platform were analyzed (C). The data are expressed as mean ± SEM (n = 10‐14 per group). *P < 0.05, **P < 0.01, compared with the DM group; # P < 0.05 compared with the DM + NC group
Next, the memory performance of rats was evaluated by probe trial testing. As indicated in Figure 2B, capacity for space memory significantly impaired in DM rats when compared with that in control ones, evidenced by decreases both in the time spent in target quadrant and in the number of times crossing the target quadrant. The time spent in target quadrant and the times across the target quadrant of DM + RAP group were much more than those of DM group, respectively, indicating that activation of autophagy prevented memory impairment in DM rats. As compared with rats in DM group, the duration of space exploration in the target quadrant and times across platform markedly increased in rats of DM + NC group, respectively. However, rats in DM + NC + CQ group exhibited less time spent in the target quadrant and less times across platform than those in DM + NC group, suggesting cotreatment CQ abrogated NC protective effects against memory impairment of DM rats. Furthermore, the results showed that 3‐MA exhibited the same opposite effects as demonstrated in the group treated with DM + NC + CQ. Taken together, these findings imply that NC can improve diabetic cognitive deficit and may be through a mechanism of facilitating autophagic flux.
3.3. NC improves neuronal cell morphology and decreases cell loss in the hippocampal CA1 area of rats with DM
Considering changes in cell morphology and number of the hippocampal CA1 neurons are closely associated with learning and memory functions; the histological features in those regions of rats were determined by H&E staining. As shown in Figure 3A, A large amount of neuron cells were observed in the hippocampal CA1 regions of rats in normal control group, which were regularly arranged with big and round nuclei. Compared with rats in normal control group, neuron cell numbers (Figure 3B) significantly reduced and cell arrangement became obviously disordered in those areas of rats with DM, suggesting that neuron cell loss and cell structure impairment were aggravated in the hippocampal CA1 region of rats with DM. In DM + RAP and DM + NC group, neuron cell morphology of those areas was better than that in DM group, and the numbers of the hippocampal CA1 neuron cells were much more than those in DM group. However, compared with DM + NC group, the structures of the hippocampal CA1 neuron cells in DM + NC + CQ group were distinctly incomplete, including unclear cell bodies and nuclear condensation, and the hippocampal CA1 neuron number decreased significantly. The changes in neuron cell morphology and cell number of DM + NC + 3‐MA group were similar to those in DM + NC + CQ group, indicating that cotreatment with autophagy inhibitor canceled NC protective effects on the hippocampal CA1 neuron cells. Collectively, these evidences suggest that NC treatment prevents neuron cell loss and cell structure impairment in the hippocampal CA1 area of diabetic rats induced by STZ, and the mechanism may be through regulating autophagic flux.
Figure 3.
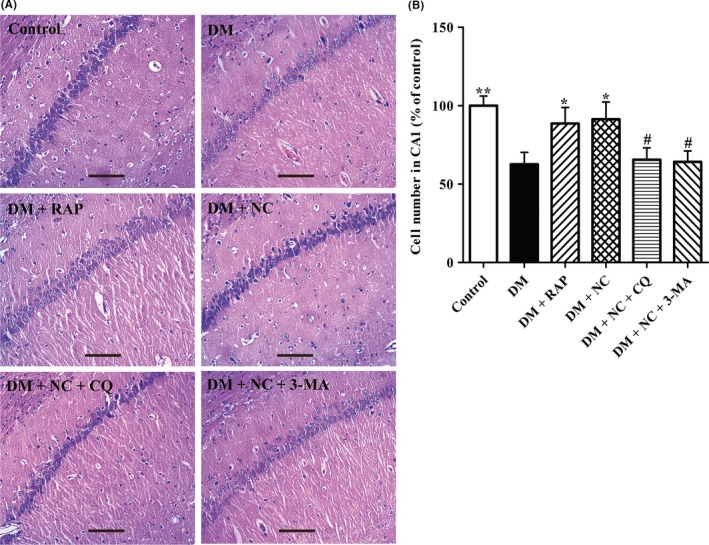
Effect of NC on the histological features in the hippocampal CA1 region of DM rats induced by STZ. DM rats were treated with RAP (1 mg/kg/day, IP), NC (100 mg/kg/day, IP), NC (100 mg/kg/day, IP) + CQ (40 mg/kg/day, IP), NC (100 mg/kg/day, IP) + 3‐MA (1.5 mg/kg/day, IP) for 35 consecutive days. H&E staining was used to measure the changes in neuronal morphology in the hippocampal CA1 area. (A) Representative light micrographs of HE‐stained neuron cells in the hippocampal CA1 area of the rats. (B) Statistical analyses for the cell number in the hippocampal CA1 region of the rats. The data are expressed as mean ± SEM (n = 3 per group). *P < 0.05, **P < 0.01, compared with the DM group; # P < 0.05 compared with the DM + NC group. Scale bar, 100 μm
3.4. NC prevents the damage to the cellular ultrastructures in the CA1 hippocampus of DM rats
To explore whether NC reverses cognitive dysfunction of DM rats via the mechanisms regulating autophagic flux, TEM and Western bolt assays were carried out to determine the impact of NC on autophagic flux in the hippocampal CA1 region of DM rats induced by STZ. We first observed the ultrastructures in the CA1 hippocampus of each group rats. As shown in Figure 4A,B, our TEM results demonstrated a distinct increase in APs in the CA1 regions of DM rats as compared with the normal control ones. As compared to control group, mitochondria of the CA1 neuron cells in the DM group showed pathological changes, including swelling, cristae fragmented, or vacuolization. Furthermore, TEM results indicated that both cotreatment with RAP and NC led to the improved ultrastructures in the CA1 hippocampus of DM rats, evidenced by decreases in the number of accumulation of APs and damaged mitochondria. However, cotreatment with CQ reversed NC protective effects on neuron cells in the CA1 hippocampus of DM rats, characterized by increased amount of APs and swollen mitochondria. As compared with DM + NC group, APs significantly reduced in the CA1 hippocampus of DM + NC + 3‐MA group, but the number of impaired mitochondria remarkably elevated. These results indicated that NC may function as an autophagic flux activator to improve the cellular ultrastructures in the CA1 hippocampus of rats with DM.
Figure 4.
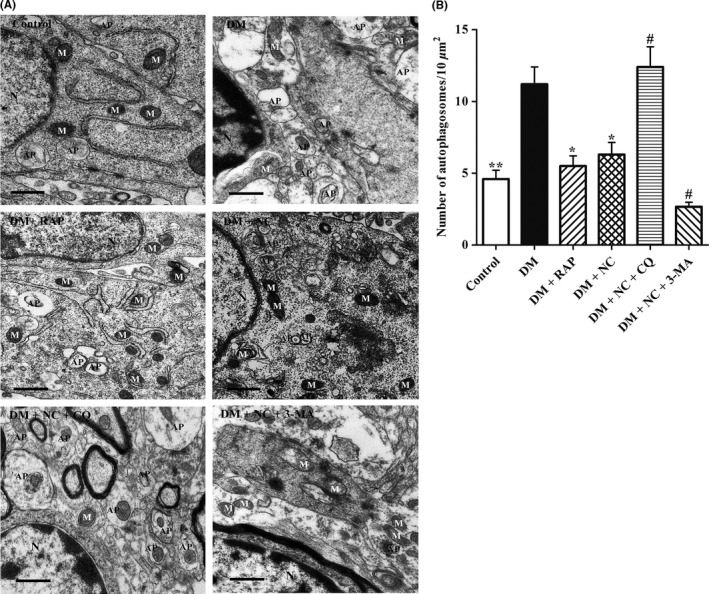
Effect of NC on the ultrastructures in the CA1 hippocampus of DM rats induced by STZ. Diabetic rats were treated with RAP (1 mg/kg/day, IP), NC (100 mg/kg/day, IP), NC (100 mg/kg/day, IP) + CQ (40 mg/kg/day, IP), NC (100 mg/kg/day, IP) + 3‐MA (1.5 mg/kg/day, IP) for 35 consecutive days. TEM was used to detect the changes in neuronal ultrastructures in rat hippocampal CA1 area. (A) Representative electron micrographs of mitochondria morphology and autophagosomes in the hippocampal CA1 area of the rats. N: Nucleus, M: Mitochondria, AP: Autophagosome. Scale bar, 500 nm. (B) Statistical analyses for the autophagosome number in the hippocampal CA1 region of the rats. The data are expressed as mean ± SEM (n = 3 per group). *P < 0.05, **P < 0.01, compared with the DM group; # P < 0.05 compared with the DM + NC group
3.5. NC prevents autophagic flux impairment in the CA1 hippocampus of DM rats
To further confirm whether NC exerts its protective roles against learning and cognitive impairment induced by DM via facilitating autophagic flux, the expressions of two biomarkers of autophagy, LC3‐II and p62 proteins in CA1 regions were detected by Western blot assay. As shown in Figure 5A,B, there were base‐level LC3‐II and p62 protein expressions in the normal control group, indicating neuron cells in CA1 areas exhibit basal autophagy. However, as compared with rats in the normal control group, the expressions of LC3‐II and p62 protein were significantly increased in DM group, implying autophagic flux dysfunction occurred. As expected, LC3‐II and p62 protein levels in NC + DM group were much lower than those in DM group, respectively. Notably, cotreatment NC with CQ further elevated the expressions of those two proteins in CA1 regions of DM rats. The expressions of LC3‐II in DM + NC + 3‐MA group significantly lowered as compared with that in DM + NC group, whereas the p62 protein expression obviously upregulated. Since the accumulation of p62 protein is positively associated with impaired autophagic flux, these results suggested that NC could rescue DM‐induced autophagic flux impairment in CA1 hippocampus of rats.
Figure 5.
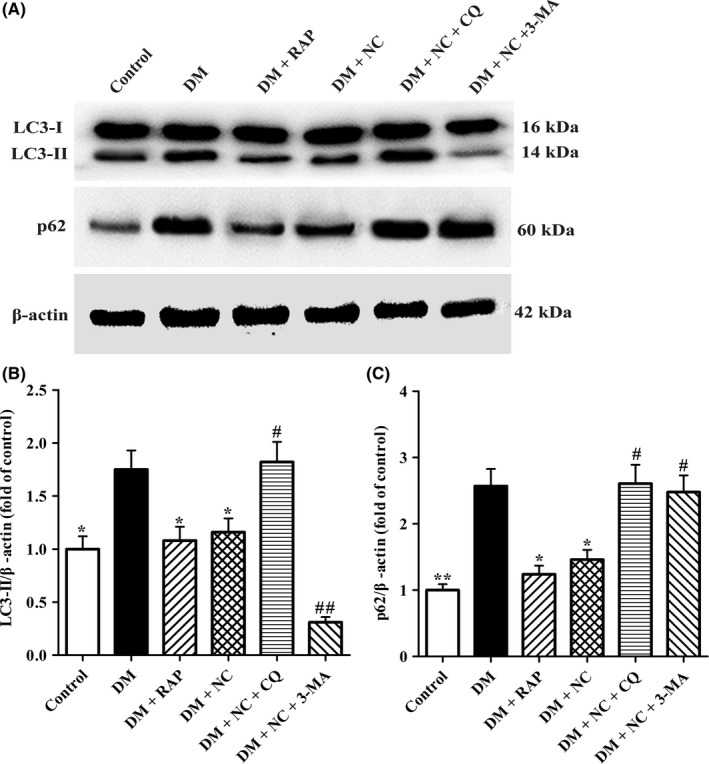
Effect of NC on the autophagic flux in the CA1 hippocampus of DM rats induced by STZ. Diabetic rats were treated with RAP (1 mg/kg/day, IP), NC (100 mg/kg/day, IP), NC (100 mg/kg/day, IP) + CQ (40 mg/kg/day, IP), NC (100 mg/kg/day, IP) + 3‐MA (1.5 mg/kg/day, IP) for 35 consecutive days. (A) Western blot was performed to detect autophagic flux‐associated proteins of LC3‐I (16 kDa), LC3‐II (14 kDa), and p62 (62 kDa) in the hippocampal CA1 area of rats. (B and C) The relative optical density values of LC3‐II and p62 proteins were quantified by the NIH Image J software. β‐actin was used as control for protein loading. The data are expressed as mean ± SEM (n = 4 per group). *P < 0.05, **P < 0.01, compared with the DM group; # P < 0.05, ## P < 0.05 compared with the DM + NC group
3.6. NC reverses DM‐induced changes in Bax, Cyt‐c, cleaved Caspase‐3, and Bcl‐2 protein expression in CA1 hippocampus tissues of rats
Considered that elevated Bax, Cyt‐c, and activated Caspase‐3 and lowered Bcl‐2 protein levels play a critical role in neuron cell death, in the present study, Western blot assay was performed to evaluate the effects of NC on the expression of the above mentioned 4 proteins. As shown in Figure 6, compared with control group, Bax, Cyt‐c, and cleaved Caspase‐3 protein expressions in DM group increased dramatically, while Bcl‐2 protein levels decreased significantly. The expressions of Bax, Cyt‐c, and cleaved Caspase‐3 declined significantly both in DM + RAP group and DM + NC group, while the expression of Bcl‐2 elevated distinctly both in the two groups, indicating that autophagy inducer RAP and NC treatment effectively reversed Bax, Cyt‐c, cleaved Caspase‐3, and Bcl‐2 protein expressions in DM rats. However, the expressions of Bax, Cyt‐c, cleaved Caspase‐3 in DM + NC + CQ group and DM + NC + 3‐MA group upregulated obviously as compared with those in DM + NC group, respectively, whereas Bcl‐2 expression downregulated significantly. Taken together, these data confirmed that NC reversed the expressions of Bax, Cyt‐c, cleaved Caspase‐3, and Bcl‐2 proteins in DM rats via facilitating autophagic flux.
Figure 6.
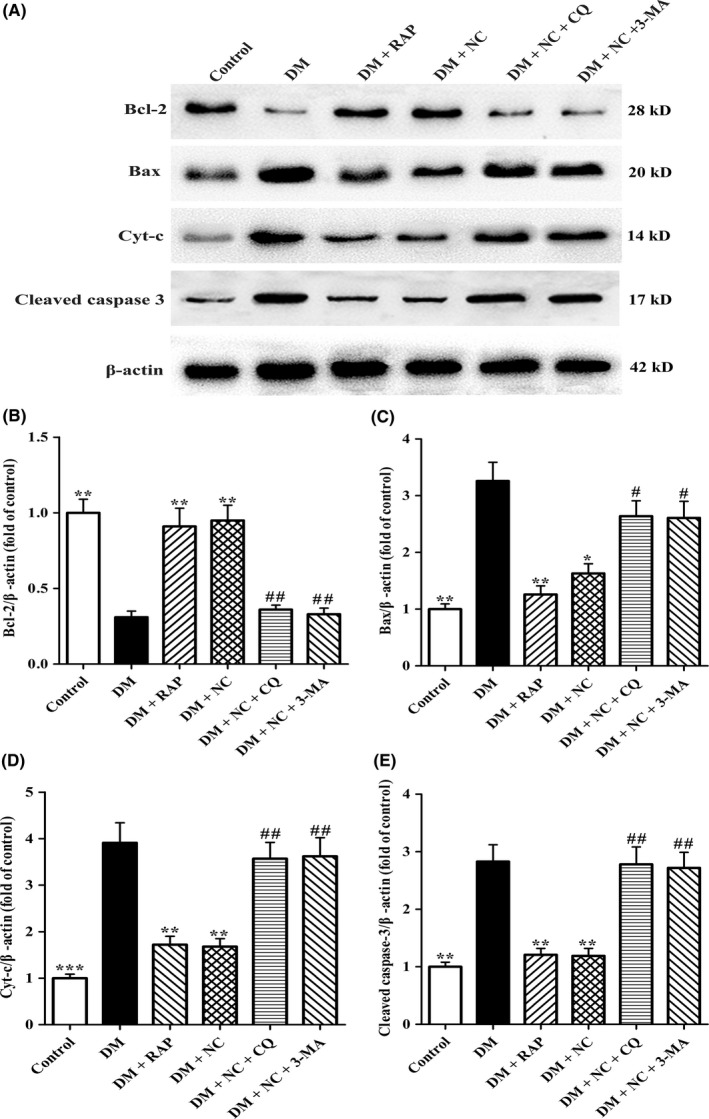
Effect of NC on apoptosis‐associated proteins Bcl‐2, Bax, Cyt‐c, and Caspase‐3 in the CA1 hippocampus of DM rats induced by STZ. Diabetic rats were treated with RAP (1 mg/kg/day, IP), NC (100 mg/kg/day, IP), NC (100 mg/kg/day, IP) + CQ (40 mg/kg/day, IP), NC (100 mg/kg/day, IP) + 3‐MA (1.5 mg/kg/day, IP) for 35 consecutive days. (A) Western blot analysis was performed to detect apoptosis‐associated proteins of Bcl‐2 (28 kD), Bax (20 kD), Cyt‐c (14 kD), and cleaved Caspase‐3 (17 kD) in the hippocampal CA1 region of rats. (B, C, D, and E) Densitometry analysis of Bcl‐2 (28 kD), Bax (20 kD), Cyt‐c (14 kD), and cleaved Caspase‐3 (17 kD) protein levels were performed by the NIH Image J software. β‐actin was used as control for protein loading. The data are expressed as mean ± SEM (n = 4 per group). *P < 0.05, **P < 0.01, ***P < 0.01, compared with the DM group; # P < 0.05, ## P < 0.05 compared with the DM + NC group
3.7. NC suppressed the hyperactivation of the mTOR/p70S6K signaling in CA1 hippocampus of DM rats
Among several signaling pathways implicated in autophagy, mTOR/p70S6K signaling is a critical one that regulates autophagy. To further reveal the underlying mechanism by which NC reversed DM‐induced autophagic flux impairment in CA1 hippocampus of rats, the phosphorylation levels of mTOR and p70S6K were measured by Western blot analysis. As shown in Figure 7A‐C, p‐mTOR and p‐p70S6K levels were significantly elevated in DM group as compared with those in control group, implying that DM leads to mTOR signaling pathway hyperactivation. In contrast, the expressions of p‐mTOR and p‐p70S6K proteins in DM + RAP group and DM + NC group were significantly lower than those in DM group, indicating that cotreatment with NC remarkably suppresses the activation of mTOR/p70S6K pathway. Collectively, these results suggest that NC restored autophagic flux of diabetic rats in CA1 hippocampus maybe via inhibiting mTOR/p70S6K signaling.
Figure 7.
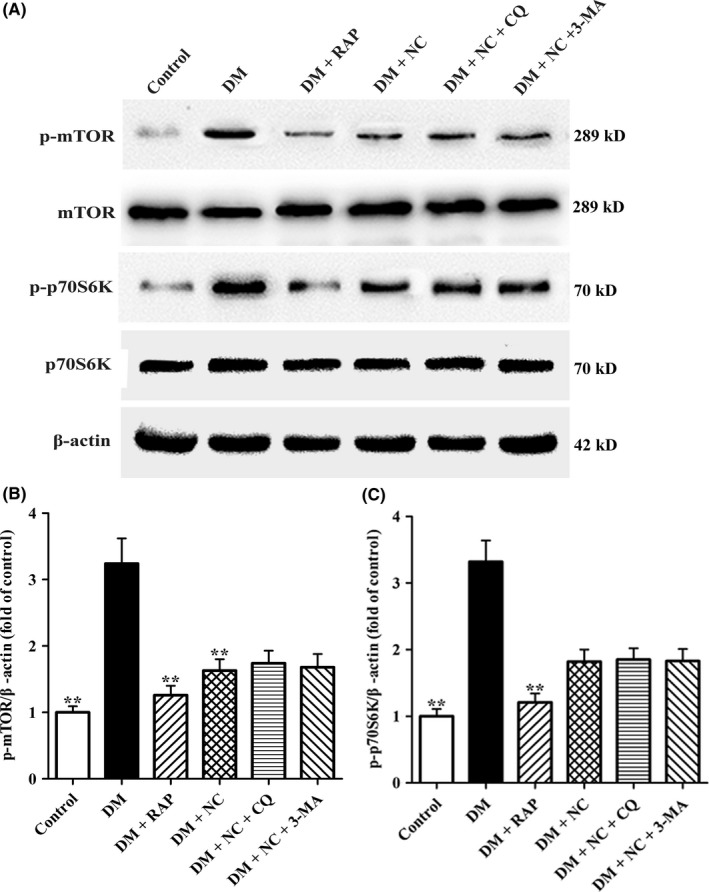
Effect of NC on the expressions of mTOR, and p70S6K in the CA1 hippocampus of DM rats. Diabetic rats were treated with RAP (1 mg/kg/day, IP), NC (100 mg/kg/day, IP), NC (100 mg/kg/day, IP) + CQ (40 mg/kg/day, IP), NC (100 mg/kg/day, IP) + 3‐MA (1.5 mg/kg/day, IP) for 35 consecutive days. (A) The expressions of mTOR, p‐mTOR (Ser2448), p70S6K, and p‐p70S6K (Thr389) in the hippocampal CA1 region of rats were determined by Western blot analysis. (B) and (C) The p‐mTOR, and p‐p70S6K protein levels were quantified by the NIH Image J software. β‐actin was used as control for protein loading. The data are expressed as mean ± SEM (n = 4 per group). **P < 0.01, compared with the DM group
4. DISCUSSION
In the present work, we investigated the influence of DM on the cognitive function and autophagic flux in the CA1 hippocampus, and mechanisms by which NC attenuated cognitive deficits in DM rats induced by STZ. As expected, our data indicated that DM rats exhibited cognitive dysfunction and autophagic flux disruption in the CA1 hippocampus. NC treatment significantly restored the CA1 hippocampal autophagic flux and thereby attenuating the CA1 neuron cell loss and cognitive impairment. Moreover, the present study demonstrated that NC reserved the impairment of autophagy and cognitive function in DM rats maybe via inhibiting the mTOR signaling pathway. These findings suggested that NC can be exploited as a potential therapeutic reagent for the improvement of DM‐related cognitive deficits.
Accumulating evidences have confirmed that DM is a major pathogenesis factor for cognitive deficits.4, 5, 26 Curcumin has been shown to be an effective agent for attenuating diabetic encephalopathy and improving surgery‐induced cognitive dysfunction in mice .17, 18, 27 In this study, we first explored the effects of the curcumin derivative NC on learning and memory performances of rats with DM induced by STZ. Our results revealed that the declined cognitive functions in DM rats were significantly reserved by NC treatment, as evidenced by decreases in escape latency as well as increases in times across target quadrant in MWM test, and elevated DI in the novel object recognition test. These data assured that NC is able to improve cognitive decline in rats with DM.
CA1 region, a major functional area of hippocampus, is essential for learning and memory.28 Considering neuron cell loss and cell structure damage in this area contribute to impairment of cognitive abilities, we then investigated the influence of NC on morphologic changes in the CA1 the hippocampus and the underlying mechanism in DM rats. Our HE results indicated that the cell number of the CA1 hippocampal neurons was decreased and the structure was damaged in the DM group. Meanwhile, TEM observations showed that CA1 neuronal ultrastructures were impaired in DM rats, manifested by accumulation of APs, damaged mitochondria, and decreased organelles. These findings are in line with previous studies that hippocampal neuronal loss and structure impairment may cause the cognitive deficits in DM animals.5, 6, 7 Interestingly, our results implied that autophagic flux blockage in the CA1 hippocampus may be responsible for structure damage to this region and impairment of cognitive function in DM rats, which indicated by the evidences that CQ worsened the CA1 injury and cognitive function decline, while RAP ameliorated the CA1 impairment and cognitive function deficits in DM rats. These data indicate that autophagy plays protective roles in preventing cognitive deficits in DM. As expected, treatment with NC significantly prevented neuron loss and neuronal structure damage in the hippocampal CA1 regions of DM rats. However, these beneficial effects of NC were abolished by cotreatment with 3‐MA or CQ, suggesting that facilitating autophagic flux is involved in NC improving cognitive performances of DM rats.
Autophagy impairment has been found in diabetic nephropathy, diabetic cardiomyopathy, and hypothalamus of diabetic mice.13, 29 In the present study, we further explored autophagic flux changes in hippocampus of diabetic rats. The results revealed that both LC‐3II and p62 protein levels elevated in DM rats. Moreover, TEM results indicated that AP aggregation remarkably increased in the CA1 hippocampus of diabetic rats as compared to the control ones. In general, the accumulated APs and heightened p62 protein expression are two markers for autophagic flux suppression.30 Therefore, these data showed that autophagic flux was impaired in hippocampus of diabetic rats.
Given that autophagic flux dysfunction in the hippocampus may contribute to neurodegenerative diseases, improving autophagic flux in these areas is hence considered as a potential therapeutic approach to cognitive decline.31, 32 In the present study, we found that NC restored autophagic flux in hippocampus CA1 region of DM rats. Our Western blotting results revealed that NC treatment profoundly elevated LC3‐II protein level and lowered p62 protein expression in the hippocampus of DM rats. However, cotreatment with NC and CQ dramatically increased p62 level and further heightened LC3‐II expression. Furthermore, our TEM results confirmed that NC improved autophagic flux in DM rats, as indicated by cotreatment with CQ caused a significant increase in APs. These results are in consistent with our preceding findings that NC can rescue autophagic flux impairment in THP‐1 cells.22 Taken together, these results showed that NC can reverse autophagic flux impairment in hippocampus of DM rats induced by STZ.
Hyperactivation of the mTOR/p70S6K signaling contributes to autophagic flux impairment in pathogenesis of Alzheimer disease and Parkinson's disease.33, 34 In this pathway, p70S6K is a critical element for mTOR‐downstream signaling. Activation of mTOR results in p70S6K phosphorylation and thereby negatively regulated autophagic flux. RAP, an inducer of autophagy by inhibiting the activation of mTOR, can improve cognitive deficits of AD mice. In this work, the results indicated that autophagic flux disruption occurred in hippocampus of DM rats via activation of mTOR/p70S6K signaling, as evidenced by elevated protein levels of p‐mTOR and p‐p70S6K. As expected, RAP or NC treatment significantly attenuated activation of mTOR/p70S6K and impairment of autophagic flux in DM rats, as confirmed by decreases in levels of p‐mTOR, p‐p70S6K and p62 proteins, and accumulation of APs in hippocampus of DM rats. These results clarified that NC reversed autophagic flux impairment in hippocampus of DM rats via inhibiting mTOR/p70S6K pathway. Considering there was no significant difference in p‐mTOR, p62 and LC3‐II expressions, and escape latency as well as times across target quadrant between in RAP treatment group and NC treatment group, we concluded that NC may have the same roles as RAP in these aspects.
Autophagy is essential mechanism for neuronal cell survival by degrading and recycling damaged organelles.35 Consequently, disrupted autophagic flux results in intracellular accumulation of damaged organelles, such as mitochondria. Transmission electron microscopy is one of the best approaches to evaluate the degradation of impaired mitochondria via autophagy.30 Our TEM results showed that the accumulation of damaged mitochondria and APs containing undegraded materials significantly increased in DM rats than those in control ones. Considering the impaired mitochondrial augments the leakage of Cyt‐c and ultimately promotes neuronal cell apoptosis, we analyzed Bcl‐2, Bax and Cyt‐c protein levels, and the activation of the Caspase‐3. Our results showed that the expressions of Bax, Cyt‐c, and cleaved Caspase‐3 proteins were obviously elevated while Bcl‐2 expression notably lowered. The decreased Bcl‐2/Bax ratio will promote permeability of mitochondria and the release of Cyt‐c.36 The released Cyt‐c causes the activation of Caspase‐3, thereby inducing apoptosis of hippocampus neurons in DM rats. These results are in line with the previous reports that damaged mitochondria contributes to apoptosis in hippocampus neuronal of DM rats.37 Notably, our study indicated that NC treatment markedly decreased the levels of Cyt‐c, Bax, and activated Caspase‐3 proteins, while obviously increased Bcl‐2 expression. These results showed that NC exerted its beneficial effects against DM‐induced neuronal apoptosis by facilitating autophagic flux to eliminate the damaged mitochondria and thus preventing Cyt‐c release into cytosol and the activation of Caspase‐3 in this pathophysiology.
Taken together, our study manifested that mTOR/p70S6K signaling was activated, resulting in autophagic flux impairment, damaged mitochondria accumulation, Caspase‐3 activation and neuronal apoptosis, and cognitive deficit in DM rats. Importantly, NC treatment restored the CA1 hippocampal autophagic flux through inhibiting the hyperactivation of mTOR/p70S6K signaling in DM rats, which promotes elimination of damaged mitochondria, and prevents neuronal apoptosis and ultimately attenuates cognitive deficits. Therefore, NC may be provided a new therapeutic candidate for the prevention and treatment of diabetic cognitive dysfunction.
CONFLICT OF INTEREST
The authors declare no conflict of interest, and all the authors listed have approved the manuscript that is enclosed.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (grants no. 81670268, 81500349), the Natural Science Foundation of Hunan Province, China (Grant No. 2016JJ2112), Graduate Student Scientific Research Innovation Project of Hunan Province (Grant No. CX2017B548), Aid Program for Chinese Medicine of Human Province (Grant No. 201524).
Gu H‐F, Li N, Tang Y‐L, et al. Nicotinate‐curcumin ameliorates cognitive impairment in diabetic rats by rescuing autophagic flux in CA1 hippocampus. CNS Neurosci Ther. 2019;25:430–441. 10.1111/cns.13059
The first two authors contributed equally to this work.
Contributor Information
Duan‐Fang Liao, Email: dfliao@hnucm.edu.cn.
Xin‐Ping OuYang, Email: y1655@163.com.
REFERENCES
- 1. Buse JB, Ginsberg HN, Bakris GL, et al. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation. 2007;115(1):114‐126. [DOI] [PubMed] [Google Scholar]
- 2. Carney EF. Prevention: intensive exercise associated with reduced risk of diabetic nephropathy in patients with type 1 diabetes mellitus. Nat Rev Nephrol. 2015;11(4):198. [DOI] [PubMed] [Google Scholar]
- 3. Zenker J, Poirot O, de Preux Charles AS, et al. Altered distribution of juxtaparanodal kv1.2 subunits mediates peripheral nerve hyperexcitability in type 2 diabetes mellitus. J Neurosci. 2012;32(22):7493‐7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strachan MW, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7(2):108‐114. [DOI] [PubMed] [Google Scholar]
- 5. Cai XJ, Wang L, Hu CM. Effects of GABAB receptor activation on spatial cognitive function and hippocampal neurones in rat models of type 2 diabetes mellitus. Biosci Rep. 2018;38(1):BSR20171184. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6. Sadeghi A, Hami J, Razavi S, Esfandiary E, Hejazi Z. The effect of diabetes mellitus on apoptosis in hippocampus: cellular and molecular aspects. Int J Prev Med. 2016;7(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baydas G, Nedzvetskii VS, Nerush PA, Kirichenko SV, Yoldas T. Altered expression of NCAM in hippocampus and cortex may underlie memory and learning deficits in rats with streptozotocin‐induced diabetes mellitus. Life Sci. 2003;73(15):1907‐1916. [DOI] [PubMed] [Google Scholar]
- 8. Betin VM, Singleton BK, Parsons SF, Anstee DJ, Lane JD. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy. 2013;9(6):881‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Monastyrska I, Klionsky DJ. Autophagy in organelle homeostasis: peroxisome turnover. Mol Aspects Med. 2006;27(5–6):483‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou X, Ikenoue T, Chen X, Li L, Inoki K, Guan KL. Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy. Proc Natl Acad Sci USA. 2009;106(22):8923‐8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lim J, Kim HW, Youdim MB, Rhyu IJ, Choe KM, Oh YJ. Binding preference of p62 towards LC3‐ll during dopaminergic neurotoxin‐induced impairment of autophagic flux. Autophagy. 2011;7(1):51‐60. [DOI] [PubMed] [Google Scholar]
- 12. Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci USA. 2015;112(52):15790‐15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jing YH, Zhang L, Gao LP, et al. Autophagy plays beneficial effect on diabetic encephalopathy in type 2 diabetes: studies in vivo and in vitro. Neuro Endocrinol Lett. 2017;38(1):27‐37. [PubMed] [Google Scholar]
- 14. Wang B, Huang J, Li J, Zhong Y. Control of macrophage autophagy by miR‐384‐5p in the development of diabetic encephalopathy. Am J Transl Res. 2018;10(2):511‐518. [PMC free article] [PubMed] [Google Scholar]
- 15. Li Z, Hao S, Yin H, Gao J, Yang Z. Autophagy ameliorates cognitive impairment through activation of PVT1 and apoptosis in diabetes mice. Behav Brain Res. 2016;305:265‐277. [DOI] [PubMed] [Google Scholar]
- 16. Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin‐induced autophagy. Autophagy. 2007;3(6):635‐637. [DOI] [PubMed] [Google Scholar]
- 17. Sevastre‐Berghian AC, Făgărăsan V, Toma VA, et al. Curcumin reverses the diazepam‐induced cognitive impairment by modulation of oxidative stress and ERK 1/2/NF‐κB pathway in brain. Oxid Med Cell Longev. 2017;2017:3037876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hamaguchi T, Ono K, Yamada M. Review: curcumin and Alzheimer's disease. CNS Neurosci Ther. 2010;16(5):285‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Narlawar R, Baumann K, Schubenel R, Schmidt B. Curcumin derivatives inhibit or modulate beta‐amyloid precursor protein metabolism. Neurodegener Dis. 2007;4(2–3):88‐93. [DOI] [PubMed] [Google Scholar]
- 20. Di Martino R, Bisi A, Rampa A, Gobbi S, Belluti F. Recent progress on curcumin‐based therapeutics: a patent review (2012–2016). Part II: curcumin derivatives in cancer and neurodegeneration. Expert Opin Ther Pat. 2017;27(8):953‐965. [DOI] [PubMed] [Google Scholar]
- 21. Zhang CP, Sun SW, Gong YZ et al. PCSK9/LDLR pathway mediates curcumin trinicotinate promoting lipid uptake of HepG2. Progress in Biochemistry and Biophysics. 2015;42(9):825‐832. [Google Scholar]
- 22. Gu HF, Li HZ, Tang YL, Tang XQ, Zheng XL, Liao DF. Nicotinate‐curcumin impedes foam cell formation from THP‐1 cells through restoring autophagy flux. PLoS One. 2016;11(4):e0154820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kong FJ, Wu JH, Sun SY, Ma LL, Zhou JQ. Liraglutide ameliorates cognitive decline by promoting autophagy via the AMP‐activated protein kinase/mammalian target of rapamycin pathway in a streptozotocin‐induced mouse model of diabetes. Neuropharmacology. 2018;131:316‐325. [DOI] [PubMed] [Google Scholar]
- 24. Nikiforuk A, Hołuj M, Kos T, Popik P. The effects of a 5‐HT5A receptor antagonist in a ketamine‐based rat model of cognitive dysfunction and the negative symptoms of schizophrenia. Neuropharmacology. 2016;105:351‐360. [DOI] [PubMed] [Google Scholar]
- 25. Ma LY, Lv YL, Huo K, et al. Autophagy‐lysosome dysfunction is involved in Aβ deposition in STZ‐induced diabetic rats. Behav Brain Res. 2017;320:484‐493. [DOI] [PubMed] [Google Scholar]
- 26. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61(5):661‐666. [DOI] [PubMed] [Google Scholar]
- 27. Wu X, Chen H, Huang C, et al. Curcumin attenuates surgery‐induced cognitive dysfunction in aged mice. Metab Brain Dis. 2017;32(3):789‐798. [DOI] [PubMed] [Google Scholar]
- 28. Schlichting ML, Zeithamova D, Preston AR. CA1 subfield contributions to memory integration and inference. Hippocampus. 2014;24(10):1248‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gonzalez CD, Lee MS, Marchetti P, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7(1):2‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agarwal S, Tiwari SK, Seth B, et al. Activation of autophagic flux against xenoestrogen Bisphenol‐A‐induced hippocampal neurodegeneration via AMP kinase (AMPK)/mammalian target of Rapamycin (mTOR) pathways. J Biol Chem. 2015;290(34):21163‐21184. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Wang BJ, Her GM, Hu MK, et al. ErbB2 regulates autophagic flux to modulate the proteostasis of APP‐CTFs in Alzheimer's disease. Proc Natl Acad Sci USA. 2017;114(15):E3129–E3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perluigi M, Di Domenico F, Butterfield DA. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis. 2015;84:39‐49. [DOI] [PubMed] [Google Scholar]
- 34. Maiese K. Targeting molecules to medicine with mTOR, autophagy and neurodegenerative disorders. Br J Clin Pharmacol. 2016;82(5):1245‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barnett A, Brewer GJ. Autophagy in aging and Alzheimer's disease: pathologic or protective? J Alzheimers Dis. 2011;25(3):385‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Putcha GV, Moulder KL, Golden JP, et al. Induction of BIM, a proapoptotic BH3‐only BCL‐2 family member, is critical for neuronal apoptosis. Neuron. 2001;29(3):615‐628. [DOI] [PubMed] [Google Scholar]
- 37. Zhang QJ, Li J, Zhang SY. Effects of TRPM7/miR‐34a gene silencing on spatial cognitive function and hippocampal neurogenesis in mice with type 1 diabetes mellitus. Mol Neurobiol. 2018;55(2):1568‐1579. [DOI] [PubMed] [Google Scholar]