

Summary
Autophagy is an essential cellular process concern with cellular homeostasis down‐regulated by mTOR, whose activity can be modulated by rapamycin, a kind of lipophilic macrolide antibiotic, through forming a complex with immunophilin FKBP12 essential for mTOR regulation to induce autophagy. Therefore, rapamycin is normally used as a neuron protective agent. The immunophilin FKBP12 binding ligand FK506 is well known as an immunosuppressive agent by inhibiting the calcineurin expression. In this study, we synthesized a series of modified compounds based on the FKBP12 binding moiety to as same as the binding structure of rapamycin and FK506 particularly. We removed the other binding regions of the complex that has the property of immunosuppression. We found that a novel small molecule named TH2849 from these derivative compounds has a significant binding connection with mTOR by comparing to calcineurin. The effects of TH2849 on calcineurin/NFAT were not as significant as FK506, and weak effects on IL2/p34cdc2/cyclin signaling pathway were also found. Moreover, TH2849 also shows mitochondrial protective effect through stabilizing the mitochondrial structure and transmembrane potential (ΔΨm) and could rescue dopaminergic neurons in MPTP‐treated zebrafishes as well as mice models with less immunosuppressive effect. Our present study shows that TH2849 works as a neuroprotective agent possibly by inducing autophagy and low immunosuppressive effect.
Keywords: autophagy, FK506, low immunosuppression, mitochondria protection, neurodegenerative diseases, rapamycin
1. INTRODUCTION
Huntington's disease (HD) and Parkinson's disease (PD) are the groups of chronic progressive neurodegenerative diseases affecting the central nervous system (CNS) caused by accumulation of abnormal intracellular aggregation of proteins in brain.1, 2 Huntington's disease (HD) is an autosomal‐dominant neurodegenerative disorder induced by expansion of a CAG trinucleotide repeat in the huntingtin (HTT) gene (>35 repeats), which encodes an abnormal polyglutamine (polyQ) tract in the N‐terminus of the HTT protein.3 Parkinson's disease (PD) is characterized by loss of pigmental neurons and the presence of Lewy bodies due to mutants of α‐synuclein, such as A53T and A30P.4 The aggregation of proteins is positively correlated with toxicity, which could lead to neuron necrosis. Moreover, the accumulation of aggregated and toxic protein complex leads to central neurodegeneration.
It has been reported that the autophagy is a cellular process of self‐digesting5 initiating from an isolation membrane, which can engulf the cytosolic components including long‐lived or aggregate‐prone proteins, superfluous or damaged organelles, and invading pathogens, and then deliver these to lysosomes for degradation.6 Therefore, a suitable way to protect neuron from impairment is to regulate the aberrant aggregation of the protein in CNS or brain by induction of autophagy.
The target of rapamycin (mTOR) is the major inhibitory signal that can shut off autophagy in the presence of abundant nutrients, growth factors, and a major representative component of autophagy cell cycle process. Rapamycin is a ligand to immunophilin FKBP12, which has been documented for binding an immunosuppressive agent FK506 with high affinity.7 The rapamycin‐FKBP12 form a complex that directly interacts with mTOR,8 blocking its activity, which leads to autophagy induction. Previous studies have reported that rapamycin could modulate the levels of both soluble mutant huntingtin proteins and the aggregated forms in the HD model of Drosophila and mouse.9 Also, the animal studies with rapamycin treatment have been shown that the mutant form of α‐synuclein is associated with PD.10
However, though rapamycin has the potential to cure neurodegenerative diseases, its role as an immunosuppressive agent largely constrained its application of an effective neuron protective drug.11 In this study, not only we introduced a series of novel rapamycin and FK506 derivative compounds based on the FKBP12 binding moiety by imitating the binding structure of rapamycin and FK506 particularly, but also we screened a compound, TH2849, from these derivatives that can work as a neuroprotective agent probably by inducing autophagy with low immunosuppressive effect.
2. MATERIALS AND METHODS
2.1. Reagents
TH series compounds were designed by Doctor Jiahe Li and synthesized by SZ Tianhe Company (Shenzhen, China), and all the substances were dissolved in DMSO. FK506, rapamycin, MPTP, and MPP+ were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Ionomycin was purchased from Merck. Antibodies against LC3, mTOR, p‐mTOR (Ser2448), p70S6K, p‐p70S6K (Ser371), ULK1, p‐ULK1 (Ser 757), and NF‐κB were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against tyrosine hydroxylase (TH) and Tom20 were from Santa Cruz Biotechnologies (Delaware Ave Santa Cruz, CA, USA). Antibodies against the β‐actin were obtained from Abmart (Arlington, MA, USA). JC‐1, DCFH‐DA, and recombinant human IL‐2 were from Beyotime Biotechnology (Shanghai, China).
2.2. Plasmids, siRNA, and transfection
The EGFP‐LC3 plasmid was kindly provided by Professor Marja Jäättelä. EGFP‐HDQ23, EGFP‐HDQ74, and EGFP‐A53T α‐synuclein plasmids were kindly provided by Professor David C. Rubinsztein. The NFATc1 cDNA was amplified using PCR primers and cloned into the pEGFP‐C1 vectors. All plasmid constructs were confirmed by automated DNA sequencing. Cells were transiently transfected with expression plasmids and reporter vectors using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Chemically synthesized siRNAs specifically targeting FKBP12, as previously reported12, were purchased from GenePharma (GenePharma Co., Ltd., Shanghai, China).
2.3. Cell culture
U251 and PC12 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, Carlsbad, CA, USA) with 10% heat‐inactivated fetal bovine serum (FBS, Gibco) at 37°C in 5% CO2. CTLL‐2 cells were maintained in RPMI 1640 medium with 10% heat‐inactivated fetal bovine serum (FBS, Gibco) at 37°C in 5% CO2.
2.4. Autophagy assays
The cells were transfected with EGFP‐LC3 with Lipofectamine 2000 with different treatments for 24 hours, and then, the cells were fixed in 4% paraformaldehyde. The cells with fluorescent dots representing EGFP‐LC3 translocation were counted by confocal microscopy as described previously.13 Confocal microscopy was performed on an FLUO‐VIEW laser scanning confocal microscope (Olympus, FV1000, Olympus Optical, Tokyo, Japan) in sequential scanning mode using a 60× objective. LC3II levels were detected using the antibody against LC3.
2.5. Mutant huntingtin protein aggregation assay
After 48 hours posttransfection with EGFP‐HDQ74, the cells were left untreated or subjected to different treatments as indicated and then fixed in 4% paraformaldehyde. Cells with EGFP‐HDQ74 aggregation were counted by confocal microscopy as described previously.14
2.6. Clearance of mutant A53T α‐synuclein assay
The assay to measure the clearance of mutant A53Tα‐synuclein was performed as described previously.14 Briefly, cells were transfected with A53T α‐synuclein for 24 hours and then left untreated or subjected to different treatments as indicated. Clearance of soluble A53T α‐synuclein was detected using the anti‐EGFP antibody.
2.7. Immunoblotting
Immunoblotting analysis was performed as previously described. For abbreviation, cell lysates were resolved on 8%‐12% SDS‐PAGE and analyzed by immunoblotting using primary antibodies, followed by enhanced chemiluminescence (ECL) detection (Thermo Fisher Scientific, Waltham, MA, USA).
2.8. Computer modeling
The AutoDock Vina 1.1.2 was used for all dockings in this study.15 The X‐ray structures of the FKBP12‐rapamycin‐mTOR complex (PDB code: 1FAP) and FKBP12‐FK506‐calcineurin complex (PDB code: 1TCO) were chosen as receptors in docking simulations. The ligand and receptor were prepared with MGLTools 1.5.2. In general, the docking parameters for Vina were kept to their default values. The center of docking grid is located on the centroid of the corresponding ligand. The top 10 poses of each compound were reserved for the binding mode analysis. The final structures were analyzed using PyMOL.16
2.9. Zebrafish PD model and whole‐mount in situ hybridization
Zebrafish were maintained as previously described.17 Embryos were collected and dechorionated at 3‐5 hours postfertilization. Dechorionated embryos were transferred to 6‐well plates. After 48 hours, the embryos were pretreated with 1 µmol/L rapamycin, 0.1 µmol/L of FK506, 1 µmol/L 2849, and 1 µmol/L 2451 for 1 hours and then co‐treated with 30 µg/mL of MPTP for 48 hours. Embryos were fixed in 4% paraformaldehyde overnight. Digoxigenin‐labeled antisense RNA probes were generated in vitro using the zebrafish dat cDNAs as templates with RNA polymerase (Promega, Madison, WI, USA). RNA whole‐mount in situ hybridizations were performed as described by Westfield.17 After in situ hybridization, embryos were transferred into glycerol, equilibrated for 10 minutes, and then placed on a slide and flattened softly using a coverslip to disperse the neurons. The number of dat‐positive neurons in the diencephalon region was counted.
2.10. MPTP treatments in mice
The Administrative Committee on Animal Research in the Tsinghua University Shenzhen Graduate School approved all the protocols for the proposed animal experiments. The mice used in this study were 8‐week‐old male C57BL/6 mice from the Institute of Laboratory Animal Sciences, Chinese Academy of Medical Science. The mice were divided into five groups of 6 mice (saline, MPTP, MPTP/rapamycin, MPTP/FK506, MPTP/TH2849, and MPTP/TH2451) and were subjected to an acute regimen. The mice from each group received one intraperitoneal injection of MPTP‐HCL (18 mg/kg) in saline or saline every 2 hours for a total of four doses over an 8‐hours period in 1 day as described previously.18, 19 The mice received two intraperitoneal injection of rapamycin (10 mg/kg), FK506 (12 mg/kg), TH2849 (10 mg/kg), and TH2451 (10 mg/kg) in vehicle (4% ethanol, 5% Tween 80%, and 5% PEG400) every 12 hours for 2 days before the first MPTP/saline injection and continued for 4 days after the last MPTP/saline injection as described previously.18, 19 The mice were killed, and their brains were used for biochemical analysis.
2.11. Immunohistochemistry and Immunofluorescence
Immunohistochemically analysis was performed as previously described. For abbreviation, the brain frozen sections were incubated with primary antibody against tyrosine hydroxylase (TH). The biotinylated secondary antibody and avidin‐conjugated peroxidase and 3, 3 diaminobenzidine tetrachloride (DAB) substrate were used to detect the primary antibody, and hematoxylin was used as counterstain. For Immunofluorescence, the TRITC secondary antibody was used to detect the primary antibody. The total number of TH‐positive SNPC neuron on each section was counted in various groups.
2.12. Statistics analysis
Differences between groups were compared by Student's t test (*P < 0.05; **P < 0.01; ***P < 0.001). The data were presented as means ± SE.
3. RESULTS
3.1. Screening Rapamycin and FK506 derivatives potentially to regulate autophagy induction
Rapamycin and FK506 both have similarity in the ring structure with a high binding affinity to the immunophilin FKBP12. This moiety composed of a C1 ester, pipecolinyl ring, and C8 and C9 carbonyls (FKBP12 binding moiety).20 After successful synthesis of this moiety structure, we renamed it as TH2451 as shown in Figure 1A. We modified TH2451 based on their rudimentary structure by a series of ramification. Autophagy is an essential cellular process concern to maintain the cellular fluid balance and eliminate the aggregate‐prone and toxic within the cell to play a key role in the various stages of tumorigenesis.21 Therefore, to examine whether these derivatives have the ability to induce autophagy, we investigated the autophagosome (LC3‐positive vesicle) formation after treatment with the rapamycin, FK506, and TH compounds for 24 hours. We found that the substantial autophagosomes were induced with the treatment of rapamycin, TH2451, TH2849, TH2287, and TH3263 in the U251 and PC12 cells (Figure 1B,C). Furthermore, we examined the protein levels of p62, a substrate of autophagy, and LC3II, which is produced by the cleavage of the C‐terminal LC3 proteins when autophagy initiates.22 Our result suggests that treating U251 cells with rapamycin, TH2849, and TH2287 could dramatically decrease the level of p62 protein but increase the level of LC3‐II protein (Figure 1D–F). Altogether, our results implied that TH2849 and TH2287 derivatives may have a strong effect in the regulation of autophagy as same as rapamycin.
Figure 1.
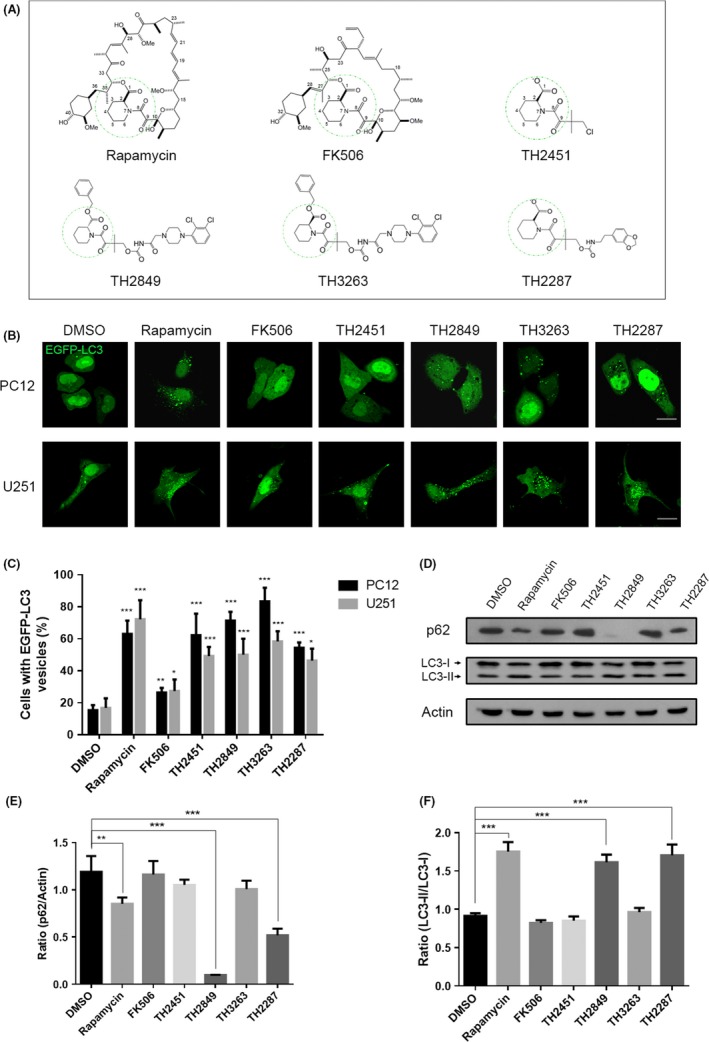
1 Screening potentially autophagy induction of TH compounds. (A) Structures of rapamycin, FK506, and TH compounds (TH2451, TH 2849, TH3263, and TH2287). Representative images (B) and quantification (C) of PC12 and U251 cells with EGFP‐LC3 vesicles (autophagosomes). The EGFP‐LC3 transfected cells were treated with 1‰ DMSO, 1 μmol/L rapamycin, 10 μmol/L FK506, 1 μmol/L TH compounds for 24 h as indicated. (D) Representative Western blotting image of p62 and LC3 I/II levels after 1‰ DMSO, 1 μmol/L rapamycin, 10 μmol/L FK506, 1 μmol/L TH compounds treatment for 24 h. Quantification of relative p62 (E) and LC3 I/II (F) expression levels in (D). All the substances were dissolved in DMSO. Scale bars: 10 μm. Data are shown as the mean ± SEM *P < 0.05, **P < 0.01, ***P < 0.001 compared to DMSO control, n = 3
3.2. TH derivative compounds modulate the protein aggregate‐prone and toxic effects in vitro
Next, we examined to assess the elimination efficacy of abnormal protein aggregation by TH compounds. The huntingtin exon 1 with the 74Q mutation (HDQ74) and A53T α‐synuclein (a mutant of α‐synuclein) were used to simulate HD and PD.14, 23, 24 EGFP‐HDQ74 transfected U251 cells were used to simulate HD, and A53T α‐synuclein transfected PC12 cells were applied to develop PD. Our results showed that by comparing EGFP‐HDQ23 with normal repeats of CAG (repeats of CAG <35), EGFP‐HDQ74 with the abnormal repeats of CAG (repeats of CAG >35) developed the aggregated proteins within U251 cells causing cell death. Meanwhile, we found that treatment of the EGFP‐HDQ74 transfected cells with rapamycin, FK506, TH2451, TH2849, TH2287, and TH3263 all could considerably diminish the number of cells containing aggregated EGFP‐HDQ74 proteins (Figure 2A). Similarly, in the cell model of PD, we investigated the effect of rapamycin, FK506, and TH compounds on decreasing the protein levels of the A53T α‐synuclein (Figure 2B,C). These results suggested that TH2451, TH2849, TH2287, and TH3263 all could promote the clearance of aggregate‐prone and toxic proteins in HD and PD in vitro.
Figure 2.
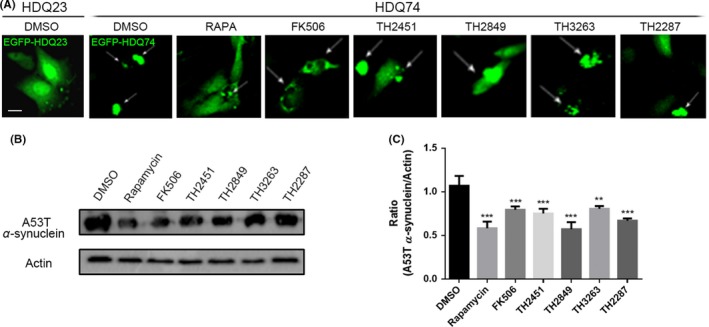
Screening potentially autophagy induction of TH compounds. (A) Representative images of EGFP‐HDQ74 aggregate‐containing U251 cells (white arrows). The EGFP‐HDQ74 transfected cells were treated with 1‰ DMSO, 1 μmol/L rapamycin, 10 μmol/L FK506, 1 μmol/L TH compounds for 48 h as indicated. Representative Western blotting image (B) and quantification (C) of A53T α‐synuclein in EGFP‐A53T α‐synuclein transfected PC12 cells with the same treatment in (A). All the substances were dissolved in DMSO. Scale bars: 10 μm. Data are shown as the mean ± SEM *P < 0.05, **P < 0.01, ***P < 0.001 compared to DMSO control, n = 3
3.3. Computer model simulates different TH compounds binding with FKBP12, mTOR, and calcineurin
Immunophilin FKBP12 is a receptor that can be bound by rapamycin and FK506 with significantly triggering a downstream reaction. However, TH compounds binding to moiety structure to form a complex with FKBP12 still need confirmation. Therefore, computer modeling of the binding of the TH compounds with FKBP12 was then carried out. As shown in Figure 3, all TH compounds, rapamycin, and FK506 shared the same binding site with FKBP12. Rapamycin‐FKBP12 and FK506‐FKBP12 complexes have been reported to bind to mTOR to trigger autophagy, and calcineurin to cause immunosuppression, respectively.25, 26 Therefore, we have shown the binding models of TH2849 and TH2287 complex with mTOR or calcineurin of each in the presence of TH2451 (TH2451 was marked to fix the main structure, which keeps the same binding mode with FKBP12 whenever co‐binding to mTOR or to calcineurin) (Figure 3). We found that both TH2849 and TH2287 use the basic structure of TH2451 to bind FKBP12. Meanwhile, the two phenyl ring of TH2849 can form the π‐π interaction with the residue Y194 of mTOR compared with only one ring of TH2287, in which situation we can infer that the interaction of TH2849‐mTOR complex is more significant than the TH2287 with mTOR. As for the binding models with calcineurin of TH2287 and TH2849, the steric excursion of the F356 and W352 residues of calcineurin made the two phenyl ring of TH2849 dogged, which could obstruct the binding between TH2849 and calcineurin. However, the binding model of calcineurin with TH2287 remains elusive, and the distance is good for a hydrophobic interaction which may stabilize the complex (Figure 3). Furthermore, we found that TH2287 shows a strong binding combination to calcineurin as compared to TH2849.
Figure 3.
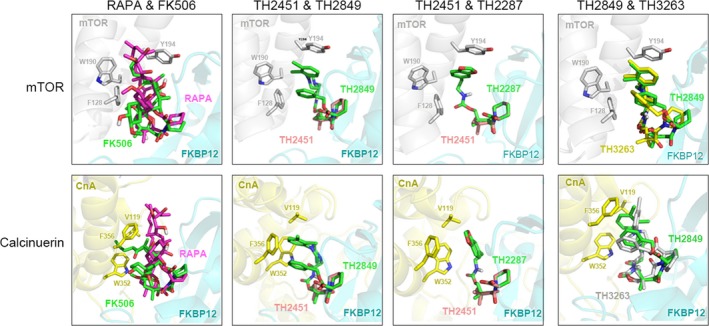
Computer modeling of the molecular models of the complexes of rapamycin (RAPA), FK506, TH compounds and FKBP12 binding with mTOR and calcineurin, respectively
Next, we compared the difference in the binding models with mTOR or calcineurin between TH2849 and TH3263, which share similar structure but different chiral carbon atom. These results demonstrated that TH3263 analogous to TH2849 can bind to both FKBP12‐mTOR and FKBP12‐calcineurin complexes (Figure 3). However, when we analyzed the binding model of TH3263 with FKBP12‐mTOR complex, we found that the TH3263 displayed the similar binding mode of action with TH2849, displayed a π‐π interaction with the residue Y194 of mTOR, and TH3263 failed to match the structure of TH2451 to bind to FKBP12 perfectly. Moreover, in the model of FKBP12‐calcineurin complex, TH3263 seems to have a broader space than TH2849 to spread its phenyl ring and make interactions with W352 and F356 of calcineurin (Figure 3). So we found a better interaction of TH3263 with calcineurin rather TH2849. These data suggested that the TH2849‐FKBP12 complex rather than TH2287‐FKBP12 or TH3263‐FKBP12 complex has the best structure to effectively bind mTOR, but the most inappropriate steric configuration to interact with calcineurin.
3.4. TH2849 may induce autophagy through inhibition of mTOR activation and activation of ULK1
The target of rapamycin (mTOR)regulation is essential for autophagy induction through suppressing the ULK1 complex.27 During starvation or growth factor withdrawal, mTOR is inactivated, and autophagy initiates. The previous studies have been reported that the rapamycin can modulate the activation of mTOR and induce autophagy.28 Moreover, to investigate the effect of TH compounds on mTOR, we detected the alteration of p70S6K with the treatment of different TH compounds in PC12 cells. The p70S6K is well‐known substrate for mTOR and remains phosphorylated by mTOR under nutrient‐rich condition. During the mTOR inhibition by well‐known antagonist including rapamycin, p70S6K dephosphorylates. Next, we incubated the PC12 cells with rapamycin, FK506, TH2451, TH2849, TH2287, and TH3263 under normal culture condition, and rapamycin, TH2451, and TH2849 showed a strong inhibitory effect on the phosphorylation of p70S6K, by comparing with FK506, TH2287, and TH3263 (Figure 4A,B).
Figure 4.
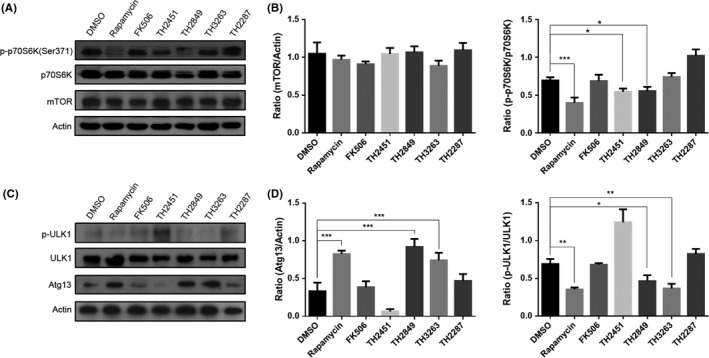
TH compounds possibly induce autophagy through inhibiting mTOR and activating ULK1. Representative Western blotting image of mTOR, p70S6K, p‐p70S6K (Ser371) (A) and ULK1, p‐ULK1 (Ser 757), Atg13 (C). MCF‐7 cells were treated with 1‰ DMSO, 1 μmol/L rapamycin, 10 μmol/L FK506, 1 μmol/L TH compounds for 24 h as indicated. Quantification of relative mTOR, p70S6K, p‐p70S6K (B) expression levels in (A) and ULK1, p‐ULK1 (Ser 757), Atg13 (D) expression levels in (B). All the substances were dissolved in DMSO. Scale bars: 10 μm. Data are shown as the mean ± SEM *P < 0.05, **P < 0.01, ***P < 0.001 compared to DMSO control, n = 3
Previous studies27 have shown that ULK1 enhances the mTOR activity. When under nutrient condition, ULK1 is phosphorylated by mTOR, which leads the dissociation of ULK1 with Atg13.27 However, during starvation or stimulation with mTOR inhibitor, ULK1 is dephosphorylated and forms a complex with Atg13, FIP200, and ATG101, which initiates the autophagy.29 Therefore, we observed the phosphorylation state of ULK1 and ATG13 in PC12 cells treated with rapamycin, FK506, and different TH compounds. As a result, rapamycin, TH2849, and TH3263 potently inhibited the phosphorylation of ULK1 (Ser 757) and Atg13, by comparing with FK506, TH2287, and TH3263 (Figure 4C,D). These data suggested that TH2451 and TH2849 potentially induce autophagy most likely due to their suppression of the activation of mTOR, which subsequently dephosphorylates ULK1 and results in ULK1 complex formation.
3.5. TH2849 induces autophagy by forming a complex with FKBP12 without interfering to the calcineurin/NFAT and IL2/p34cdc2/cyclin A signal pathway
According to the prediction of computer modeling, TH2849 has a binding ability to FKBP12. Therefore, we conducted biological experiment to confirm it. By knocking down FKBP12 with siRNA, we found that the autophagy failed to be activated though with treatment of rapamycin, TH2451, and TH2849 in PC12 cells (Figure 5A–C). These data demonstrated that TH2849 and TH2451 may induce autophagy through binding FKBP12.
Figure 5.
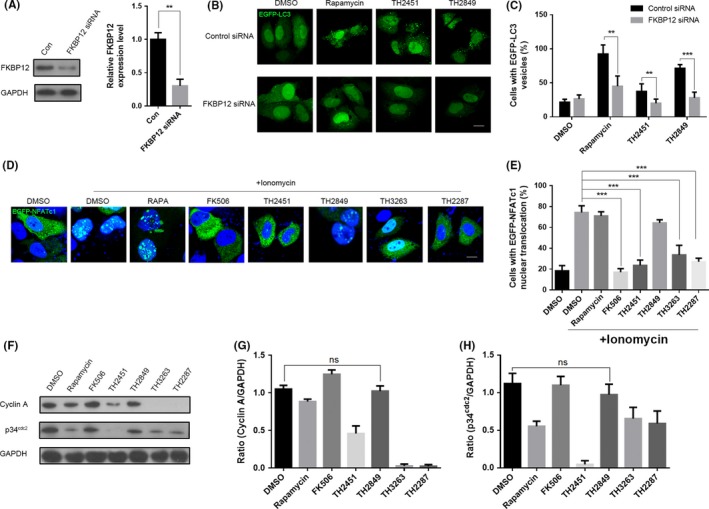
TH2849 potentially induces autophagy by forming a complex with FKBP12 without interfering to the calcineurin/NFAT and IL2/p34cdc2/cyclin A signal pathway. (A) PC12 cells were pretreated with siRNA against FKBP12 for 48 h, and then, the protein and mRNA levels were examined. Representative images (B) and quantification (C) of PC12 cells with EGFP‐LC3 vesicles (autophagosomes) in the presence or absence of siRNA against FKBP12. PC12 cells co‐transfected with siRNA against FKBP12 and EGFP‐LC3 were treated with 1‰ DMSO, 1 μmol/L rapamycin, 10 μmol/L FK506, 1 μmol/L TH 2451 and 1 μmol/L TH 2849 for 24 h. (D) Representative images of distribution of EGFP‐NFATc1. The EGFP‐NFATc1 transfected MCF‐7 cells were treated with 2 μmol/L ionomycin or co‐treated with 1‰ DMSO, 1 μmol/L rapamycin, 1 μmol/L FK506, TH2451, TH 2849, TH3263 and TH2287 for 2 h as indicated. (E) Quantification of nucleus translocated EGFP‐NFATc1 cells in (D). (F) Representative Western blotting image of p34cdc2 and cyclin A levels. Factor‐deprived CTLL‐2 cells were cultured for 14 h in basic medium only, 50 units/mL IL‐2 with 1‰ DMSO, 1 μmol/L rapamycin, 1 μmol/L FK506, TH2451, TH 2849, TH3263, and TH2287 were added respectively during the last 1 hour. Quantification of relative p34cdc2 (G) and cyclin A (H) expression levels in (F). All the substances were dissolved in DMSO. Scale bars: 10 μm. Data are shown as the mean ± SEM *P < 0.05, **P < 0.01, ***P < 0.001 compared to DMSO control, n = 3.
Meanwhile, due to the nonspecific interaction between TH2849 and calcineurin calculated by the bioinformatics model, we next investigated the role of TH2849 on calcineurin/NFAT signal pathway which is essential for immune system activation. Calcineurin is a calcium/calmodulin‐dependent serine‐threonine protein phosphatase.30 Previous studies have been reported that the calcineurin activation cause by the high level of cytoplasmic calcium which leads to the transcription factor of activated T cell (NFATc) dephosphorylated and translocation into the nucleus. This will up‐regulate the transcription of interleukin genes such as IL‐2, IL‐4, and IL‐6, which play vital role in the stimulation of T‐helper lymphocytes and other cytokines.31 The FK506‐FKBP12 complex inhibits the calcineurin; therefore, FK506 has been broadly applied as an immunosuppressive drug.32 We managed to detect the nuclear translocation of NFATc1 to value the contribution of TH2849 in suppressing calcineurin/NFAT signal pathway. After incubation with different compounds and stimulation with ionomycin in the EGFP‐NFATc1 transfected CTLL‐2 cells, the confocal images showed that the nucleus translocation of NFATc1 was not significantly blocked by rapamycin and TH2849 comparing with FK506, TH2451, TH3263, and TH2287 (Figure 5D,E).
It has been reported that rapamycin works as a immunosuppressive mainly counting on this IL2/p34cdc2/cyclin A signal pathway drives stimulated T cells through a late G1‐phase into S‐phase.33, 34, 35 During late G1‐phase, two cyclin‐dependent kinases, p34cdc2 and p33cdk2, participate in the sequence of events contributing to S‐phase commitment and then help to activate different cyclins such as cyclin A, cyclin D, and cyclin E.36, 37 To testify whether our TH compounds have the same immunosuppressive mechanism, we determined the protein level of p34cdc2 and cyclin A with different TH compound treatment in CTLL‐2 cells under IL‐2 stimulation. We found that the expression of both p34cdc2 and cyclin A reduced with the incubation of rapamycin, TH2451, TH3263, and TH2287. Conversely, cells treated with FK506 and TH2849 showed little changes in the expression of both p34cdc2 and cyclin A (Figure 5F,G,H). Altogether, these results implied that TH2849 has shown significantly trigger autophagy through binding FKBP12, unable to suppress normal immune function, and showed lower inhibitory effects on IL2/p34cdc2/cyclin A and calcineurin/NFAT signal pathways, which are the major cause of immunosuppression to rapamycin and FK506, respectively.
3.6. TH2849 could ameliorate mitochondrial damage caused by MPP+ in vitro
Mitochondrial damage plays a key role in the development of neurodegenerative diseases, including PD and HD.3 Neurotoxins, such as 1, 2, 3, 6‐tetrahydropyridine (MPTP) which is converted into MPP+ by monoamine oxidase B to cause mitochondrial damage in dopaminergic neurons, have been used to achieve PD‐like syndrome.18, 38 Next, we examined the TH2849 protective mechanism to relieve PD from fragmentation; firstly, we used the antibody against mitochondrial outer membrane protein Tom 20 to observe mitochondrial fragmentation. When U251 cells are treated with MPP+ for 24 hours, by confocal study we found that co‐treated U251 cells with rapamycin, TH2849, and TH2451 significantly modulated the Tom 20 by mitochondrial fragmentation (Figure 6A). Further, we examined the mitochondrial transmembrane potential (ΔΨm) by JC‐1. We found that co‐incubation of U251 cells with rapamycin or TH2849 could effectively sustain the normal mitochondrial transmembrane potential (ΔΨm) even in the presence of MPP+ (Figure 6B). Similarly, ROS level and the activation level of nuclear translocation of NF‐κB were also maintained in common condition in TH2849 or TH2451 treatment group when MPP+ existed (Figure 6C,D). These data suggested that TH2849 could reduce the mitochondrial damage caused by MPP+ in vitro through stabilizing the mitochondrial structure and transmembrane potential (ΔΨm), which indicated its possible role in PD treatment.
Figure 6.
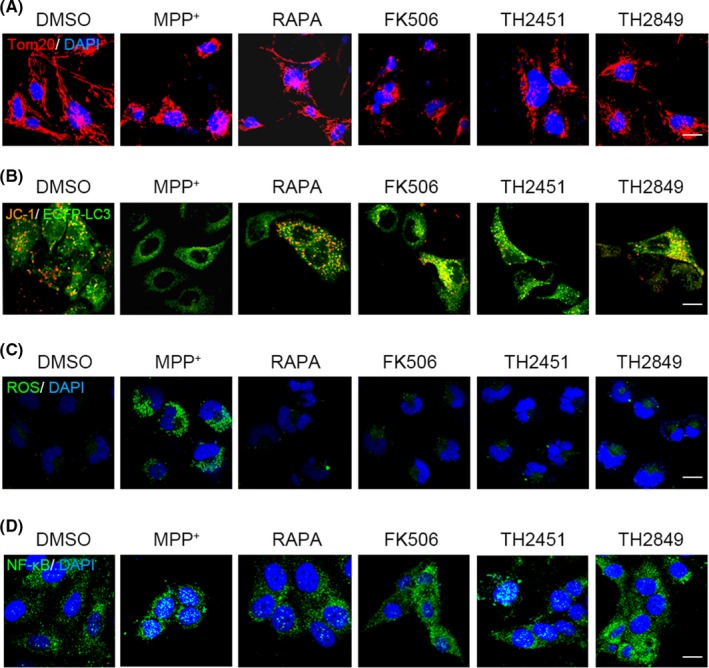
TH2849 protects mitochondrial from damage induced by MPP+. Representative confocal image of MDA‐MB‐231 cells incubated with treated with 1‰ DMSO, 10 μmol/L MPP+ alone, or co‐treated with 1 μmol/L rapamycin, 10 μmol/L FK506, 1 μmol/L TH 2451, and 1 μmol/L TH 2849 for 24 h, and then, the immunofluorescence experiment was performed with the primary antibody against (A) Tom20 and (D) NF‐Κb, or the fluorogenic probe DCFH‑DA was used to detect the ROS levels (C). B, EGFP‐LC3 transfected PC12 cells were treated with 10 μmol/L MPP+ alone or co‐treated with 1‰ DMSO, 1 μmol/L rapamycin, 10 μmol/L FK506, 1 μmol/L TH 2451, and 1 μmol/L TH 2849 for 24 h. Mitochondrial transmembrane potential (ΔΨm) was detected by JC‐1. All the substances were dissolved in DMSO. Scale bars: 10 μm. n = 3
3.7. TH2849 shows protective mechanism in MPTP‐induced PD model
To investigate the TH2849 neuron protective effect in vivo, we developed the acute injury PD models in zebrafishes and mice by MPTP. When the zebrafishes or mice were treated with MPTP, the significantly dopaminergic neurons loss was found (Figure 7A,B). Meanwhile, we also found that TH2849 could rescue dopaminergic neurons in MPTP‐treated zebrafishes and mice. The number of dopaminergic neurons (TH‐positive cells) in the substantia nigra pars compacta (SNPC) of MPTP‐treated zebrafishes and mice significantly increased in rapamycin, FK506, and TH2849 treatment groups (Figure 7A,B). Interestingly, although treating MPTP‐induced PD model mice with either rapamycin or FK506 or TH2849 all recovered the damaging dopaminergic neurons, we did not tested the suppression of IL‐2 and the IL‐6 levels in mice treated with TH2849, which had been shown that there was no immunosuppression effect induced by TH2849 compared with FK506 and rapamycin (Figure 7C,D). These results demonstrated that TH2849 could improve the neurodegenerative disease pathogenesis condition in MPTP‐treated zebrafishes and mice models through protection of dopaminergic neurons, which cannot inhibit normal immune system function in the meantime.
Figure 7.
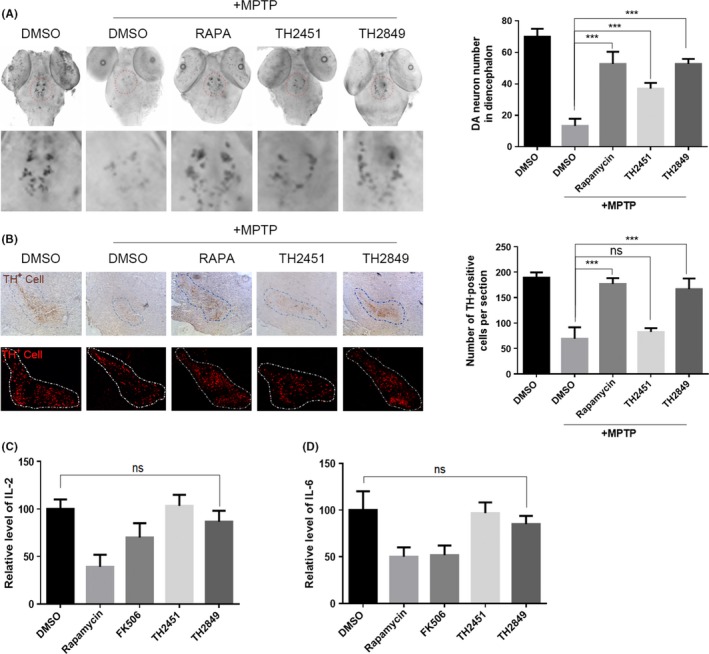
TH2849 rescues MPTP‐induced dopaminergic neuronal death in zebrafish and mouse PD model. A, Representative images and quantification of dopaminergic neurons in zebrafish detected with dat probe. RNA whole‐mount in situ hybridization of three dpf embryos pretreated with 1‰ DMSO, 1 μmol/L rapamycin, 1 μmol/L TH2849, and 1 μmol/L TH2451, respectively for 5 h and then co‐treated with 30 μg/mL MPTP incubated for 48 h. B, Representative images and quantification of dopaminergic neurons in mice detected with TH antibody. For acute regimen treatment, the mice from each group received one intraperitoneal injection of MPTP‐HCl (18 mg/kg) in saline, or saline every 2 h for a total of four doses over an 8‐h period in 1 d. For protection treatment, the mice received two intraperitoneal injection of 1‰ DMSO (10 mg/kg), rapamycin (10 mg/kg), FK506 (12 mg/kg), TH2849 (10 mg/kg), and TH2451 (10 mg/kg) every 12 h for 2 d before the first MPTP/DMSO injection and continued for 4 d after the last MPTP/DMSO injection. The mice received two intraperitoneal injection of 1‰ DMSO (10 mg/kg), rapamycin (10 mg/kg), FK506 (12 mg/kg), TH2849 (10 mg/kg), and TH2451 (10 mg/kg) every 12 h for 6 d, and then, the blood serum was collected and used to detect the IL‐2 (C) and IL‐6 (D). All the substances were dissolved in DMSO. Scale bars: 10 μm. Data are shown as the mean ± SEM *P < 0.05, **P < 0.01, ***P < 0.001 compared to DMSO control, n = 5
4. DISCUSSION
Previous studies have shown that the clearance of mutant huntingtin proteins (HDQ74) and mutant α‐synuclein (A53T α‐synuclein) is largely mediated by autophagy.14 Up‐regulation of autophagy is a capable way to cure neurodegenerative diseases by inhibiting the mTOR; especially, rapamycin could efficiently up‐regulate autophagy and eliminate both of the levels of soluble and the aggregated mutant huntingtin protein in Drosophila and mouse models of HD.9 The treatment of mice expressing the mutant form of α‐synuclein associated with PD with rapamycin also could regulate the neuronal toxicity.10 Although, up‐regulation of autophagy through mTOR‐dependent pathway is a promising strategy for the treatment of neurodegenerative diseases; however, rapamycin might not be suitable for a long‐term use in human body11 because mTOR is the most important signal pathway in controlling the cell growth, which regulates the ribosome biogenesis, protein translation. Moreover, rapamycin is widely used as a immunosuppressive agent, which significantly down‐regulates IL2/p34cdc2/cyclin A signal pathway, impairs wound healing, and displays immunosuppressive effects .8, 11, 39
It has been reported that the rapamycin inhibits mTOR through binding FKBP12. FK506 also can bind FKBP12 but has very different downstream mechanism, which is the inhibitor of calcineurin/NFAT signal pathway rather than mTOR pathway.32 Calcineurin is a calcium/calmodulin‐dependent serine‐threonine protein phosphatase. Whenever antigen‐presenting cells interact with the receptor on T cells, calcium level will rise in the both cells mentioned above, which will continuously trigger calcineurin through activating calmodulin and the regulatory subunit binding to the catalytic subunit,30 eliciting nuclear factor of activated T cell (NFATc) translocating into the nucleus by dephosphorylating NFATc, where it up‐regulates the transcription of interleukin genes such as IL‐2, IL‐4, and IL‐6. In this study, we found that FK506 shows significant effects on neuron to protect from degradation. We also found that FK506 works as an immunosuppressive agent due to suppressing the calcineurin/NFAT signal pathway, which supports the published data.39
Our results showed that the rapamycin and FK506 all have high affinity to bind FKBP12 on account of sharing a similar large ring structure, which consists of the C1 ester, the pipecolinyl ring, and the C8 and C9 carbonyls. We synthesized a rapamycin and FK506 renamed as TH compounds, and then, we screened out four TH compounds (TH2451, TH2849, TH2287, and TH3263) from these derivatives. Next, we examined and found that the derivative potentially possessed the rapamycin‐like autophagy induction as well as the clearance ability of aggregate‐prone proteins such as HDQ74 and A53T α‐synuclein proteins. Furthermore, having identified TH compounds potentially with the rapamycin‐like autophagy property, we listed the computer binding models of these four selected TH compounds (TH2451, TH2849, TH2287, and TH3263) with mTOR or calcineurin that have same binding site with FKBP12 like rapamycin and FK506. The TH derivatives had the strongest binding force with mTOR due to the π‐π interaction with the residue Y194 of mTOR from two phenyl rings of its structure, but the steric excursion of the F356 and W352 residues of calcineurin obstructs the binding between TH2849 and calcineurin. That indicated TH2849 has the most appropriate steric configuration to activate rapamycin‐like autophagy without the immunosuppression effect of FK506.
Next, we found byknocking down FKBP12 with siRNA, the autophagy induced by rapamycin, TH2451, and TH2849 was modulated, which testified that rapamycin, TH2451, and TH2849 really function as autophagy inducers by binding FKBP12. In addition, the TH‐mTOR forming complex has an effect on mTOR‐associated autophagy pathway. Previous studies have shown that autophagy can be suppressed through phosphorylation ULK1 and Atg13, which blocks the formation of the ULK1 complex.27, 40, 41, 42 Our results of immunoblotting of two mTOR substrates P70S6K and ULK1, TH2451, and TH2849 could effectively induce autophagy by the activation of mTOR which subsequently dephosphorylates ULK1 and results in ULK1 complex formation.
In terms ofthe immunosuppression effect of these TH compounds, as predicted by the computer modeling, the immunofluorescence result showed that TH2849 did not have any inhibition function on nuclear translocation of NFATc1, which means it would not impact calcineurin/NFAT signal pathway compared with FK506. Meanwhile, we continued to investigate the effect of TH2849 on IL2/p34cdc2/cyclin A signal pathway, which is the key mechanism why rapamycin can be applied as an immunosuppressive agent.33, 34, 35 Normally, the stimulation of interleukin‐2 (IL‐2) results in T cells through a late G1‐phase into S‐phase.43 Two cyclin‐dependent kinases, p34cdc2 and p33cdk2, help to activate different cyclins such as cyclin A, cyclin D, and cyclin E.36, 37 However, unlike rapamycin, we did not found a significant effect of TH2849 on the IL2/p34cdc2/cyclin A signal pathway. But the reason accounting for this remains elusive.
Mitochondrial is a critical organelle for regulation of cell death and damaging of mitochondrial caused by mutations of mitochondrial DNA and oxidative stress leading to disease pathogenesis.3 The genes leading to PD pathogenesis also implicate associated with mitochondria mutated gene include α‐synuclein, parkin, DJ‐1, PINK1, LRRK1, and HTRA2 directly or indirectly affect mitochondrial. By treatment of the cells with MPP+, a series of impacts, such as the mitochondrial damage and fragmentation, loss of membrane potential, and production of ROS, can be induced. Our data showed that TH2849 could efficiently protect the cells from the mitochondrial damage induced by MPP+ in vitro.
In vivo study had also been carried out in this manuscript based on MPTP‐induced zebrafishes and mice PD models. The loss of dopaminergic neurons in brain is the major cause of PD. Our data showed that TH2849 could protect against dopaminergic neuron death both in MPTP‐induced zebrafishes and in mice models. TH2849 could rescue the loss of dopaminergic neurons apparently in MPTP‐induced zebrafishes and mice models. Our results indicated that TH2849 might inhibit neurodegenerative disease pathogenesis probably through autophagy induction and removal of toxic proteins and damaged mitochondrial in vivo.
But we also notice that the interaction between TH2849 and mTOR is not as specific as that between rapamycin and mTOR. After all, rapamycin has the strongest effect on eliciting autophagy and neuroprotection in our experiments. Some researchers argue that neuroprotection is associated with immunophilins other than FKBP12, notably FKBP52 and FKBP38,44, 45 which we guess may be the potential reason why our TH compounds cannot reach the perfect neuroprotection performance. Furthermore, from the literature we found other structure‐based rapamycin derivative design approach, such as ILS‐920 and WYE‐592, two semi‐synthetic analogs of rapamycin that were also nonimmunosuppressive.46 These two analogs showed unanticipated selectivity for FKBP52 (over 200‐fold selectivity for FKBP52 versus FKBP12); thus, they owned better neuron protection effect compared with rapamycin. But it should also be noticed that these two compounds were only reported to be effective on ischemic stroke and that little is known about its therapeutic effect on several typical neurodegenerative diseases, such as PD, AD, and HD.
5. CONCLUSIONS
In this study, we screened out a compound TH2849 which could retain the neuron protective effect of rapamycin but without serious immunosuppression. TH2849 can interact with FKBP12 similar to rapamycin and FK506 by comparing all synthesized TH compounds; TH2849 owns the most obvious effect on the clearance of aggregate‐prone and toxic proteins due to potentially rapamycin‐like autophagy property. This autophagy activation resulted from the formation of TH2849‐FKBP12 complex, which could bind mTOR effectively to trigger mTOR‐associated autophagy cascade reaction. Meanwhile, due to its special steric structure, TH2849 does not strongly inhibit neither calcineurin/NFAT nor IL2/p34cdc2/cyclin A signal pathways, which implied its less side effect on immunosuppression. Furthermore, in vitro models of MPP+‐induced PD showed that TH2849 could rescue cell death through relieving mitochondrial damage, and in vivo results of MPTP‐induced zebrafishes and mice PD models demonstrated that the loss of dopaminergic neurons in brain could be stopped by TH2849 with low immunosuppressive effect (Figure 8).
Figure 8.
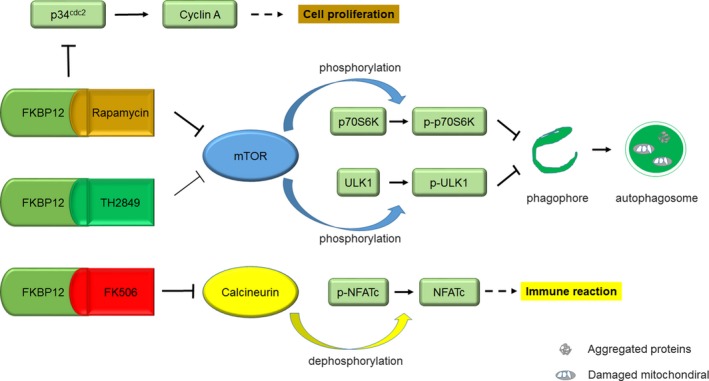
Model of potentially autophagy induction and neuron protection but less immunosuppression by TH2849
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the grants Shenzhen Science and Technology Program, JCYJ20170412153453623, JCYJ20130402145002433.
Ding L, Nan W‐H, Zhu X‐B, et al. Rapamycin and FK506 derivative TH2849 could ameliorate neurodegenerative diseases through autophagy with low immunosuppressive effect. CNS Neurosci Ther. 2019;25:452–464. 10.1111/cns.13062
Ding and Nan are contributed equally to this work.
Contributor Information
Han‐Bing Zhong, Email: shixj@sz.tsinghua.edu.cn, Email: zhonghb@sustc.edu.cn.
Xiao‐Jun Shi, Email: shixj@sz.tsinghua.edu.cn, Email: zhonghb@sustc.edu.cn.
REFERENCES
- 1. Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4(1):49‐60. [DOI] [PubMed] [Google Scholar]
- 2. Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis. 2011;43(1):17‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787‐795. [DOI] [PubMed] [Google Scholar]
- 4. Goedert M. Alpha‐synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2(7):492‐501. [DOI] [PubMed] [Google Scholar]
- 5. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728‐741. [DOI] [PubMed] [Google Scholar]
- 7. Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12‐rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;239–41. [DOI] [PubMed] [Google Scholar]
- 8. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926‐1945. [DOI] [PubMed] [Google Scholar]
- 9. Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate‐prone proteins. Hum Mol Genet. 2005;15(3):433‐442. [DOI] [PubMed] [Google Scholar]
- 10. Malagelada C, Jin ZH, Jackson‐Lewis V, Przedborski S, Greene LA. Rapamycin Protects against Neuron Death in In Vitro and In Vivo Models of Parkinson's Disease. J Neurosci. 2010;30(3):1166‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8(2):108‐117. [DOI] [PubMed] [Google Scholar]
- 12. Romano S, D'Angelillo A, Pacelli R, Staibano S, De Luna E, Bisogni R, et al. Role of FK506‐binding protein 51 in the control of apoptosis of irradiated melanoma cells. Cell Death Differ. 2010;17(1):145‐157. [DOI] [PubMed] [Google Scholar]
- 13. Hoyer‐Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin‐dependent kinase kinase‐beta, and Bcl‐2. Mol Cell. 2007;25(2):193‐205. [DOI] [PubMed] [Google Scholar]
- 14. Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR‐independent autophagy pathway. Nat Chem Biol. 2008;4(5):295‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeLano W. The PyMOL Molecular Graphics System. DeLano Scientific. 2008. [Google Scholar]
- 17. Ren G, Xin S, Li S, Zhong H, Lin S. Disruption of LRRK2 does not cause specific loss of dopaminergic neurons in zebrafish. PloS One. 2011;6(6):e20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liberatore GT, Jackson‐Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5(12):1403‐1409. [DOI] [PubMed] [Google Scholar]
- 19. Wu DC, Jackson‐Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22(5):1763‐1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP‐12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229(1):105‐124. [DOI] [PubMed] [Google Scholar]
- 21. Song X, Yuan Z, Yuan H, Wang L, Ji P, Jin G, et al. ATG12 expression quantitative trait loci associated with head and neck squamous cell carcinoma risk in a Chinese Han population. Mol Carcinog. 2018. [DOI] [PubMed] [Google Scholar]
- 22. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rubinsztein DC. Lessons from animal models of Huntington's disease. Trends Genet. 2002;18(4):202‐209. [DOI] [PubMed] [Google Scholar]
- 24. Ostrerova‐Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T α‐synuclein mutation increases iron‐dependent aggregation and toxicity. J Neurosci. 2000;20(16):6048‐6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70‐kDa S6 kinase and is activated by inhibition of FKBP12–rapamycin associated protein. Proc Natl Acad Sci U S A. 1999;96(8):4438‐4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, et al. X‐ray structure of calcineurin inhibited by the immunophilin‐immunosuppressant FKBP12‐FK506 complex. Cell. 1995;82(3):507‐522. [DOI] [PubMed] [Google Scholar]
- 27. Kim J, Kundu M, Viollet B, Guan K‐L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585‐595. [DOI] [PubMed] [Google Scholar]
- 29. Ganley IG, Lam dH, Wang J, Ding X, Chen S, Jiang X. ULK1· ATG13· FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297‐12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80(4):1483‐1521. [DOI] [PubMed] [Google Scholar]
- 31. Stern JB, Smith KA. Interleukin‐2 induction of T‐cell G1 progression and c‐myb expression. Science. 1986;233:203‐207. [DOI] [PubMed] [Google Scholar]
- 32. Mukai H, Kuno T, Chang C‐D, Lane B, Luly JR, Tanaka C. FKBP12‐FK506 Complex Inhibits Phosphatase Activity of Two Mammalian Isoforms of Calcineurin Irrespective of Their Substrates or Activation Mechanisms1. J Biochem. 1993;113(3):292‐298. [DOI] [PubMed] [Google Scholar]
- 33. Kahan BD, Camardo JS. Rapamycin: Clinical Results and Future Opportunities1. Transplantation. 2001;72(7):1181‐1193. [DOI] [PubMed] [Google Scholar]
- 34. Morice W, Wiederrecht G, Brunn G, Siekierka J, Abraham R. Rapamycin inhibition of interleukin‐2‐dependent p33cdk2 and p34cdc2 kinase activation in T lymphocytes. J Biol Chem. 1993;268(30):22737‐22745. [PubMed] [Google Scholar]
- 35. Morice W, Brunn G, Wiederrecht G, Siekierka J, Abraham R. Rapamycin‐induced inhibition of p34cdc2 kinase activation is associated with G1/S‐phase growth arrest in T lymphocytes. J Biol Chem. 1993;268(5):3734‐3738. [PubMed] [Google Scholar]
- 36. Dulic V, Lees E, Reed SI. Association of Human Cyclin E with a Periodic G~ 1‐S Phase Protein Kinase. Science. 1992. [DOI] [PubMed] [Google Scholar]
- 37. Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, et al. Formation and activation of a cyclin E‐cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257(5077):1689‐1695. [DOI] [PubMed] [Google Scholar]
- 38. Jackson‐Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi D‐K, et al. Blockade of microglial activation is neuroprotective in the 1‐methyl‐4‐phenyl‐1, 2, 3, 6‐tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22(5):1763‐1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pritchard DI. Sourcing a chemical succession for cyclosporin from parasites and human pathogens. Drug Discov Today. 2005;10(10):688‐691. [DOI] [PubMed] [Google Scholar]
- 40. Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR‐mediated autophagy regulation. Mol Biol Cell. 2009;20(7):2004‐2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297‐12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981‐1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246(4930):603‐608. [DOI] [PubMed] [Google Scholar]
- 44. Gold BG, Densmore V, Shou W, Matzuk MM, Gordon HS. Immunophilin FK506‐binding protein 52 (not FK506‐binding protein 12) mediates the neurotrophic action of FK506. J Pharmacol Exp Ther. 1999;289(3):1202‐1210. [PubMed] [Google Scholar]
- 45. Edlich F, Weiwad M, Wildemann D, Jarczowski F, Kilka S, Moutty M‐C, et al. The specific FKBP38 inhibitor N‐(N′, N′‐dimethylcarboxamidomethyl) cycloheximide has potent neuroprotective and neurotrophic properties in brain ischemia. J Biol Chem. 2006;281(21):14961‐14970. [DOI] [PubMed] [Google Scholar]
- 46. Ruan B, Pong K, Jow F, Bowlby M, Crozier RA, Liu D, et al. Binding of rapamycin analogs to calcium channels and FKBP52 contributes to their neuroprotective activities. Proc Natl Acad Sci. 2008;105(1):33‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]