

Abstract
Background
Tuberculosis causes more deaths than any other infectious disease globally. Bacillus Calmette‐Guérin (BCG) is the only available vaccine, but protection is incomplete and variable. The modified Vaccinia Ankara virus expressing antigen 85A (MVA85A) is a viral vector vaccine produced to prevent tuberculosis.
Objectives
To assess and summarize the effects of the MVA85A vaccine boosting BCG in humans.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register; Central Register of Controlled Trials (CENTRAL); MEDLINE (PubMed); Embase (Ovid); and four other databases. We searched the WHO ICTRP and ClinicalTrials.gov. All searches were run up to 10 May 2018.
Selection criteria
We evaluated randomized controlled trials of MVA85A vaccine given with BCG in people regardless of age or HIV status.
Data collection and analysis
Two review authors independently assessed the eligibility and risk of bias of trials, and extracted and analyzed data. The primary outcome was active tuberculosis disease. We summarized dichotomous outcomes using risk ratios (RR) and risk differences (RD), with 95% confidence intervals (CI). Where appropriate, we combined data in meta‐analyses. Where meta‐analysis was inappropriate, we summarized results narratively.
Main results
The search identified six studies relating to four Phase 2 randomized controlled trials enrolling 3838 participants. Funding was by government bodies, charities, and philanthropic donors. Five studies included infants, one of them infants born to HIV‐positive mothers. One study included adults living with HIV. All trials included authors from Oxford University who led the laboratory development of the vaccine. Participants received intradermal MVA85A after BCG in some studies, and before selective deferred BCG in HIV‐exposed infants.
The largest trial in 2797 African children was well conducted with low risk of bias for most parameters. Risk of bias was uncertain for selective reporting because there were no precise case definition endpoints for active tuberculosis published prior to the trial analysis.
MVA85A added to BCG compared to BCG alone probably has no effect on the risk of developing microbiologically confirmed tuberculosis (RR 0.97, 95% CI 0.58 to 1.62; 3439 participants, 2 trials; moderate‐certainty evidence), or the risk of starting on tuberculosis treatment (RR 1.10, 95% CI 0.92 to 1.33; 3687 participants, 3 trials; moderate‐certainty evidence). MVA85A probably has no effect on the risk of developing latent tuberculosis (RR 1.01, 95% CI 0.85 to 1.21; 3831 participants, 4 trials; moderate‐certainty evidence). Vaccinating people with MVA85A in addition to BCG did not cause life‐threatening serious adverse effects (RD 0.00, 95% CI –0.00 to 0.00; 3692 participants, 3 trials; high‐certainty evidence). Vaccination with MVA85A is probably associated with an increased risk of local skin adverse effects (3187 participants, 3 trials; moderate‐certainty evidence), but not systemic adverse effect related to vaccination (144 participants, 1 trial; low‐certainty evidence). This safety profile is consistent with Phase 1 studies which outlined a transient, superficial reaction local to the injection site and mild short‐lived symptoms such as malaise and fever.
Authors' conclusions
MVA85A delivered by intradermal injection in addition to BCG is safe but not effective in reducing the risk of developing tuberculosis.
1 May 2019
Up to date
All studies incorporated from most recent search
All published trials found in the last search (10 May, 2018) were included.
Keywords: Humans; BCG Vaccine; Tuberculosis Vaccines; HIV Seropositivity; HIV Seropositivity/complications; HIV Seropositivity/immunology; Primary Prevention; Randomized Controlled Trials as Topic; Tuberculosis; Tuberculosis/prevention & control; Vaccines, DNA
Plain language summary
MVA85A vaccine as a booster to BCG for prevention of tuberculosis
What is the aim of this review?
The aim of this Cochrane review was to evaluate the effectiveness and safety of using MVA85A in addition to BCG compared to using BCG alone for prevention of tuberculosis.
Key messages
MVA85A in addition to BCG showed no added benefit to BCG in prevention of acquiring tuberculosis.
What was studied in the review?
Tuberculosis is an infectious airborne disease which affects the lungs and other organs in the body. It can either be active when a person shows signs and symptoms or has confirmatory tests for tuberculosis or latent when a person has inhaled the bacteria before but does not show signs and symptoms of sickness. Currently, there is only one vaccine licensed for prevention of this disease, which is called BCG. However, the ability for the BCG vaccine to prevent tuberculosis differs in different settings and patient groups resulting in tuberculosis still remaining a problem worldwide despite children being immunized. MVA85A is a vaccine that was investigated for prevention of tuberculosis with the hope that when used in addition to BCG it will improve prevention of people getting tuberculosis.
What are the main results of this review?
After examining the research published up to 10 May 2018, we included six study findings from four randomized controlled trials (clinical trials where people are randomly put into one of two or more treatment groups), enrolling 3838 children and adults. Based on these studies of mostly children and adults living in Africa, MVA85A added to BCG compared to BCG alone probably has no effect on the risk of developing active tuberculosis defined as microbiologically confirmed tuberculosis (moderate‐certainty evidence) or the risk of starting on tuberculosis treatment (moderate‐certainty evidence). MVA85A has no effect on the risk of developing latent tuberculosis (moderate‐certainty evidence). MVA85A does not cause any life‐threatening serious side effects (highly‐certainty evidence). There were more local skin reactions in people vaccinated with MVA85A, however, there was no increase in overall side effects in people given MVA85A.
How up‐to‐date is this review?
The review authors searched for studies that have been published up to May 2018.
Summary of findings
Summary of findings for the main comparison. MVA85A compared to placebo for preventing tuberculosis.
| MVA85A compared to placebo for preventing tuberculosis | ||||||
| Patient or population: HIV‐positive and ‐negative adults and children Setting: South Africa, Senegal Intervention: MVA85A Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (trials) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with MVA85A | |||||
| Active tuberculosis: confirmed by culture or Xpert® MTB/RIF longest reported follow‐up | 17 per 1000 | 16 per 1000 (10 to 28) | RR 0.97 (0.58 to 1.62) | 3439 (2 RCTs) | ⊕⊕⊕⊝ Moderatea,b,c | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of developing active tuberculosis. |
| Active tuberculosis: started on tuberculosis treatment | 102 per 1000 | 112 per 1000 (94 to 136) | RR 1.10 (0.92 to 1.33) | 3687 (3 RCTs) | ⊕⊕⊕⊝ Moderatea,c,d | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of needing to start tuberculosis treatment. |
| Latent tuberculosis | 114 per 1000 | 115 per 1000 (97 to 138) | RR 1.01 (0.85 to 1.21) | 3831 (4 RCTs) | ⊕⊕⊕⊝ Moderatec,d,e | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of developing latent tuberculosis. |
| Serious adverse effects | 1 per 1000 | 1 per 1000 (0 to 4) | RD 0.00 (–0.00 to 0.00)f | 3692 (3 RCTs) | ⊕⊕⊕⊕ High | Vaccinating people with MVA85A in addition to BCG did not cause life‐threatening serious adverse effects. |
| Adverse effects of any severity (local reactions of the skin) | Vaccination with MVA85A was associated with more reactions at the site of the injection.g | — | 3187 (3 RCTs) | ⊕⊕⊕⊝ Moderateh,i,j | Vaccinating people with MVA85A in addition to BCG probably increased the risk of having an adverse reaction related to vaccination at the site of the injection. | |
| Adverse effects of any severity (systemic symptoms) | Adverse events reported included malaise, lethargy, fever, and vomiting although differences between groups were not significant at a 95% CI level.g | — | 144 (1 RCT) |
⊕⊕⊝⊝ Lowk,l,m | Vaccinating people with MVA85A in addition to BCG may not have been associated with an increase in adverse effects related to vaccination. | |
| Adverse events of any severity | 808 per 1000 | 849 per 1000 (824 to 873) | RR 1.05 (1.02 to 1.08) | 3836 (4 RCTs) | ⊕⊕⊕⊕ Highn | Vaccination with MVA85A alone slightly increased the risk of having an adverse event. |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BCG: Bacillus Calmette‐Guérin; CI: confidence interval; RCT: randomized controlled trial; RD: risk difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aNot downgraded for risk of bias. The largest trial was at unclear risk of bias due to selective reporting; however, the outcomes presented were unlikely to be affected by this (Tameris 2013). bDowngraded by one level for imprecision. Few events and wide CIs containing clinically appreciable benefit and harm. cNot downgraded for indirectness. The only trial in HIV‐positive adults was stopped early meaning it was underpowered to detect efficacy (Ndiaye 2015). Therefore, evidence of efficacy is more generalizable to infants; however, results in adults were consistent with little or no effect being seen across all endpoints. dDowngraded by one level for imprecision. Broad CI containing little or no effect and clinically appreciable harm. eNot downgraded for risk of bias. The largest trial was at unclear risk of bias due to selective reporting; however, the outcome of latent tuberculosis was unlikely to be affected by this (Tameris 2013). fRisk difference presented as explained in our result section. gExtensive investigation of the vaccine in Phase 1 studies outlined in the Background of this review outlined "a transient, superficial reaction local to the injection site and mild short‐lived viral symptoms" consistent with the findings reported in the Phase 2 trials. hDowngraded by one level for imprecision. Broad CIs containing clinically appreciable benefit and harm. iNot downgraded for risk of bias. The largest study reported local adverse events and defined these as solicited by the vaccine (Tameris 2013). jNot downgraded for heterogeneity. While there might be some heterogeneity between the included trials in terms of time of outcome collection, the outcomes are consistent in favour to placebo as shown in Analysis 1.5. kDowngraded by one level for risk of bias. There were some deficiencies in the trial reporting these outcomes. lAdditional safety data from Phase 1 studies in 712 participants did not show any adverse effect signals (see section in Background of this review). mDowngraded by one level for imprecision. Few events reported in the largest trial (Tameris 2013), data not disaggregated in the second largest trial (Ndiaye 2015). nNot downgraded for inconsistency. I2 value of 37% judged to be non‐significant heterogeneity.
Background
Description of the condition
Tuberculosis is an infectious disease caused by Mycobacterium tuberculosis. It was estimated that 10 million people developed tuberculosis in 2017. Tuberculosis now ranks first, followed by HIV, as the leading cause of death from an infectious disease worldwide killing an estimated 1.6 million people in 2017, including 300,000 people living with HIV. Over 95% of these people were living in low‐ and middle‐income countries (WHO 2018).
Tuberculosis can be classed as active when people experience signs or symptoms of tuberculosis or have radiological evidence of it. Tuberculosis can also be classified as latent tuberculosis infection (LTBI) where immunological evidence of previous exposure to M tuberculosis exists without clinical or radiological evidence of the disease (CDC 2000). Of healthy adults with immunological evidence of previous exposure to M tuberculosis, the overall lifetime risk of progressing to active disease if not treated for the infection is 5% to 10% (Harries 2006). Often this happens months or years after the initial infection in response to a weakening of the body's immune system. The probability of developing active disease is higher in HIV‐positive people, people with diabetes, and young children (Baker 2011; Perez‐Velez 2012; Tiemersma 2011). Fifty percent of infants with evidence of LTBI will progress to active disease if untreated (Marais 2004). People with LTBI require early diagnosis and treatment to reduce the pool of active tuberculosis cases. This is particularly important in high‐risk groups, such as those coinfected with HIV (Sharma 2012). Tuberculosis can be treated with long courses of multiple antibiotics, but the rise of HIV and spread of multidrug‐resistant tuberculosis (MDR‐TB) means that tuberculosis is still one of the largest threats to public health worldwide (WHO 2018). Structural determinants such as rapid urbanization of populations and economic inequalities, social determinants such as poverty and poor housing, alongside biological factors such as HIV and drug‐resistant strains of tuberculosis play a vital role in the spread of tuberculosis through vulnerable populations (Daftary 2012).
The Bacillus Calmette‐Guérin (BCG) vaccine is currently the only available vaccine. Epidemiological studies indicate that it has a protective effect against tuberculosis disease in children, particularly against the more severe forms of the disease such as tuberculosis meningitis or miliary tuberculosis (Roy 2014). The effectiveness of BCG differs greatly depending on the site of infection. It has consistent protection against tuberculous meningitis and miliary disease in children but variable protection against pulmonary tuberculosis (Abubakar 2013; Colditz 1995). As a result, despite many areas achieving high coverage of BCG vaccination, the disease remains a problem, and a new tuberculosis vaccine remains an important global research priority (WHO 2018).
Previously it has been impossible to ascertain reliably whether the BCG vaccine protected against active disease or infection with M tuberculosis. This was due to the tuberculin skin test being unable to distinguish between cases of LTBI and people who had been vaccinated with BCG (Roy 2014). Therefore, the development and use of interferon γ release assays (IGRA), which can distinguish between tuberculosis infection and vaccination, has proved useful. This has allowed researchers to establish that BCG vaccination reduces the risk of Mycobacterium infection in some settings (Eisenhut 2009).
Description of the intervention
Many researchers and policy makers emphasize that a new effective vaccine could be a major contribution to tuberculosis control and elimination as a public health problem (de Cassan 2010). There are 12 vaccine candidates in clinical trials: eight in Phase 2 or Phase 3, and four in Phase 1. They include candidates to prevent the development of tuberculosis, and candidates to help improve the outcomes of treatment for tuberculosis disease (WHO 2018).
The modified Vaccinia Ankara virus‐expressing antigen 85A (MVA85A) is a viral vector vaccine based on the modified Vaccinia Ankara (MVA) virus. MVA is an attenuated virus that does not replicate in human tissue and, as such, has been used as a platform to encode multiple antigens and allowing development of multivalent vaccines (Altenburg 2014). In this case, MVA has had pieces of DNA from M tuberculosis inserted into it, so that it expresses the antigen 85A. This antigen complex is an enzyme that is involved in the cell wall biosynthesis of M tuberculosis and constitutes a vital part of the way in which the bacteria forms its outer mycomembrane. This is important for the viability of the mycobacterium and works as an effective barrier to drug therapies by repelling some antibiotics and preventing them from entering the cell (Favrot 2013).
Immunological studies have shown that a prime boost strategy, where MVA85A is used to boost the effects of BCG, is effective in expanding immune responses specific to M tuberculosis (Beveridge 2007). Thus, MVA85A was proposed primarily as a booster to people already vaccinated with BCG (Tameris 2013). Further studies have assessed MVA85A in other regimens including in combination with other viral vector vaccines (Sheehan 2015).
How the intervention might work
MVA85A is the first vaccine since 1968 to be tested in efficacy trials (Tameris 2013). It has been tried with a promise of prolonged antimycobacterial immunity in human UK trials (McShane 2004), and in tuberculosis endemic areas (Hawkridge 2008). The intention is that MVA85A would boost the immune response to tuberculosis above that which is afforded by vaccination with BCG (Roy 2014). MVA85A is administered as a single intradermal dose in people who have already received BCG vaccine (Tameris 2013). Other routes have been studied in animal studies, such as intravenous administration (Romano 2006), and are being considered in humans (Satti 2014).
The researchers who developed the vaccine evaluated its effects in animals and conducted Phase 1 studies in humans. Early literature and reviews by the team noted the vaccine was safe and produced an immune response in several populations (McShane 2004; Rowland 2012).
One independent systematic review of the animal studies, carried out by some members of this Cochrane Review team, raised questions about whether these animal studies provided evidence of efficacy in the various animal models used (Kashangura 2015), when clinical and pathological endpoints were examined in a variety of animal models subjected to challenge studies. This has led to a debate about the reporting of animal studies, in particular the lack of published protocols so that the question being tackled in an animal study is made clear in advance (Cohen 2018). These studies administered BCG, BCG and MVA85A, or no vaccine. Afterwards, animals were exposed to tuberculosis challenge. Clearly progression to clinical trial is not solely based on evidence derived from preclinical efficacy studies, and MVA85A was evaluated in a number of trials in humans before proceeding to an efficacy study (McShane 2018). However, preclinical studies remain an important component of the tuberculosis vaccine development paradigm (Barker 2012; McShane 2014).
The systematic review of animal studies pointed out that there was one study in macaques where more monkeys required euthanasia in the MVA85A plus BCG vaccine group than the BCG control group (Kashangura 2015). This led to considerable controversy as to whether the publication of the results were delayed (Cohen 2018). The findings from this study could be the result of chance; or because the vaccine impaired functional immunity; or the result of a separate adverse effect. The vaccine development team then carried out a relatively large number of safety studies in humans; and, in their words, "none of the 14 trials of MVA85A in over 400 humans (the target species) before the infant efficacy trial showed a safety signal" (McShane 2018). The standard approach for Cochrane Reviews within the Cochrane Infectious Diseases Group is to only summarize efficacy trials. However, as the primary concern of the studies included in this review was safety, we summarized the considerable number of Phase 1 studies that the researchers carried out to exclude severe adverse effects attributable to the vaccine in humans in this 'Background' section of the review. We searched registered clinical trial databases (ClinicalTrials.gov, World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP), Pan African Trials Registry, EU Clinical Trials Register) in June 2017 and summarized the Phase 1 studies identified in Table 2. We found 21 separate studies as registered (prospectively and retrospectively) dating from 2003 with the most recent studies scheduled to complete follow‐up in 2018. In addition, we found an existing narrative review of Phase 1 studies (Rowland 2012), which summarized Phase 1 safety data relating to selected trials including unpublished data and compared this to selected trials in yellow fever and BCG.
1. Summary of Phase 1 studies.
| NCT trial number | Route | Dates | Intervention and schedule details | Country | Participants (age) | HIV | Adverse events | Reference |
| NCT00423566 | ID | 2002–2003 | MVA85A; 1 dose | UK | 14 adults (18–45 years) | –ve | 7 trials (112 participants); combined in 1 report: no serious AE attributable to the vaccine | McShane 2004; Rowland 2012 |
| NCT00423839 | ID | 2003–2005 | MVA85A; 1 dose, 2 doses (5 × 107 pfu) |
Gambia | 21 adults | NR | No serious AE attributable to the vaccine | Brookes 2008; Ibanga 2006; Owiafe 2012 |
| NCT00427830 | ID | 2003–2005 | MVA85A; 1 dose (5 × 107 pfu) | UK | 21 adults | –ve | No serious AE attributable to the vaccine | McShane 2004; Pathan 2012; Rowland 2012; Tanner 2014; Whelan 2009 |
| NCT00427453 | ID | 2003–2005 | MVA85A; 1 dose (5 × 107 pfu) | UK | 10 adults | –ve | No serious AE attributable to the vaccine | Pathan 2012; Rowland 2012 |
| NCT00456183 | ID | 2005–2007 | MVA85A, (5 × 107 pfu) | UK | 12 adults with latent tuberculosis | –ve | No vaccine‐related serious AEs 7 trials (112 participants; data combined in 1 report) |
Rowland 2012; Sander 2009; Tanner 2014 |
| NCT00465465 | ID | 2005–2007 | MVA85A; 1 dose (1 × 108 pfu for 12 participants, and 1 × 107 pfu for 12 participants) | UK | 24 adults | –ve | No serious AE attributable to the vaccine | Griffiths 2011; Matsumiya 2013; Pathan 2007; Rowland 2012 |
| NCT00460590 | ID | 2005–2008 | MVA85A (5 × 107 pfu) | South Africa | 36 adults and adolescents | –ve | No vaccine‐related serious AEs | Hawkridge 2008; Scriba 2010; Tameris 2014; Tanner 2014 |
| NCT00480454 | ID | 2006–2009 | MVA85A; 1 dose MVA85A (2.5 × 107 pfu, 5 × 107 pfu) Groups
|
The Gambia | 214 infants (4 months) | NR | No serious AE judged to be related to the vaccine | Odutola 2012; Ota 2011 |
| NCT00395720 | ID | 2006–2010 | MVA85A; 1 dose (5 × 107pfu for 10 participants, and 1 × 108 pfu for 10 participants) | UK | 20 adults | +ve | No serious AE attributable to the vaccine | Minassian 2011 |
| NCT00480558 | ID | 2007–2011 | MVA85A; 1 dose (5 × 107 pfu) 4 groups with background of
|
South Africa | 48 adults (18–50 years) | +ve | No vaccine‐related serious AEs | Scriba 2012; Tanner 2014; Tameris 2014 |
| NCT00653770 | ID | 2007–2010 | FP85A, MVA85A (5 × 107 pfu) | UK | 31 adults | –ve | No serious AE attributable to the vaccine | Rowland 2013 |
| NCT00548444 | ID | 2007–2010 | MVA85A; 1 dose (1 × 108 pfu), administered as 2 injections (5 × 107 pfu each injection) |
UK | 12 adults | –ve | 7 trials (112 participants); data combined in 1 report: no serious AE attributable to the vaccine | Porter (unpublished data: source Rowland 2012) |
| NCT00731471 | ID | 2008–2011 | MVA85A; 2 doses (spaced by 6–12 months) (1 × 108 pfu) | Senegal | 24 adults | +ve | No serious AE attributable to the vaccine | Dieye 2013 |
| NCT01181856 | ID IM |
2010–2011 | MVA85A; 1 dose (1 × 108 pfu) | UK | 24 adults | –ve | No serious AE attributable to the vaccine | Matsumiya 2013; Meyer 2013 |
| NCT01194180 | ID | 2010–2012 | MVA85A, BCG; 1 dose (1 × 108 pfu) Group A: BCG naive, no MVA85A Group B: BCG naive, MVA85A Group C: BCG vaccinated, no MVA85A Group D: BCG vaccinated, MVA85A. |
UK | 49 adults recruited; 48 completed study | –ve | No serious AE attributable to the vaccine | ; Harris 2014b; Matsumiya 2013 |
| NCT01497769 | Aerosol ID |
2011–2013 | MVA85A; 1 dose: 1 × 108, 1 × 107 pfu | UK | 24 adults | –ve | No vaccine related serious adverse effects. | Satti 2014 |
| NCT01683773 | ID | 2012–2014 | AERAS‐402 MVA85A; Group A: 2 doses AERAS‐402 then MVA85A Group B: 1 dose AERAS‐402 then MVA85A |
UK | 40 adults | –ve | No vaccine related serious AEs | Sheehan 2015 |
| NCT01879163 | ID | 2013–2014 | MVA85A IMX313; Group A: low‐dose MVA85A‐IMX313 (1 × 107 pfu) Group B: dose MVA85A‐IMX313 (5 × 107 pfu) Group C: MVA85A (5 × 107 pfu) |
UK | 30 BCG vaccinated adults | –ve | No vaccine‐related serious AE | Minhinnick 2016 |
| NCT01829490 | IM | 2013–2016 | MVA85A, ChAdOx1 85A; Group A: 1 dose ChAdOx1 85A Group B: 1 dose ChAdOx1 85A then MVA85A Group C: 2 doses ChAdOx1 85A then MVA85A (1 × 108 pfu) |
UK | 42 adults | –ve | No data reported yet | No publication NCT01829490 |
| NCT01954563 | Aerosol ID |
2013–2016 | MVA85A; Group 1: aerosol then ID Group 2: ID then aerosol Group 3: ID then ID (5 × 107 pfu) |
UK | 37 adults | –ve | No data reported yet |
Manjaly 2016 (conference abstract) |
| NCT02532036 | Aerosol ID |
2015–2018 | MVA85A; 1 × 107 pfu aerosol inhaled, 5 × 107 aerosol and ID |
UK | 15 adults | –ve | No data reported yet | NCT02532036 |
–ve: negative; +ve: positive; AE: adverse event; ART: antiretroviral therapy; BCG: bacillus Calmette‐Guérin; EPI: Expanded Programme on Immunization; ID: intradermal; IM: intramuscular; MTB: Mycobacterium tuberculosis; NR: not reported; pfu: plaque‐forming unit.
The 21 studies included 712 participants investigated from 2002 with follow‐up expected to be completed by 2018. The studies covered a diverse population in the UK, South Africa, Senegal, and The Gambia with HIV‐positive and HIV‐negative people as well as infants, children, and adults. Intramuscular, intradermal, and aerosolized delivery routes were all investigated. The summary showed most of the adverse effects related to vaccination were mild and were contained locally to the injection site. There were very few serious adverse effects; erythema and mild pain were the most common adverse effects of the vaccine.
Why it is important to do this review
Summarizing the evidence to date will be useful to the public, scientists, and to others interested in innovation in tuberculosis as a case study from laboratory development to field testing. If critical appraisal and systematic review of this vaccine in humans shows no clear effect, this raises questions about any further testing. However, as of November 2017, there were ongoing studies looking at aerosolized delivery of the vaccine (NCT01954563; NCT02532036). In 2017, studies were published that addressed the immunogenicity of the candidate tuberculosis vaccine MVA85A in Schistosomiasis‐infected teenagers (Wajja 2017), and a further efficacy study in HIV‐exposed infants (Nemes 2018).
Objectives
To assess and summarize the effects of the MVA85A vaccine boosting BCG in humans.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs) that include measures of clinical efficacy (Phase 2 clinical trials).
Types of participants
Any person regardless of age or HIV status.
Types of interventions
Intervention
MVA85A vaccine regardless of vaccination schedule, dosage, route, or formulation given with BCG.
Control
BCG alone, or Candin® (Candida albicans skin test antigen).
Types of outcome measures
Primary outcomes
-
Active tuberculosis, defined by:
clinical signs and symptoms plus confirmation by microscopy, culture, or Xpert® MTB/RIF (an automated nucleic‐acid amplification test);
treatment commenced for tuberculosis.
Secondary outcomes
Latent tuberculosis, diagnosed by IGRA or Mantoux without clinical or radiological evidence of active disease.
Adverse outcomes
Adverse effects of any severity, defined as "an adverse event for which the causal relation between the intervention and the event is at least a reasonable possibility" (Loke 2011).
Serious adverse effects, defined as an adverse event attributable to the intervention "leading to death, are life threatening, requires inpatient hospitalisation or prolongation of existing hospitalisation, or result in persistent or significant disability or incapacity" (ICH 1994).
Adverse events of any severity, defined as "any untoward medical occurrence that may present during treatment with a pharmaceutical product but which does not necessarily have a causal relationship with this treatment" (WHO‐ART 2008).
Abnormal haematological tests during the follow‐up period after being vaccinated.
Abnormal biochemical tests during the follow‐up period after being vaccinated.
Search methods for identification of studies
We conducted the literature search up to the 10 May 2018 and identified potential studies regardless of language or publication status (published, unpublished, in press, and in progress).
Electronic searches
We searched the following databases using the search terms and strategy described in Appendix 1: the Cochrane Infectious Diseases Group Specialized Register (10 May 2018); the Cochrane Central Register of Controlled Trials (CENTRAL, 2018, Issue 4, published in the Cochrane Library); MEDLINE (PubMed, 1966 to 10 May 2018); Embase (Ovid, 1947 to 10 May 2018); Science Citation Index‐Expanded, Social Sciences Citation index, conference proceedings (Web of Science, 1900 to 10 May 2018); and CINAHL (EBSCOHost (1982 to 10 May 2018). We also searched the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/en/), and ClinicalTrials.gov (clinicaltrials.gov/ct2/home), for trials in progress, up to 10 May 2018, using MVA85A, "modified vaccinia virus Ankara", Ag85A, "Antigen 85A", and tuberculosis OR tuberculosis as search terms.
Searching other resources
We searched the proceedings and abstracts of the following tuberculosis conferences: Union World Conference on Lung Health, European Respiratory Society, and the International Conference of the American Thoracic Society (ATS), from 2012 to 2018. We also handsearched reference lists of relevant papers.
Data collection and analysis
Selection of studies
Two review authors independently screened all abstracts retrieved by the search strategy above using predefined eligibility criteria designed and piloted by the review authors. We excluded clearly irrelevant studies. We searched for multiple publications using studies from the same data set. We retrieved full‐text copies for all trials thought to be potentially relevant. Two review authors (SoJ and SaJ) independently assessed all identified trials for inclusion in the review using the predefined inclusion criteria.
We resolved any disagreements in assessment through discussion. In cases of unresolved differences, a third review author adjudicated. We kept records of the initial results and the changes after discussion. We also kept a list all studies excluded after full‐text assessment in the Characteristics of excluded studies table. We illustrated the study selection process in a PRISMA diagram (Figure 1).
1.

Study flow diagram.
Data extraction and management
We designed and piloted data extraction forms. Two review authors independently performed data extraction. We gathered information from each included trial separately on trial characteristics. These included:
study setting, design, study duration, population sample size, and power calculations;
baseline characteristics of study population including age, sex, weight, prematurity, HIV, other comorbidity, whether breastfeeding, race, HIV status, antiretroviral therapy (ART), CD4 count, and viral load;
intervention and control group vaccine dosages, routes of administration, and times of vaccination;
time of outcome measure after administering MVA85A;
duration of follow‐up, withdrawals from the study, and reasons for withdrawal.
All outcomes were dichotomous, so we tabulated the numbers of participants who developed tuberculosis or an adverse event (n) with the total sample size number (N) in each comparison group. We documented the different definitions of outcomes in the trials for further consideration and only combined data from endpoints that were similar across studies.
Assessment of risk of bias in included studies
We assessed risk of bias for RCTs using the Cochrane 'Risk of bias' tool (Higgins 2011). Two review authors independently assessed studies for risk of bias. We resolved any disagreement through discussion and, where necessary, through consultation with a third review author.
We assessed sequence generation (if predictable method used) and allocation concealment for selection bias and detection bias by looking at blinding methods. We also considered both the intention of blinding and the success of blinding for each outcome. If there was no description of the procedure, for example how randomization was done, we marked it as unclear.
In addition, we examined the objectivity of outcome measures, use of intention‐to‐treat (ITT) analysis, loss to follow‐up, and selective outcome reporting to assess the risk of bias in included studies. We assessed whether outcome measures were specified a priori and whether the published endpoints matched those specified in study protocols.
We assessed incomplete outcome data in each included trial to determine the proportion of missing results and whether it affected the results in terms of event risk and effect size. We assessed if reasons for missing data were related to adverse events or death from MVA85A and if missing data were balanced in the two experimental groups to have an overall decision on risk associated with incomplete outcome data.
We assessed other dimensions to risk of bias, including conflicts of interest, large differences in baseline characteristics, and early cessation of the trial.
We assessed the included trials for risk of bias of adverse events by examining if monitoring was active or passive; whether participants and outcome assessors were blinded; whether the outcome data reporting was complete; whether all participants were included; and whether data analysis was independent of pharmaceutical companies (Table 3; Bukirwa 2014). We also looked at the times when data were collected in comparison to when they were reported. All this information was included under overall study assessment of blinding, selective outcome reporting, incomplete outcome data, or other biases.
2. Adverse events risk of bias assessment methods.
| Criterion | Assessment | Explanation |
| Participant‐reported symptoms | ||
| Was monitoring active or passive? | Active Passive Unclear |
We classified monitoring as 'active' when authors reviewed participants at set time points and enquired about symptoms. |
| Was blinding for participants and outcome assessors adequate? | Adequate Inadequate Unclear |
We classified blinding as 'adequate' when both participants and outcome assessors were blinded to the intervention group, and the methods of blinding (including use of a placebo) were described. |
| Was outcome data reporting complete or incomplete? | Complete Incomplete |
We classified outcome data reporting as 'complete' when data were presented for all the time points where it was collected. |
| Were all participants included in reporting? | Yes No |
We reported the percentage of randomized participants included in adverse event reporting. |
| Was the analysis independent of study sponsor? | Yes No Unclear |
We classified the analysis of trials sponsored by pharmaceutical companies as independent of the sponsor when it was clearly stated that the sponsor had no input to the trial analysis |
| Laboratory tests | ||
| Number of tests undertaken | — | We extracted the type and number of laboratory tests were taken. |
| Timing of tests: was number and timing of tests adequate? | Adequate Inadequate |
We classified the number and timing of tests as 'adequate,' when tests were taken at baseline, plus 2 other time points within the first week after treatment, plus the last day of the study. We classified the number of test taken as 'inadequate,' if either the laboratory controls in the first week or controls at 4 weeks were not performed. |
| Reporting of test results: was reporting of test results complete? | Complete Incomplete |
We classified reporting as 'complete' when test results of all time points were reported. For the trials with inadequate number of tests taken, we considered completeness of reporting as inconsequential, and therefore did not record a judgement. |
| Independence of data analysis: was data analysis independent? | Yes No Unclear |
We classified the analysis of trials sponsored by pharmaceutical companies as independent of the sponsor when it is clearly stated that the sponsor had no input to the trial analysis. |
Adapted from Bukirwa 2014.
Measures of treatment effect
We analysed all data using Review Manager 5 (Review Manager 2014). We pooled dichotomous data using risk ratios (RR) with their corresponding 95% confidence intervals (CI). When inappropriate due to a small number of events in each group, we presented the pooled data using risk difference (RD) with their 95% CI.
Unit of analysis issues
For included studies that had multiple intervention arms, we included data from these studies by splitting the control group so that participants were only included in the meta‐analysis once.
Dealing with missing data
In our protocol, we anticipated that if the amount of incomplete outcome data was such that the trials were thought to be at a high risk of bias, we may have used imputation and perform sensitivity analyses to investigate the impact of these missing data. However, we identified no studies where missing data affected our ability to measure outcomes. Therefore, we used available‐case analysis, as planned in our protocol.
Assessment of heterogeneity
We assessed extracted data from included trials to find key differences in population groups, study setting, intervention and control groups, dosages and route of vaccine administration, or timing between BCG and boosting. We assessed degree of risk of bias, when and how the outcome was measured, and variation in treatment effects.
We determined the level of heterogeneity by inspecting forest plots for overlapping CIs. We judged a Chi2 P value significance level of 0.1 or less as likely heterogeneity. An I2 statistic value of less than 40% was regarded as not showing any significant heterogeneity.
Assessment of reporting biases
There was an insufficient number of trials included and so we were unable to assess for publication bias using funnel plots or Egger regression.
Data synthesis
We used the fixed‐effect Mantel‐Haenszel model for meta‐analysis where there was little heterogeneity. The intention for meta‐analysis of adverse outcomes was limited to three to five of the most frequent adverse effects and all those that were considered to be serious. However, due to different methods of monitoring adverse effects that in turn lead to different results where meta‐analysis could not be performed, we gave a narrative report.
Subgroup analysis and investigation of heterogeneity
We intended to explore heterogeneity by: subgroup by children and adults; background prevalence of tuberculosis (or tuberculosis incidence in the control group); HIV status; and geographical location. However, there were not enough trials to explore such subgroups when we found high heterogeneity.
We considered random‐effects meta‐analysis if subgroup analysis did not explain the heterogeneity. We applied the I2 statistic according to guidance of: less than 40% as not significant heterogeneity; 30% to 60% representing moderate heterogeneity; 50% to 90% representing substantial heterogeneity; and 75% to 100% considerable heterogeneity (Higgins 2011). We regarded a Chi2 P value significance level of 0.1 or less and an I2 statistic greater than 40% as showing significant heterogeneity, in which case we either considered a random‐effects model or did not perform meta‐analysis.
Sensitivity analysis
We did not perform sensitivity analysis for imputed data, risk of bias, or any other peculiarities between the trials identified during the review process.
Certainty of the evidence
We assessed the certainty of the evidence using the GRADE approach (Schünemann 2013). We constructed a 'Summary of findings' table, which outlines the main review findings alongside the certainty of the evidence.
Results
Description of studies
Results of the search
We identified 153 records, with 152 records remaining after removing duplicates. We excluded 118 records based on title and abstract and assessed the full text of 34 articles. We excluded 28 full‐text articles. Six articles fulfilled the eligibility criteria and were included in the review. See Figure 1 for the flow diagram of inclusion and exclusion of studies in the review.
Included studies
Six studies (3838 participants) that met our inclusion criteria reported findings from four Phase 2 clinical trials (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Andrews 2017 and Bunyasi 2017 presented data based on the Tameris 2013 clinical trial. The six included studies are described in the Characteristics of included studies table.
Setting and time
All took place in South Africa involving rural and urban areas between 2008 and 2015, with one trial that took place at two sites: South Africa and Senegal (Ndiaye 2015).
Source of funding
Aeras sponsored five trials (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). The University of Oxford sponsored one trial (Scriba 2011). The Wellcome Trust funded all the trials. Other funders were Oxford Emergent Tuberculosis Consortium (OETC) for Ndiaye 2015 and Tameris 2013, the European and Developing Countries Clinical Trials Partnership and the Bill and Melinda Gates Foundation for Ndiaye 2015, the UK Medical Research Council for Nemes 2018, and the EuropeAID European Commission for Scriba 2011. Andrews 2017 and Bunyasi 2017 conducted further follow‐up based on the participants enrolled in Tameris 2013, and mentioned that there was no specific additional funding for the analysis performed.
Participants
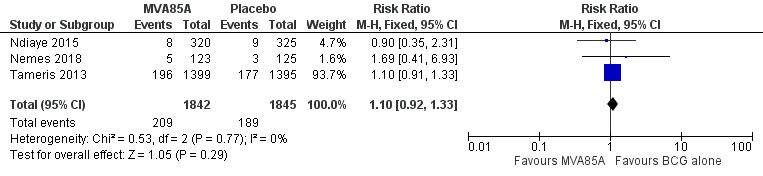
Five trials included infants (Andrews 2017; Bunyasi 2017; Nemes 2018; Scriba 2011; Tameris 2013). One trial assessed the efficacy and safety of the vaccine in adults with HIV (Ndiaye 2015). Tameris 2013 and Scriba 2011 recruited infants who were HIV‐negative, while Nemes 2018 assessed the vaccine in newborns of HIV‐positive mothers. None of the trials reported other morbidities. In Tameris 2013, 412 (29.4%) participants in the intervention group and 268 (26.4%) participants in the control group were preterm.
Interventions
Intervention
All the infants in the intervention groups received a single dose of intradermal MVA85A. In the trial recruiting adults, the 324 adults allocated in the intervention group received a second dose (booster) of intradermal vaccine six months after the first dose (Ndiaye 2015). The vaccine was given at a dose of 1 × 108 plaque‐forming units (pfu) in Ndiaye 2015, Nemes 2018, and Tameris 2013. Scriba 2011 assessed three different doses of the vaccine by giving a dose of 2.5x107 pfu, 5x107 pfu and 1x 108 pfu to 36 participants in each of the three groups. All the infants in Scriba 2011 and Tameris 2013 received the BCG vaccine in the first four weeks of life, prior to receiving the MVA85A vaccine, as an inclusion criteria. Nemes 2018 gave the MVA85A vaccine to the neonates in the first 96 hours of life, with no prior administration of BCG, and gave BCG at eight weeks of age only to HIV‐negative infants. Ndiaye 2015 did not mention whether the adults they recruited received BCG.
Comparator
Five trials gave Candida skin test antigen (Candin®) as a placebo, using the same route (intradermal) and schedule (one or two doses) as for the intervention group in each of the trial (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). Scriba 2011 gave the infants in the comparator group one dose of pneumococcal 7‐valent conjugate vaccine by the intramuscular route.
Outcomes
Three studies reported different endpoints as measures of tuberculosis disease (Ndiaye 2015; Nemes 2018; Tameris 2013). These are compared in Table 4.
3. Differences in tuberculosis endpoint assessment.
| Study | Endpoint 1 | Endpoint 2 | Endpoint 3 |
| Tameris 2013 | Any of the following criteria.
|
"Included all infants who met endpoint 1 criteria; had marginally less stringent criteria to define TB infection and household exposure." Any of the following numerical categories.
|
All participants placed on treatment for TB by a health professional with the intent of treating TB regardless of whether they have met the other efficacy endpoints. |
| Andrews 2017 | Revised endpoint 1 from Tameris 2013 that removed QFT conversion from the diagnostic criteria to avoid bias towards association with QFT status. | Not used | Not used |
| Bunyasi 2017 | Not used | Not used | Same definition as for Tameris 2013. |
| Ndiaye 2015 | Any of the following numerical categories.
|
Any of the following numerical categories:
|
Same definition as for Tameris 2013. |
| Scriba 2011 | Not applicable | Not applicable | Not applicable |
| Nemes 2018 | Outcomes not specified in the methods section. In results, authors specified that 8 participants were diagnosed as TB: "of whom one was M.tb [Mycobacterium tuberculosis] culture positive and 7 were diagnosed on clinical/ radiographic grounds and TB contact history. Two of the TB cases were QFT positive." |
Not used | Not used |
AFB: acid‐fast bacilli; CSF: cerebrospinal fluid; CT: computerized tomography; QFT: quantiFERON; TB: tuberculosis. aIn Tameris 2013, endpoint 2: criteria in bold indicate where different from endpoint 1.
All the included studies reported data on latent tuberculosis (or tuberculosis infection) to assess either efficacy or safety outcomes. Four trials looked at safety outcomes, including adverse effects of any severity, serious adverse effects, and adverse events of any severity (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Tameris 2013 collected data on biochemical or haematological blood test findings but did not report this element of their primary outcome. Ndiaye 2015 collected data on blood tests but did not report disaggregated findings. Only Scriba 2011 and Nemes 2018 reported on blood test data collected.
Length and method of follow‐up
Scriba 2011 followed up participants for 24 weeks, Nemes 2018 for 52 weeks, Ndiaye 2015 for at least six months after the last participant was enrolled, and Tameris 2013 for up to 39 months. Andrews 2017 was an observational follow‐up study of the participants enrolled in Tameris 2013; authors analysed the data collected at day 336 after the intervention and at the end of the study, which ranged from six to 24 months after day 336. Bunyasi 2017 followed the participants recruited in Tameris 2013 for a median of five years.
Investigators of five studies used diary cards to record adverse events during the seven days following vaccination (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Scriba 2011; Tameris 2013); Nemes 2018 did not mention this. Researchers performed blood investigations at several intervals in all trials, to detect adverse events and to assess immunogenicity. Ndiaye 2015 and Tameris 2013 performed active follow‐up every three months to identify signs, symptoms, or exposure to tuberculosis that merited further investigation, while this was done at irregular but planned intervals in Scriba 2011 and Nemes 2018. The long‐term follow‐up study was based on passive surveillance based on the electronic tuberculosis register database (Bunyasi 2017).
Excluded studies
We excluded 28 studies from the review, with the reasons for exclusion listed in the Characteristics of excluded studies table.
Studies awaiting classification
We did not identify any studies that are awaiting classification.
Ongoing studies
We did not identify any ongoing studies.
Risk of bias in included studies
See Characteristics of included studies table for the assessment of the risk of bias for each included study. See Figure 2 and Figure 3 for the risk of bias summaries.
2.
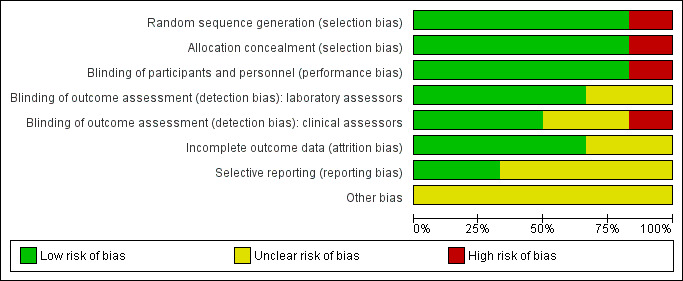
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Five trials were at low risk of selection bias (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). They reported adequate sequence generation and methods of allocation concealment. Scriba 2011 used systematic allocation at a 3:1 ratio allowing predictability of the sequence (high risk of bias).
Blinding
Three studies had adequate blinding of participants, study personnel, laboratory assessors, and clinical assessors and were at low risk for performance and detection bias in all domains (Ndiaye 2015; Nemes 2018; Tameris 2013). Five studies reported blinding of participants and study personnel (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). Scriba 2011, an open‐label trial with different routes of administration for placebo and vaccine, had low risk of detection bias for laboratory assessors as outcomes were objective and high risk of detection bias for subjective assessments by clinicians. Two studies were at unclear risk of detection bias for laboratory assessors and clinicians (Andrews 2017; Bunyasi 2017). Andrews 2017 did not provide any details on blinding, while Bunyasi 2017 reported on post‐trial data and had no information on how data was collected from registers.
Incomplete outcome data
Four trials reported details of all randomized participants (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Only a few participants randomized were not included in the analysis, without resulting in a disbalance between the intervention and control groups. Indeed, three participants were randomized in the control group in Tameris 2013, but not included in the efficacy analysis (two of them were not included either in the safety analysis), while five participants were randomized (four in the intervention group and one in the control group), but not included in the efficacy analysis in Ndiaye 2015. As a result we considered these studies at low risk of attrition bias. There were no details of how many of each group came from the 119 participants excluded from Tameris 2013 for analysis in Bunyasi 2017. Andrews 2017 and Bunyasi 2017 had an unclear risk of attrition bias as these were follow‐up studies from Tameris 2013, and there were unclear discrepancies with those reported previously.
Selective reporting
Nemes 2018 was prospectively registered and appeared free of selective outcome reporting as ascertained from data in trial registers and reports of trials. We also judged Scriba 2011 at low risk of reporting bias, with all the outcomes reported in their methods section presented in the results.
Four studies were at unclear risk of bias due to selective reporting (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Tameris 2013). There were multiple instances where predefined endpoints were poorly defined or were deviated from in the final reported results as laid out in Table 5.
4. Differences between details of studies published prior to commencement and reported outcomes.
| Study | Protocol | Published findings | Differences between protocol and published findings | ||
| Stated outcomes published prior to commencement of trial that differ to published outcomes | Measurement of outcome as stated a priori | Measurement of outcome as stated in published findings | Reported findings | ||
| Andrews 2017 | No protocol published. | ||||
| Bunyasi 2017 | No protocol published (extended post‐trial follow‐up of Tameris 2013). | ||||
| Ndiaye 2015 | Adverse events: blood tests a | "Percentage of participants with adverse events" AEs measured up to day 28 SAEs measured up to 6 months. |
"Phlebotomy for routine haematological and biochemical analysis was done at screening, before booster vaccination, and on days 7 and 28 after each vaccination." | "Routine haematological and biochemical test results did not differ between study groups (data not shown)." | Haematological and biochemical blood tests not outlined as a measure of safety in the study protocol. Blood test findings reported unclearly. |
| Nemes 2018 | Safety | Clinicaltrials.gov – local, regional, and systemic AEs and SAEs which would be reported as cumulative 12‐month incidences. | "Infants followed for safety end points at weeks 1, 4, 6, and 8 after MVA85A/control vaccination and thereafter, at weeks 9, 12, and 16 (corresponding to weeks 1, 4, and 8 following delayed BCG vaccination at 8 weeks of age), and at week 52." | Reported total events for AEs per group after MVA85A and before BCG and for whole follow‐up period. Data including for laboratory AEs were not disaggregated as prespecified. | Data including for laboratory AEs were not disaggregated as prespecified. |
| Scriba 2011 | Safetya | Local and systemic AEs for the first week. | Diary cards | Local and systemic AEs reported on ≥ 1 day of the first 7 days after MVA85A vaccination. | None |
| Blood tests (days 7, 28) | Biochemical and haematological tests (days 7, 28) | Reported number and percentages of participants with abnormal results and reported that, "all except one patient that had elevated liver enzymes remained unresolved by day 28." | |||
| Immunology | ESAT‐6/CFP‐10 | Infants converted – suggestive of TB infection but seemed to be reported as safety data not efficacy. | |||
| Tameris 2013 | Safety profile – AEsa | AEs measured up to day 28 SAEs measured throughout follow‐up. |
Collected data on solicited and unsolicited local and systemic AEs. Active surveillance for SAEs. |
AEs broken down by type of event and reported in supplementary material. Only local events at the injection site were considered to be related to the vaccine. | Causal relationship with AEs other than local injection site reactions was not reported. |
| Safety profile – blood testsa | Testing up to 28 days postvaccination. | "Peripheral blood for routine haematological and biochemical tests was taken at screening and on day 7 and day 28 after vaccination in an initial safety cohort of at least 330 infants." | Not reported | Primary outcome not reported | |
| Efficacy of MVA85Ab | Using an endpoint derived from epidemiological cohort surveys in BCG vaccinated infants. | Not reported – simply stated clinical endpoints 'developed.' | Composite clinical endpoints 1, 2, 3 (see Table 4) Microbiologically confirmed cases reported in appendix. |
The "primary efficacy endpoint" was measured using an endpoint not derived from cohort studies. The endpoint definition differed from all other implied or reported ways of measuring efficacy in the other studies. The point estimate showed clinically significant benefit for endpoint 1 (no benefit seen at the 95% confidence level). This endpoint was reported as the main efficacy finding. All other point estimates show no clinically significant benefit or harm. |
|
AE: adverse events; BCG: bacillus Calmette‐Guérin; ESAT‐6/CFP‐10: early secretory antigenic‐6/culture filtrate protein‐10; SAE: severe adverse events; TB: tuberculosis. aPrimary outcomes as outlined in study protocols. bSecondary outcomes as outlined in study protocols.
Description of Tameris 2013 published prior to commencement of the trial (NCT00953927) stated that the authors intended to report endpoints of clinical disease based on "observational cohort studies." This was subsequently changed following the publication of the trial in October 2013 to include "clinically‐derived tuberculosis diagnostic criteria." The main trial reports adapting the primary elements proposed in a consensus statement (Graham 2012). There was no record of the change in approach from empirically derived endpoints to endpoints developed by the investigators in the study protocol.
Tameris and colleagues reported on three outcomes with complex definitions (Table 4).
Endpoint one, described as "primary efficacy endpoint," comprising nine criteria, which included a binary measure of quantiFERON conversion.
Endpoint two, described as "exploratory efficacy endpoint," comprising nine criteria.
Endpoint three, described as "exploratory efficacy endpoint," which was defined as "all participants placed on treatment for tuberculosis."
The difference between endpoints one and two, which varied in the direction of the point estimate of the effect, was 5 mm on a tuberculin skin test or household contact with acid‐fast bacilli (AFB) smear‐positive person (Table 4). The process of defining these three endpoints was unexplained, and it is unclear why these specific definitions were used. These endpoint definitions were only used in this trial and not in subsequent studies.
In a subsequent critique, Behr and colleagues noted that the outcomes reported in the trial did not include the simple measure of a positive microbiological endpoint (Behr 2013). The endpoint used in the abstract was endpoint one, which authors have settled as primary efficacy outcome, while endpoints two and three were reported as exploratory outcomes. The complexity of the definitions and the analysis in Behr's paper pointed to the risk of selective reporting. This may not have been intentional, but arose with post‐hoc approaches with different approaches to expressing the results, but could be excluded if outcomes were precisely and clearly defined a priori. The only information publicly available prior to the trial commencing were broad descriptions of the outcome. Hence for selective reporting the classification was unclear.
Andrews 2017 was at unclear risk of reporting bias as this was a nested observational study and there was no prespecified study protocol. Ndiaye 2015 was at unclear risk of reporting bias as the authors commented that there were no differences between biological and haematological tests; however, no data or how these data were analysed to come to this conclusion were reported.
Other potential sources of bias
We considered that the risk of other potential biases was unclear in all included studies. We were concerned as a number of the authors were involved in the private company manufacturing the vaccine or were patent holders for MVA85A. In these circumstances, it would be good practice for this to be declared in the publication. Only one study declared no conflicts in relation to patent holding (Scriba 2011).
Two trials reported a role of funders in design, data analysis, and manuscript writing (Ndiaye 2015; Tameris 2013), and one study had employees of the funder involved in manuscript writing (Andrews 2017). Ndiaye 2015 calculated incident tuberculosis cases from day 28 after vaccination versus from day 0 in Tameris 2013. This was likely to be due to the risk of pre‐existing undiagnosed tuberculosis being inappropriately counted as developing following the intervention. If participants are not followed from the start of the intervention then a period of follow‐up has been excluded, and participants who experienced the outcome soon after intervention will be missing from analyses. We considered this to be of unclear risk of bias as it is unclear if this impacted on outcomes.
Adverse events
For adverse events, we conducted additional assessments on adequacy of safety monitoring and completeness of reporting for participant‐reported outcomes and laboratory tests taken (Table 6). Four trials reported on safety outcomes (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Monitoring of participant‐reported outcomes was active in all trials and blinding was adequate in two trials (Nemes 2018; Tameris 2013). All trials reported specified timing of data collection but only one study reported under some of the days (Scriba 2011). None of the trials completely reported outcomes on prespecified time points including for laboratory results. All trials reported all participants who received intervention per‐protocol. Timing of taking laboratory tests was inadequate in Scriba 2011 and Tameris 2013 as there was no clear indication of tests being taken at the end of the study.
5. Summary of monitoring and reporting of adverse events.
| Study | Participant reported adverse events | Outcome data reporting | Laboratory tests | ||||||||
| Monitoring active or passive | Blinding of participants or outcome assessors | Times data collected | Times data reported | Complete/not complete | Percentage of participants reported on | Analysis independent of study sponsor | Number of tests taken | Timing of tests and adequacy | Complete reporting of test results | Independence of data analysis | |
| Scriba 2011 | Active | Inadequate | 60 min, D 2, 7, 28, 84, and 168 | D 7, 28 | Incomplete | 100% | Unclear | Biochemistry and haematology | Inadequate | Inconsequential | Unclear |
| Ndiaye 2015 | Active | Inadequate | D 7, 28, and 84 after boost 3 monthly until end of study | NR | Incomplete | 99.8% | No | Haematology, chemistry, virological markers | Adequate | Incomplete | No |
| Tameris 2013 | Active | Adequate | Baseline, D 7 and 28, throughout up to D 84 | NR | Incomplete | 99.9% | No | Biochemistry and haematology | Inadequate | Incomplete | No |
| Nemes 2018 | Active | Adequate | Week 1, 4, 6, 8, 16, and 52 | NR | Incomplete | 85.9% | Unclear | Not specified | Adequate | Incomplete | Unclear |
D: day; min: minute; NR: not reported.
Effects of interventions
See: Table 1
See Table 1.
Active tuberculosis
Studies varies in the way they defined active tuberculosis (see section "description of studies" (Table 4)). Tameris 2013 and Ndiaye 2015 reported hierarchical endpoints including microbiologically confirmed tuberculosis, composite clinical definitions, and participants starting on tuberculosis treatment, with no significant effect consistently seen across endpoints (Analysis 2.1; Analysis 2.2; Table 4; Table 7).
2.1. Analysis.

Comparison 2 Comparison of endpoints, Outcome 1 Tameris 2013: incidence of tuberculosis (TB) according to post‐hoc endpoints.
2.2. Analysis.

Comparison 2 Comparison of endpoints, Outcome 2 Ndiaye 2015: incidence of TB according to post hoc defined endpoints.
6. Results of the different endpoints of active tuberculosis.
| Active TB | Tameris 2013 | Andrews 2017 | Bunyasi 2017 | Ndiaye 2015 | Scriba 2011 | Nemes 2018 | ||||||
| MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | |
| Endpoint 1a | 32/1399 (2.3%) | 39/1395 (2.8%) | 58/2797 (2.1%) with NDD | N/A | N/A | 6/320 (1.9%) | 9/325 (2.8%) | N/A | N/A | 5/123 (4.1%) | 3/125 (2.4%) | |
| Endpoint 2a | 55/1399 (3.9%) | 52/1395 (3.7%) | N/A | N/A | N/A | N/A | 6/320 (1.9%) | 9/325 (2.8%) | N/A | N/A | N/A | N/A |
| Endpoint 3a | 196/1399 (14.0%) | 177/1395 (12.6%) |
N/A | N/A | 3.3/100 pyo (95% CI 2.9 to 3.9) |
3.0/100 pyo (95% CI 2.6 to 3.5) | 8/320 (2.5%) | 9/325 (2.8%) | N/A | N/A | N/A | N/A |
CI: confidence interval; N/A: not applicable; NDD: no disaggregated data; pyo: person‐years of observation; TB: tuberculosis. aSee Table 4 for description of endpoints.
Tameris 2013 reported three endpoints in their main manuscript, with endpoint one described as their primary efficacy endpoint (RR 0.82, 95% CI 0.52 to 1.30, point estimate favouring MVA85A). A fourth endpoint was described in the supplementary material, taking into account the microbiologically confirmed cases of tuberculosis. Other outcomes (endpoint two, endpoint three, and endpoint four of microbiologically confirmed cases) were not statistically different, although their point estimate favoured placebo (endpoint two: RR 1.05, 95% CI 0.73 to 1.53; endpoint 3: RR 1.10, 95% CI 0.91 to 1.33; endpoint four (microbiologically confirmed): RR 1.10, 95% CI 0.60 to 2.00; Analysis 2.1; Figure 4).
4.
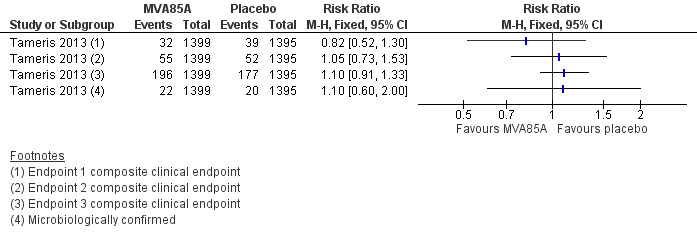
Forest plot of comparison: 2 Comparison of endpoints, outcome: 2.1 Tameris 2013: incidence of tuberculosis according to post‐hoc endpoints.
Two studies reported no effect of MVA85A on cases of active tuberculosis confirmed by culture or Xpert® MTB/RIF (RR 0.97, 95% CI 0.58 to 1.62; 3439 participants, two trials) (Analysis 1.1; Figure 5; Ndiaye 2015; Tameris 2013).
1.1. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 1 Active tuberculosis (TB): confirmed by culture or Xpert® MTB/RIF longest reported follow‐up.
5.
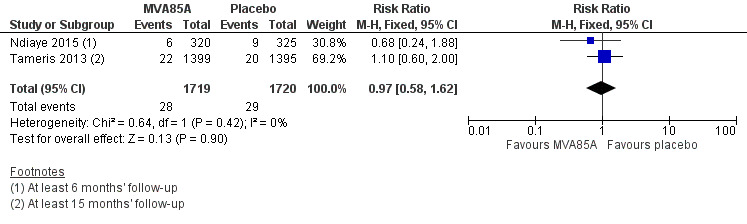
Forest plot of comparison: 1 MVA85A Vs Placebo, outcome: 1.1 Tuberculosis confirmed by culture or Xpert® MTB/RIF longest reported follow‐up.
Three studies (Ndiaye 2015; Nemes 2018; Tameris 2013) reported no effect of MVA85A on cases of active tuberculosis when considering patients started on tuberculosis treatment (RR 1.10, 95% CI 0.92 to 1.33; 3687 participants, 3 trials; Analysis 1.2; Figure 6).
1.2. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 2 Active TB: started on TB treatment.
6.
Forest plot of comparison: 1 MVA85A versus placebo, outcome: 1.2 Active tuberculosis: started on tuberculosis treatment.
Nemes 2018 reported active tuberculosis as defined by participants starting tuberculosis treatment. One participant in this trial was diagnosed by culture; however, the authors did not report what intervention this participant received.
Latent tuberculosis
Four studies reported no effect of MVA85A on cases of latent tuberculosis (RR 1.01, 95% CI 0.85 to 1.21; 3831 participants, four trials; Analysis 1.3).
1.3. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 3 Latent TB.
Scriba 2011 was underpowered and not designed to detect measures of efficacy. However, they reported latent tuberculosis, presumably as a measure of safety, as this outcome was poorly defined a priori.
Adverse effects
Four studies reported effects of any severity (Table 8). We presented the effect of the estimates for adverse effects of any severity with disaggregated (Analysis 1.4; Figure 7) and aggregated data (Analysis 1.5) to provide detailed information as provided by the study authors. However, we did not perform meta‐analysis of the estimates due to high heterogeneity. Local reactions of the skin at the injection site was the most common adverse effect associated with the vaccine MVA85A, this was reported in three studies, with the three studies showing direction towards more adverse effects in the intervention group (3187 participants; Nemes 2018; Scriba 2011; Tameris 2013). However, only one study reported systemic symptoms defined as fever, lethargy, malaise, and vomiting (144 participants; Scriba 2011). Therefore, we chose to report adverse effects of any severity disaggregated by local reactions of the skin and systemic symptoms in our Table 1 as different amount of information is provided for each group (Scriba 2011).
7. Adverse effects of the MVA85A vaccine.
| Study | MVA85A | Placebo | Breakdown | Author conclusions | ||||
| Number of participants with ≥ 1 event caused by the intervention | Total participants | Number of participants with ≥ 1 event caused by the control | Total participants | Detailed AEs | MVA85A | Placebo | ||
| Ndiaye 2015 | 318 | 324 | 307 | 325 | Solicited AEsa | 288 | 235 | "Solicited adverse events were more common in MVA85A group and most were local injection site reactions." |
| Nemes 2018 | 105b | 123 | 30b | 125 | Not detailed | N/A | N/A | "Infants in MVA85A arm were more likely to experience an AE than in control arm. Injection site reactions were more frequent in MVA85A recipients and mild." |
| Scriba 2011 | 106c | 108c | 6 | 36 | Injection sited | 106 | 6 | "Desquamation significantly increased with greater vaccine dose." |
| Malaise | 6 | 1 | ||||||
| Lethargy | 6 | 2 | ||||||
| Tactile fever | 18 | 0 | ||||||
| Documented fever | 13 | 2 | ||||||
| Vomiting | 6 | 2 | ||||||
| Elevated liver enzyme levels | 13 | 4 | ||||||
| Increased white cell count | 0 | 1 | ||||||
| Tameris 2013 | Local 1251e | 1399 | Local 628e | 1396 | Not detailed | 1251 | 628 | None |
AE: adverse event; N/A: not applicable. aIncluded injection reactions, mild influenza‐like symptoms, and regional lymphadenopathy. bAuthors of the study reported 105 participants with at least one adverse effect in the vaccine group and 30 participants in the placebo group, where causal relationship was defined as definite. cAggregated between three groups receiving different doses. dIncluded desquamation (scaling), pain, redness, and swelling. eAuthors of the study reported local and systemic adverse events. Authors specified in their protocol that, "Solicited adverse events of local injection site reactions will be considered causally related to study vaccine (adverse reaction)." Therefore, we reported such adverse events as adverse effects. Causal relationship with other adverse events was not reported.
1.4. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 4 Adverse effects of any severity.
7.
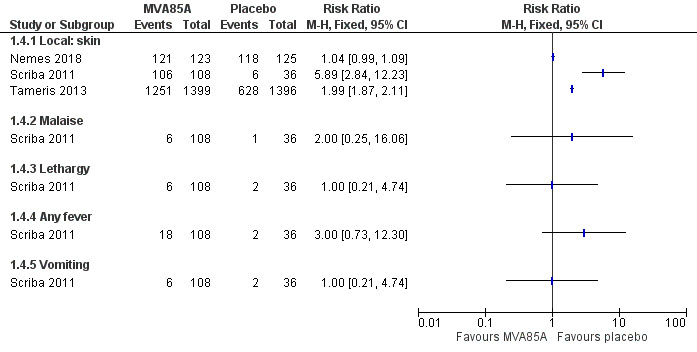
Forest plot of comparison: 1 MVA85A versus placebo, outcome: 1.4 Adverse effects of any severity.
1.5. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 5 Adverse effects of any severity: aggregated.
Three studies reported no increase in the risk of experiencing a serious adverse effect attributable to MVA85A (3692 participants; Analysis 1.6). Nemes 2018 reported serious adverse events and specified that none of them were related to the investigational product. Therefore, we classified this as no serious adverse effects following the definition of our review.
1.6. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 6 Serious adverse effects.
Adverse events of any severity
Four studies reported a small increase in the risk of experiencing an adverse event of any severity following vaccination with MVA85A (RR 1.05, 95% CI 1.02 to 1.08; 3836 participants; Analysis 1.7; Table 9). Adverse effects related to the vaccine and adverse events not attributed to the vaccine were conflated in the largest trial. No disaggregated data were available.
1.7. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 7 Adverse events of any severity.
8. Adverse events summary table.
| Study | Adverse events of any severity | |||
| MVA85A | Placebo | |||
| Tameris 2013 | 1120/1399 (80.1%) | 1059/1396 (75.9%) | ||
| Andrews 2017 | NR | NR | ||
| Bunyasi 2017 | NR | NR | ||
| Ndiaye 2015 | 321/324 (99.1%) |
312/325 (96%) |
||
| Scriba 2011 | 2.5 × 107 pfu = 35 μL |
5 × 107 pfu = 70 μL |
1 × 108 pfu = 135 μL |
1/36 |
| 1/36 | 3/36 | 6/36 | ||
| Nemes 2018 | Mild 122/123 (99.2%) |
121/125 (96.8) |
||
| Moderate 62/123 (50.4%) |
54/125 (3.6%) |
|||
| Severe 11/123 (8.9%) |
14/125 (11.2%) |
|||
NR: not reported; pfu: plaque‐forming unit.
Abnormal haematological and biochemical tests
Three studies reported abnormal haematological or biochemical laboratory tests. The percentage of those with elevated liver enzymes ranged from 2.8% to 25% in the three different groups reported in Scriba 2011 and there was a dose–response effect of MVA85A. However, none of the doses showed a significant increase at a 95% CI. Ndiaye 2015 reported that routine haematological and biochemical test results did not differ between study groups but disaggregated data were not reported. Nemes 2018 reported no difference between groups in the percentage of people with abnormal biochemical tests (11.4% versus 10.4%), but disaggregated data were not reported. The largest study performed haematological and biochemical tests but did not report data (Tameris 2013). We summarized the report and findings of abnormal haematological and biochemical tests in Table 10, and presented the effect of estimate for abnormal biochemical tests only (Analysis 1.8), as only one study reported disaggregated data for abnormal haematological tests.
9. Abnormal haematological and biochemical tests.
| Study | Haematological blood tests | Biochemical blood tests | ||||
| MVA85A | Placebo | MVA85A | Placebo | |||
| Tameris 2013 | NR | NR | NR | NR | ||
| Andrews 2017 | NR | NR | NR | NR | ||
| Bunyasi 2017 | NR | NR | NR | NR | ||
| Ndiaye 2015 | NRa | NRa | NRa | NRa | ||
| Scriba 2011 | 0/108b | 1/36b | 2.5 × 107 pfu = 35 μL |
5 × 107 pfu = 70 μL |
1 × 108 pfu = 135 μL |
4/36 (11%) |
| 1/36 (2.8%) |
3/36 (8.3%) |
9/36 (25%) |
||||
| Nemes 2018 | NR | NR | 14/123 (11.4%) | 13/125 (10.4%) |
||
NR: not reported; pfu: plaque‐forming unit. aAuthors stated that routine haematological and biochemical test results did not differ between study groups but did not present data. bOne participant had increased white cell count concurrently with an increase in alanine aminotransferase during an episode of gastroenteritis. Authors did not describe any other case of abnormal haematological test in the rest of the participant, although it was not stated explicitly.
1.8. Analysis.

Comparison 1 MVA85A versus placebo, Outcome 8 Abnormal biochemical tests.
Discussion
Summary of main results
Vaccinating people with MVA85A in addition to BCG:
probably makes little or no difference to the risk of developing active tuberculosis (moderate‐certainty evidence);
probably makes little or no difference to the risk of needing to start tuberculosis treatment (moderate‐certainty evidence);
probably does not have an important effect on the risk of developing latent tuberculosis (moderate‐certainty evidence);
does not cause life‐threatening serious adverse effects (high‐certainty evidence);
probably increases the risk of having an adverse reaction related to vaccination at the site of the injection (moderate‐certainty evidence);
may not be associated with an increase in systemic adverse effects related to vaccination (low‐certainty evidence).
Vaccination with MVA85A alone slightly increases the risk of having an adverse event (high‐certainty evidence).
Overall completeness and applicability of evidence
This review included trials from two countries in Africa. No studies that measured efficacy of the MVA85A vaccine have been carried out elsewhere. The review included studies on HIV‐positive adults, HIV‐negative infants, and infants exposed to HIV. It would be reasonable to generalize the results of these findings to other populations of HIV‐negative infants. The early cessation of the only trial in HIV‐positive adults, resulting in reduced follow‐up from two years to minimum six months and a reduction of study sample size from 1200 to 625, led this study to be underpowered for evaluation of efficacy (Ndiaye 2015). This may have limited the certainty of any inferences made to adults with HIV at high risk of contracting tuberculosis in terms of efficacy of MVA85A in this population. The effect of tuberculosis vaccination would be very similar regardless of geographical variation. Data from this review consistently showed no effect of the vaccine. As such, it is reasonable to generalize these findings to broader populations. For safety outcomes, the Phase 1 studies that we summarized in the Background section and Table 2, included adults, children, and infants from the UK and three African countries. Most of the adverse effects related to vaccination were mild and were contained locally to the injection site. This supports the trial findings summarized in this review.
Certainty of the evidence
Overall the included studies were well‐conducted. For most of our outcomes, there were few events and broad CIs for the pooled estimates of effect which contained clinically appreciable benefit and harm or no effect (see Table 1).
In the largest trial, the main reported endpoint (endpoint one) point estimate was in the direction of benefit of the vaccine on tuberculosis disease (Analysis 2.1; Tameris 2013). Whether this was due to the definition of endpoints or due to statistical heterogeneity was unclear. To minimize the impact of this inconsistency we presented results for cases diagnosed microbiologically and cases defined by being started on treatment. This was felt to reflect the most specific measure of efficacy and a measure of the real‐world situation. As a result of this, the methodological uncertainties surrounding case definition did not reduce our confidence in the effect estimates.
Failure to follow‐up participants from the start of intervention for efficacy measures in Ndiaye 2015 risked biasing outcomes. While it is plausible that participants with undiagnosed active tuberculosis would be inappropriately picked up, it is also plausible that participants who hypothetically could have developed tuberculosis immediately after vaccination would be excluded from analysis. However, the potential impact of this was unclear and as such we did not downgrade due to risk of bias for efficacy outcomes including this study.
In terms of latent tuberculosis, using the online calculator at www.sealedenvelope.com/power/binary‐noninferior/ at a significance level of 5% and with 80% power at a failure rate of 11% and a non‐inferiority limit of 5% a sample size per group of 484 would be sufficient to demonstrate non‐inferiority. Therefore, in terms of risk of developing latent tuberculosis where we had high certainty evidence that MVA85A had no important effect in reducing risk, we are confident that future trials are unlikely to change this result as we had 3831 participants in the analysis versus a minimum number of 484 participants required in each group.
Regarding the safety outcomes, the summary of findings from the Phase I trials for MVA85A performed in adults, adolescents, and infants with 712 participants showed that most of the adverse effects related to vaccination were mild and were contained locally to the injection site, and none of the trials reported a serious adverse event attributable to the vaccine. This supports the certainty of the evidence found in this review.
Potential biases in the review process
We followed standard methods in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The Cochrane Infectious Disease Group Information Specialist performed a comprehensive literature search with no restriction in language to identify all eligible studies, thus it is unlikely that we missed any large studies. We were unable to formally assess publication bias as fewer than 10 studies met our inclusion criteria.
Agreements and disagreements with other studies or reviews
No previous systematic reviews have been undertaken looking at the effects of MVA85A.
There has been much debate over the contribution of animal studies to the progression of MVA85A vaccine to trial (Cohen 2018; McShane 2018). We systematically assessed Phase 1 and 2 data and we found no difference in tuberculosis incidence in any population, and no increase in the risk of serious adverse effects attributable to the vaccine. There was a small increase in the risk of experiencing any adverse event.
The findings of this review are consistent in that MVA85A is not efficacious for preventing tuberculosis and that there is no evidence that the MVA85A vaccine caused any serious harm to participants in the trials during its investigation.
Authors' conclusions
Implications for practice.
MVA85A in conjunction with Bacillus Calmette‐Guérin (BCG) has no effect on the risk of developing active or latent tuberculosis.
Implications for research.
Researchers should define outcomes precisely before starting the trial. If composite outcomes are developed during the trial, this process needs to be transparent, clearly reported, and published prior to breaking the randomized code. Standardization of outcome measures for tuberculosis vaccine efficacy may make it easier for future researchers in the field and allow easy comparison and meta‐analysis of different study outcomes.
Acknowledgements
The Academic Editor was Dr Michael Eisenhut.
We acknowledge Taryn Young for involvement in conception of the review question, protocol development, and initial contribution to the study review. We are grateful to Vittoria Lutje, the Information Retrieval Specialist of the Cochrane Infectious Diseases Group (CIDG) for her help with the literature search strategy.
The review author team and the CIDG editorial base are supported by the Research, Evidence and Development Initiative (READ‐It) project. READ‐It (project number 300342‐104) is funded by UK aid from the UK government; however, the views expressed do not necessarily reflect the UK government’s official policies.
Appendices
Appendix 1. Search strategies
Cochrane Central Register of Controlled Trials
#1 tuberculosis or TB:ti,ab,kw (Word variations have been searched)
#2 MeSH descriptor: [Tuberculosis] explode all trees
#3 MeSH descriptor: [BCG Vaccine] explode all trees
#4 "BCG vaccin*":ti,ab,kw (Word variations have been searched)
#5 bacill* Calmette‐Guerin
#6 #1 or #2 or #3 or #4 or #5
#7 "antigen 85A" or Ag85A or "modified vaccinia ankara" or MVA85A
#8 MVA85*
#9 #7 or #8
#10 #9 and #6
MEDLINE (PubMed)
| #12 | Search #7 and #11 |
| #11 | Search ((#8) OR #9) OR #10 |
| #10 | Search "drug therapy" [Subheading] |
| #9 | Search randomized or placebo or randomly or trial or groups Field: Title/Abstract |
| #8 | Search "Randomized Controlled Trial" [Publication Type] OR "Controlled Clinical Trial" [Publication Type] |
| #7 | Search #3 and #6 |
| #6 | Search 4 or 5 |
| #5 | "antigen 85A" OR Ag85A OR "modified vaccinia ankara" OR MVA85A Field: Title/Abstract |
| #4 | "antigen 85A, Mycobacterium tuberculosis" [Supplementary Concept] or "MVA 85A" [Supplementary Concept]) |
| #3 | Search 1 or 2 |
| #2 | (("BCG Vaccine"[Mesh]) OR (“bcg vaccin*” or “bacille Calmette‐Guérin” )Field: Title/Abstract |
| #1 | "Tuberculosis"[Mesh] or (tuberculosis or TB) Field: Title/Abstract |
Embase
1 (tuberculosis or tuberculous or TB).mp.
2 tuberculosis/
3 1 or 2
4 BCG vaccine/ or BCG vaccin*.mp. or BCG vaccination/
5 3 or 4
6 MVA85A.mp.
7 antigen 85A.mp.
8 Ag85A.mp.
9 modified vaccinia virus ankara.mp.
10 modified vaccine ankara.mp.
11 6 or 7 or 8 or 9 or 10
12 5 and 11
13 (randomized or randomised or placebo or double‐blind* or single‐blind*).mp.
14 randomized controlled trial/ or controlled clinical trial/
15 crossover procedure/
16 13 or 14 or 15
17 12 and 16
CINAHL (EBSCOHost)
| # | Search terms |
| S1 | TX ( tuberculosis or TB or BCG ) |
| S2 | TX ( (MVA85A or "antigen 85A" or "modified vaccinia ankara" ) |
| S3 | TX ( (randomized trial or controlled trial or placebo or double‐blind* or single‐blind* ) |
| S4 | S1 AND S2 AND S3 |
Web of Science
| # 2 |
TOPIC: (tuberculosis or TB or BCG) ANDTOPIC: (MVA85A or "antigen 85A" or "modified vaccinia ankara") ANDTOPIC: (randomized trial or controlled trial or placebo or double‐blind* or single‐blind*) Timespan=All years Search language=Auto |
| # 1 |
TOPIC: (tuberculosis or TB or BCG) ANDTOPIC: (MVA85A or "antigen 85A" or "modified vaccinia ankara") Timespan=All years |
Data and analyses
Comparison 1. MVA85A versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Active tuberculosis (TB): confirmed by culture or Xpert® MTB/RIF longest reported follow‐up | 2 | 3439 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.58, 1.62] |
| 2 Active TB: started on TB treatment | 3 | 3687 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.92, 1.33] |
| 3 Latent TB | 4 | 3831 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.85, 1.21] |
| 4 Adverse effects of any severity | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Local: skin | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Malaise | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Lethargy | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Any fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Vomiting | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse effects of any severity: aggregated | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6 Serious adverse effects | 3 | 3692 | Risk Difference (M‐H, Fixed, 95% CI) | 0.00 [‐0.00, 0.00] |
| 7 Adverse events of any severity | 4 | 3836 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [1.02, 1.08] |
| 8 Abnormal biochemical tests | 2 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.60, 1.97] |
Comparison 2. Comparison of endpoints.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tameris 2013: incidence of tuberculosis (TB) according to post‐hoc endpoints | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Ndiaye 2015: incidence of TB according to post hoc defined endpoints | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Andrews 2017.
| Methods | Study objective: to investigate the relation between QFT conversion interferon‐γ values and risk of subsequent active TB disease and of QFT reversion. This is a follow‐up study of the Tameris 2013 trial. Study design: observational follow‐up study based on a parallel‐group, randomized, placebo‐controlled double‐blind Phase 2b trial. Study duration: 41 months Length of follow‐up: ≥ 15 months after enrolment, and up to 41 months (based on the Tameris 2013 trial) Follow‐up method: no additional data for this study than described in the Tameris 2013 trial. Losses to follow‐up: 285/2797 children from Tameris 2013 to enrolment for this study analysis at day 336; 467/2512 children from day 336 until the end of the study. Power calculation: not relevant for this observational follow‐up study. |
|
| Participants | Number: 2512/2797 participants enrolled in Tameris 2013 were quantiFERON‐negative at enrolment and had another quantiFERON done at day 336 and were therefore enrolled for this study analysis. No disaggregated data on age and sex between intervention and control groups among these 2512 participants. Target group: infants aged 4–6 months Inclusion criteria
Exclusion criteria
HIV status: negative Other comorbidities: none reported Preterms:
|
|
| Interventions | Intervention group
Control group
|
|
| Outcomes | Outcomes included in this review
Outcomes not included in this review:
|
|
| Notes | Country: South Africa Setting: rural, near Cape Town Background prevalence of TB: extremely high. The overall incidence of TB in South Africa in 2011 was estimated to be almost 1%, and the incidence of TB in children aged < 2 years was about 3% at the trial site. Study dates: enrolment 15 July 2009 to 4 May 2011 and follow‐up until 60 days after the 25 October 2012 Study sponsor: Aeras Other funders: Wellcome Trust and Oxford Emergent TB Consortium (OETC). No additional funding than from the Tameris 2013 trial was obtained for the analysis of these data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from report: "Young children were randomly assigned (1:1) using independently generated sequences with block sizes of four to receive one dose of the vaccine MVA85A or Candida spp skin test antigen (placebo control)." Comment: an independent statistician prepared the randomization schedule as reported in the trial where the data came from that is referenced above (Tameris 2013). |
| Allocation concealment (selection bias) | Low risk | Comment: voice response system adequately concealed allocation of intervention as reported in Tameris 2013. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: same as in Tameris 2013. It did not affect long‐term follow‐up. Quote from Tameris 2013: "Parents or legal guardians of study participants, study staff administering vaccine or undertaking follow up clinical assessments and laboratory staff were masked to intervention group assignment." "Doses were prepared and labelled in masked syringes by an unmasked study pharmacist." |
| Blinding of outcome assessment (detection bias): laboratory assessors | Unclear risk | Comment: no information on whether laboratory assessors were blinded. |
| Blinding of outcome assessment (detection bias): clinical assessors All outcomes | Unclear risk | Quote from report: "Study clinicians were not masked to QFT values, but strict case definitions were used that excluded QFT results." Comment: although clinicians were not masked to QFT values, relevant outcome of conversion is objective and authors used strict case definitions. May not necessarily affect incidence in the two groups as there were no QFT differences between placebo and MVA85A at 336 days (baseline). No details on whether they were masked to group (MVA85A or placebo). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote from report: "Among the 2797 young children enrolled in the MVA85A trial (Tameris 2013) 2772 (99%) young children had a negative QFT at enrolment, five (<1%) had no quantitative results available, and 20 (1%) had an indeterminate result. 1399 young children were allocated to MVA85A and 1398 were allocated to placebo. Among those 2772 young children with a negative QFT at baseline, 2512 (91%) had a QFT done at the day 336 visit." Comment: no imputation Of above 2512, 172 positive and 13 indeterminate. Numbers of negative and converted did not add up to the initial study group. |
| Selective reporting (reporting bias) | Unclear risk | Comment: outcome/objective of this study not seen in protocol for trial (MVA85A 020 TRIAL). Could not find separate protocol for Andrews trial. |
| Other bias | Unclear risk | Comment: employees and beneficiaries of funders were involved in design, analysis, and manuscript writing. This study was a follow‐up of children enrolled in Tameris 2013 trial. |
Bunyasi 2017.
| Methods | Study objective: to evaluate the long‐term effectiveness of infant MVA85A vaccination against TB. This is a long‐term follow‐up study of the Tameris 2013 trial. Study design: retrospective passive follow‐up of the randomized controlled trial Study duration: 22 months for enrolment in the original trial Length of follow‐up: median of 5 years' follow‐up Follow‐up method: passive surveillance based on the electronic TB register database. Losses to follow‐up: there was some inconsistency between the number of participants included for this long‐term follow‐up study and the number of participants who were lost to follow‐up at an early point in the original trial (Tameris 2013). Power calculation: not relevant for this observational follow‐up study |
|
| Participants | Number: 2794 in the Tameris 2013 trial, 2678 included in this long‐term follow‐up analysis Median age: 4.8 years (IQR 4.4 to 5.2) at the end of the extended follow‐up period, comparable across intervention and control groups with no detailed data given in the manuscript. Target group: infants aged 4–6 months Inclusion criteria for the base trial
Exclusion criteria for the base trial
HIV status: negative Other comorbidities: none reported Preterms in the initial sample size of the base trial
|
|
| Interventions | Intervention group
Control group
|
|
| Outcomes | Outcomes included in this review
Outcomes not included in this review
|
|
| Notes | Country: South Africa Setting: rural, near Cape Town Background prevalence of TB: extremely high. The overall incidence of TB in South Africa in 2011 was estimated to be almost 1%, and the incidence of TB in children aged < 2 years was about 3% at the trial site. Study dates: enrolment from 15 July 2009 to 4 May 2011 and follow‐up to 2014 Study sponsor: Aeras Other funders: Wellcome trust and Oxford Emergent TB Consortium (OETC). No additional funding than from the Tameris 2013 trial was obtained for the analysis of these data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from Tameris 2013: "We randomly allocated infants in a 1.1 ratio with a block size of 4 using interactive voice /online response system…" "An independent statistician prepared the randomisation schedule." |
| Allocation concealment (selection bias) | Low risk | Comment: voice response system adequately concealed allocation of intervention as reported in Tameris 2013. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: same as in Tameris 2013. It did not affect long term follow‐up. Quote from Tameris 2013: "Parents or legal guardians of study participants, study staff administering vaccine or undertaking follow‐up clinical assessments and laboratory staff were masked to intervention group assignment." "Doses were prepared and labelled in masked syringes by an unmasked study pharmacists." |
| Blinding of outcome assessment (detection bias): laboratory assessors | Unclear risk | Not applicable. |
| Blinding of outcome assessment (detection bias): clinical assessors All outcomes | Unclear risk | Quote from report: "We also obtained post‐trial data from a regional electronic TB register (ETR) (2012–2014). Comment: no information on how data were collected from this register. Clinical diagnosis of TB was also a subjective outcome. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote from report: "199 participants discontinued FU [follow‐up] early." Comment: 119 participants were excluded from Tameris 2013 for analysis in the current study. No details on how many participants there were from each group of the study. |
| Selective reporting (reporting bias) | Unclear risk | Comment: raw data not reported. Only reported incidence rate ratios. There were no disaggregated data on missing data for each group. Number of participants with TB were not reported per group. Only incidence per year. |
| Other bias | Unclear risk | Quote from report: "The authors received no specific funding for this work," "Conflicts of interest: none declared." "Study is a follow up to Tameris 2013 where the trial sponsor contributed to study design, data interpretation, and writing of the manuscript." |
Ndiaye 2015.
| Methods | Study objective: to assess the safety, immunogenicity, and efficacy of MVA85A vaccine in adults HIV‐positive. Study design: multicentre randomized double‐blind placebo‐controlled trial, Phase 2 Study duration: 46 months (from August 2011 to May 2014) Length of follow‐up: ≥ 6 months after enrolment Follow‐up method
Losses to follow‐up: 14 participants. 5/324 (1.5%) in the intervention group and 9/326 (2.7%) in the control group. Additionally, 3 participants in the intervention group and 2 in the control group withdrew consent; and 2 participants in the intervention group and 4 in the control group died before the end of the study. In total, 325/649 participants completed the study. Power calculation: the sample size calculation was planned to detect active TB. However, after the Tameris 2013 efficacy data were revised, the authors changed the trial design with safety as the primary objective. A smaller sample size was considered and follow‐up was shortened. Therefore, the present trial was underpowered to detect an effect on active TB. |
|
| Participants | Number: 649 (292 from Cape Town, 358 from Dakar).
Target group: adults Inclusion criteria
Exclusion criteria
HIV status: positive Other comorbidities: none reported Preterms: not mentioned |
|
| Interventions | Intervention group
Control group
|
|
| Outcomes | Outcomes included in this review
Outcomes not included in this review
|
|
| Notes | Countries: South Africa and Senegal Setting: Cape Town (South Africa) and Dakar (Senegal), urban Background prevalence of TB
Study dates: 4 August 2011 to 24 April 2013 for enrolment, with follow‐up until 19 May 2014 Study sponsor: Aeras. Collaborators: University of Oxford and European and Developing Countries Clinical Trials Partnership (EDCTP) (IP.2007.32080.002) Funders: Bill & Melinda Gates Foundation, Wellcome Trust, and Oxford‐Emergent Tuberculosis Consortium |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from the report: "Participants were randomly assigned (1:1) in blocks of four by a randomly generated sequence of participant identification numbers via an interactive voice response system to receive two intradermal injections of either 1 × 108 pfu MVA85A or placebo." |
| Allocation concealment (selection bias) | Low risk | Quote from the report: "A statistician uninvolved with study analyses prepared the interactive voice response system randomisation schedule." Comment: the interactive automated voice response system would make it impossible to predict the allocation sequence. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote from the report: "Participants, nurses (who were involved in assessment and follow‐up) investigators, and laboratory staff were masked to group allocation." "Doses of vaccines were prepared and labelled in masked syringes." Quote from the protocol: "The MVA85A/AERAS‐485 and the placebo will be packaged and labelled to appear indistinguishable from each other at the time of injection. Identical syringes and needles will be used for preparation and administration of injections of vaccine/placebo, and labels accompanying the syringes of prepared vaccine/placebo doses will not indicate which is in the syringe." |
| Blinding of outcome assessment (detection bias): laboratory assessors | Low risk | Comment: as quoted above and outcome objective |
| Blinding of outcome assessment (detection bias): clinical assessors All outcomes | Low risk | Quote from the protocol in supplement: "The study vaccine manager and the study monitor will be the only persons unblinded at the site during the study and must not reveal individual subject treatment assignments to any other member of the study team. The study vaccine manager must be a designated study team member who is not an employee of Aeras and who will have no other clinical or regulatory responsibilities associated with the conduct of the study during the entire study period. Unblinded study personnel must not participate in the evaluation of adverse events." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote from the report: "650 were randomly assigned; 649 were included in the safety analysis and 645 in the per‐protocol analysis." Median follow‐up for the 320 recipients of MVA85A was 655 days and for the 325 recipients of placebo was 654 days. "Other than 4 participants, all participants were included in the analysis." Comment: when authors refer to "per‐protocol analysis," this is actually regarding the analysis for the efficacy outcome. Results for per‐protocol analyses were noted to be not different from the intention‐to‐treat results that were not reported. |
| Selective reporting (reporting bias) | Unclear risk | Quote from the report: "Routine haematological and biochemical test results did not differ between study groups (data not shown)." Adverse effects solicited by the vaccine were not disaggregated by type of event. |
| Other bias | Unclear risk | Quote from the report: "The secondary outcome was the efficacy of MVA85A for the prevention of active tuberculosis in the per‐protocol population which was determined by the incidence of active tuberculosis meeting the definition of endpoint 1, calculated as the number of new cases of active tuberculosis with a date of diagnosis from 28 days after the first vaccination until the end of the study follow‐up (May 19, 2014)." Comment: the start of the intervention did not coincide with the start of follow‐up; therefore a period of follow‐up was excluded, and participants who experienced the outcome soon after intervention were missing from analyses. As such, the way in which outcomes were measured may bias effect estimates. This study was stopped early owing to data from the Tameris 2013 trial. As such, it was underpowered to measure efficacy outcomes. Quote: "Aeras was the trial sponsor and contributed to study design and data analysis." Comment: impact of sponsor involvement in analysis of results unclear |
Nemes 2018.
| Methods | Study objective: to assess safety and immunogenicity of MVA85A vaccination in newborns of HIV‐positive mothers, followed by selective deferred BCG vaccination at 8 weeks for HIV‐negative infants. Study design: double‐blind, randomized controlled trial Study duration: not mentioned Length of follow‐up: 52 weeks Follow‐up method
Losses to follow‐up: 9 participants (3 in the intervention group, 6 in the control group) Power calculation: the sample size had 90% probability of detecting a serious adverse event with a true occurrence rate of 1.5% in infants receiving MVA85A vaccine and 80% power to detect a 15% difference in the rate of non‐serious adverse events (20% compared to 35%) between the 2 study groups (P < 0.05). |
|
| Participants | Number: 248
Target group: infants of HIV‐positive mothers Inclusion criteria
Exclusion criteria
HIV status: infants of HIV‐positive mothers
Other comorbidities: none reported Preterms: median gestational age
|
|
| Interventions | Intervention group
Control group
|
|
| Outcomes | Outcomes included in this review:
Outcomes not included in this review
|
|
| Notes | Country: South Africa Setting: urban (Cape Winelands east district and Khayelitsha) Background prevalence of TB: not mentioned Study dates: not reported. According to Clinicaltrial.gov, the study started in October 2012, and was completed in October 2015. Study sponsor: Aeras. Other funders: UK Medical Research Council, Department for International Development, and Wellcome Trust Joint Global Health Trials programme and AERAS. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from supplementary: "Assignment to study arm was double‐blinded and based on a random number sequence prepared by an independent statistician." |
| Allocation concealment (selection bias) | Low risk | Quote from supplementary: "The study pharmacist, the only unblinded member of the study team, controlled the numbered sealed envelopes containing randomization arm and sequential 3‐digit enrolment number." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: from the statement that the pharmacist was the only unblinded member in the team we assumed everyone else was blinded and it was effective. |
| Blinding of outcome assessment (detection bias): laboratory assessors | Low risk | Comment: from the statement that the pharmacist was the only unblinded member in the team we assumed everyone else was blinded and it was effective. |
| Blinding of outcome assessment (detection bias): clinical assessors All outcomes | Low risk | Comment: from the statement that the pharmacist was the only unblinded member in the team we assumed everyone else was blinded and it was effective. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: minimal attrition and balance between groups; 16 in MVA85A group and 19 in control group as set out in figure 1b. |
| Selective reporting (reporting bias) | Low risk | Comment: reported everything they set out in protocol. Additionally reported QFT conversion and incident TB disease; outcomes were not specified in protocol but of importance to mention. |
| Other bias | Unclear risk | Comment: authors declared no conflict of interest. 1 author declared that they were patent holders for MVA85A and were responsible for its development in Scriba 2011. |
Scriba 2011.
| Methods | Study objective: to assess the safety of and to characterize the T‐cell response induced by 3 doses of the candidate vaccine, MVA85A, in BCG‐vaccinated infants from a setting where TB was endemic. Study design: open‐label, Phase 2a safety, immunogenicity, and dose‐finding study Study duration: 23 months Length of follow‐up: 168 days (24 weeks) Follow‐up method
Losses to follow‐up: none Power calculation: not mentioned |
|
| Participants | Number: 144
Target group: infants aged 5–12 months Inclusion criteria
Exclusion criteria
HIV status: negative Other comorbidities: none reported Preterms: not mentioned |
|
| Interventions | Intervention group
Control group
|
|
| Outcomes | Outcomes included in this review
Outcomes not included in this review
|
|
| Notes | Country: South Africa Setting: Cape Town, urban Background prevalence of TB: extremely high (incidence of 1%) Study dates: February 2008 to December 2009 (according to data published in clinicaltrial.gov, not mentioned in the paper) Study sponsor: University of Oxford. Funders: EuropeAID European commission, Wellcome trust |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote from the report: "The aim was to enroll 144 infants into 3 consecutive vaccine dose groups of 48, who would be systematically allocated at a 3:1 ratio to receive either MVA85A (groups 1–3) or placebo." Comment: randomization method predictable. |
| Allocation concealment (selection bias) | High risk | Quote from the report: "…systematically allocated at a 3:1 ratio…" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote from report: "MVA85A, contract manufactured at Impfstoffwerk Dessau‐Tornau (Biologika), was administered intradermally over the deltoid region of the arm contralateral to where BCG was administered." Prevenar was administered intramuscularly. Comment: open label with 2 different routes of administration. Subjective outcomes, so could influence participants when reporting the symptoms. |
| Blinding of outcome assessment (detection bias): laboratory assessors | Low risk | Comment: open label, but with no repercussion on objective laboratory outcomes. |
| Blinding of outcome assessment (detection bias): clinical assessors All outcomes | High risk | Comment: open label, with high repercussion on subjective clinical outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No attrition |
| Selective reporting (reporting bias) | Low risk | Comment: all the outcomes mentioned in the methods section were reported in the results. |
| Other bias | Unclear risk | Quote from report: "…are named inventors on a composition of matter patent for MVA85A filed by the University of Oxford and are shareholders in a joint venture formed for the further development of this vaccine." Comment: unknown role of funders in the elaboration of the study and 2 authors with potential conflict of interest. |
Tameris 2013.
| Methods | Study objective: to assess safety, immunogenicity, and efficacy of MVA85A against TB and Mycobacterium tuberculosis infection in infants. Study design: parallel‐group, randomized, placebo‐controlled double‐blind Phase 2b trial Study duration: 39 months Length of follow‐up: ≥ 15 months after enrolment, and up to 39 months Follow‐up method
Losses to follow‐up
Power calculation: given a TB cumulative incidence of 3% over 18 months in the control group, 1392 participants per treatment group (2784 participants total) would be required to demonstrate positive efficacy when the true efficacy of MVA85A/AERAS‐485 was approximately 60%. An estimate of 7.5% of participants lost to follow‐up in each treatment group was assumed over 18 months. |
|
| Participants | Number: 2797
Target group: infants aged 4–6 months Inclusion criteria
Exclusion criteria
HIV status: negative Other comorbidities: none reported Preterms
|
|
| Interventions | Intervention group
Control group
|
|
| Outcomes | Outcomes included in this review
Outcomes not included in this review
|
|
| Notes | Country: South Africa Setting: rural, near Cape Town Background prevalence of TB: extremely high. The overall incidence of TB in South Africa in 2011 was estimated to be almost 1%, and the incidence of TB in children aged < 2 years was about 3% at the trial site. Study dates: enrolment 15 July 2009 to 4 May 2011 and follow‐up to 25 October 2012 Study sponsor: Aeras. Collaborators: University of Oxford and University of Cape Town. Funders: Aeras, Wellcome trust and Oxford Emergent tb consortium (OETC) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from the report: "We randomly allocated infants in a 1.1 ratio with a block size of 4 using interactive voice /online response system." "An independent statistician prepared the randomisation schedule." |
| Allocation concealment (selection bias) | Low risk | Comment: voice response system adequately concealed allocation of intervention. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote from the report: "Parents or legal guardians of study participants, study staff administering vaccine or undertaking follow up clinical assessments and laboratory staff were masked to intervention group assignment." "Doses were prepared and labelled in masked syringes by an unmasked study pharmacist." Comment: syringes had equal amount of placebo and control. Quote from the protocol: "packaged and labelled to appear indistinguishable to each other." |
| Blinding of outcome assessment (detection bias): laboratory assessors | Low risk | Comment: laboratory staff were masked to intervention group assignment. |
| Blinding of outcome assessment (detection bias): clinical assessors All outcomes | Low risk | Comment: staff undertaking clinical assessments were masked to intervention group assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote from the report: "The number of participants discontinuing the study did not differ between the two treatment groups." 1126 infants (5%) were lost to follow‐up, 11 died (< 1%), and 62 (2%) had consent withdrawn. Comment: reasons for missing outcome data balanced between the 2 groups and proportion of missing data not enough to have a clinically relevant impact on the intervention effect estimate. Per‐protocol analysis was done and only 1 person was excluded from analysis from the placebo group due to dose deviation. |
| Selective reporting (reporting bias) | Unclear risk | Quote from study description from clinical trials.gov: "Adverse events and clinically relevant laboratory results for the safety cohort will be summarized to examine the relationship between treatment group and key safety endpoints including number (percentage) of solicited and spontaneous adverse events, rates of reactogenicity, and number (percentage) of subjects with newly abnormal post‐vaccination laboratory values based on predefined neonatal toxicity criteria." Comment: data were collected; however, no summary provided on biochemical or haematological adverse effects. Unclear if endpoints were specified a priori as endpoint definition was only published alongside the trial and approach outlined a priori on clinical trial registry was amended. The differences between endpoint point 1 and 2 were 5 mm on TST; 2 positive smears compared to 1 positive smear and residence in household with positive AFB member. These endpoints were significantly different from the endpoints used in 2 other trials that included efficacy measures (Ndiaye 2015; Nemes 2018). |
| Other bias | Unclear risk | Quote from the report: "Aeras was the trial sponsor. Aeras and the Oxford‐Emergent Tuberculosis Consortium (OETC) contributed to study design, data interpretation, and writing of the manuscript." Comment: impact of sponsor involvement on study findings unclear. |
AFB: acid‐fast bacilli; ART: antiretroviral therapy; ATT: antituberculosis therapy; BCG: Bacillus Calmette‐Guérin; cART: combination antiretroviral therapy; CFP‐10: culture filtrate protein‐10; cfu: colony‐forming unit; ELISA: enzyme‐linked immunosorbent assay; ELISPOT: enzyme‐linked immune absorbent spot; EPI: Expanded Programme on Immunization; ESAT‐6: early secretory antigenic‐6; FP: floating point; IQR: interquartile range; MVA: modified Vaccinia Ankara; PBMC: peripheral blood mononuclear cell; PCR: polymerase chain reaction; pfu: plaque‐forming unit; PMTCT: prevention of mother‐to‐child transmission; PPD: purified protein derivative; QFT: QuantiFERON‐TB Gold In‐Tube; SD: standard deviation; TB: tuberculosis; TST: tuberculin skin test; WHO: World Health Organization.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Brookes 2008 | Different study design |
| Bunyasi 2015 | Different outcomes measured |
| Dieye 2013 | Different study design |
| Harris 2011 | Different study design |
| Harris 2014a | Different study design |
| Hawkridge 2008 | Different study design |
| Matsumiya 2014a | Different outcomes measured |
| Matsumiya 2014b | Different outcomes measured |
| Matsumiya 2014c | Different outcomes measured |
| McShane 2004 | Different study design |
| Meyer 2013 | Different study design |
| Minassian 2011 | Different study design |
| Minhinnick 2016 | Different study design |
| Mulenga 2015 | Different intervention |
| Odutola 2012 | Different study design |
| Ota 2011 | Different study design |
| Pathan 2007 | Different study design |
| Pathan 2012 | Different study design |
| Rowland 2012 | Different study design |
| Rowland 2013 | Different study design |
| Sander 2009 | Different study design |
| Satti 2014 | Different study design |
| Scriba 2010 | Different study design |
| Scriba 2012 | Different study design |
| Sheehan 2015 | Different study design |
| Tameris 2014 | Measured different outcomes |
| Tanner 2014 | Different study design |
| Whelan 2009 | Different study design |
Differences between protocol and review
Changes to the author team: Taryn Young stepped down from the review author team.
We intended to pilot data extraction forms; however, given the small number of included studies we assessed the appropriateness of the form during the actual data extraction.
In our protocol, we mentioned that the control for the type of intervention would be "BCG alone." However, we did include in our review studies that they used Candin® as control intervention, as this is currently used in control groups for randomized controlled trials assessing MVA85A.
We encountered multiple different definitions of active tuberculosis in different trials. We took the approach of defining active tuberculosis as confirmed by culture and participants starting on tuberculosis treatment to allow a consistent approach across the included studies.
We reported adverse effects of any severity disaggregated by local reactions of the skin and systemic symptoms and we gave justification for this decision in the result section.
The initial risk of bias for adverse event assessment tool had three options to assess completeness of reporting of participant‐reported outcomes. The options complete/incomplete/unclear were reduced to complete/incomplete as there was no difference between the options incomplete and unclear reporting.
The detailed subgroup analysis prespecified in the protocol was not done due to too few studies.
Contributions of authors
RK drafted the review, screened abstracts, extracted data, analysed results, and wrote the final review.
SoJ (Sophie Jullien) drafted the review, screened abstracts, extracted data, analysed results, and wrote the final review.
PG contributed to the methods, coherence, and writing of the final review.
SaJ (Samuel Johnson) co‐ordinated the review, screened abstracts, extracted data, performed analysis of data, and helped draft the final review.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK.
External sources
-
Department for International Development, UK.
Project number 300342‐104
Declarations of interest
RK has no known conflicts of interest.
SoJ worked for the CIDG from September 2015 to April 2016.
PG is the Director of the Research, Evidence and Development Initiative (READ‐It) project (project number 300342‐104) and CIDG Co‐ordinating Editor.
SaJ worked for the CIDG from January 2017 to July 2018.
None of the review authors receive salary, payment, academic fees, or academic status related to vaccine development.
These authors contributed equally to this work
These authors contributed equally to this work
Unchanged
References
References to studies included in this review
Andrews 2017 {published data only}
- Andrews JR, Nemes E, Tameris M, Landry BS, Mahomed H, McClain JB, et al. Serial QuantiFERON testing and tuberculosis disease risk among young children: an observational cohort study. Lancet Respiratory Medicine 2017;5(4):282‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bunyasi 2017 {published data only}
- Bunyasi EW, Luabeya AK, Tameris M, Geldenhuys H, Mulenga H, Landry BS, et al. Impact of isoniazid preventive therapy on the evaluation of long‐term effectiveness of infant MVA85A vaccination. International Journal Tuberculosis and Lung Disease 2017;21(7):778‐83. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ndiaye 2015 {published data only}
- Ndiaye BP, Thienemann F, Ota M, Landry BS, Camara M, Dièye S, et al. Safety, immunogenicity, and efficacy of the candidate tuberculosis vaccine MVA85A in healthy adults infected with HIV‐1: a randomised, placebo‐controlled, phase 2 trial. Lancet Respiratory Medicine 2015;3(3):190‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nemes 2018 {published data only}
- Nemes E, Hesseling A, Tameris M, Mauff K, Downing K, Mulenga H, et al. Safety and immunogenicity of newborn MVA85A vaccination and selective, delayed Bacille Calmette‐Guerin (BCG) for infants of HIV infected mothers: a Phase 2 randomized controlled trial. Clinical Infectious Diseases 2018;66(4):554‐63. [DOI: 10.1093/cid/cix834] [DOI] [PMC free article] [PubMed] [Google Scholar]
Scriba 2011 {published data only}
- Scriba TJ, Tameris M, Mansoor N, Smit E, Merwe L, Mauff K, et al. Dose‐finding study of the novel tuberculosis vaccine, MVA85A, in healthy BCG‐vaccinated infants. Journal of Infectious Diseases 2011;203(12):1832‐43. [DOI: 10.1093/infdis/jir195] [DOI] [PubMed] [Google Scholar]
Tameris 2013 {published data only}
- Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo‐controlled phase 2b trial. Lancet 2013;381(9871):1021‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Brookes 2008 {published data only}
- Brookes RH, Hill PC, Owiafe PK, Ibanga HB, Jeffries DJ, Donkor SA, et al. Safety and immunogenicity of the candidate tuberculosis vaccine MVA85A in West Africa. PLOS One 2008;3(8):e2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bunyasi 2015 {published data only}
- Bunyasi EW, Tameris M, Geldenhuys H, Schmidt BM, Luabeya AK, Mulenga H, et al. Evaluation of Xpert(R) MTB/RIF assay in induced sputum and gastric lavage samples from young children with suspected tuberculosis from the MVA85A TB vaccine trial. PLOS One 2015;10(11):e0141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dieye 2013 {published data only}
- Dieye TN, Ndiaye BP, Dieng AB, Fall M, Britain N, Vermaak S, et al. Two doses of candidate TB vaccine MVA85A in antiretroviral therapy (ART) naive subjects gives comparable immunogenicity to one dose in ART plus subjects. PLOS One 2013;8(6):e67177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harris 2011 {published data only}
- Harris S, Meyer J, Satti I, Kosotv M, Rowland R, Poulton ID, et al. Comparison of the safety and immunogenicity of a candidate TB vaccine, MVA85A, given by the intramuscular or intradermal route in BCG‐vaccinated adults. Annual Congress of the British‐Society‐for‐Immunology. 2011; Vol. 135:55.
Harris 2014a {published data only}
- Harris SA, Meyer J, Satti I, Marsay L, Poulton ID, Tanner R, et al. Evaluation of a human BCG challenge model to assess antimycobacterial immunity induced by BCG and a candidate tuberculosis vaccine, MVA85A, alone and in combination. Journal of Infectious Diseases 2014;209(8):1259‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hawkridge 2008 {published data only}
- Hawkridge T, Scriba TJ, Gelderbloem S, Smit E, Tameris M, Moyo M, et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. Journal of Infectious disease 2008; Vol. 198, issue 4:544‐52. [DOI] [PMC free article] [PubMed]
Matsumiya 2014a {published data only}
- Matsumiya M. Immune response to MVA85A or placebo in BCG‐vaccinated South African infants at days 0 and 28 post‐vaccination. Arrayexpress‐repository, V1 2014. [https://www.ebi.ac.uk/arrayexpress/experiments/E‐GEOD‐56561]
Matsumiya 2014b {published data only}
- Matsumiya M, Harris SA, Satti I, Stockdale L, Tanner R, O’Shea MK, et al. Inflammatory and myeloid‐associated gene expression before and one day after infant vaccination with MVA85A correlates with induction of a T cell response. Biomed Central Infectious Diseases 2014; Vol. 14:314. [DOI] [PMC free article] [PubMed]
Matsumiya 2014c {published data only}
- Matsumiya M. GSE56559: immune response to MVA85A or placebo in BCG‐vaccinated South African infants at day 1 post‐vaccination. Arrayexpress‐repository, V1 2014. [https://www.ebi.ac.uk/arrayexpress/experiments/E‐GEOD‐56559]
McShane 2004 {published data only}
Meyer 2013 {published data only}
- Meyer, J, Harris, S. A, Satti I, Poulton ID, Poyintz HC, Tanner R, et al. Comparing the safety and immunogenicity of a candidate TB vaccine MVA85A administered by intramuscular and intradermal delivery. Vaccine 2013; Vol. 31, issue 2013:1026‐33. [DOI] [PMC free article] [PubMed]
Minassian 2011 {published data only}
- Minassian, AM, Rowland R, Beveridge NE, Poulton ID, Satti I, Harris S, et al. A Phase I study evaluating the safety and immunogenicity of MVA85A, a candidate TB vaccine, in HIV‐infected adults. BrItish Medical Journal 2015; Vol. 1:e000223. [DOI] [PMC free article] [PubMed]
Minhinnick 2016 {published data only}
- Minhinnick A, Satti I, Harris S, Wilkie M, Sheehan S, Stockdale L, et al. A first‐in‐human phase 1 trial to evaluate the safety and immunogenicity of the candidate tuberculosis vaccine MVA85A‐IMX313, administered to BCG‐vaccinated adults. Vaccine Vol. 34, issue 2016:1412‐21. [DOI] [PMC free article] [PubMed]
Mulenga 2015 {published data only}
- MulengaH, Tameris MD, Luabeya KK, Geldenhuys H, Scriba TJ, Hussey GD, et al. The Role of Clinical Symptoms in the Diagnosis of Intrathoracic Tuberculosis in Young Children. Pediatric Infectious Diseases Journal Vol. 34:1157‐62. [DOI] [PMC free article] [PubMed]
Odutola 2012 {published data only}
Ota 2011 {published data only}
Pathan 2007 {published data only}
- Pathan AA, Sander CR, Fletcher HA, Poulton I, Alder NC, Beveridge NE, et al. Boosting BCG with recombinant modified vaccinia ankara expressing antigen 85A: different boosting intervals and implications for efficacy trials. PLOS One 2007;2(10):e1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pathan 2012 {published data only}
- Pathan AA, Minassian AM, Sander CR, Rowland R, Porter DW, Poulton ID, et al. Effect of vaccine dose on the safety and immunogenicity of a candidate TB vaccine, MVA85A, in BCG vaccinated UK adults. Vaccine 2012;30(38):5616‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rowland 2012 {published data only}
- Rowland R, Brittain N, Poulton ID, Minnassain AM, Sander C, Porter DW, et al. A review of the tolerability of the candidate TB vaccine, MVA85A compared with BCG and Yellow Fever vaccines, and correlation between MVA85A vaccine reactogenicity and cellular immunogenicity. Trials in vaccinology 2012;1:27‐35. [Google Scholar]
Rowland 2013 {published data only}
- Rowland R, Pathan AA, Satti I, Poulton ID, Matsimuya MM, Whitaker M, et al. Safety and immunogenicity of an FP9‐vectored candidate tuberculosis vaccine (FP85A), alone and with candidate vaccine MVA85A in BCG‐vaccinated healthy adults: a phase I clinical trial. Human Vaccines and Immunotherapeutics 2013;9(1):50‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sander 2009 {published data only}
- Sander CR, Pathan AA, Beveridge NE, Poulton I, Minassian A, Alder N, et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in Mycobacterium tuberculosis‐infected individuals. American Journal of Respiratory and Critical Care Medicine 2009;179:724‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Satti 2014 {published data only}
- Satti I, Meyer J, Harris SA, Thomas ZM, Griffiths K, Antrobuset RD, et al. Safety and immunogenicity of a candidate tuberculosis vaccine MVA85A delivered by aerosol in BCG‐vaccinated healthy adults: a phase 1, double‐blind, randomised controlled trial. Lancet Infectious diseases 2014;14:939‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scriba 2010 {published data only}
- Scriba TJ, Tameris M, Mansoor N, Thomas ZM, Griffiths K, Antrobus RD, et al. Modified vaccinia Ankara‐expressing Ag85A, a novel tuberculosis vaccine, is safe in adolescents and children, and induces polyfunctional CD4+ T cells. European Journal of Immunology 2010;40:279‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scriba 2012 {published data only}
- Scriba TJ, Tameris M, Smit E, Merwe l, Jane Hughes E, Kadira B, et al. A phase IIa trial of the new tuberculosis vaccine, MVA85A, in HIV‐ and/or Mycobacterium tuberculosis‐infected adults. American Journal of Respiratory and Critical Care Medicine 2012;185(7):769‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sheehan 2015 {published data only}
- Sheehan S, Harris SA, Satti I, Hokey DA, Dheenadhayalan V, Stockdale L, et al. A Phase I, Open‐Label Trial, Evaluating the Safety and Immunogenicity of Candidate Tuberculosis Vaccines AERAS‐402 and MVA85A, Administered by Prime‐Boost Regime in BCG‐Vaccinated Healthy Adults. PLOS One 2015;10(11):e0141687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tameris 2014 {published data only}
- Tameris M, Geldenhuys H, Luabeya AK, Smit E, Hughes JE, Vermaak S, et al. The candidate TB vaccine, MVA85A, induces highly durable Th1 responses. PLOS One 2014;9(2):e87340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanner 2014 {published data only}
- Tanner R, Kakalacheva K, Miller E, Pathan AA, Chalk R, Sander CR, et al. Serum indoleamine 2,3‐dioxygenase activity is associated with reduced immunogenicity following vaccination with MVA85A. Biomed Central Infectious Diseases 2014;14:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Whelan 2009 {published data only}
- Whelan KT, Pathan AA, Sander CR, Fletcher HA, Poulton I, Alder NC, et al. Safety and immunogenicity of boosting BCG vaccinated subjects with BCG: comparison with boosting with a new TB vaccine, MVA85A. PLOS One 2009;10(11):e5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Abubakar 2013
- Abubakar I, Pimpin L, Ariti C, Beynon R, Mangtani P, Sterne JA, et al. Systematic review and meta‐analysis of the current evidence on the duration of protection by bacillus Calmette‐Guérin vaccination against tuberculosis. Health Technology Assessment 2013;17(37):1‐372, v‐vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
Altenburg 2014
- Altenburg AF, Kreijtz JH, Vries RD, Song F, Fux R, Rimmelzwaan GF, et al. Modified vaccinia virus ankara (MVA) as production platform for vaccines against influenza and other viral respiratory diseases. Viruses 2014;6(7):2735‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Baker 2011
- Baker MA, Harries AD, Jeon CY, Hart JE, Kapur A, Lönnroth K, et al. The impact of diabetes on tuberculosis treatment outcomes: a systematic review. BMC Medicine 2011;9:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barker 2012
- Barker L, Hessel L, Walker B. Rational approach to selection and clinical development of TB vaccine candidates. Tuberculosis 2012;92(Suppl 1):S25‐9. [DOI] [PubMed] [Google Scholar]
Behr 2013
- Behr MA, Schwartzman K, Pai M. Tuberculosis vaccine trials. Lancet 2013;381(9885):2252‐3. [DOI] [PubMed] [Google Scholar]
Beveridge 2007
- Beveridge NE, Price DA, Casazza JP, Pathan AA, Sander CR, Asher TE, et al. Immunisation with BCG and recombinant MVA85A induces long‐lasting, polyfunctional Mycobacterium tuberculosis‐specific CD4+ memory T lymphocyte populations. European Journal of Immunology 2007;37(11):3089‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bukirwa 2014
- Bukirwa H, Unnikrishnan B, Kramer CV, Sinclair D, Nair S, Tharyan P. Artesunate plus pyronaridine for treating uncomplicated Plasmodium falciparum malaria. Cochrane Database of Systematic Reviews 2014, Issue 3. [DOI: 10.1002/14651858.CD006404.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2000
- Centers for Disease Control and Prevention. Diagnostic standards and classification of tuberculosis in adults and children. This official statement of the American Thoracic Society and the Centers for Disease Control and Prevention was adopted by the ATS Board of Directors, July 1999. This statement was endorsed by the Council of the Infectious Disease Society of America. American Journal of Respiratory and Critical Care Medicine 2000;161(4 Pt 1):1376‐95. [DOI] [PubMed] [Google Scholar]
Cohen 2018
- Cohen D. Oxford TB vaccine study calls into question selective use of animal data. BMJ 2018;360:j5845. [DOI] [PubMed] [Google Scholar]
Colditz 1995
- Colditz GA, Berkey CS, Mosteller F, Brewer TF, Wilson ME, Burdick E, et al. The efficacy of bacillus Calmette‐Guérin vaccination of newborns and infants in the prevention of tuberculosis: meta‐analyses of the published literature. Paediatrics 1995;96(1 Pt 1):29‐35. [PubMed] [Google Scholar]
Daftary 2012
- Daftary A, Padayatchi N. Social constraints to TB/HIV healthcare: accounts from coinfected patients in South Africa. AIDS Care 2012;24(12):1480‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
de Cassan 2010
- Cassan SC, Pathan AA, Sander CR, Minassian A, Rowland R, Hill AV, et al. Investigating the induction of vaccine‐induced Th17 and regulatory T cells in healthy, Mycobacterium bovis BCG‐immunized adults vaccinated with a new tuberculosis vaccine, MVA85A. Clinical and Vaccine Immunology 2010;17(7):1066‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Eisenhut 2009
- Eisenhut M, Paranjothy S, Abubakar I, Bracebridge S, Lilley M, Mulla R, et al. BCG vaccination reduces risk of infection with Mycobacterium tuberculosis as detected by gamma interferon release assay. Vaccine 2009;27(44):6116‐20. [DOI] [PubMed] [Google Scholar]
Favrot 2013
- Favrot L, Grzegorzewicz AE, Lajiness DH, Marvin RK, Boucau J, Isailovic D, et al. Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nature Communications 2013;4:2748. [DOI: 10.1038/ncomms3748] [DOI] [PMC free article] [PubMed] [Google Scholar]
Graham 2012
- Graham SM, Ahmed T, Amanullah F, Browning R, Cardenas V, Casenghi M, et al. Evaluation of tuberculosis diagnostics in children: 1. Proposed clinical case definitions for classification of intrathoracic tuberculosis disease. Consensus from an expert panel. Journal of Infectious Diseases 2012;205(2):S199‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Griffiths 2011
- Griffiths KL, Pathan AA, Minassian AM, Sander CR, Beveridge NE, Hill AV, et al. Th1/Th17 cell induction and corresponding reduction in ATP consumption following vaccination with the novel Mycobacterium tuberculosis vaccine MVA85A. PLOS One 2011;6(8):e23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harries 2006
- Harries AD, Dye C. Tuberculosis. Annals of Tropical Medicine and Parasitology 2006;100(5‐6):415‐31. [DOI] [PubMed] [Google Scholar]
Harris 2014b
- Harris SA, Satti I, Matsumiya M, Stockdale L, Chomka A, Tanner R, et al. Process of assay selection and optimization for the study of case and control samples from a phase IIb efficacy trial of a candidate tuberculosis vaccine, MVA85A. Clinical and Vaccine Immunology 2014;21(7):1005‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org. Chichester (UK): Available from www.handbook.cochrane.org..
Ibanga 2006
- Ibanga HB, Brookes RH, Hill PC, Owiafe PK, Fletcher HA, Lienhardt C, et al. Early clinical trials with a new tuberculosis vaccine, MVA85A, in tuberculosis‐endemic countries: issues in study design. Lancet Infectious Diseases 2006;6(8):522‐8. [DOI] [PubMed] [Google Scholar]
ICH 1994
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Clinical safety data management: definitions and standards for expedited reporting E2A.1994. ICH harmonised tripartite guideline. Current Step 4 version dated 27 October 1994. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2A/Step4/E2A_Guideline.pdf (accessed 23 August 2017).
Kashangura 2015
- Kashangura R, Sena ES, Young T, Garner P. Effects of MVA85A vaccine on tuberculosis challenge in animals: systematic review. International Journal of Epidemiology 2015;44(6):1970‐81. [DOI: 10.1093/ije/dyv142] [DOI] [PMC free article] [PubMed] [Google Scholar]
Loke 2011
- Loke YK, Price D, Herxheimer A. Chapter 14: Adverse effects. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Manjaly 2016
- Manjaly TZ, Satti I, Wilkie M, Harris S, Riste M, Hamidi A, et al. A phase I trial evaluating aerosol administration of a candidate TB vaccine, MVA85A, as a way to induce potent local cellular immune responses and avoid anti‐vector immunity. American Journal of Respiratory and Critical Care Medicine 2016;193:A5487. [Google Scholar]
Marais 2004
- Marais BJ, Gie RP, Schaaf HS, Hesseling AC, Obihara CC, Starke JJ, et al. The natural history of childhood intra‐thoracic tuberculosis: a critical review of literature from the pre‐chemotherapy era. International Journal of Tuberculosis and Lung Disease 2004;8(4):392‐402. [PubMed] [Google Scholar]
Matsumiya 2013
- Matsumiya M, Stylianou E, Griffiths K, Lang Z, Meyer J, Harris SA, et al. Roles for Treg expansion and HMGB1 signaling through the TLR1‐2‐6 axis in determining the magnitude of the antigen‐specific immune response to MVA85A. PLOS One 2013;8(7):e67922. [DOI] [PMC free article] [PubMed] [Google Scholar]
McShane 2014
- McShane H, Williams A. A review of preclinical animal models utilised for TB vaccine evaluation in the context of recent human efficacy data. Tuberculosis 2014;94(2):105‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
McShane 2018
- McShane H, Hill A, Hatherill M, Tameris M, Shea J, Ginsberg A. Helen McShane and colleagues reply to Deborah Cohen. BMJ 2018;360:k236. [DOI] [PubMed] [Google Scholar]
NCT00395720
- NCT00395720. The safety and immunogenicity of a TB vaccine; MVA85A, in healthy volunteers who are infected with HIV. clinicaltrials.gov/ct2/show/NCT00395720 (first received 25 August 2017).
NCT00423566
- NCT00423566. A phase I study of the safety and immunogenicity of a recombinant MVA vaccine encoding a secreted antigen from M. tuberculosis, antigen 85A, delivered intradermally by a needle injection in healthy volunteers. clinicaltrials.gov/ct2/show/NCT00423566 (first received 25 August 2017).
NCT00423839
- NCT00423839. A phase I study of the safety and immunogenicity of MVA85A in healthy Gambian volunteers. clinicaltrials.gov/ct2/show/NCT00423839 (first received 25 August 2017).
NCT00427453
- NCT00427453. A phase I study of the safety and immunogenicity of a recombinant MVA vaccine encoding a secreted antigen from M. tuberculosis, antigen 85A, delivered intradermally by a needle injection in healthy volunteers who have received BCG immunisation 1 month previously. clinicaltrials.gov/ct2/show/NCT00427453 (first received 25 August 2017).
NCT00427830
- NCT00427830. A phase I study of the safety and immunogenicity of a recombinant MVA vaccine encoding a secreted antigen from M. tuberculosis, antigen 85A, delivered intradermally by a needle injection in healthy volunteers who have previously received BCG. clinicaltrials.gov/ct2/show/NCT00427830 (first received 25 August 2017).
NCT00456183
- NCT00456183. Safety and immunogenicity of MVA85A in volunteers latently infected with TB. clinicaltrials.gov/ct2/show/NCT00456183 (first received 25 August 2017).
NCT00460590
- NCT00460590. Safety and immunogenicity of MVA85A, in healthy volunteers in Cape Town. clinicaltrials.gov/ct2/show/NCT00460590 (first received 25 August 2017).
NCT00465465
- NCT00465465. A study of 2 doses of a new TB vaccine, MVA85A, in healthy volunteers previously vaccinated with BCG. clinicaltrials.gov/ct2/show/NCT00465465 (first received 25 August 2017).
NCT00480454
- NCT00480454. Safety, immunogenicity, and impact of MVA85A, on the immunogenicity of the EPI vaccines. clinicaltrials.gov/ct2/show/NCT00480454 (first received 25 August 2017).
NCT00480558
- NCT00480558. A study of MVA85A, in asymptomatic volunteers infected with TB, HIV or both. clinicaltrials.gov/ct2/show/NCT00480558 (first received 25 August 2017).
NCT00548444
- NCT00548444. T‐cell turnover following vaccination with MVA85A. clinicaltrials.gov/ct2/show/NCT00548444 (first received 25 August 2017).
NCT00653770
- NCT00653770. A phase I study to assess the safety and immunogenicity of tuberculosis (TB) vaccine candidates FP85A and MVA85A. clinicaltrials.gov/ct2/show/NCT00653770 (first received 25 August 2017).
NCT00731471
- NCT00731471. A phase I study of a new tuberculosis (TB) vaccine, MVA85A, in healthy volunteers with HIV. clinicaltrials.gov/ct2/show/NCT00731471 (first received 25 August 2017).
NCT00953927
- NCT00953927. A study of MVA85A in healthy infants. clinicaltrials.gov/ct2/show/NCT00953927 (first received 25 August 2017).
NCT01181856
- NCT01181856. Safety of tuberculosis vaccine, MVA85A, administered by the intramuscular route and the intradermal route. clinicaltrials.gov/ct2/show/NCT01181856 (first received 25 August 2017).
NCT01194180
- NCT01194180. A BCG challenge model study to assess anti‐mycobacterial immunity induced by BCG and a candidate TB vaccine, MVA85A. clinicaltrials.gov/ct2/show/NCT01194180 (first received 25 August 2017).
NCT01497769
- NCT01497769. Safety of tuberculosis vaccine, MVA85A, administered by the aerosol route and the intradermal route. clinicaltrials.gov/ct2/show/NCT01497769 (first received 25 August 2017).
NCT01683773
- NCT01683773. Safety study of tuberculosis vaccines AERAS‐402 and MVA85A. clinicaltrials.gov/ct2/show/NCT01683773 (first received 25 August 2017).
NCT01829490
- NCT01829490. Safety study of ChAdOx185A vaccination with and without MVA85A boost in healthy adults. clinicaltrials.gov/show/NCT01829490 (first received 11 April 2013).
NCT01879163
- NCT01879163. Phase I trial evaluating safety and immunogenicity of MVA85A‐IMX313 compared to MVA85A in BCG vaccinated adults. clinicaltrials.gov/ct2/show/NCT01879163 (first received 25 August 2017).
NCT01954563
- NCT01954563. Study evaluating aerosol and intradermal administration of a candidate tuberculosis (TB) vaccine, MVA85A, as a way to increase immune response and avoid anti‐vector immunity. clinicaltrials.gov/ct2/show/NCT01954563 (first received 25 August 2017).
NCT02532036
- NCT02532036. MVA85A aerosol versus intramuscular vaccination in adults with latent Mycobacterium tuberculosis (M. tb) Infection. clinicaltrials.gov/show/NCT02532036 (first received 25 August 2015).
Owiafe 2012
- Owiafe P, Hill P, Ibanga HB, Brookes RH, McShane H, Sutherland JS, et al. Differential cytokine levels in adults induced by a novel candidate TB boost vaccine, MVA85A‐according to previous BCG vaccination status. Journal of Vaccines & Vaccination 2012;3(7):158. [Google Scholar]
Perez‐Velez 2012
- Perez‐Velez CM, Marais BJ. Tuberculosis in children. New England Journal of Medicine 2012;367(4):348‐61. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Romano 2006
- Romano M, D'Souza S, Adnet PY, Laali R, Jurion F, Palfiet K, et al. Priming but not boosting with plasmid DNA encoding mycolyl‐transferase Ag85a from Mycobacterium tuberculosis increases the survival time of Mycobacterium bovis BCG vaccinated mice against low dose intravenous challenge with M. Tuberculosis H37Rv. Vaccine 2006;24:3353‐64. [DOI] [PubMed] [Google Scholar]
Roy 2014
- Roy A, Eisenhut M, Harris RJ, Rodrigues LC, Sridhar S, Habermann S, et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta‐analysis. BMJ 2014;349:g4643. [DOI: 10.1136/bmj.g4643] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A. GRADE handbook for grading quality of evidence and strength of recommendations. Available from guidelinedevelopment.org/handbook October 2013.
Sharma 2012
- Sharma SK, Mohanan S, Sharma A. Relevance of latent TB infection in areas of high TB prevalence. Chest 2012;142(3):761‐73. [DOI] [PubMed] [Google Scholar]
Tiemersma 2011
- Tiemersma EW, Werf MJ, Borgdorff MW, Williams BG, Nagelkerke NJ. Natural history of tuberculosis: duration and fatality of untreated pulmonary tuberculosis in HIV negative patients: a systematic review. PLOS One 2011;6(4):e17601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wajja 2017
- Wajja A, Kizito D, Nassanga B, Nalwoga A, Kabagenyi J, Kimuda S, et al. The effect of current Schistosoma mansoni infection on the immunogenicity of a candidate TB vaccine, MVA85A, in BCG‐vaccinated adolescents: an open‐label trial. PLOS Neglected Tropical Diseases 2017;11(5):e0005440. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2018
- World Health Organization. Global tuberculosis report 2018. www.who.int/tb/publications/global_report/en/ (accessed 3 January 2018).
WHO‐ART 2008
- Uppsala Monitoring Centre. WHO adverse reaction terminology (WHO‐ART). www.who‐umc.org/vigibase/services/learn‐more‐about‐who‐art/ (accessed 23 August 2017).
References to other published versions of this review
Kashangura 2018
- Kashangura R, Jullien S, Garner P, Young T, Johnson S. MVA85A vaccine to enhance BCG for preventing tuberculosis. Cochrane Database of Systematic Reviews 2018, Issue 1. [DOI: 10.1002/14651858.CD012915] [DOI] [PMC free article] [PubMed] [Google Scholar]