

Abstract
A ribonucleotide reductase (RNR) from Flavobacterium johnsoniae (Fj) differs fundamentally from known (subclass a-c) class I RNRs, warranting its assignment to a new subclass, Id. Its β subunit shares with Ib counterparts the requirements for manganese(II) and superoxide (O2–) for activation, but it does not require the O2–-supplying flavoprotein (NrdI) needed in Ib systems, instead scavenging the oxidant from solution. Although Fj β has tyrosine at the appropriate sequence position (Tyr 104), this residue is not oxidized to a radical upon activation, as occurs in the Ia/b proteins. Rather, Fj β directly deploys an oxidized dimanganese cofactor for radical initiation. In treatment with one-electron reductants, the cofactor can undergo cooperative three-electron reduction to the II/II state, in contrast to the quantitative univalent reduction to inactive “met” (III/III) forms seen with I(a-c) βs. This tendency makes Fj β unusually robust, as the II/II form can readily be reactivated. The structure of the protein rationalizes its distinctive traits. A distortion in a core helix of the ferritin-like architecture renders the active site unusually open, introduces a cavity near the cofactor, and positions a subclass-d-specific Lys residue to shepherd O2– to the Mn2II/II cluster. Relative to the positions of the radical tyrosines in the Ia/b proteins, the unreactive Tyr 104 of Fj β is held away from the cofactor by a hydrogen bond with a subclass-d-specific Thr residue. Structural comparisons, considered with its uniquely simple mode of activation, suggest that the Id protein might most closely resemble the primordial RNR-β.
Graphical Abstract
INTRODUCTION:
DNA replication is among the most central of life processes. In all known organisms, the four 2’-deoxyribonucleoside triphosphate substrates for DNA synthesis are provided by the action of a single type of enzyme, ribonucleotide reductase (RNR), which reduces the 2’ carbon of the corresponding ribonucleotides.1, 2 This chemically difficult dehydroxylation reaction proceeds by a mechanism that is largely conserved among all known RNRs from different organisms. It involves removal of the 3’ hydrogen atom of the substrate3 by a thiyl radical, which is generated early in each turnover by transient one-electron oxidation of a cysteine (Cys) residue.4, 5 Three distinct strategies for generation of this Cys• have been discovered and are the basis for the division of known RNRs into three classes, I-III.6 In the class III enzymes, found in obligatorily and facultatively anaerobic microbes, the Cys• is generated by a main-chain glycyl radical.7 This radical is installed upon the RNR by an activating enzyme that uses an iron-sulfur cluster as its cofactor and S-adenosyl-L-methionine as its substrate (a radical-SAM enzyme).8 The glycyl radical is quite unstable in the presence of O2, reacting by a pathway that leads to oxidative cleavage of the protein backbone.7 Although competing theories have since been advanced,9 Reichard originally suggested that the modern class III enzymes most closely resemble the primordial RNR that first evolved in the anoxic world.6 In this scenario, the subsequent advent of oxygenic photosynthesis and resultant accumulation of O2 in the atmosphere could have challenged the capacity of existing life forms to replicate and mandated the rapid evolution of a new means to generate the Cys•. Classes I and II might then have arisen from separate, rapid evolutionary responses that preserved deoxynucleotide production in the face of the threat posed by increasing O2 levels.6 On the path to class II, rescue of the O2-compromised class III-like enzyme would have involved acquisition of an alkylcob(III)alamin cofactor to serve in the manner that 5’-deoxyadenosylcob(III)alamin now functions in the modern class II enzymes. In the genesis of class I, this hypothesis would hold, a separate protein with a redox-active cofactor capable of extracting an electron from the Cys residue would have been recruited to rescue the failing class III-like enzyme. The recruited protein would have become the cofactor subunit, β, and the rescued class III-like enzyme the catalytic subunit, α, of the modern-day class I RNRs.
RNR β subunits are members of the ferritin-like protein structural superfamily made up primarily of non-heme di-iron oxygenases and oxidases.10, 11 The RNR βs use their reduced dimetal clusters and either O2 or a reduced form thereof to produce a stable, one-electron oxidant that generates the Cys• in α12 The identities of the transition metal ions, the activating O2 species, and the stable oxidant further segregate known class I RNRs into three subclasses, a-c. Members of the Ia subclass, which includes the Homo sapiens enzyme targeted by a number of cancer drugs, are activated by reaction of the Fe2II/II-β complex with O2 (Scheme 1A).13, 14 An Fe2III/IV intermediate in this reaction15, 16 oxidizes a tyrosine (Tyr) residue near the cofactor to a stable radical (Tyr•) as the metal cluster is reduced to its Fe2III/III state.17, 18 During catalysis, the Tyr• in β reversibly oxidizes the Cys in α to generate the Cys•.19, 20 For the Escherichia coli (Ec) class Ia enzyme, it was shown that a chain of three additional Tyr residues (and possibly a tryptophan) connecting the α and β active sites mediates this radical translocation in a conformationally-gated, multistep electron-transfer process that involves transient pathway Tyr radicals.20 These aromatic residues are strictly conserved across the entire class, and the radical-translocation mechanism is also likely to be conserved.21 A model of the heterotetrameric Ec RNR holoenzyme (α2β2), generated from the individual experimental structures of its homodimeric α2 and β2 subunits,22, 23 suggested that the distance between the Cys and Tyr• is > 35 Å,22, 23 and measurements of inter-subunit distances have been consistent with this model.20, 24, 25
Scheme 1.

Metallocofactor activation pathways in class I RNRs.
Class Ib RNRs have a dimanganese cofactor that generates the Tyr• (Scheme 1B). The Mn2II/II cluster is unreactive toward O2 itself.12, 26 A flavoprotein activator, NrdI, that can form a complex with β reduces O2 to superoxide (O2–), which diffuses through a channel connecting the NrdI and β active sites to oxidize the Mn2II/II cluster.26, 27 This reaction produces a Mn2III/IV intermediate, which generates the Tyr• as it decays to the stable Mn2III/III state.28 The Ib RNRs that have been studied in vitro are also active, albeit less so, in their Fe2III/III-Tyr• forms characteristic of the Ia enzymes.12, 29
Class Ic RNRs use the dimetal cluster itself, rather than a Tyr• produced by it, for Cys• production.30–33 Work on the subclass-founding Chlamydia trachomatis (Ct) enzyme showed that it can employ a MnIV/FeIII cofactor,31, 34 with the MnIV oxidant in metal site 135–37 (the site closer to the Tyr• in the I-a/b proteins38). The MnII/FeII-β complex reacts with O2 to form a MnIV/FeIV intermediate that is reduced by one electron to the active state (Scheme 1C).33, 39 H2O2 can also convert the MnII/FeII-β complex to the active MnIV/FeIII state in two steps via MnIII/FeIII and MnIV/FeIV intermediates.40 At the position of the radical-harboring tyrosines of the Ia/b proteins, the Ic β subunits instead have phenylalanine;30, 41 the absence of the key Tyr is the most obvious hallmark of this subclass.42 Although both Fe2II/II and Mn2II/II complexes of the Ct β subunit can also form and the Fe2II/II complex reacts rapidly with O2,39 only the heterodinuclear cluster can support turnover.31
The β subunits in all three known subclasses generate a potent oxidant, protect it from reaction with cellular reductants, and control its reduction by the Cys residue in α in response to binding of the substrate.20 This conformationally gated, long-distance, inter-subunit, radical-translocation process seems too complex to have emerged en bloc in the initial evolutionary event, raising questions about the nature of the cofactor in the original class I RNR β, the sequence of steps through which the three known strategies for Cys• generation evolved, and whether other mechanisms might remain to be discovered.9, 42 A sequence analysis published shortly after the identification of the Mn/Fe cofactor in Ct RNR noted an invariant correlation between the identities of the residue corresponding to the Tyr• oxidant of the class Ia/b βs and the first metal-ligand residue in the amino acid sequence.30, 42 In the sequences that were known at that time, all β subunits having the radical-harboring Tyr also have aspartate (Asp) at the first ligand position (Asp/Tyr), whereas all sequences with phenylalanine (Phe) in place of the Tyr (the Ic βs) have glutamate (Glu) as the first ligand (Glu/Phe). We identified more than 260 sequences deposited since that time that break this correlation by pairing Glu with Tyr. Sequence-similarity analyses suggested that these Glu/Tyr βs make up their own cluster, separate from the three clusters comprising the subclass a-c β subunits (Fig. S1), and that the partner β subunits segregate similarly (Fig. S2). This analysis suggested that the presumptive RNRs might employ a different cofactor, activation mechanism, or both, perhaps defining a new subclass.
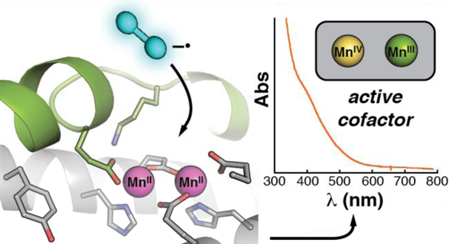
To evaluate this possibility, we examined the enzyme from Flavobacterium johnsoniae (Fj), an organism that has been extensively studied as a model for bacterial gliding and is amenable to genetic manipulation.43 We show here that Fj β does indeed employ a cofactor and activation mechanism different from those of its subclass a-c counterparts. Whereas reaction of its Fe2II/II complex with O2 is sluggish and fails to activate the protein for nucleotide reduction, its Mn2II/II complex efficiently scavenges superoxide from solution, generating a pair of stable, oxidized (formally III/IV) dimanganese clusters and activating the protein by more than 200-fold (Scheme 1D). Despite its possession of a tyrosine residue (Tyr104) aligning with the Tyr• initiators of the class Ia/b β subunits, no tyrosyl radical is formed upon activation of Fj β. Instead, the oxidized dimanganese species themselves serve as initiator, as shown by the decay of both complexes upon treatment of the Fj holoenzyme with a radical-trapping mechanism-based inactivator. Structural features not present in the subclass a-c proteins but shared by Fj β and a second recently characterized β subunit (from Leeuwenhoekiella blandensis, Lb)44 in the same sequence cluster explain the novel activation mechanism of the Fj protein. Together with results from this other recent study, which the authors judged to be “suggestive of a novel high-valent homodimeric Mn-cofactor,”44 our results establish that the Fj and Lb enzymes (and presumably the other orthologs with Glu/Tyr β subunits) do indeed constitute a new subclass, d, of class I RNRs.
EXPERIMENTAL DETAILS:
Over-expression of Wild-Type (wt) and Variant Fj RNR β.
N-terminally His6-tagged Fj α and β were overexpressed from the plasmid pET28a in E. coli BL21 (DE3) cells. Details of the construction of the plasmid vectors can be found in the Supporting Information. In order to produce β protein that was either metal-depleted or loaded with FeII or MnII, growths were carried out by one of the following procedures (1–4). Procedures 1 and 2 produced metal-depleted β protein. In both procedures, 1 L cultures of the over-producing strains were grown at 37 °C in rich Luria-Bertani (LB) medium (20 g/L yeast extract, 35 g/L tryptone, 5 g/L sodium chloride, pH 7.4) supplemented with 50 µg/ml kanamycin to an OD600 of ~ 0.8. Flasks were cooled to ~ 4 °C by incubation on ice for 1 h, and expression was induced by addition of IPTG to a final concentration of 0.2 mM. In procedure 1, 1,10-phenanthroline was added to a final concentration of 0.5 mM at the time of induction, whereas the chelator was omitted in procedure 2. In the growth procedures 3 and 4, Fj RNR β was over-expressed in M9 minimal medium [50 mM Na2HPO4, 20 mM KH2PO4, 10 mM NaCl, 20 mM NH4Cl, 0.5% glucose, 0.1 mM CaCl2, 2 mM MgSO4, 2 µM Fe(NH4)2(SO4)2] supplemented with either MnCl2 (procedure 3) or Fe(NH4)2(SO4)2 (procedure 4). Single colonies were grown overnight at 37 °C with shaking in 0.50 L of this medium supplemented with 50 µg/mL kanamycin. A 40 mL aliquot of the starter culture was used to inoculate 1 L of the same medium supplemented with 50 µg/mL kanamycin, and the culture was incubated at 37 °C with shaking to an OD600 of ~ 0.8. The culture was cooled to 4 °C by incubation on ice for 1 h. Protein expression was induced by addition of 0.2 mM IPTG. Cultures were also supplemented with either 250 µM MnCl2 (procedure 3) or 250 µM Fe(NH4)2(SO4)2 (procedure 4) at the time of induction. In all growth procedures, cultures were subsequently incubated for 20 h at 18 °C with shaking at 200 rpm. The cells were harvested by centrifugation at 12,000g for 30 min at 4 °C, frozen in liquid N2, and stored at −80 °C until they were used. A typical yield of wet cell mass was ~ 3,~ 8, ~7 and ~6 g/L culture medium in procedures 1 and 4, respectively.
Purification of the Fj β subunit.
Purification of Fj β was carried out at 4 °C. In a typical purification, frozen cell paste was re-suspended in 5 g/mL buffer A [100 mM sodium HEPES, pH 7.6, 300 mM NaCl, 10% (v/v) glycerol] containing 10 mM imidazole and 45 μg/mL PMSF. The suspended cells were lysed by use of a microfluidizer (Microfluidics M110EH-30, Newton, MA) at 20,000 psi over 5 min. The supernatant was collected after centrifugation at 26,000g for 1 h. The supernatant was loaded onto a NiII-NTA column (~ 1 mL resin per 10 mL supernatant) in buffer A. The column was washed with six column volumes of buffer A containing 10–20 mM imidazole and protein was eluted with six column volumes of buffer A containing 250 mM imidazole. Fractions containing the protein, as identified by denaturing polyacrylamide gel electrophoresis (SDS-PAGE) with coomassie staining, were combined and the protein was concentrated in a 15 mL centrifugal filter unit with 30 kDa molecular weight cutoff (Amicon ultra-15, Millipore). The imidazole was removed from the protein by chromatography on a Sephadex G-50 column in buffer A. Concentrations of the RNR subunits were determined spectrophotometrically by using computationally derived molar absorption coefficients (ε280) of 46,870 M−1cm−1 for the Fj β monomer. Minor changes to the purification protocol for protein used in crystallography, XAS, and titration experiments can be found in the Supporting Information.
The β protein purified from rich LB medium in the absence of phenanthroline (procedure 2) was found to contain ~ 0.2 equiv bound Mn and ~ 0.35 equiv bound Fe per monomer, as determined by inductively coupled plasma atomic absorption spectroscopy (ICP-AES). The protein was depleted of metal ions using previously described reductive chelation and EDTA dialysis steps,31 with the following changes: the dialysis steps were carried out at 4 °C and the protein was separated from the FeII-(ferrozine)3 using a Sephadex G-50 column. β protein treated in this manner was found to contain ~ 0.03 equiv Mn and ~ 0.01 equiv Fe per monomer, as determined by ICP-AES. The protein was frozen in liquid N2 and stored at – 80 °C until it was used. Yields of β protein were ~ 5, 65 (30 after ferrozine chelation), 50, and 20 mg/L culture medium in procedures 1-4, respectively. The protein was found to be ~ 95% pure, as determined by observation of a single band in SDS-PAGE with coomassie staining.
Over-expression and Purification of Fj RNR α Subunit.
The plasmid encoding the His6-tagged Fj RNR α subunit was used to transform Ec Rosetta (DE3) cells. Cultures were grown in 1 L flasks of rich LB medium supplemented with 50 µg/mL kanamycin and 25 µg/mL chloramphenicol at 37 °C with shaking at 200 rpm to an OD600 of ~ 0.8. Cultures were induced and harvested as described above for Fj β. A typical yield of wet cell paste was ~ 8 g/L culture medium. Purification of the β subunit was carried out at 4 °C. In a typical purification, frozen cell paste was re-suspended in 5 g/mL buffer A containing 10 mM imidazole, 45 μg/ml PMSF and 10 mM 2-mercaptoethanol. The cells were lysed as described above for preparation of β, and the supernatant was collected after centrifugation at 26,000g for 1 h. Streptomycin sulfate was added to the supernatant to a final concentration of 5% (w/v), and the mixture was centrifuged at 26,000g for 30 min. The clarified cell lysate was loaded onto a NiII-NTA column (~ 1 mL resin per 10 mL supernatant) in buffer A with 10 mM 2-mercaptoethanol. The column was washed with six column volumes buffer A containing 10–20 mM imidazole and 10 mM 2-mercaptoethanol, and was then washed with another six column volumes buffer A containing 250 mM imidazole and 10 mM 2-mercaptoethanol to elute the protein. Fractions containing the eluted protein were pooled, and the protein was concentrated in a 15 mL centrifugal filter unit with a 30 kDa molecular weight cutoff (Amicon ultra-30, Millipore). The imidazole was removed from the protein by chromatography on Sephadex G-50 in buffer A containing 10 mM DTT. Concentrated protein was frozen in liquid N2 and stored at – 80 °C until it was used. A typical yield was ~ 90 mg/L culture medium. The protein was found to be ~ 95% pure as judged by observation of a single band in SDS-PAGE (12% gel) with coomassie staining.
Determination of Protein Concentration:
Concentrations of the RNR subunits were determined spectrophotometrically by using the computationally derived molar absorption coefficients (ε280) of 82,320 M−1cm−1 for the Fj α monomer and 46,870 M−1cm−1 for the Fj β monomer.
Determination of Metal Content in Protein Preparations by ICP-AES.
Samples for ICP-AES analysis were prepared from the preparations of Fj RNR α and β subunits and Vu SOD. Aliquots of 5 mL containing 10 µM protein (monomer) and 3.5% (v/v) nitric acid were centrifuged at 3,500g for 10 min at room temperature to pellet the denatured protein. The metal content was determined by using a Perkin-Elmer Optima 5300 inductively coupled plasma emission spectrometer at the Penn State University Institute of Energy and the Environment (PSIEE) Laboratory. Single element standards of Mn and Fe (purchased from High Purity Standards, Charleston, SC) were used for calibration. Results of representative determinations are presented in Table S1.
Activation of Metal-depleted Fj β and Determination of Effective Stoichiometry.
All quoted RNR α and β concentrations are of the monomer, and all equivalencies of other reaction components (e.g., MnII, HQ, NQ) are given on a per monomer basis. Unless otherwise noted, all steps in the β-activation reaction were carried out in air at 4 °C. The desired quantity (0 – 2.5 equiv) of MnII was added to an aliquot of the metal-depleted wt or variant Fj RNR β protein (0.8 – 1.0 mM) from a buffered solution of MnCl2 (10 mM in 100 mM sodium HEPES, 300 mM NaCl, 10% (v/v) glycerol, pH 7.6). After mixing, the solution was incubated for ~10 min. The MnII loaded β was then treated with the desired quantity (0 – 2 equiv) of either NQ or HQ. The resultant solution was then aerated by pipetting over a period of ~ 10 min in the case of NQ or ~ 40 min in the case of HQ (the latter compound reacts less rapidly with dissolved O2). The HQ or NQ was removed by 5 rounds of spin-concentration/dilution. In experiments examining suppression of Fj β activation by Vu SOD (Fig. 1D), O2 was initially removed from the metal-depleted β protein. The SOD was then added to the protein to 0.32 mM, and the solution was incubated for 20 min at ambient temperature (to allow for scavenging of O2–) before 1.5 equiv MnII was added. The desired quantity of NQ was then added with subsequent aeration as above. Dilution of the β protein into the nucleotide reduction reaction resulted in a final Vu SOD concentration of 40 µM, thus also allowing for scavenging of O2– produced from dissolved O2.
Fig. 1. Manganese dependence, and requirements for in vitro activation, of Fj RNR β.

(A) Comparison of the catalytic activities of β preparations (i) isolated from over-producing Escherichia coli grown in minimal medium supplemented with 50 µM MnCl2 (gray), (ii) depleted of metals by chelation and treated aerobically with 1.5 MnII/β (orange), and (iii) depleted of metals and then treated aerobically with 1.5 MnII/β followed by 1.0 HQ/β (blue). (B) Determination of the MnII equivalence point in activation of metal-depleted β with MnII, O2 (excess), and NQ (1/β). (C) Determination of the NQ equivalence point in activation of the metal-depleted β with 1.5 MnII/β, O2 (excess) and NQ. (D) Suppression of activation in activity assays with the high β concentration (100 µM) by inclusion of 40 µM Vu SOD in the treatments with 1.5 MnII/β O2 (excess), and NQ.
RNR Activity Assays.
Activity assays were carried out with 1–100 µM β monomer, 10 equiv α (relative to β), 2 mM CDP, 2 mM ATP, 10 mM DTT, and 12.5 mM MgSO4 in 100 mM sodium HEPES (pH 7.6), 300 mM NaCl, and 10 % (v/v) glycerol (assay buffer) at room temperature (21 ± 2 °C ) under an air atmosphere. Fj RNR α subunit was present at 10 equiv relative to the β subunit to ensure that β limited the measured activity. In reactions with Vu SOD present, all components except the MnII-treated β were mixed, and the solution was incubated for ~ 5 min to allow time for scavenging of ambient O2– by the Vu SOD. All reactions were initiated by addition of the desired quantity of activated β (prepared as described above) and terminated at desired time points by treatment of a 50 µL aliquot with 5 µL 2 M formic acid. The acid-quenched samples were centrifuged at 17,000g for 10 min, and the supernatants from the acid-quenched samples were filtered through 10 kDa Nanosep (Pall Corporation, Port Washington, NY) centrifugal filters. The sample filtrate was diluted by a factor of 20 with water and analyzed by LC-MS (Agilent 1200 series LC system coupled to Agilent 6410 QQQ mass spectrometer, Agilent Technologies, Santa Clara, CA) equipped with an Agilent 1.8 µm Extended-C18 (50 × 4.6 mm) column. Details of the LC-MS method can be found in the Supporting Information.
EPR Spectroscopy.
Continuous-wave (CW) EPR spectra were acquired at 5 – 90 K on a Bruker ESP300 spectrometer equipped with a continuous flow cryostat (Oxford Instruments) and an ITC300 temperature controller (Oxford Instruments). Measurements were carried out using two different types of rectangular resonators: a conventional Bruker ER/4102 ST resonator, which operates in the TE102 (9.48 GHz) perpendicular mode, and a Bruker dual mode resonator (ER 4116 DM), which can switch from operation in the TE102 (~ 9.63 GHz) perpendicular mode to the TE012 parallel mode (~ 9.38 GHz). The microwave frequency was measured with a 5350B Hewlett Packard frequency counter. Custom-made quartz tubes (Quartz Scientific, Inc., Fairport Harbor, Ohio) of uniform inner and outer diameter were used in all experiments to ensure accurate spin quantification.
X-ray Absorption Spectroscopy.
250 μL aliquots of 2.3 mM activated β, isolated from growth procedure 1, were loaded into Mössbauer cells sealed with 38 μm Kapton tape and frozen in liquid N2. To make the MnII control, a small portion of β was removed from the activation mixture following the addition of MnII and prior to the addition of NQ. Details on the preparation of the activated β and the Mn2III/III Bs NrdF complex for XAS can be found in the Supporting Information. All XAS data were acquired at beamline 7–3 at SSRL, except for the data on the Mn2III/III Bs NrdF complex, which were collected at beamline 9–3. For all experiments, the ring was operating at 3 GeV and 500 mA. A Si(220) monochromator (φ=90°) was used for energy selection, and the beam was detuned by 50% to remove harmonic contamination. On beamline 9–3, a 10 keV cutoff mirror was also used. The first inflection point of a Mn foil placed upstream of the sample was used for energy calibration (6539.0 eV). Samples were placed at 45° relative to the incident beam and maintained below 15 K using an Oxford liquid helium flow cryostat; Kα fluorescence was measured using a 30 element (7–3) or a 100 element (9–3) germanium detector. Damage was assessed for all samples. A full-length XAS scan was carried out on a given sample spot, and the same spot was then subjected to a rapid edge-only scan. If the edge was observed to shift, the scan was shortened and/or the incident beam was attenuated by the insertion of aluminum Reynolds filters upstream of the sample. For activated Fj β, only a very subtle difference between the two spectra was noted (Fig. S9), indicating that the sample was not damaged during scanning. Damage was not observed at any point in the experiment for the Mn2II/II species. The final scan for activated Fj β was performed with the incident intensity reduced by a factor of ~ 4; it covered the energy ranges of 6315 – 6525 eV (10 eV steps), 6525 – 6570 eV (0.25 eV steps), and 6570 – 7109 eV (0.06 k steps) and resulted in 562 s of exposure. For the Mn2III/III standard, a similar scan was performed: 6310 – 6520 eV (10 eV steps), 6520 – 6570 eV (0.4 eV steps), and 6570 – 7097 eV (0.07 k steps) for 170 s. XAS spectra were calibrated and averaged using EXAFSPAK (George, G. N. EXAFSPAK; SSRL, SLAC, Stanford University: Stanford, CA) and normalized by setting the post-edge intensity to unity using BlueprintXAS.45
Reductive Titration of Oxidized Fj β with Sodium Dithionite.
An anoxic solution of 0.3 mM Fj β, prepared via growth procedure 1 and activated as described above, was titrated with a freshly prepared solution of sodium dithionite in assay buffer. The dithionite concentration was determined by titration against a 1.0 mM solution of potassium ferricyanide (ε280 = 1020 M−1 cm−1). The dithionite solution used in the titrations was between 5 and 12 mM, and serial additions of ~ 0.25 molar equivalents (0.5 electron equivalents) relative to the Mn2 cofactor concentration were delivered via syringe (Hamilton Company, Reno NV). The Mn2 cofactor concentration varied from 0.15–0.225 mM and was determined independently in each replicate by ICP-AES. Methyl viologen (MV) was included in the protein solution at a concentration of 12 μM as a redox mediator and indicator (see below). After each addition, the solution was mixed by pipetting and allowed to reach equilibrium for ~ 5 min, during which time reaction progress was monitored by repetitive acquisition of its UV-visible absorption spectrum. The endpoint of the titration was signaled by the first appearance of the distinctive absorption spectrum of the methyl viologen radical, which indicated that uptake of electrons by the Fj β cofactor had reached completion.
Discovery of the Presence of Two Oxidized Dimanganese Complexes in Activated Fj β: Resolution and Simulation of their CW EPR Spectra.
EPR spectra were simulated using the MATLAB (Mathworks) based program Easyspin (http://easyspin.org/) simulation software and Kazan viewer, a suite of utilities (written by Drs. Alexey Silakov of Penn State University and Boris Epel of the University of Chicago) that runs within MATLAB. The X-band CW EPR spectra of activated Fj β revealed two multiline signals associated with distinct Mn2 clusters with Stot = ½ ground state. These signals were simulated by using the following spin Hamiltonian:
in which the first term describes the electron Zeeman effect of the Stot = ½ ground state and the remaining two terms describe the hyperfine coupling with the two Mn sites. The quantities gtot, A1, and A2 are given with respect to the total spin of the ground state, Stot = ½, and can be related to the intrinsic quantities by standard spin projection techniques.46
The complexity of the EPR spectra and the large number of parameters mandated a multi-step strategy for the analysis. In the first step, we interrogated the temperature dependence of the multi-line spectrum. This step revealed the presence of (at least) two constituent complexes, 1 and 2, with Stot = ½ ground state and different electronic relaxation rates. Complex 1 relaxes more rapidly, and the temperature dependence of its spectrum afforded an estimate of the exchange coupling constant, J, for this species (vide infra).47
EPR Characterization of 2.
At temperatures exceeding 30 K, broadening of the signal of 1 simplified the overall spectrum and exposed a well-defined 16-line signal associated with 2. The spectrum is similar to those observed previously for other Mn2III/IV complexes and observable up to 60 K. To simulate this component, parameters similar to those reported for the III/IV state of the ferritin-like dimanganese catalase and the Tyr•-generating Mn2III/IV complex in Ec class Ib RNR β were used as initial estimates (Table S3). In these well-characterized systems, the gtot-tensors are nearly isotropic, precluding resolution of principal components in spectra acquired with low microwave frequencies (e.g., X- and Q-band). For this reason, and so as not to further increase the already large number of adjustable parameters in the simulation, we assumed axial symmetries for both the hyperfine and g-tensors. A global least-squares search using the simplex algorithm implemented in Easyspin was carried out to identify the parameters that give the best agreement between the experimental and simulated spectra. In a final step, the three components of A1, A2, and gtot were allowed to vary independently so as to achieve a better fit. The analysis converged on the parameters given in Table S2. These parameters match well with those reported for the other known Mn2III/IV complexes (Table S3).
EPR Characterization of 1.
At temperatures ≤ 10 K, a second multi-line component, which has greater intensity, is superimposed upon the signal from 2. The spectrum of the associated complex, 1, was resolved by subtracting the contribution of 2 from the spectrum recorded at 10 K. For this purpose, a reference spectrum of 2 was obtained by recording the EPR spectrum of the sample at 30 K. At this temperature, the signal arising from 1 is broadened nearly into the baseline, whereas the features arising from 2 remain as sharp as at 10 K (they are not broadened due to relaxation). The relative intensity of the spectrum of 2 to be subtracted from the 10 K experimental spectrum was estimated on the basis of the sharp highest-field line of the signal of 2 (g ~ 1.716), which is resolved from all features of the spectrum that arise from complex 1. In this region, the features of 1 are much more broad, allowing for a straightforward estimation of the relative intensity of the Mn2III/IV-associated signal used for subtraction (± 5%). The reference spectrum of 1 was simulated in the same stepwise fashion as was done for the spectrum of 2 (see above). This procedure yielded the parameters quoted in Table S2.
Microwave power saturation data (Fig. S12) acquired at 5.1 – 9.8 K were obtained by recording the sample signal intensity versus the microwave power at each selected temperature. The power at half-saturation (P½) at each temperature was obtained by a least-square-fitting to the data of the function:, in which K is a proportionality constant and b is the so-called inhomogeneity factor, assumed to be 1 (inhomogeneous limit).48 The natural logarithm of P½ of the signal was plotted versus the reciprocal temperature (1/T) in the temperature range 5.1 – 9.8 K and the function P½ = A·T + B/[exp(− Δ/T)-1)] was fit to the data47, in which Δ is the energy of the first excited spin state with Stot = 3/2. Using the isotropic Heisenberg exchange coupling term (Ĥex = J S1∙S2) the energy difference to the first excited state with Stot = 3/2 is given as Δ = 3/2J, yielding J ~ 22 cm−1. The power saturation data at temperatures as low as 5.1 K suggest the presence of a third species with relaxation properties similar to those of 1. However, the complexity of the spectra precluded resolution of the EPR properties of this possible third component.
Reaction of Fj RNR with N3-UDP Monitored by EPR Spectroscopy.
The N3-UDP reactions were carried out in a buffer of 50 mM sodium HEPES (pH 7.6), 120 mM NaCl, and 3 % (v/v) glycerol in the absence of O2 at room temperature in a total reaction volume of 0.28 mL. EPR samples had final concentrations of 0.70 mM α (DTT was removed in the absence of O2 prior to the reaction), 1 mM ATP and 0.9 mM N3-UDP. Control samples lacked N3-UDP. After thorough mixing by repeated pipetting, reactions were initiated at 21 ± 2 °C by addition of β (to 0.35 mM) that had previously been subjected to the activation procedure [carried out either in the absence (Fig. 3B) or in the presence (Fig. S13) of Vu SOD] followed by removal of NQ from activated β using concentration and re-dilution. These solutions were incubated at 21 ± 2 °C for an additional 90 s with continuous mixing. During this incubation, samples were transferred to quartz EPR tubes and frozen in liquid N2 after 90 s.
Fig. 3. EPR spectroscopy probing the nature of the oxidized dimanganese cofactor in Fj RNR and its role in catalysis.

(A) Resolution of the spectra of the two complexes in the active state (blue spectra) by their different temperature dependencies, and simulation of these spectra (red spectra) to extract insight into their structures. Details of sample preparation, spectrometer conditions, and simulation parameters are provided in the SOM. (B) Partial decay of the EPR spectra of both oxidized dimanganese species (red spectra, I and III) upon reaction of Fj RNR with the radical-trapping mechanism-based inactivator, 2’-azido-2’-deoxyuridine 5’-phosphate (blue spectra, II and IV). The feature associated with N• is indicated by an asterisk.
General Crystallographic Methods.
All crystallographic datasets were collected at the Life Sciences Collaborative Access Team (LS-CAT) and the National Institute of General Medical Science and National Cancer Institute Collaborative Access Team (GM/CA-CAT) beamlines at the Advanced Photon Source (Argonne National Laboratory). Mn anomalous scattering datasets were collected at the Berkeley Center for Structural Biology at the Advanced Light Source (Lawrence Berkeley National Laboratory) or at GM/CA-CAT. All diffraction datasets were processed with the HKL2000 package49 and, unless stated otherwise, phases were obtained by molecular replacement with the program PHASER50 within the CCP4 software package.51 Model building and refinement were carried out with Coot52 and Refmac5,53 respectively. PHENIX was used for metal ion occupancy refinement.54 Structures were validated and analyzed for Ramachandran parameters with the Molprobity server.55 Figures were prepared with the PyMOL molecular graphics software package (Schrödinger, LLC).
Fj MnIIβ to MnII Crystallized Under Anoxic Conditions. Fj
β grown by procedure 1 (phenanthroline added during growth) was purified by NiII-NTA chromatography as described above, with minor changes to the lysis step (see Supporting Information). The protein was further purified on a DEAE Sepharose anion exchange column (GE Healthcare), followed by a size exclusion step (HiLoad 16/600 Superdex 200, GE Healthcare). Purified Fj β was exchanged into 50 mM sodium HEPES pH 7.6, 50 mM NaCl and concentrated to 10 mg/mL in an 10 kDa MWCO centrifugal filter device (Amicon Ultra, Millipore).
Crystals of the apo β were grown from 10 mg/mL solutions in 50 mM sodium HEPES pH 7.6, 50 mM NaCl by the hanging drop vapor diffusion method at room temperature with 0.2 M magnesium acetate and 20% (w/v) PEG 3350 as the precipitating solution. Triangular prism-shaped crystals appeared within 1–2 days. A crystallization tray containing these samples was transferred to an anoxic glovebox (Coy Laboratory Products) and incubated for 1 week. All subsequent crystal-handling steps were performed inside the glovebox. Prior to data collection, the crystals were soaked for 3–5 minutes in an O2-free solution containing 1 mM MnCl2, 20 mM HQ, 50 mM sodium HEPES, pH 7.6, 300 mM NaCl, and 30 % (v/v) glycerol. Samples were mounted on rayon loops and flash-frozen by direct plunge into liquid N2. Data collection and refinement statistics are given in Table S5. Phases were obtained by molecular replacement using the coordinates of a previously obtained structure of Fj β obtained under aerobic conditions (see Supporting Information).
The final model consists of residues 1–300 for chain A, residues 1–300 for chain B, four MnII ions, and 68 water molecules (PDB accession code 6CWP). Of the residues modeled, 97.4% are in allowed or preferred regions indicated by Ramachandran statistics. Anomalous difference Fourier maps collected at the MnII absorption edge (6.65 keV) exhibit strong density at the MnII sites, of which each was modeled at full occupancy. Electron density was not observed for any of the residues associated with the N-terminal tag or intervening linker in chain A, but the thirteen residues that immediately precede the N-terminal Met of Fj β could be modeled in chain B (Figs. S25, S26). Continuous electron density was not observed for the final 24 residues at the C-terminus of the protein in either chain. However, a small portion of the C-terminus of chain A could be modeled in a crystallization contact (Fig. S25).
Sequence-Similarity-Network (SSN) Analysis to Resolve Id RNR Sequences and Phylogenetic Analysis.
A BLAST search against proteins in the RefSeq database with the sequence of the β subunit of Fj class Id ribonucleotide reductase [GenBank Identifier (GI): 500937268] as query identified 265 sequences with attributes that appear to be structural hallmarks of the new Id subclass (vide infra). This set was further curated to include only 259 non-redundant sequences. The least similar sequence in this set has 37% similarity to Fj β. For the phylogenetic analysis, 31 Ia, 31 Ib, 30 Ic, and 34 Id sequences were selected. In selection of the Ia β sequences, only those encoded near a gene for a YfaE-like [2Fe-2S] ferredoxin, which has been implicated in the cofactor maintenance pathway, were selected. Ib sequences were selected only when encoded near a NrdI-like flavodoxin, which has been shown to provide the O2– for cofactor oxidation.28 Selection of class Ic representatives required only the Glu/Phe combination and co-clustering with the archetypal Ct Ic β in SSNs (vide infra). Sequences were aligned with the MUSCLE (multiple sequence comparison by log-expectation) software package (http://www.ebi.ac.uk/Tools/msa/muscle/). Maximum-likelihood unrooted trees were computed with RaxML56 using the LG model, with a gamma model for the rate of heterogeneity and considering invariant sites and empirical base frequencies. The best-fit phylogenetic model for β subunit evolution was additionally evaluated with ProtTest 2.4,57 which we used to select the model with the smallest Akaike information criterion (AIC). The bootstrap values, which reflect the confidence level for the nodes (reliability), are reported for the major branches. The scale bar represents the number of substitutions per site. Trees were rooted using the R2-like oxidase Rv0233 sequence as the outgroup (Figure 5, main text). Other phylogenetic analyses have concluded that the R2-like oxidases diverged from the lineage that includes class I RNRs prior to any branching within the RNR subclade.58 We also computed trees using alternative, but still structurally-related, groups of proteins as outgroups. Trees obtained with the aldehyde deformylating oxygenases (ADOs; Fig. S24) or the α subunits of the hydroxylase components of soluble methane monooxygenases (sMMOHαs; Fig. S25) as the outgroups have similar topologies with respect to the segregation of the class I RNRs (vide infra). All trees show divergence of class I RNRs from the R2lox group prior to any branching within the RNR subfamily and support the choice of these sequences as a valid outgroup.
Fig. 5. Active site structures and preferred hypothetical molecular phylogeny for RNRs from the four known class I subclasses.

(A) A divergent conformation and interaction with residues near metal site 1 for Tyr 104 in Fj β (top left) relative to the radical-harboring Tyr and hallmark Phe of the class Ia-c βs (top right and bottom). Note the unique hydrogen bond of Tyr 104 with the second-sphere Thr 191, a position that is conserved as either Thr or Ser in all members of the new Id subclass. In Fj β, this hydrogen bond supplants those between the corresponding tyrosines and first carboxylate-ligand residues in the Ia/b βs, thereby orienting the Tyr 104 side-chain away from metal 1 and potentially explaining its failure to undergo oxidation to a radical. The structures in the other three panels are of the Ec Ia (Fe2II/II complex, PDB accession code 1PIY; top right), Ec Ib (Mn2II/II complex, PDB accession code 39N3; bottom right) and Ct Ic (MnII/FeII complex, PDB accession code 4M1I; bottom left) β proteins. (B) Rooted tree showing the molecular phylogeny validated by the structural comparisons in A. The methods used in the analysis are described in the SI. The numbers shown at the nodes are bootstrap values and correspond to level of confidence in a given node. The bar at the top gives the evolutionary distance scale in units of substitutions per site. This tree was rooted by using a set of R2-like oxidases as the outgroup.
RESULTS AND DISCUSSION:
Activity of Fj β with, and apparent activation by, manganese.
The α and β subunits of the Fj RNR were produced in Escherichia coli BL21(DE3) with a 20-residue (His)6-containing affinity tag appended to their N-termini and purified by immobilized metal ion affinity chromatography, as described in the Supporting Information. The β subunit isolated from cultures grown in minimal medium supplemented with 250 µM MnCl2 emerged with primarily manganese bound, typically ~ 1.6 Mn/β (see the SI for metal analysis). Because Fj is an environmental microbe, we assayed these preparations at 21 ± 2 °C, rather than the 37 °C used in prior studies of RNRs that function in warm-blooded animals. In the presence of CDP, ATP, excess (10 equiv) Fj RNR α, and dithiothreitol (DTT) as the reductant (see Supporting Information for the assay procedure and Fig. S3 for assay validation), the preparations exhibited catalytic rate constants (v/β = 1.2 ± 0.1 s−1) within the range of values reported for representative class Ia-c RNRs (v/β = 1.25 – 5 s−1),19, 26, 37, 39, 59 and the expected proportionality between rate of dCDP production and enzyme concentration was observed for β concentrations between 1 and 100 µM (Fig. 1A, gray). The β subunit could be obtained in a metal-depleted form, either by culturing the over-producing strain in Luria-Bertani (LB) medium supplemented at the onset of β expression with the iron chelator, phenanthroline, or by removing metals from the protein produced in LB (without phenanthroline) by a previously described reductive-chelation procedure followed by dialysis against EDTA.31 Both procedures yielded protein with diminished activity (0.01 – 0.15 s−1). These preparations were markedly (5–10–fold) activated by addition of only MnII in the presence of O2. When catalytic activity was examined at [β] ≤ 10 µM, preparations reconstituted in this manner exhibited v/β values exceeding 1 s−1 (Fig. 1A, orange), similar to the activities of preparations isolated with ~ 1.6 equiv Mn already bound. Activation by sole addition MnII (in the presence of O2) to apo β has not previously been seen for a class I RNR. The Ia and Ic enzymes require iron for activity, whereas the Mn-dependent class Ib RNRs require the flavoprotein activator, NrdI, to generate the cluster oxidant, O2–, and present it to the cofactor in β12 In general, the nrdI gene is found immediately adjacent to the nrdE and nrdF genes encoding the Ib RNR α and β subunits, respectively. There is no recognizable nrdI homologue in the Fj genome, nor in any of the > 260 other genomes harboring members of the putative new Id subclass, in the vicinity of the genes for the RNR subunits. In addition, by contrast to the Ia-c proteins that have been studied,12 the Fe2II/II complex of Fj β reacts very sluggishly with O2 (Figs. S4 and S5). Its apparent in vitro activation by just MnII and O2, without need of a NrdI homolog, and the modest O2 reactivity of its Fe2II/II complex thus distinguish the Fj protein from other known RNR βs.
Involvement of superoxide in activation of Fj β.
In nucleotide-reduction assays with ten times the concentration of the MnII/O2-activated Fj β (100 µM) and proportionally greater [α] (1 mM), rates of dCDP production were greater by only a factor of two (or less), resulting in v/β values diminished by at least a factor of five (Fig. 1A, orange). This unusual concentration dependence suggested the requirement for an additional factor, present at ≤ 20 µM ambient concentration, for full activation of the metal-depleted protein. The analogy to the class Ib RNRs in the use of manganese (only) suggested that this factor could be O2–, which, for the case of Fj β might be scavenged from solution rather than supplied by NrdI. To evaluate this possibility, we tested whether inclusion of a superoxide dismutase (SOD) in assays of the Fj RNR might prevent its activation by aerobic addition of MnII. We selected an SOD from Vigna unguiculata (Vu), because it uses iron rather than manganese (which would have interfered in EPR analysis described below) and for which an expression vector was available as a kind gift from Prof. José Morán at Univ. Pública de Navarra. Inclusion of 5 µM Vu SOD during both the MnII/O2 treatment of metal-depleted β and the nucleotide-reduction reaction with α diminished activity by a factor of ~ four for the case of reactions at low enzyme concentration (10 µM β, 100 µM α). Conversely, inclusion of a compound that can reduce O2 to O2–, either 1,4-dihydroxybenzene (hydroquinol or HQ; see Scheme S1 for analysis of all possible outcomes), 1,4-dihydroxynaphthalene (naphthoquinol or NQ)60, 61 or a reduced flavin,62 in the MnII/O2 treatment of the metal-depleted Fj β resulted in full activation to v/β = 1.3 ± 0.1 s−1 at the higher enzyme concentration (Fig. 1A, blue) and, for the case of NQ (the only compound tested), was able to overcome the suppressive effect of the Vu SOD at the lower enzyme concentration. These compounds activated Fj β only when O2 was also simultaneously present. The MnII-β protein could also be activated by addition of a solution of potassium superoxide and the crown ether, 1,4,7,10,13,16-hexaoxacyclooctadecane, in dimethylsulfoxide,63 although this procedure was neither as effective nor as reproducible as in situ O2– generation with a quinol. Activation by either HQ/O2 or superoxide itself was suppressed by inclusion of a high concentration of Vu SOD (see below). These results implicate O2– as the species that activates the MnII complex of Fj β.
More quantitative analysis of the activation process was undertaken at high Fj β concentration, under which conditions the dynamic range of resultant activity in the absence and presence of a superoxide-generating compound was the greatest. In terms of accuracy in activity determinations, these conditions were suboptimal, because they resulted in the conversion of a large fraction of the CDP substrate to dCDP product in the shortest accessible reaction time and gave product-versus-time curves that were exponential rather than linear. Nevertheless, fitting of these progress curves by an exponential function afforded rates that were proportional to those obtained at diminished enzyme concentrations with more experimentally accessible steady-state regimes (Figure S3). In activation of the protein with excess NQ and O2, maximum activity (v/β ~ 1.3 s−1) required ~ 1.5 ± 0.2 MnII/β (Fig. 1B). The least active preparations of metal-depleted β (those from which Mn was removed by dialysis against EDTA) were activated almost 200-fold by this treatment. With excess MnII and O2, ~ 1 NQ/β yielded maximal activation (Fig. 1C). Vu SOD present at 320 µM in the optimized reconstitution reaction and 40 µM in the nucleotide reduction reaction suppressed activity by ~ 95% (Fig. 1D).
Oxidation of the dimanganese cluster but not Tyr 104 in activation of Fj β.
The nature of its cofactor and its O2–-mediated in vitro assembly were probed by electron paramagnetic resonance (EPR) spectroscopy. Addition of MnII to preparations of β from which metal had been depleted by production in the presence of phenanthroline resulted in the development of a broad, featured g ~ 2 EPR resonance (Fig. 2A, black). Hyperfine coupling to 55Mn (with nuclear spin quantum number, I, of 5/2) is not evident in this spectrum because it is so broad. It is markedly different from the narrower, isotropic, sextet spectrum of MnII in solution,64, 65 confirming that the metal-depleted Fj β can take up the added MnII. In titrations of the protein with MnII monitored by EPR, this spectrum grew in homogeneously to a maximum MnII/β ratio of ~ 1.8; at ratios greater than 1.8, the signal of free MnII began to grow in (Figure S6). For this reason, and to obviate the complication of removing variable contributions from free MnII, we elected to use lesser MnII/β equivalencies in many of the EPR experiments described below. The MnII/β equivalence points determined from the EPR-monitored and activity-monitored titrations thus agree well and establish that the cofactor is dinuclear. The spectrum and temperature dependence thereof (Figure S7) are different from those previously reported for antiferromagnetically coupled Mn2II/II complexes of class Ia and Ib β proteins26, 28, 65 and suggest that the two MnII ions in Fj β are less strongly coupled..
Fig. 2. Formation of an oxidized dimanganese cofactor in Fj RNR.

(A) Changes in the g ~ 2 region of the EPR spectrum associated with conversion of the reduced, Mn2II/II form of the Fj β cofactor (black spectrum) to the oxidized, active state (blue spectra), and requirement for the simultaneous presence of both O2 and a superoxide-generating quinol (gray spectra). (B) Comparison of the manganese x-ray absorption edges in oxidized (blue line) and reduced (black line) Fj β to those of the oxidized active states of class Ib (red line) and Ic (yellow line) βs. (C) Quantification by reductive titration with sodium dithionite in the presence of the redox mediator/indicator, methyl viologen (MV), of the oxidizing equivalents stored in Fj β upon cofactor activation.
Activation by treatment of the Mn2II/II-β complex with O2 and either HQ or NQ resulted in decay of the MnII-associated EPR signal and development of a complex, multi-line spectrum centered at g ~ 2 (Fig. 2A, light blue and dark blue lines). As in the experiments monitoring enzymatic activity, the EPR-detected conversion required the simultaneous presence of both components, O2 and quinol: treatment with either component in the absence of the other left the spectrum of the Mn2II/II-β complex nearly unchanged (Fig. 2A, compare black and gray). The need for their simultaneous presence again reflects reduction of O2 by the quinol and addition of the O2– product to the Mn2II/II cluster. The resultant multi-line EPR signal is associated with oxidized Mn complexes with a total-electron-spin ground state of Stot = ½. Because its development correlates with acquisition of catalytic activity, the multi-line spectrum could arise from the active cofactor. This situation would be drastically different from that seen in activation of the class Ib enzymes by reaction of the Mn2II/II-β with O2– supplied by NrdI.12, 66 In that case, an EPR-silent Mn2III/III cluster is produced along with a magnetically coupled EPR-active Tyr•. Importantly, even though Fj β does have a Tyr at the position (104) that aligns with the radical-harboring Tyr residues of the class Ia/b RNRs, no EPR signal attributable to a Tyr• was seen to develop in the activation reaction, and the Tyr 104 → Phe variant (Y104F) developed the same multi-line spectrum (Fig. S8). Intriguingly, despite the absence of a Tyr• in the activated wild-type protein, the Y104F variant exhibited drastically diminished activity: less than one turnover was observed after a 20-min incubation. Tyr 104 in Fj β thus has an important role in catalysis, but one that is distinct from the role of the corresponding (radical-harboring) tyrosines in the Ia/b enzymes.
Manganese oxidation states in the activated Fj β.
The average oxidation state of the manganese in the activated Fj β was probed by Mn K-edge X-ray absorption spectroscopy (XAS). Control samples of Mn2II/II-β (prior to activation) exhibited an edge energy and shape consistent with the known +II oxidation state (Fig. 2B, black line). Upon activation by the O2/NQ treatment, the edge shifted to higher energy (Fig. 2B, blue line), as expected for oxidation of the Mn2 cluster. Consistent with EPR analysis of samples prepared similarly (Fig. 2A), the absence of a shoulder at ~ 6552 eV on the rising edge in the activated samples ruled out the presence of significant residual Mn2II/II complex. The ~ +3 eV shift in edge energy does not, a priori, quantify the change in oxidation state associated with activation, both because the product is heterogeneous (see below) and because details of coordination geometry and electronic structure can also impact the edge position.67 However, comparison to corresponding spectra obtained on the closely related Mn-containing clusters in other class I RNR βs, for which the Mn oxidation states are known, afforded an estimate of the average Mn oxidation state in the activated Fj β samples. The absorption edge for the Fj β sample was seen to fall between those of the Mn2III/III complex of the Bacillus subtilis (Bs) class Ib β28 and the MnIV/FeIV activation intermediate in the Ct class Ic β68Fig. 2B red and yellow lines), suggesting an average oxidation state between +III and +IV for the Mn in activated Fj β.
Anoxic titrations of the activated protein with a standardized solution of the strong reductant, sodium dithionite, in the presence of the redox mediator/indicator, methyl viologen (MV), provided support for the XAS-based estimate of the average manganese oxidation state. Serial additions of the reductant led to progressive loss of the ultraviolet-visible absorption feature (Fig. 2C), multi-line EPR signal (Fig. S10), and catalytic activity (Fig. S11A) and growth of the EPR signal of the Mn2II/II form (Fig. S11B). Only after complete reduction, which required 2.89 ± 0.14 electrons per Mn2 cluster (mean and standard deviation from six independent trials), did the indicator undergo reduction to its intensely colored radical form (MV•) (Fig. 2A, brown). Similar stoichiometries have been reported for the active forms of the class Ia-c βs12 and reflect storage of three oxidizing equivalents at the cofactor sites upon activation. For the case of Fj β, the oxidation of the Mn2II/II cluster formally to Mn2III/IV would most simply rationalize this result, although formation of a species with one or more oxidizing equivalent on a ligand and the Mn2 cluster in a correspondingly lower oxidation state is formally possible.
Cooperative three-electron reduction of the Fj Mn2III/IV cofactor.
Two notable features of these reductive titrations are (1) that complete loss of activity required more than two electron equivalents and (2) that the EPR signal of the fully reduced (Mn2II/II) state grew in linearly, without any lag (Fig. S11B). These observations suggest that the Mn2III/IV cofactor of Fj β is prone to cooperative three-electron reduction, rather than well separated, sequential univalent reductions. In the latter case, one electron equivalent would have completely reduced (to Mn2III/III) and inactivated the entire population, and no fully-reduced complex would have appeared at low electron equivalencies. In fact, the subclass a-c β proteins behave more similarly to this extreme.12, 37 This propensity to form the fully reduced (Mn2II/II) state may ensure that, upon reductive inactivation, the cofactor is rapidly returned to its superoxide-reactive form, poised for reactivation. It could thus make the activity of the Fj β protein unusually robust and independent of the specific accessory factors known to participate in reactivation of other RNR β subunits (termed cofactor maintenance).
Two oxidized dimanganese complexes in activated Fj β.
X-band EPR spectra of the activated Fj β acquired at different temperatures revealed that the multi-line signal actually comprises two components, reflecting the presence of two different complexes (Fig. 3A), hereafter 1 and 2. At temperatures ≥ 30 K, the spectrum of 1 becomes very broad as a consequence of fast relaxation. The broadening of its derivative features exposes the 16-line signal of 2, which remains sharp at elevated temperatures (Fig. 3A, II, blue) and resembles spectra previously reported for Mn2III/IV complexes known or thought to have bridging oxo ligands.69–71 Among these complexes is the Mn2III/IV intermediate that generates the Tyr• during activation of the class Ib RNR-β from Bs.28, 69–71 The oxo bridges mediate strong antiferromagnetic coupling between the MnIII (S = 2) and MnIV (S = 3/2) ions and give well-separated Stot = ½ electron-spin ground states with EPR signatures that are evident at elevated temperatures, as observed here for 2. Indeed, this signal can be simulated well (Fig. 3A, II, red) with g-values and hyperfine-coupling parameters (legend to Fig. 3 and Table S2) similar to those reported for the other (µ-oxo)-Mn2III/IV complexes.28, 69, 70
The spectrum of 1 (Fig. 3A, I-II, blue) was resolved by subtraction of the contribution of 2 (Fig. 3A, II, blue) from the experimental spectrum of the activated protein acquired at 10 K (Fig. 3A, I). This spectrum is also centered at g ~ 2, thus revealing a Stot = ½ ground state also for 1, but exhibits greater width and a more complex 55Mn hyperfine pattern than the spectrum of 2. The resolved spectrum can be simulated (Fig. 3A, I-II, red) with spin Hamiltonian parameters provided in Table S2. From the power and temperature dependencies of its EPR signal (Fig. S12), an estimate of the exchange coupling constant, J, of ~ 22 cm−1 (considering the exchange coupling interaction as Hex = J•S1•S2) was obtained for 1. The spin-Hamiltonian parameters and the modest antiferromagnetic ex- change coupling suggest that 1 could be a Mn2II/III complex;69 in this scenario, the XAS data and reductive titrations would imply that an unusual ligand set stores two oxidizing equivalents and shifts the X-ray K-edge to an anomalously high energy (Fig. 2B). A more likely explanation is that 1 is, in fact, a Mn2III/IV cluster with an unusual geometric and/or electronic structure. The modest coupling between its two Mn ions would suggest that it either lacks a µ-oxo ligand or has a coordination geometry that diminishes the efficacy of the bridge as an exchange mediator.72–74
Evidence for radical initiation by the oxidized dimanganese cluster(s).
The radical-trapping mechanism-based inactivator, 2’-azido-2’-deoxyuridine 5’-diphosphate (N3-UDP), was deployed to determine which, if either, of the two oxidized dimanganese clusters in the activated Fj β is the functional cofactor (Fig. 3B). Past work on Ia and Ic RNRs showed that the substrate analog is processed by an altered mechanism that results in irreversible reduction of the Cys•-generating oxidant in β (the Tyr• or MnIV/FeIII cofactor) and accumulation of a meta-stable nitrogen-centered radical (N•) in α31, 75, 76 Treatment of active Fj β with N3-UDP also led to production of the N•, as shown by its characteristic EPR signal (Fig. 3B, asterisk; Fig. S13). Formation of the N• was associated with partial decay of the EPR signals of both 1 and 2 (Fig. 3B, Fig. S14). This result is consistent with radical initiation by both complexes, at least for a single-turnover of the stoichiometric inactivator. However, we consider it likely that one of the two species is primarily or exclusively responsible for the protein’s robust catalytic activity.
X-ray crystallographic characterization of Fj Mn2II/II-β.
A 1.9-Å-resolution x-ray crystal structure of the Mn2II/II complex of Fj β (PDB accession code 6CWP) was obtained by soaking crystals of the metal-depleted protein (obtained by expression in the presence of 1,10-phenanthroline) with excess MnII in the absence of O2 (Fig. 4A-C). These conditions ensured that a homogeneous structure that is representative of the pre-activated state of the enzyme. The protein is a homodimer (β2) (Fig. S15), as are all other structurally characterized class I RNR βsubunits.12 In each monomer, a fully occupied Mn2II/II site is coordinated by the expected two His and four Glu motif (Fig. 4C, Figs. S16-S17) contributed by the four core α-helices of the all-helical topology that defines the ferritin-like superfamily.11, 77 Typically, enclosure of the cofactor within this helical core affords complete shielding from the solvent, a property that is expected to be crucial for the stability of the Tyr• and MnIV/FeIII radical initiators of the class Ia-c RNRs. The crystal structure of Fj Mn2II/II-β2 reveals a restructuring of the core α-helix (residues 50–82) that provides ligand Glu 67 of the characteristic Glu-Tyr pair. The backbone atoms of the six residues following this ligand (68–73) transition from the usual, canonical α-helical structure to a more extended conformation that resembles a β-strand (Fig. S18). The associated deviation in secondary structure opens a ~ 300 Å3 cavity that potentially exposes both metal ions to solvent (Fig. 4B). The unfolded helix accommodates, in addition to the aforementioned cavity, a short hydrophilic channel that is lined by ordered water molecules (Fig. 4D). A similar water-lined channel is a defining structural feature of the class Ib β subunits27, 78 and was proposed to conduct superoxide, generated by the flavoprotein activase, NrdI, to the Mn2 cofactor site in β Fig. S19).28 A hydrophilic route to the metal binding site might also be required in the Fj protein for the observed scavenging of free O2–. The unusual structural element in Fj β enables Lys 71 to insert its β-NH3+ functional group into a hydrogen-bond brace formed by the backbone carbonyl and side chain O atoms of ligands Glu 67 and Glu 97 at Mn site 1 (Fig. 4C). As a consequence, the cationic side chain of Lys 71 is close to the cofactor, ~ 4.2 Å from Mn1. A positively charged residue in the vicinity of the cofactor has not been observed in any other class Ia-c RNR β subunit studied to date (Fig. S21), although this feature was also recently noted in the aforementioned lower-resolution structure with partial CaII-occupancy (physiologically irrelevant) Lb β from the same sequence cluster.44 In fact, our network analysis indicated that Lys 71 is universally conserved across the new Id subclass (Fig. S20). The alteration in local electrostatic potential expected to accompany the approximation of the Lys 71 ammonium group and N-terminal dipole of the interrupted helix, together with the uniquely more open Mn2II/II site, may enable the assembly of the active cofactor by direct capture of O2–. A similar structural motif is an essential feature of certain SOD enzymes.79 Additionally, the open nature of the metal site potentially rationalizes the ease of cofactor reduction without accumulation of an intermediate Mn2III/III (met) state.
Fig. 4. A cavity and solvent channel associated with a disrupted core α-helix revealed by x-ray crystal structures of Fj RNR β.

(A) Comparison of the overall folds of Fj β (left) and Escherichia coli (Ec) class Ia β (PDB accession code 1PIY; right). Core alpha helix 1 (orange) in Fj β undergoes a structural transition near the MnII-binding site. Pink spheres are MnII ions and green spheres are FeII ions. (B) Right panel: a model of the internal cavity and solvent channel that are both accommodated by the disruption to core helix 1 (orange); Left panel: the cavity potentially exposes both metal ions (purple) to solvent. (C) Close-up of the metal binding site in the structure of the Mn2II/II state of Fj β from anoxically prepared crystals revealing a proximal Lys residue in the cavity and an exogenous ligand bound at site 1 (modeled as a water molecule, w1, shown as red sphere). (D) A surface-bound divalent metal ion (MgII, green sphere) is linked via the solvent channel to the cofactor site.
The crystal structure also reveals that Tyr 104 of the new class Id Fj β is displaced from the conserved position of its counterpart in the Ia/b β proteins, in which the Tyr residue harbors a stable radical (Fig. 5A). In the reported structures of Ia/b β s with either MnII or FeII bound, the unique Asp ligand (the first ligand residue in the sequence) is monodentate to the metal ion in site 1, while its other oxygen accepts a hydrogen-bond from the phenolic hydroxyl group of the radical-harboring Tyr.27, 80 The proximity of the Tyr and Asp residues is enforced, in part, by the steric demands of the β-branched side chain of a conserved Ile residue (Fig. 5A, right panels). In the Ia/b β proteins, the residue appears to crowd the Tyr and its phenolic oxygen toward metal site 1, resulting in a 4–5 Å separation. This configuration may enable proton-transfer steps that are coupled to both its oxidation to Tyr• during the activation process and its subsequent transient reduction during catalysis.19, 81 Tyr 104 in Fj β is rotated away from Mn1, with its side chain hydroxyl group 7.5 Å away and lacking a direct hydrogen-bond connection to the cofactor (Fig. 5A, top left). The residue instead interacts with the side-chain hydroxyl group of Thr 191, aligning with the conserved Ile residues that crowd the Tyr in the Ia/b proteins. The β proteins that are identified by the Glu/Tyr pair and Lys 71 equivalent as members of the new Id subclass all have either Thr or Ser at this position (Figs. S20-21). The sequestration of the Tyr 104 side chain via a neutral, second-sphere hydrogen-bonding partner could rationalize both the failure of the residue to be oxidized to Tyr• and the production of the same pair of oxidized Mn2 complexes in the Y104F variant. However, it is not obvious how Tyr 104 could, from this location, play an essential role in nucleotide reduction. It is thus plausible that a conformational change driven by interaction of β with α alters the disposition of the side chain relative to the cofactor during turnover.
Possible evolutionary paths to the four subclasses, a-d.
The subclass-d-founding Fj and Lb RNRs join the class Ic Ct enzyme as cases in which an oxidized dimetal cluster acts directly as radical initiator rather than merely as generator of the catalytic Tyr•, as occurs in the longer-known Ia/b enzymes.12, 31 It further exemplifies a distinct type of dimetal cluster with the capacity for the tightly controlled, bidirectional radical transfer that is necessary for robust, efficient catalytic nucleotide reduction within the α subunit. Given the indispensability of the metal cluster in all known cases, either for initiating turnover or in the pre-catalytic activation step, the original class I enzyme would almost certainly have employed a metal-based oxidant. But from which metal(s) was it composed?
Relative to the other class I RNRs, activation of the Id Fj β is uniquely simple. It appears to entail the simple addition of O2– to the Mn2II/II cluster. Already redox balanced for formation of the one-electron-oxidizing cofactor, the reaction does not require complex electron trafficking, as occurs in formation of the Tyr•-generating Fe2III/IV intermediate from the Fe2II/II cluster and O2 during activation of Ia βs82 and decay of the MnIV/FeIV intermediate during activation of the Ct Ic β39 There is currently no indication that an accessory protein factor, such as the NrdI protein that activates Ib βs,12 is required to activate the class Id protein. It is possible that an as-yet-undiscovered dedicated superoxide donor for Fj β exists in vivo, however, the capacity to be activated in vitro without such a source is notable and distinctive. By its use of a homodinuclear metallocofactor, Fj β avoids the imperative faced by the Mn/Fe-dependent Ic βs to direct two different divalent transition metal ions into their proper sites.37 Might this simplest activation mechanism and cofactor structure of the Id RNR resemble those of the original class I enzyme that was spawned by rising levels of O2? One might hope to address this question by phylogenetic analysis of the known RNRs, but it was not possible to define with confidence the evolutionary path to the four known subclasses. Rooted trees reflecting either initial divergence of the subclass a and b lineage from the c and d lineage (Figs. S22-23) or initial divergence of the class Id lineage from that leading to the subclass a-c enzymes (Fig. 5B) were obtained, depending on the choice of outgroup.
From a structural perspective, an initial divergence of the class Id lineage is favored. A comparison of the oxidant sites (Fig. 5A) shows that the class Ia-c βs are much more similar to one another in this region than they are to Fj β, particularly in the rotamer of the Tyr equivalent (Phe 127) and presence of the Ile residue that impacts the position of this Tyr/Phe. Its early divergence from the class Ia-c lineage might suggest that the modern-day Id subclass would retain more aspects of the original β cofactor structure and radical-initiation mechanism, including, perhaps, the identities of the metal ions and activating oxidant as well as the conformation, environment, and role of the key Tyr. In this scenario, structural adjustments early along the divergent a-c lineage would have set the stage for production and stabilization of the Tyr• along the a/b branch, and later substitution of this Tyr for Phe would have been a crucial event in emergence of subclass c. This analysis suggests that, rather than using O2 itself, the original class I RNR might have exploited its reduction product, superoxide, to perpetuate DNA synthesis under the threat of a class-III-like enzyme succumbing to rising levels of O2.6 Evolution of the O2-activated a-c subclasses, with their more complex iron- or flavoprotein-dependent activation mechanisms, would then have come later, perhaps driven by the emergence of oxidant-defense enzymes that scavenged superoxide and impeded activation of the initial, class Id-like enzyme.
Supplementary Material
ACKNOWLEDGMENTS:
This work was supported by grants from the Searle Scholars Program (to A.K.B.) and the NIH (GM100011 and GM119707 to A.K.B. and GM111978 to M.-E.P.). C.J.P. and E.J.B. are supported by the NIH via National Research Service Awards (GM113389–01 and GM116353–01). Portions of this work were conducted at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. We also acknowledge the resources of the Berkeley Center for Structural Biology at the Advanced Light Source (ALS), which is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The ALS is a Department of Energy Office of Science User Facility under Contract No. DE-AC02–05CH11231. Additional work was performed at the Advanced Photon Source (APS), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. GM/CA at APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by an NIH–Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289–01A1). Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We thank J. Morán (Univeristy Pública de Navarra) for the gift of the expression vector for Vu SOD and J. Stubbe (Massachusetts Institute of Technology) for materials and helpful discussions.
REFERENCES
- [1].Nordlund P, and Reichard P (2006) Ribonucleotide reductases, Annu Rev Biochem 75, 681–706. [DOI] [PubMed] [Google Scholar]
- [2].Stubbe J (2000) Ribonucleotide reductases: the link between an RNA and a DNA world?, Curr Opin Struct Biol 10, 731–736. [DOI] [PubMed] [Google Scholar]
- [3].Stubbe J, and Ackles D (1980) On the mechanism of ribonucleoside diphosphate reductase from Escherichia coli. Evidence for 3’-C--H bond cleavage, J Biol Chem 255, 8027–8030. [PubMed] [Google Scholar]
- [4].Mao SS, Yu GX, Chalfoun D, and Stubbe J (1992) Characterization of C439SR1, a mutant of Escherichia coli ribonucleotide diphosphate reductase: evidence that C439 is a residue essential for nucleotide reduction and C439SR1 is a protein possessing novel thioredoxin-like activity, Biochemistry 31, 9752–9759. [DOI] [PubMed] [Google Scholar]
- [5].Licht S, Gerfen GJ, and Stubbe J (1996) Thiyl radicals in ribonucleotide reductases, Science 271, 477–481. [DOI] [PubMed] [Google Scholar]
- [6].Reichard P (1993) From RNA to DNA, why so many ribonucleotide reductases?, Science 260, 1773–1777. [DOI] [PubMed] [Google Scholar]
- [7].Sun X, Harder J, Krook M, Jornvall H, Sjöberg B-M, and Reichard P (1993) A possible glycine radical in anaerobic ribonucleotide reductase from Escherichia coli: nucleotide sequence of the cloned nrdD gene, Proc Natl Acad Sci U S A 90, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, and Miller NE (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods, Nucleic Acids Res 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lundin D, Berggren G, Logan DT, and Sjöberg B-M (2015) The origin and evolution of ribonucleotide reduction, Life (Basel) 5, 604–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Krebs C, Bollinger JM Jr., and Booker SJ (2011) Cyanobacterial alkane biosynthesis further expands the catalytic repertoire of the ferritin-like ‘di-iron-carboxylate’ proteins, Curr Opin Chem Biol 15, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Andrews SC (2010) The ferritin-like superfamily: evolution of the biological iron storeman from a rubrerythrin-like ancestor, Biochim Biophys Acta 1800, 691–705. [DOI] [PubMed] [Google Scholar]
- [12].Cotruvo JA Jr., and Stubbe J (2011) Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo, Annu Rev Biochem 80, 733–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Atkin CL, Thelander L, Reichard P, and Lang G (1973) Iron and free radical in ribonucleotide reductase. Exchange of iron and Mössbauer spectroscopy of the protein B2 subunit of the Escherichia coli enzyme, J Biol Chem 248, 7464–7472. [PubMed] [Google Scholar]
- [14].Tong WH; Chen S; Lloyd SG; Edmondson DE; Hunynh BH; Stubbe J (1996) Mechanism of assembly of the diferric cluster-tyrosyl radical cofactor of Escherichia coli ribonucleotide reductase from the diferrous form of the R2 subunit, J Am Chem Soc 118, 2107–2108. [Google Scholar]
- [15].Doan PE, Shanmugam M, Stubbe J, and Hoffman BM (2015) Composition and structure of the inorganic core of relaxed intermediate X(Y122F) of Escherichia coli ribonucleotide reductase, J Am Chem Soc 137, 15558–15566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Burdi D, Sturgeon BE, Tong WH, Stubbe J, and Hoffman BM (1996) Rapid freeze-quench ENDOR of the radical X intermediate of Escherichia coli ribonucleotide reductase using 17O2, (H2O)-17O, and (H2O)-2H, J Am Chem Soc 118, 281–282. [Google Scholar]
- [17].Bollinger JM Jr., Edmondson DE, Huynh BH, Filley J, Norton JR, and Stubbe J (1991) Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase, Science 253, 292–298. [DOI] [PubMed] [Google Scholar]
- [18].Bollinger JM Jr., Tong WH, Ravi N, Huynh BH, Edmondson DE, and Stubbe J (1994) Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of Escherichia coli ribonucleotide reductase 2. Kinetics of the excess Fe2+ reaction by optical, EPR, and Mössbauer spectroscopies, J Am Chem Soc 116, 8015–8023. [Google Scholar]
- [19].Stubbe J, Nocera DG, Yee CS, and Chang MCY (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer?, Chem Rev 103, 2167–2201. [DOI] [PubMed] [Google Scholar]
- [20].Minnihan EC, Nocera DG, and Stubbe J (2013) Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase, Acc Chem Res 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dassama LMK, Jiang W, Varano PT, Pandelia M-E, Conner DA, Xie J, Bollinger JM Jr., and Krebs C (2012) Radical-translocation intermediates and hurdling of pathway defects in “super-oxidized” (Mn(IV)/Fe(IV)) Chlamydia trachomatis ribonucleotide reductase, J Am Chem Soc 134, 20498–20506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Uhlin U, and Eklund H (1994) Structure of ribonucleotide reductase protein R1, Nature 370, 533–539. [DOI] [PubMed] [Google Scholar]
- [23].Nordlund P, and Eklund H (1993) Structure and function of the Escherichia coli ribonucleotide reductase protein R2, J Mol Biol 232, 123–164. [DOI] [PubMed] [Google Scholar]
- [24].Livada J, Martinie RJ, Dassama LMK, Krebs C, Bollinger JM Jr., and Silakov A (2015) Direct measurement of the radical translocation distance in the class I ribonucleotide reductase from Chlamydia trachomatis, J Phys Chem B 119, 13777–13784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bennati M, Weber A, Antonic J, Perlstein DL, Robblee J, and Stubbe J (2003) Pulsed ELDOR spectroscopy measures the distance between the two tyrosyl dadicals in the R2 subunit of the E. coli ribonucleotide reductase, J Am Chem Soc 125, 14988–14989. [DOI] [PubMed] [Google Scholar]
- [26].Cotruvo JA Jr., and Stubbe J (2010) An active dimanganese(III)-tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase, Biochemistry 49, 1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Boal AK, Cotruvo JA Jr., Stubbe J, and Rosenzweig AC (2010) Structural basis for activation of class Ib ribonucleotide reductase, Science 329, 1526–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cotruvo JA Jr., Stich TA, Britt RD, and Stubbe J (2013) Mechanism of assembly of the dimanganese-tyrosyl radical cofactor of class Ib ribonucleotide reductase: Enzymatic generation of superoxide is required for tyrosine oxidation via a Mn(III)Mn(IV) intermediate, J Am Chem Soc 135, 4027–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Huque Y, Fieschi F, Torrents E, Gibert I, Eliasson R, Reichard P, Sahlin M, and Sjöberg BM (2000) The active form of the R2F protein of class Ib ribonucleotide reductase from Corynebacterium ammoniagenes is a diferric protein, J Biol Chem 275, 25365–25371. [DOI] [PubMed] [Google Scholar]
- [30].Högbom M, Stenmark P, Voevodskaya N, McClarty G, Gräslund A, and Nordlund P (2004) The radical site in chlamydial ribonucleotide reductase defines a new R2 subclass, Science 305, 245–248. [DOI] [PubMed] [Google Scholar]
- [31].Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak MA, Krebs C, and Bollinger JM Jr. (2007) A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase, Science 316, 1188–1191. [DOI] [PubMed] [Google Scholar]
- [32].Jiang W, Yun D, Saleh L, Bollinger JM Jr., and Krebs C (2008) Formation and function of the manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase, Biochemistry 47, 13736–13744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jiang W, Hoffart LM, Krebs C, and Bollinger JM Jr. (2007) A manganese(IV)/iron(IV) intermediate in assembly of the manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase, Biochemistry 46, 8709–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jiang W, Bollinger JM Jr., and Krebs C (2007) The active form of Chlamydia trachomatis ribonucleotide reductase R2 protein contains a heterodinuclear Mn(IV)/Fe(III) cluster with S = 1 ground state, J Am Chem Soc 129, 7504–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Andersson CS, Öhrström M, Popović-Bijelić A, Gräslund A, Stenmark P, and Högbom M (2012) The manganese ion of the heterodinuclear Mn/Fe cofactor in Chlamydia trachomatis ribonucleotide reductase R2c is located at metal position 1, J Am Chem Soc 134, 123–125. [DOI] [PubMed] [Google Scholar]
- [36].Dassama LMK, Boal AK, Krebs C, Rosenzweig AC, and Bollinger JM Jr. (2012) Evidence that the beta subunit of Chlamydia trachomatis ribonucleotide reductase is active with the manganese ion of its manganese(IV)/iron(III) cofactor in site 1, J Am Chem Soc 134, 2520–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dassama LMK, Krebs C, Bollinger JM Jr., Rosenzweig AC, and Boal AK (2013) Structural basis for assembly of the Mn(IV)/Fe(III) cofactor in the class Ic ribonucleotide reductase from Chlamydia trachomatis, Biochemistry 52, 6424–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nordlund P, Sjöberg B-M, and Eklund H (1990) Three-dimensional structure of the free radical protein of ribonucleotide reductase, Nature 345, 593–598. [DOI] [PubMed] [Google Scholar]
- [39].Jiang W, Saleh L, Barr EW, Xie J, Gardner MM, Krebs C, and Bollinger JM Jr. (2008) Branched activation- and catalysis-specific pathways for electron relay to the manganese/iron cofactor in ribonucleotide reductase from Chlamydia trachomatis, Biochemistry 47, 8477–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jiang W, Xie J, Nørgaard H, Bollinger JM Jr., and Krebs C (2008) Rapid and quantitative activation of Chlamydia trachomatis ribonucleotide reductase by hydrogen peroxide, Biochemistry 47, 4477–4483. [DOI] [PubMed] [Google Scholar]
- [41].Roshick C, Iliffe-Lee ER, and McClarty G (2000) Cloning and characterization of ribonucleotide reductase from Chlamydia trachomatis, J Biol Chem 275, 38111–38119. [DOI] [PubMed] [Google Scholar]
- [42].Bollinger JM Jr., Jiang W, Green MT, and Krebs C (2008) The manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase: structure, assembly, radical initiation, and evolution, Curr Opin Struct Biol 18, 650–657. [DOI] [PubMed] [Google Scholar]
- [43].McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, Rhodes RG, Goltsman E, Wang W, Xu J, Hunnicutt DW, Staroscik AM, Hoover TR, Cheng YQ, and Stein JL (2009) Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis, Appl Environ Microbiol 75, 6864–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Grinberg IR, Lundin D, Hasan M, Crona M, Jonna VR, Loderer C, Sahlin M, Markova N, Borovok I, Berggren G, Hofer A, Logan DT, and Sjöberg B-M (2018) Novel ATP-cone-driven allosteric regulation of ribonucleotide reductase via the radical-generating subunit, eLife 7, e31529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Delgado-Jaime MU, Mewis CP, and Kennepohl P (2010) Blueprint XAS: a Matlab-based toolbox for the fitting and analysis of XAS spectra, J Synchrotron Radiat 17, 132–137. [DOI] [PubMed] [Google Scholar]
- [46].Bencini A, and Gatteschi D (1990) Electron paramagnetic resonance of exchange-coupled systems, Springer-Verlag, Berlin. [Google Scholar]
- [47].Pearce LL, Kurtz DM Jr., Xia Y-M, and Debrunner PG (1987) Reduction of the binuclear iron site in octameric methemerythrins. Characterization of intermediates and a unifying reaction scheme., J Am Chem Soc 109, 7286–7293. [Google Scholar]
- [48].Rutter R, Hager LP, Dhonau H, Hendrich M, Valentine M, and Debrunner P (1984) Chloroperoxidase compound I: Electron paramagnetic resonance and Mössbauer studies, Biochemistry 23, 6809–6816. [DOI] [PubMed] [Google Scholar]
- [49].Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [50].McCoy AJ, Grosse-Kunstleve RW, Storoni LC, and Read RJ (2005) Likelihood-enhanced fast translation functions, Acta Crystallogr D 61, 458–464. [DOI] [PubMed] [Google Scholar]
- [51].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments, Acta Crystallogr D 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics, Acta Crystallogr D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [53].Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr D 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [54].Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, and Adams PD (2008) Automated structure solution with the PHENIX suite, Methods Mol Biol 426, 419–435. [DOI] [PubMed] [Google Scholar]
- [55].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr D 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Abascal F, Zardoya R, and Posada D (2005) ProtTest: selection of best-fit models of protein evolution, Bioinformatics 21, 2104–2105. [DOI] [PubMed] [Google Scholar]
- [58].Lundin D, Poole AM, Sjöberg B-M, and Högbom M (2012) Use of structural phylogenetic networks for classification of the ferritin-like superfamily, J Biol Chem 287, 20565–20575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Salowe SP, and Stubbe J (1986) Cloning, overproduction, and purification of the B2 subunit of ribonucleoside-diphosphate reductase, J Bacteriol 165, 363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].O’Brien PJ (1991) Molecular mechanisms of quinone cytotoxicity, Chem Biol Interact 80, 1–41. [DOI] [PubMed] [Google Scholar]
- [61].Cadenas E, Boveris A, Ragan CI, and Stoppani AO (1977) Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria, Arch Biochem Biophys 180, 248–257. [DOI] [PubMed] [Google Scholar]
- [62].Massey V (1994) Activation of molecular oxygen by flavins and flavoproteins, J Biol Chem 269, 22459–22462. [PubMed] [Google Scholar]
- [63].Cotruvo JA Jr., and Stubbe J (2011) Escherichia coli class Ib ribonucleotide reductase contains a dimanganese(III)-tyrosyl radical cofactor in vivo, Biochemistry 50, 1672–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Reed GH, and Markham GD (1984) EPR of Mn(II) complexes with enzymes and other proteins, In Biological Magnetic Resonance (Berliner LJ, Ed.), pp 73–142, Plenum Press, New York. [Google Scholar]
- [65].Pierce BS, Elgren TE, and Hendrich MP (2003) Mechanistic implications for the formation of the diiron cluster in ribonucleotide reductase provided by quantitative EPR spectroscopy, J Am Chem Soc 125, 8748–8759. [DOI] [PubMed] [Google Scholar]
- [66].Cox N, Ogata H, Stølle P, Reijerse E, Auling G, and Lubitz W (2010) A tyrosyl-dimanganese coupled spin system is the native metalloradical cofactor of the R2F subunit of the ribonucleotide reductase of Corynebacterium ammoniagenes, J Am Chem Soc 132, 11197–11213. [DOI] [PubMed] [Google Scholar]
- [67].Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, and Solomon EI (1997) A multiplet analysis of Fe K-edge 1s → 3d pre-edge features of iron complexes, J Am Chem Soc 119, 6297–6314. [Google Scholar]
- [68].Martinie RJ, Blaesi EJ, Krebs C, Bollinger JM Jr., Silakov A, and Pollock CJ (2017) Evidence for a di-mu-oxo diamond core in the Mn(IV)/Fe(IV) activation intermediate of ribonucleotide reductase from Chlamydia trachomatis, J Am Chem Soc 139, 1950–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Teutloff C, Schafer KO, Sinnecker S, Barynin V, Bittl R, Wieghardt K, Lendzian F, and Lubitz W (2005) High-field EPR investigations of MnIIIMnIV and MnIIMnIII states of dimanganese catalase and related model systems, Magn Reson Chem 43, S51–S64. [DOI] [PubMed] [Google Scholar]
- [70].Khangulov S, Sivaraja M, Barynin VV, and Dismukes GC (1993) The dimanganese(III,IV) oxidation state of catalase from Thermus thermophilus: electron nuclear double resonance analysis of water and protein ligands in the active site, Biochemistry 32, 4912–4924. [DOI] [PubMed] [Google Scholar]
- [71].Christou G (1991) Mixed valence manganese carboxylates of various nuclearities, In Mixed Valency Systems: Applications in Chemistry, Physics, and Biology (Prassides K, Ed.), pp 371–376, Kluwer Acadmic Publishers, Dordrecht. [Google Scholar]
- [72].Baldwin MJ, Stemmler TL, Riggs-Gelasco PJ, Kirk ML, Penner-Hahn JE, and Pecoraro VL (1994) Structural and magnetic effects of successive protonations of oxo bridges in high-valent manganese dimers, J Am Chem Soc 116, 11349–11356. [Google Scholar]
- [73].Orio M, Pantazis DA, Petrenko T, and Neese F (2009) Magnetic and spectroscopic properties of mixed valence manganese(III,IV) dimers: a systematic study using broken symmetry density functional theory, Inorg Chem 48, 7251–7260. [DOI] [PubMed] [Google Scholar]
- [74].Pantazis DA, Krewald V, Orio M, and Neese F (2010) Theoretical magnetochemistry of dinuclear manganese complexes: broken symmetry density functional theory investigation on the influence of bridging motifs on structure and magnetism, Dalton Trans 39, 4959–4967. [DOI] [PubMed] [Google Scholar]
- [75].Skoog L, Bjursell G, Thelander L, Hagerstrom T, Hobbs J, and Eckstein F (1977) 2’-Deoxy-2’-azidocytidine, a new inhibitor of DNA replication in mammalian cells, Eur J Biochem 72, 371–378. [DOI] [PubMed] [Google Scholar]
- [76].Fritscher J, Artin E, Wnuk S, Bar G, Robblee JH, Kacprzak S, Kaupp M, Griffin RG, Bennati M, and Stubbe J (2005) Structure of the nitrogen-centered radical formed during inactivation of E. coli ribonucleotide reductase by 2’-azido-2’-deoxyuridine-5’-diphosphate: trapping of the 3’-ketonucleotide, J Am Chem Soc 127, 7729–7738. [DOI] [PubMed] [Google Scholar]
- [77].Cooley RB, Arp DJ, and Karplus PA (2010) Evolutionary origin of a secondary structure: pi-helices as cryptic but widespread insertional variations of alpha-helices that enhance protein functionality, J Mol Biol 404, 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Eriksson M, Jordan A, and Eklund H (1998) Structure of Salmonella typhimurium NrdF ribonucleotide reductase in its oxidized and reduced forms, Biochemistry 37, 13359–13369. [DOI] [PubMed] [Google Scholar]
- [79].Strange RW, Yong CW, Smith W, and Hasnain SS (2007) Molecular dynamics using atomic-resolution structure reveal structural fluctuations that may lead to polymerization of human Cu-Zn superoxide dismutase, Proc Natl Acad Sci U S A 104, 10040–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Voegtli WC, Sommerhalter M, Saleh L, Baldwin J, Bollinger JM Jr., and Rosenzweig AC (2003) Variable coordination geometries at the diiron(II) active site of ribonucleotide reductase R2, J Am Chem Soc 125, 15822–15830. [DOI] [PubMed] [Google Scholar]
- [81].Wörsdörfer B, Conner DA, Yokoyama K, Livada J, Seyedsayamdost M, Jiang W, Silakov A, Stubbe J, Bollinger JM Jr., and Krebs C (2013) Function of the diiron cluster of Escherichia coli class Ia ribonucleotide reductase in proton-coupled electron transfer, J Am Chem Soc 135, 8585–8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, and Bollinger JM Jr. (2000) Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical., J Am Chem Soc 122, 12195–12206. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.