

Summary
Aims
Neural stem cells (NSCs) in the adult mammalian spinal cord are activated in response to spinal cord injury (SCI); however, mechanisms modulating this process are not clear. Here, we noticed SCI elevated expression of vascular endothelial growth factor (VEGF) and we aimed to validate the roles of VEGF in NSCs activation after SCI and investigated the related signals during the process.
Methods
In vitro we detected whether VEGF promoted spinal cord NSCs proliferation and investigated the involved signals; In vivo, we injected VEGF into rat spinal cord to check the NSCs activation.
Results
In vitro, VEGF triggered spinal cord NSCs proliferation and maintained self‐renewal. Further investigations demonstrated VEGF transactivated epidermal growth factor receptor (EGFR) through VEGF receptor 2 (VEGFR2) to promote spinal cord NSCs proliferation. In vivo, we injected VEGF into spinal cord by laminectomy to confirm the roles of VEGF‐VEGFR2‐EGFR signals in NSCs activation. VEGF significantly elevated the number of activated NSCs and increased EGFR phosphorylation. In contrast, intraspinal injection of specific inhibitors targeting EGFR and VEGFR2 decreased NSCs activation after SCI. Our results demonstrate that VEGF‐VEGFR2‐EGFR axis is important for NSCs activation after SCI, providing new insights into the mechanisms of spinal cord NSCs activation postinjury.
Keywords: epidermal growth factor receptor, neural stem cells activation, spinal cord injury, vascular endothelial growth factor, VEGF receptor 2
1. INTRODUCTION
Neural stem cells (NSCs) in the adult mammalian central nervous system (CNS) have now been found in two principal regions, the brain and the spinal cord.1, 2, 3 In the mammalian adult brain, NSCs reside in specialized microenvironments, located in the lateral ventricle’s subventricular zone (SVZ) and in the subgranular zone (SGZ) of the hippocampal dentate gyrus.4 The adult mammalian spinal cord also harbors ciliated ependymal cells lining the central canal, which have stem cell potential in vitro. Most adult spinal cord NSCs remain quiescent under normal conditions, but following spinal cord injury (SCI) the rarely dividing ependymal cells can become activated and migrate from the central canal to sites of injury.5, 6 However, the mechanisms that are involved in these processes are yet to be revealed.
The transition of spinal cord NSCs from quiescent to proliferative cells is associated with changes in the microenvironment after SCI. Previous report shows vascular integrity improves neuroregeneration, implying the important roles of vascular in neuroregeneration.7, 8 In adults, NSCs are located near vascular endothelial cells that provide oxygen to neural cells, and proliferate in small clusters around dividing capillaries in the CNS.9 After injury, events such as hypoxic conditions, cytokine, and growth factor secretion contribute to NSCs proliferation.10 Vascular endothelial growth factor A (VEGF‐A, VEGF in short), a member of VEGF family and first identified as a regulator of angiogenesis, can be easily induced by hypoxic conditions.11 In the nervous system, VEGF exerts a protective effect on neurons and promotes neurite extension in spinal cord explants, possibly by stabilizing and upregulating MAP2 (microtubule‐associated protein 2) expression.12 Importantly, VEGF has mitotic effects on NSCs through interactions with VEGF receptors (VEGFRs) and enhances neurogenesis in the hippocampus,13, 14, 15 whereas NSCs‐specific loss of VEGF results in impaired stem cell maintenance.16 Furthermore, VEGF is upregulated following CNS injury.17, 18, 19 Nevertheless, very little is known about the contribution of VEGF to spinal cord NSCs activation following SCI.
Epidermal growth factor receptor (EGFR, also known as ErbB1) was the first identified receptor of the ErbB family. Currently, the ErbB family includes EGFR, ErbB2, ErbB3, and ErbB4. EGFR can be transactivated by several signaling molecules, including G‐protein‐coupled receptor (GPCR)‐mediated signaling, protein kinase C (PKC), and nonreceptor tyrosine kinases such as Src and Pyk2.20, 21, 22 In the nervous system, EGFR signaling promotes the proliferation and survival of NSCs and inhibits neuronal differentiation.23, 24, 25 In particular, EGFR interacts with some pathways to promote NSCs proliferation in vivo.26, 27 Notably, EGFR expression is upregulated under hypoxic conditions generated by spinal or peripheral nervous injury.28, 29 Here, we demonstrate that VEGF promotes spinal cord NSCs activation through EGFR transactivation following SCI. Our results reveal that VEGF may act as a driving force for NSCs activation, and provide new insights into the mechanisms of NSCs activation following SCI.
2. MATERIALS AND METHODS
2.1. Animals and ethics statement
Female Sprague Dawley (SD) rats (210‐230 g) were purchased from Vital River (China) and housed in temperature‐controlled (24°C) and humidity‐controlled (50%) animal quarters with a 12‐hour light/dark cycle with free access to food and water. All animal experiments were performed in accordance with the Chinese Ministry of Public Health Guide and US National Institutes of Health Guide for the care and use of laboratory animals. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
2.2. Animal surgery models
Surgeries were performed under strict sterile conditions.
Female adult SD rats (210‐230 g) were used for surgery. Animals were anesthetized by intraperitoneal (ip) injection of chloral hydrate (0.5 g/kg) before surgery according to previous protocol.30 Two surgical models were used: (1) complete spinal cord injury between thoracic 8 and 9 (T8‐T9); (2) intraspinal injection.
For complete spinal cord injury, the pelage on the back of the anesthetized rat was shaved, and the skin was cleaned with povidone iodine solution. A 2‐mm‐long gap was made between T8 and T9 of the spinal cord. Absorbable gelatin sponge (Xiang En) was used to control bleeding at the transection site. The rat survival rate after injury was above 80%.
For injection models, laminectomy and intraspinal injections into the same site of the spinal cord (left side of T8‐T9 in the spinal cord, at the same distance from the central canal) were performed using a needle loaded with different factors or inhibitors at 45° to the spinal cord. The outer diameter of the needle was 0.5 mm (approximately 25 G). The tip of the needle was kept inside the spinal cord for 1 minute after each injection to avoid liquid reflux and enable sufficient absorption of the factors or inhibitors. The rat survival rate was above 95%.
2.3. Experimental groups and treatment
-
1
Complete spinal cord injury between T8 and T9 (n = 35; 5 rats/group) was divided into seven groups: 0, 1, 3, 7 days postinjury, respectively; SCI group (as control), gefitinib group, and cabozantinib group.
Animals of gefitinib group and cabozantinib group were administrated with inhibitors, respectively, by intraperitoneal injection for a week before surgery (75 mg/kg, dissolved in 0.5% DMSO; 0.5% DMSO injection as control), and administration continued throughout the experiments. The 0‐day postinjury (0 dpi) group represents a model without spinal cord injury, but with a laminectomy.
-
2
Intraspinal injection (n = 30; 5 rats/group) was divided into six groups: control group; VEGF group; gefitinib group; cabozantinib group; VEGF + gefitinib group; VEGF + cabozantinib group.
Animals of inhibitors‐treated groups were administrated with two specific inhibitors (gefitinib and cabozantinib), respectively, by intraperitoneal injection for a week before surgery (75 mg/kg, dissolved in 0.5% DMSO; 0.5% DMSO injection as control), and administration continued throughout the experiments. Intraspinal injections into the spinal cord were performed using a 25‐G needle loaded with different factors (inhibitors: 75 mg/kg; VEGF: 500 ng/rat) at 45° to the spinal cord.
Each day the bladders of animals were squeezed to extrude urine twice and the animals were injected with penicillin/streptomycin once, with free access to food and water.
2.4. Tissue preparation
Spinal cord tissue was collected from around the injury site (1 cm) after cardiac puncture and fixed with 4% PFA (paraformaldehyde) overnight for immunofluorescence. Tissue was then dehydrated in 30% sucrose for 2 days. Cryosections (20 µm) were taken and used for immunofluorescence studies by incubating the sections overnight with primary antibodies before labeling with Alexa Fluor‐conjugated secondary antibodies. All of the samples were used for assays, and every experiment was repeated three times.
2.5. Spinal cord NSCs culture
Spinal cord NSCs were isolated from the spinal cord of postnatal day 3‐5 SD rats. The NSCs were cultured in suspension in complete medium containing DMEM/F12 (Invitrogen, Carlsbad, CA, USA), B27 supplement (Invitrogen), 30% glucose (Sigma, Saint Louis, MO, USA), 1.83 mg/mL heparin (Sigma), 100 U/mL penicillin/streptomycin (Invitrogen), nonessential amino acids (NEAA; Invitrogen), and sodium pyruvate (Invitrogen), which was enriched with either 20 ng/mL VEGF, 50 ng/mL VEGF, or 100 ng/mL VEGF (Peprotech, Rocky Hill, CT, USA). 20 ng/mL EGF (Peprotech) combined with 20 ng/mL bFGF (Peprotech) was used as the control. Low attachment culture dishes were used for cultures. Sphere formation and the average diameters of NSCs were assessed on days 3 and 5. The medium was replaced every 2‐3 days. After 7 days, NSCs were prepared for passaging or plating.
2.6. Differentiation of spinal cord NSCs
Spinal cord NSCs spheres were cultured for 7 days and dissociated with 0.25% trypsin in 0.02% EDTA at 37°C for 20 minutes, with mechanical agitation every 10 minutes. High‐glucose DMEM (Invitrogen) with 10% FBS was used to stop digestion before 5 × 104 cells per well were seeded in 48‐well plates coated with 100 µg/mL PLL (poly‐l‐lysine; Sigma). Cells were allowed to adhere to dishes overnight. After 24 hours, cells were washed three times with PBS to remove FBS (FBS might affect differentiation) before the addition of the differentiation medium. The differentiation medium consisted of high‐glucose DMEM, N2 supplement (Invitrogen), 30% glucose, 100 U/mL penicillin/streptomycin, NEAA, sodium pyruvate, and 3% glutamine (Invitrogen). In accordance with routine culture, half of the medium was replaced every 2 days. Immunofluorescence was performed after 7 days. Images were captured using a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
2.7. Secondary sphere formation and proliferation assays
Spheres were dissociated with 0.25% trypsin and filtered through a 40‐µm membrane to obtain single cells, which could then form secondary spheres. The single NSCs were seeded in 3.5‐cm dishes (106 cells/dish). Sphere formation and the average diameters of NSCs were assessed on day 5. The cells were passaged in low attachment plates and cultured with VEGF‐containing medium.
The proliferation rate was checked via EdU labeling. The neurospheres were dissociated into single cells with 0.25% trypsin for 20 minutes. Before seeding cells, 96‐well plates were coated with 100 µg/mL PLL (Sigma) for 30 minutes at 37°C and then washed three times with PBS. Approximately 5000 cells were seeded per well and incubated for 2 hours at 37°C with 5% CO2 before adding EdU (50 µM). The assay was performed according to the EdU kit instructions (RiboBio, Guangzhou, China).
2.8. Cell viability assay
Approximately 1 × 104 single NSCs were seeded per well in 96‐well plates with a total volume of 100 µL/well. The inhibitors gefitinib and cabozantinib both at 1 µM were added to VEGF‐containing medium to culture spinal cord NSCs for 24 hours. The control group received 0.1% DMSO. Ten microliters of WST‐1 (Beyotime, Shanghai, China) was added to each well, and the cells were cultured at 37°C in 5% CO2 for 4 hours. VEGF‐containing medium with no cells was used as a blank. After 4 hours, the plates were shaken for 1 minute to mix the solutions before detection. The plates were read at 450 nm (420‐480 nm), and the reference wavelength was 690 nm.
2.9. Immunofluorescence
Cells were fixed with 4% PFA for 30 minutes at room temperature (RT), treated with 0.3% Triton X‐100 for 10 minutes, and blocked for 1 hour with 10% BSA at RT. Cells were then incubated with primary antibodies (1:500) at 4°C overnight and subsequently incubated with Alexa Fluor 488‐conjugated secondary antibodies (Invitrogen) for 1 hour at RT. Hoechst 33342 (1 mg/mL) dye was used to stain nuclear DNA. The respective images of cells were captured using a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss) and Leica SCN400 slide scanner (Leica Microsystems, Wetzlar, Germany). The primary antibodies are listed in Supplementary Table S1.
2.10. Western blotting
Cells or spinal cord tissue was lysed into RIPA lysis (with protease inhibitors) buffer or 2% SDS for 30 minutes on ice and then centrifuged at 12 000 × g at 4°C. The spinal cord tissue of the SCI group consisted of 2 mm pieces from the front to back of the lesions (T8‐T9), and the counterpart was collected from the sham group. The tissue was subsequently pulverized in liquid nitrogen. For the phosphorylation assay, phosphorylase inhibitors (Selleck, Shanghai, China) were added into the RIPA buffer (with protease inhibitors). VEGF was added into NSCs to stimulate for 10 minutes and collected cell samples for phosphorylation assay. Proteins (80 µg for each lane) were resolved using SDS‐PAGE with loading controls on the same blot and transferred onto nitrocellulose membranes, which were blocked using 5% skimmed milk for 1 hour at RT. The membranes were incubated with primary antibodies at 4°C overnight and subsequently hybridized with secondary antibodies conjugated with horseradish peroxidase (HRP) for 2 hours. Protein binding was visualized using enhanced chemiluminescence reagent (Thermo, Waltham, MA, USA). The experiments were processed in parallel and repeated at least three times. The primary antibodies are listed in Supplementary Table S1.
2.11. Calcium imaging
Dissociated spinal cord NSCs were seeded on cover slips coated with PLL and stimulated with 100 ng/mL VEGF after starvation for 3 days. The cells were washed three times with HBSS (Invitrogen) and incubated with 5 µM Fluo‐3 AM (Beyotime) for 30 minutes at 37°C in 5% CO2. Unbound Fluo‐3 AM was removed by three wash steps. The cells were incubated with HBSS for another 30 minutes at 37°C in 5% CO2. Images were collected with a Leica SCN400 slide scanner.
2.12. Statistical analysis
Two‐tailed unpaired Student’s tests were used to test the comparisons between two groups. All error bars represent mean ± SEM. The significance level was set at P ≤ 0.05 (exact P values, see figure legends). All experiments were repeated three times.
3. RESULTS
3.1. Spinal cord NSCs activation is accompanied by elevated VEGF expression after SCI
In the intact spinal cord, NSCs are confined within the ependymal cell population and remain quiescent, and the expression of nestin is restricted.31 Our previous study shows that spinal cord NSCs are activated and proliferate robustly after SCI, using nestin immunofluorescence to represent NSCs.32 We performed severe T8‐T9 SCI rat models (n = 5 rats/group) and observed the nestin‐positive NSCs around the injury sites were significantly elevated from day 1, peaked on day 3 postinjury, and subsequently decreased on day 7 postinjury (Figure 1A), which suggests that 1 dpi (days postinjury) is when NSCs activation occurs earlier after SCI. Destruction of the vascular system was the earliest event after SCI, subsequently generating a hypoxic condition, which may contribute to spinal cord NSCs activation after injury.33 In our study, we found that Hif 1α expression, which was induced by hypoxia and has been shown to promote VEGF expression, was increased around the injured region after 3 days postinjury (n = 2; Supplementary Figure S1). Furthermore, we found that VEGF had a similar expression pattern to nestin (Figure 1B). In addition, the evaluation of protein levels of nestin and VEGF around the injured regions further confirmed that both NSCs activation and VEGF expression were elevated on day 3 post‐SCI (Figure 1C). These results imply that the elevated VEGF expression may play a role in the activation of endogenous spinal cord NSCs after SCI.
Figure 1.
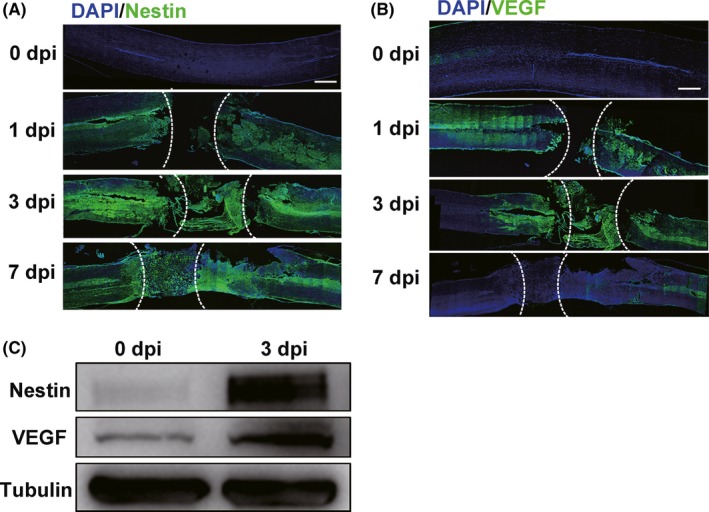
Endogenous spinal cord NSCs are activated and accompanied by elevated VEGF expression after SCI (n = 5 rats/group). A, Endogenous spinal cord NSCs are activated after SCI. Few nestin+ NSCs in 0 dpi group. SCI, spinal cord injury. dpi, days postinjury. Scale bar represents 500 µm. B, Immunofluorescence shows increased VEGF expression after SCI. Scale bar represents 500 µm. C, Western blot shows expression of both nestin and VEGF is increased after SCI (3 dpi). The lesion sites are outlined by dashed lines in (A) and (B). 0 dpi group represents models without spinal cord injury but with laminectomy
3.2. VEGF promotes the proliferation of spinal cord NSCs in vitro
The elevation of VEGF expression accompanied by NSCs activation after SCI indicates that VEGF may exert mitotic effects on spinal cord NSCs. To test this hypothesis, we investigated whether VEGF could promote spinal cord NSCs proliferation in vitro. We isolated cells from the spinal cord tissue of SD rat pups (postnatal days 3‐5) because it was difficult to gain a large number of adult spinal cord NSCs, leading to small numbers of neurospheres.34, 35 Spinal cord tissue cells were cultured in suspension with different concentrations of VEGF: 0 ng/mL (as a control), 20, 50, and 100 ng/mL. As expected, several cellular aggregates were seen on day 1 (data not shown), and these had grown into neurospheres by days 3 and 5, with increasing diameters (Figure 2A). We measured the average diameters of the spheres in response to different concentrations of VEGF at different time points. The results showed that 20 ng/mL VEGF induced significant growth of the neurospheres compared with the control group, which was further promoted by 50 and 100 ng/mL VEGF (Figure 2B). Next, we used EdU labeling to assess cell proliferation in neurospheres. The percentage of EdU+ cells was increased with increasing concentrations of VEGF (Figure 2C,D), suggesting that the cells were at a proliferative state. Furthermore, cyclin D1, a cell cycle marker, was significantly elevated in response to VEGF, indicating that the cells in the neurospheres were activated (Figure 2E).
Figure 2.
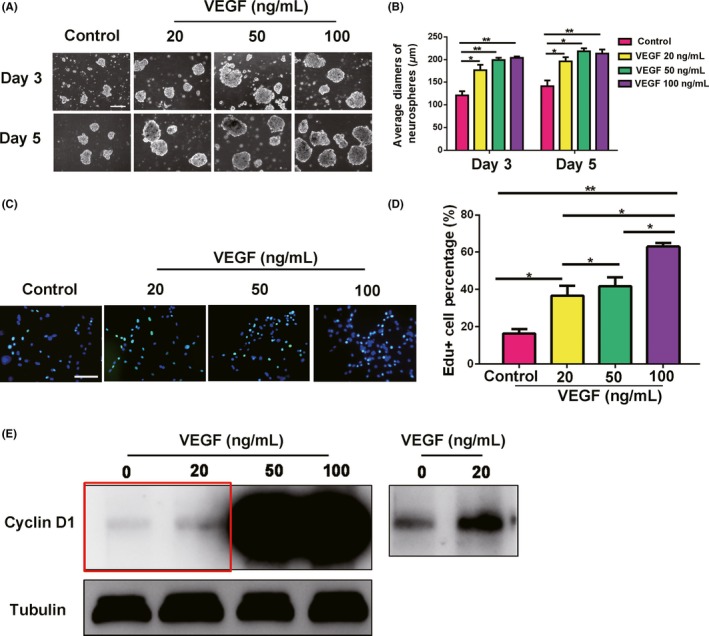
VEGF promotes the proliferation of spinal cord‐derived neurospheres in vitro. A, Different concentrations of VEGF promote the formation of spinal cord‐derived neurospheres. Scale bar represents 200 µm. B, Average diameters of spheres cultured for 3 and 5 d. P = 0.023, 0.0046, and 0.0071 for VEGF 20, 50, and 100 ng/mL, compared with control on day 3, respectively. P = 0.030, 0.013, and 0.0068 for VEGF 20, 50, and 100 ng/mL compared with control on day 5, respectively. Control group indicates 0 ng/mL VEGF. C, EdU staining showing that VEGF promotes the proliferation of spinal cord‐derived neurospheres. Scale bar represents 100 µm. D, Quantification of percentage of EdU+ cells under different concentrations of VEGF. VEGF 100 ng/mL shows the highest percentage of EdU+ cells (P = 0.00010). *P ≤ 0.05 and **P ≤ 0.01. E, VEGF promotes cyclin D1 expression in the neurospheres. The right shows the region in the red box after secondary exposure
We further identified that the cells in the spinal cord‐derived neurospheres induced by VEGF had NSCs identities. As the secondary sphere reformation assay is usually used to identify NSCs, we dissociated the neurospheres into single cells and continued to culture them in suspension for 5 days. The observation that the secondary spheres presented larger diameters with increasing VEGF concentrations might be because the secondary spheres were more proliferative (Figure 3A,B). As 100 ng/mL VEGF was able to induce NSCs proliferation more robustly than 20 and 50 ng/mL, we performed subsequent experiments using 100 ng/mL VEGF. Next, we identified the expression of NSCs markers, nestin, vimentin, and BLBP (brain lipid‐binding protein) in the neurospheres. The neurospheres cultured with VEGF displayed similar expression patterns of these NSCs markers as those in response to EGF combined with bFGF, which was the classic culture method for NSCs (Figure 3C,D). In addition, cells derived from the VEGF‐cultured spheres could also differentiate into Tuj1+ neurons and GFAP+ astrocytes, in accordance with the differentiation potential of NSCs (Figure 3E). VEGF could therefore serve as a growth factor to induce spinal cord NSCs proliferation in vitro.
Figure 3.
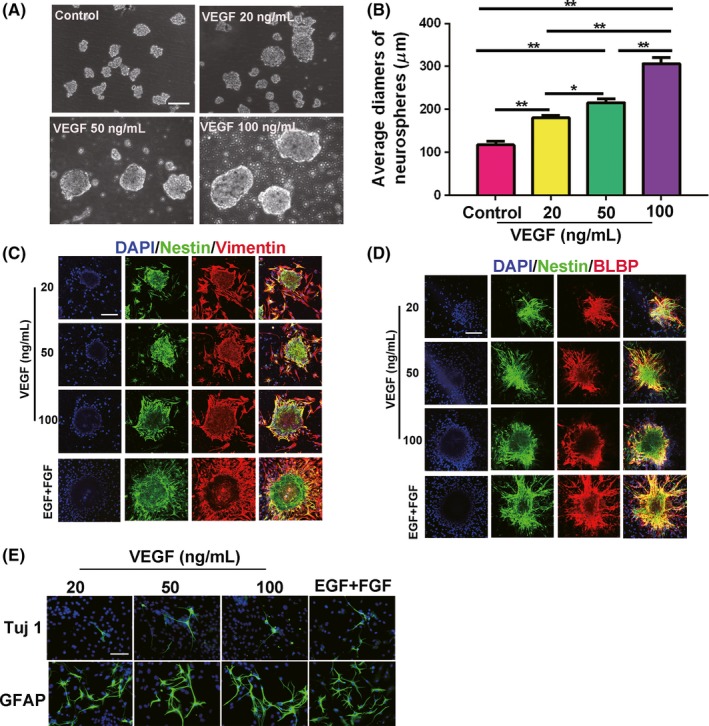
VEGF‐induced spinal cord‐derived neurospheres have self‐renewal ability and differentiation potential similar to NSCs. A, Neurospheres cultured for 5 d can form secondary spheres after being passaged in the presence of VEGF. Scale bar represents 100 µm. B, Average diameters of secondary spheres. VEGF 100 ng/mL shows the most proliferation compared with control (P = 0.0011). *P ≤ 0.05 and **P ≤ 0.01. Control, 0 ng/mL VEGF. C and D, Neurospheres induced by VEGF express NSCs markers. Nestin (green); BLBP (red); vimentin (red). Scale bar represents 200 µm. E, Neurospheres induced by VEGF differentiate into Tuj1+ neurons (green) and GFAP+ astrocytes (green). Scale bar represents 100 µm. EGF + FGF group is used as the positive control in (C‐E)
3.3. Transactivation of EGFR by VEGF promotes the proliferation of spinal cord NSCs
EGFR is a type of RTK, and it plays an important role in cell proliferation. In the hippocampus, EGFR is extensively expressed in TAPs (transit‐amplifying precursors), which are considered the most rapidly proliferating cells in the brain.36 Previous studies have shown that EGFR immunoreactivity is increased following ischemia caused by brain injury.37 In our study, we found that EGFR phosphorylation in injured spinal cord tissue was also elevated after SCI (Supplementary Figure S2). We speculated that VEGF could interact with EGFR to promote spinal cord NSCs proliferation. To verify this hypothesis, we first assessed the expression of EGFR in spinal cord NSCs in vitro. The dissociated NSCs were seeded on cover slips precoated with PLL (poly‐l‐lysine) and starved for 3 days in serum‐free medium to inhibit the in‐house phosphorylation of EGFR (pEGFR). On day 4, we checked whether VEGF could stimulate EGFR phosphorylation, with 20 ng/mL EGF as the positive control. Immunofluorescence showed that spinal cord NSCs expressed EGFR, and EGFR could be activated into pEGFR by VEGF or EGF, 10 minutes for stimulation (Figure 4A). Next, using western blotting, we further confirmed that VEGF could induce EGFR phosphorylation (Figure 4B). We employed the pEGFR‐specific inhibitor gefitinib (ZD1839, 1 µM) to efficiently block EGFR phosphorylation at tyrosine residues. Western blotting showed that VEGF increased pEGFR levels, whereas gefitinib inhibited this effect, even in the presence of VEGF (Figure 4C). These results confirmed that VEGF could activate the phosphorylation of EGFR in spinal cord NSCs. Next, we characterized the effects of gefitinib on spinal cord NSCs proliferation. Compared with the VEGF group, the spheres exhibited growth arrest and reduced average diameters in the presence of gefitinib or combined with VEGF (Figure 4D,E). These results suggest that pEGFR induced by VEGF is essential for the proliferation of spinal cord NSCs.
Figure 4.
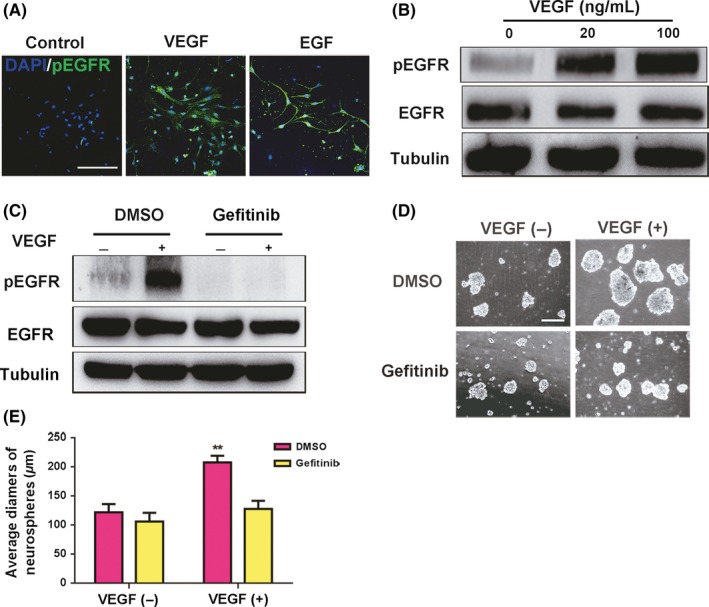
VEGF promotes the proliferation of spinal cord NSCs through EGFR phosphorylation. A, VEGF activates EGFR phosphorylation in spinal cord NSCs in vitro. EGF is used as the positive control. EGF, 20 ng/mL; VEGF, 100 ng/mL. Scale bar represents 200 µm. B, Western blotting shows that VEGF activates EGFR. VEGF, 100 ng/mL. 10 minutes for VEGF stimulation. C, Inhibitor of pEGFR, gefitinib, blocks VEGF‐induced EGFR phosphorylation. Gefitinib (dissolved in DMSO), 1 µM; VEGF, 100 ng/mL. D and E, Gefitinib decreases the proliferation of spinal cord NSCs. Gefitinib, 1 µM; VEGF, 100 ng/mL. Scale bar represents 200 µm. P = 0.026
3.4. VEGF transactivates the EGFR signal through VEGFR2
VEGF has two receptors, VEGFR1 and VEGFR2. First, we determined the expression of VEGFRs in spinal cord NSCs. Our results showed that spinal cord NSCs expressed VEGFR2 but not VEGFR1, and that VEGFR2 could be activated when VEGF was added in vitro (Figure 5A). In vivo results further validate the expression of VEGFR2, but not VEGFR1 in central canal and VEGFR2 could also be phosphorylated after injury (Supplementary Figure S3). We postulated that VEGF could activate EGFR through VEGFR2. As expected, both 20 and 100 ng/mL VEGF could activate VEGFR2 and phosphorylate EGFR (Figure 5B). When the VEGFR2‐specific inhibitor cabozantinib (XL184, 1 µM) was added in our system, we found that the phosphorylation of both VEGFR2 and EGFR was inhibited (Figure 5C). The growth of spinal cord NSCs was also inhibited by cabozantinib (Figure 5D). Additionally, western blotting results showed that EGFR phosphorylation was blocked under the inhibitor gefitinib; however, phosphorylation of VEGFR2 was not inhibited (Supplementary Figure S4), and none of the inhibitors were toxic to spinal cord NSCs survival (Supplementary Figure S5). These results indicate that VEGF transactivates EGFR via the activation of VEGFR2. Furthermore, VEGF‐activated spinal cord NSCs expressed nestin, Sox2, and Notch pathway targets Hes1 and Hes5, which enabled NSCs to self‐renew. In contrast, the two inhibitors gefitinib and cabozantinib resulted in the downregulation of these markers in NSCs (Figure 5E). Taken together, our results demonstrate that VEGF promotes the self‐renewal of spinal cord NSCs through the VEGFR2‐EGFR axis.
Figure 5.
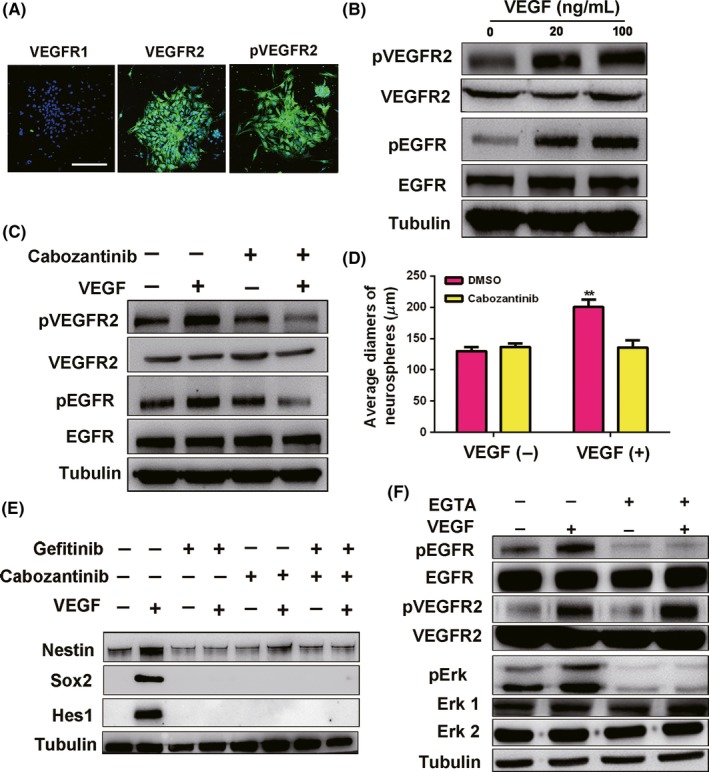
VEGFR2‐EGFR signaling maintains the self‐renewal ability of spinal cord NSCs. A, Spinal cord NSCs express VEGFR2 but not VEGFR1 in vitro. Scale bar represents 200 µm. B, VEGF at 20 and 100 ng/mL phosphorylate VEGFR2 and EGFR. C, Inhibitor of pVEGFR2, cabozantinib, blocks the phosphorylation of VEGFR2 and EGFR. Cabozantinib (dissolved in DMSO), 1 µM. D, Cabozantinib decreases the diameters of spinal cord neurospheres. Cabozantinib, 1 µM P = 0.019. E, Inhibitors of EGFR and VEGFR2 block the self‐renewal of spinal cord NSCs. Both cabozantinib and gefitinib are used at 1 µM. F, Calcium‐chelating agent EGTA inhibits VEGF‐induced pEGFR signaling. 10 min for VEGF stimulation. VEGF, 100 ng/mL. EGTA, 1.5 mM
Very little is known about the mechanisms of EGFR transactivation by VEGF. Calcium signaling was reported to play an important role in EGFR transactivation.20 Thus, we investigated whether calcium signaling was involved in the transactivation of EGFR by VEGF in NSCs activation. We used the calcium‐chelating agent EGTA to deplete intracellular calcium. As expected, EGTA blocked EGFR transactivation induced by VEGF and the pEGFR downstream pErk signal, whereas VEGFR2 was still activated (Figure 5F). Moreover, VEGF could induce the release of calcium signals, and EGTA inhibited the proliferation of spinal cord NSCs significantly in vitro (Supplementary Figure S6). These results indicate that the transactivation of EGFR by VEGF is likely mediated by calcium signaling.
3.5. VEGF activates spinal cord NSCs by VEGFR2‐EGFR signaling in vivo
To test the effects of VEGF on spinal cord NSCs in vivo, we injected VEGF into the rat spinal cord (at the same site as the previously described injections, n = 30) using a 25‐G needle at 45° to the spinal cord (Figure 6A). The proliferative effects of VEGF may be concealed by robust NSCs activation after SCI. Here, we performed injection model because injection induced less NSCs activation than SCI. On day 3 after the injection, we found that the VEGF injection group had a higher number of activated NSCs (nestin+/Ki67+) in the central canal than the control group (injection of PBS and DMSO) and inhibitors‐treated group; in addition, few nestin+ NSCs could be observed when a pVEGFR2 or pEGFR inhibitor was used together with VEGF, and injection induced a few NSCs activation in control group (Figure 6B). We then assessed the levels of pEGFR and pVEGFR2 around the injection regions. Our results showed that VEGF injection induced the phosphorylation of EGFR and VEGFR2 in the injection site (Figure 6C,D). By contrast, pVEGFR2 and pEGFR were blocked by their specific inhibitors, and the number of nestin+ NSCs was diminished in the presence of pVEGFR2 or pEGFR inhibitors (Figure 6C,D). Our results imply that VEGF activates endogenous spinal cord NSCs through VEGFR2‐EGFR signaling in vivo.
Figure 6.
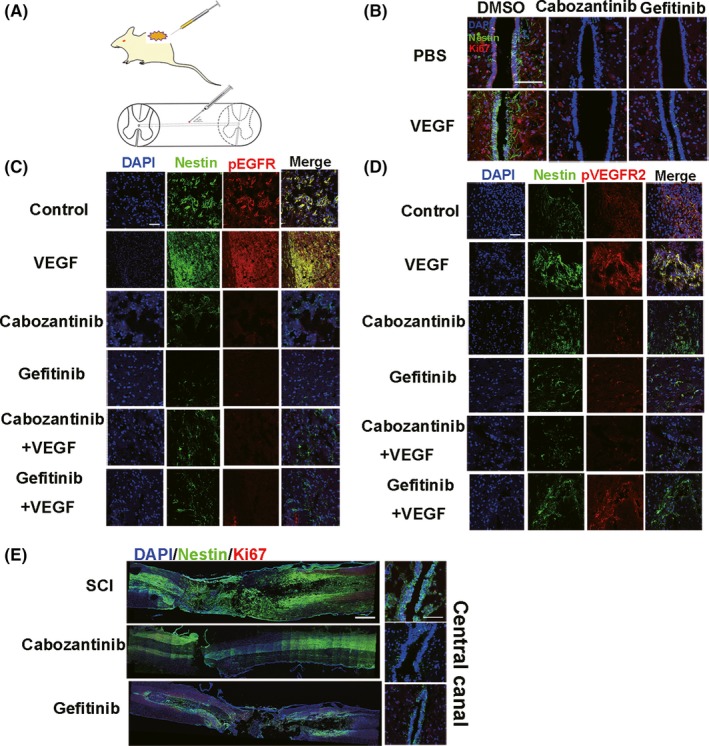
VEGF activates spinal cord NSCs through VEGFR2‐EGFR signaling on day 3 after injection in vivo (n = 5 rats/group). A, Model of the spinal cord intraspinal injection. The needle is about 25 gauge, at a 45° angle to the spinal cord (see Materials and Methods). The zigzag region indicates the injection site (T8‐T9). B, Injection of cabozantinib and gefitinib decreases the number of nestin+/Ki67+ spinal cord NSCs in the central canal. Injection of combination of PBS and DMSO as control. The results show that injection induced NSCs activation in central canal. Scale bar represents 200 µm. C, Nestin (green) and pEGFR (red) staining at the site of injection. Scale bar represents 100 µm. D, Nestin (green) and pVEGFR2 (red) staining at the site of injection. Scale bar represents 100 µm. A number of activated NSCs aggregate around the injection site (B‐D). E, Cabozantinib and gefitinib reduce nestin+/Ki67+ NSCs after SCI. Left, whole spinal section. Right, central canal; dpi, days postinjury. Nestin (green), Ki67 (red). Scale bar represents 500 µm in left and 200 µm in right panel
We demonstrated that VEGFR2‐EGFR signaling is important for the activation of spinal cord NSCs; therefore, we aimed to investigate its effects on NSCs activation after severe SCI. We found that, after severe SCI, intraspinal injection of gefitinib and cabozantinib (both at 75 mg/kg) inhibited spinal cord NSCs activation around the injection site and in the central canal, where spinal cord NSCs typically aggregated (Figure 6E). These results demonstrate that VEGFR2‐EGFR signaling is essential for the SCI‐induced activation of spinal cord NSCs.
4. DISCUSSION
The adult CNS is considered relatively static, with little cell turnover. It has been established that spinal cord NSCs can be activated in response to injury, although very little is known about the mechanisms involved in this process. Our results provided evidence of possible mechanisms of NSCs activation after injury and demonstrated that VEGF‐VEGFR2‐EGFR axis is important for SCI‐induced NSCs activation.
4.1. Driving force of spinal cord NSCs activation after injury
SCI first destructs the vascular system and rapidly generates hypoxic conditions, which favor CNS neurogenesis and pathogenesis.8 For instance, hypoxia favors radial glia expansion and induces Musashi‐1 expression, which activates endogenous NSCs in brain injury.8, 38 Importantly, VEGF can be easily induced by hypoxia and expressed in adult NSCs, and promotes NSCs proliferation through MEK/ERK‐ and PI3K/Akt‐dependent signaling in hippocampus.39 However, the roles of VEGF in spinal cord NSCs activation after SCI are not well studied. Our in vitro experiments demonstrated that VEGF maintained spinal cord NSCs proliferation and differentiation into neurons and astrocytes, although we failed to detect expression of the oligodendrocyte marker O4 under our routine differentiation protocol, because some additional essential supplements should be added to the medium to induce oligodendrocyte differentiation.40 In vivo, VEGF injection into the spinal cord activated NSCs, whereas inhibitors of VEGFR2 decreased NSCs activation after SCI.
In all, our results illustrate that hypoxia and VEGF may act as driving force of NSCs activation after injury, at least to some extent.
4.2. VEGF activates EGFR signal through VEGFR2
Our results showed VEGF activated EGFR signal through VEGFR2. Primarily we supposed that VEGF promoted the expression and secretion of EGF (epidermal growth factor) to activate EGFR. We cultured spinal cord NSCs under VEGF‐containing medium for 3‐5 days in vitro and checked the density of EGF in the supernatant by EGF ELISA. The results showed no EGF secretion by VEGF (data not shown), implying spinal cord NSCs cultured by VEGF cannot activate EGFR through EGF. Further studies such as interactions between VEGFR2 and EGFR were needed.
Previous studies have demonstrated many pathways involved in EGFR transactivation, often focusing especially on calcium signaling in neurons.20 Our results also demonstrate that calcium signaling may be involved in EGFR transactivation via VEGF. Calcium is a secondary messenger in cell signal transduction, and it might trigger other intracellular signaling cascades that are beneficial for NSCs proliferation. Further investigation is needed to explore whether other pathways are also involved in the transactivation of EGFR.
4.3. NSCs activation after SCI involves other signals and our in vivo results provide insights for mechanisms of the spinal cord NSCs activation after SCI
The presence of a small number of NSCs under gefitinib or cabozantinib in vivo indicates that other signals may also be involved in the activation of spinal cord NSCs mediated by the VEGF‐VEGFR2‐EGFR signal (Figure 6). In our results, the group that received gefitinib combined with VEGF generated a slightly higher number of NSCs than the gefitinib‐only group, indicating that VEGF‐induced activation of NSCs may not be solely dependent on the pEGFR signal. Gefitinib had very weak inhibitory effects on pVEGFR2, but cabozantinib could block pEGFR, indicating that pEGFR was a downstream effector of VEGF‐VEGFR2 signaling. Nevertheless, transactivation of EGFR might not only be controlled by VEGF, and EGFR may not be the unique stimulator of spinal cord NSCs activation after injury; other possible stimulators include EGF and FGF may contribute to NSCs activation, for example. It is worth investigating the other signals involved in activating NSCs after SCI. Discovering the mechanisms of NSCs activation after injury may help us to further understand the behavior of endogenous spinal cord NSCs.
4.4. Our results provide a possible strategy for spinal cord injury repair after SCI
Spinal cord injury is devastating owing to the loss of local axons and local glial cells. If we could induce endogenous NSCs to proliferate and differentiate into neural cells, they could perhaps contribute to improved functional recovery after SCI. Endogenous NSCs have some advantages over exogenous cell transplantation. Endogenous NSCs avoid detrimental immune responses, bypass feasibility problems of obtaining graftable cells, and circumvent ethical issues. When there is NSCs transplantation within 24 hours after an injury, a small number of the grafted cells survive owing to the inflammatory response. Our results showed expression of VEGF peaked on 3 dpi and then decreased on 7 dpi, implying prolonged VEGF expression may activate more endogenous NSCs. Furthermore, drugs‐targeted EGFR promoted SCI recovery and our results showed EGFR signal is important for NSCs activation, indicating the importance of optimal administration time of drugs. In all, our study is instructive because of manipulation of the activation of spinal cord NSCs, thus maximizing the endogenous NSCs contributions to SCI repair, and it may have therapeutic potential in the future.
5. CONCLUSION
In our study, we found spinal cord NSCs could be activated after SCI. During the process, we noticed SCI‐induced hypoxia condition which promoted VEGF expression. In vitro we demonstrated VEGF could promote spinal cord NSCs proliferation through VEGFR2‐EGFR signal. The specific inhibitors of VEGFR2 and EGFR blocked NSCs proliferation. In vivo, VEGF injection into spinal cord T8‐T9 activated more nestin+ NSCs, and inhibitors of VEGFR2 and EGFR decreased NSCs activation after SCI. Our study provides a new insight in mechanism of SCI‐induced NSCs activation and is instructive for handling endogenous NSCs which are promising on SCI therapy.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We are grateful to the members of J.D. Lab for valuable discussions. We thank Drs. Sufang Han, Yongxiang Fang, and Yi Cui for advice on animal surgery. This work was supported by grants from the Key Research Program of the Chinese Academy of Sciences (Grant No. ZDRW‐ZS‐2016‐2), National Key R&D Program of China (2016YFC1101500, 2017YFA0104700), and Youth Innovation Promotion Association CAS (2015077).
Liu S‐M, Xiao Z‐F, Li X, et al. Vascular endothelial growth factor activates neural stem cells through epidermal growth factor receptor signal after spinal cord injury. CNS Neurosci Ther. 2019;25:375–385. 10.1111/cns.13056
The first two authors contributed equally to this work.
REFERENCES
- 1. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255(5052):1707‐1710. [DOI] [PubMed] [Google Scholar]
- 2. Weiss S, Dunne C, Hewson J, et al. Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J Neurosci. 1996;16(23):7599‐7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Temple S. The development of neural stem cells. Nature. 2001;414(6859):112‐117. [DOI] [PubMed] [Google Scholar]
- 4. McKay R. Stem cells in the central nervous system. Science. 1997;276(5309):66‐71. [DOI] [PubMed] [Google Scholar]
- 5. Xu Y, Kitada M, Yamaguchi M, Dezawa M, Ide C. Increase in bFGF‐responsive neural progenitor population following contusion injury of the adult rodent spinal cord. Neurosci Lett. 2006;397(3):174‐179. [DOI] [PubMed] [Google Scholar]
- 6. Barnabe‐Heider F, Frisen J. Stem cells for spinal cord repair. Cell Stem Cell. 2008;3(1):16‐24. [DOI] [PubMed] [Google Scholar]
- 7. Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9(3):169‐181. [DOI] [PubMed] [Google Scholar]
- 8. Lange C, Turrero Garcia M, Decimo I, et al. Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. Embo J. 2016;35(9):924‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carmeliet P. Blood vessels and nerves: common signals, pathways and diseases. Nat Rev Genet. 2003;4(9):710‐720. [DOI] [PubMed] [Google Scholar]
- 10. Garcia E, Aguilar‐Cevallos J, Silva‐Garcia R, Ibarra A. Cytokine and growth factor activation in vivo and in vitro after spinal cord injury. Mediat Inflamm. 2016;2016:9476020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li YQ, James RB, Nordal RA, Su ZF, Wong CS. Hypoxia in radiation‐induced blood‐spinal cord barrier breakdown. Cancer Res. 2001;61:3348‐3354. [PubMed] [Google Scholar]
- 12. Rosenstein JM, Krum JM, Ruhrberg C. VEGF in the nervous system. Organogenesis. 2010;6(2):107‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. During MJ, Cao L. VEGF, a mediator of the effect of experience on hippocampal neurogenesis. Curr Alzheimer Res. 2006;3(1):29‐33. [DOI] [PubMed] [Google Scholar]
- 14. Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci USA. 2002;99(18):11946‐11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Segi‐Nishida E, Warner‐Schmidt JL, Duman RS. Electroconvulsive seizure and VEGF increase the proliferation of neural stem‐like cells in rat hippocampus. Proc Natl Acad Sci USA. 2008;105(32):11352‐11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kirby ED, Kuwahara AA, Messer RL, Wyss‐Coray T. Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. Proc Natl Acad Sci USA. 2015;112(13):4128‐4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oh JS, An SS, Gwak SJ, et al. Hypoxia‐specific VEGF‐expressing neural stem cells in spinal cord injury model. NeuroReport. 2012;23(3):174‐178. [DOI] [PubMed] [Google Scholar]
- 18. Chen MH, Ren QX, Yang WF, Chen XL, Lu C, Sun J. Influences of HIF‐lα on Bax/Bcl‐2 and VEGF expressions in rats with spinal cord injury. Int J Clin Exp Pathol. 2013;6(11):2312‐2322. [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood‐brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106(7):829‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koprivica V, Cho KS, Park JB, et al. EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science. 2005;310(5745):106‐110. [DOI] [PubMed] [Google Scholar]
- 21. Zwick E, Hackel P, Prenzel N, Ullrich A. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol Sci. 1999;20(10):408‐412. [DOI] [PubMed] [Google Scholar]
- 22. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G‐protein‐coupled receptors. Nature. 1996;379(6565):557‐560. [DOI] [PubMed] [Google Scholar]
- 23. Maretzky T, Evers A, Zhou W, et al. Migration of growth factor‐stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat Commun. 2011;2:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ayuso‐Sacido A, Graham C, Greenfield JP, Boockvar JA. The duality of epidermal growth factor receptor (EGFR) signaling and neural stem cell phenotype: cell enhancer or cell transformer? Curr Stem Cell Res Ther. 2006;1:387‐394. [DOI] [PubMed] [Google Scholar]
- 25. Boockvar JA, Kapitonov D, Kapoor G, et al. Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Mol Cell Neurosci. 2003;24(4):1116‐1130. [DOI] [PubMed] [Google Scholar]
- 26. Reinchisi G, Parada M, Lois P, et al. Sonic Hedgehog modulates EGFR dependent proliferation of neural stem cells during late mouse embryogenesis through EGFR transactivation. Front Cell Neurosci. 2013;7:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self‐renewal. Nature. 2010;467(7313):323‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen ZD, Xu L, Tang KK, et al. NF‐kappaB‐dependent transcriptional upregulation of cyclin D1 exerts cytoprotection against hypoxic injury upon EGFR activation. Exp Cell Res. 2016;347(1):52‐59. [DOI] [PubMed] [Google Scholar]
- 29. Joung I, Yoo M, Woo JH, Chang CY, Heo H, Kwon YK. Secretion of EGF‐like domain of heregulinbeta promotes axonal growth and functional recovery of injured sciatic nerve. Mol Cells. 2010;30(5):477‐484. [DOI] [PubMed] [Google Scholar]
- 30. Fan C, Li X, Xiao Z, et al. A modified collagen scaffold facilitates endogenous neurogenesis for acute spinal cord injury repair. Acta Biomater. 2017;51:304‐316. [DOI] [PubMed] [Google Scholar]
- 31. Milbreta U, von Boxberg Y, Mailly P, Nothias F, Soares S. Astrocytic and vascular remodeling in the injured adult rat spinal cord after chondroitinase ABC treatment. J Neurotrauma. 2014;31(9):803‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li X, Zhao Y, Cheng S, et al. Cetuximab modified collagen scaffold directs neurogenesis of injury‐activated endogenous neural stem cells for acute spinal cord injury repair. Biomaterials. 2017;137:73‐86. [DOI] [PubMed] [Google Scholar]
- 33. Madri JA. Modeling the neurovascular niche: implications for recovery fron CNS injury. J Physiol Pharmacol. 2009;60:95‐104. [PubMed] [Google Scholar]
- 34. Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisén J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96(1):25‐34. [DOI] [PubMed] [Google Scholar]
- 35. Sabelström H, Stenudd M, Frisén J. Neural stem cells in the adult spinal cord. Exp Neurol. 2014;260:44‐49. [DOI] [PubMed] [Google Scholar]
- 36. Bottai D, Fiocco R, Gelain F, et al. Neural stem cells in the adult nervous system. J Hematother Stem Cell Res. 2003;12(6):655‐670. [DOI] [PubMed] [Google Scholar]
- 37. Papavassiliou E, Gogate N, Proescholdt M, et al. Vascular endothelial growth factor (vascular permeability factor) expression in injured rat brain. J Neurosci Res. 1997;15(49):451‐460. [DOI] [PubMed] [Google Scholar]
- 38. Yagita Y, Kitagawa K, Sasaki T, et al. Differential expression of Musashi1 and nestin in the adult rat hippocampus after ischemia. J Neurosci Res. 2002;69(6):750‐756. [DOI] [PubMed] [Google Scholar]
- 39. Fournier NM, Lee B, Banasr M, Elsayed M, Duman RS. Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK‐ and PI3K/Akt‐dependent signaling. Neuropharmacology. 2012;63(4):642‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raff MC, Miller RH. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303(5916):390‐396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials