

Abstract
Ovarian cancer (OC) is the most common gynecologic malignancy with high incidence and mortality. The present study aimed to develop approaches for determining the recurrence type and identify potential miRNA markers for OC prognosis. The miRNA expression profile of OC (the training set, including 390 samples with recurrence information) was downloaded from The Cancer Genome Atlas database. The validation sets GSE25204 and GSE27290 were obtained from the Gene Expression Omnibus database. Prescreening of clinical factors was conducted using the survival package, and the differentially expressed miRNAs (DE-miRNAs) were identified using the limma package. Using the Caret package, the optimal miRNA set was selected to build a Support Vector Machine (SVM) classifier. The miRNAs and clinical factors independently related to prognosis were analyzed using the survival package, and the risk score system was constructed. Finally, the miRNA-target regulatory network was built by Cytoscape software, and enrichment analysis was performed. There were 46 DE-miRNAs between the recurrent and non-recurrent samples. After the optimal 19-miRNA set was selected for constructing the SVM classifier, 6 DE-miRNAs (miR-193b, miR-211, miR-218, miR-505, miR-508 and miR-514) independently related to prognosis were further extracted to build the risk score system. The neoplasm cancer status was independently correlated with the prognosis and conducted with stratified analysis. Additionally, the target genes in the regulatory network were enriched in the regulation of actin cytoskeleton and the TGF-β signaling pathway. The 6-miRNA signature may serve as a potential biomarker for OC prognosis, particularlyfor recurrence.
Keywords: ovarian cancer, microRNA, Support Vector Machine, risk score system, miRNA-target regulatory network, enrichment analysis
Introduction
Ovarian cancer (OC) is the most lethal gynecologic malignancy with a 5-year overall survival (OS) of ~47%, which has almost never changed over the past 20 years (1). In 2015, 1.2 million women suffered from OC, and the disease led to 161,100 deaths worldwide (2). The symptoms of OC are inconspicuous and non-specific thus most cases are diagnosed at a later stage (3). Therefore, early diagnosis and treatment of OC are critical for improving the outcomes of the disease, and prognosis mainly depends on the disease degree, tumor subtypes and medical conditions (2,4). Understanding the underlying mechanisms of OC could facilitate the development of advanced treatment approaches.
MicroRNAs (miRNAs) play important roles in OC pathogenesis and progression. By comparison of the transcriptome data from different tissues with genome-scale biomolecular networks, miR-124-3p was identified as a potential biomarker for OC (5). miR-27a is considered as an oncogene which inhibits forkhead box O1 (FOXO1) in OC (6), while miR-34a serves as a suppressor by downregulating histone deacetylase 1 (HDAC1) (7). miR-409-3p was found to enhance the cisplatin-sensitivity of OC cells by inhibiting autophagy controlled by Fip200 (8).
In addition, a growing number of studies have shown that the dysregulation of miRNAs is associated with the prognosis of OC. The miR-200 family members have been identified as prognostic indicators for the disease stage, tumor histology and survival of OC (9). For example, miR-200b-429 may be a promising marker for OC survival, and the low expression of miR-200 indicates a poor prognosis and plays a regulatory role in the tumor (10). An upregulated serum miR-221 expression level is correlated with tumor stage and grade of epithelial ovarian cancer (EOC), which serves as an independent factor for a poor prognostic in EOC (11). The serum level of miR-141 and miR-200c can distinguish OC patients from healthy controls, and they may be utilized as markers for predicting the prognosis of OC (12). A high expression level of miR-203 has been reported as a candidate marker that predicts the progression and adverse outcome of patients with EOC (13,14). Serum miR-21 expression was found to be increased in EOC patients, and it may function as a novel marker for the diagnosis and prognosis of EOC (15). The expression of miR-150 is higher in primary serous OC than in omental metastases, and its lower expression is associated with shorter progression free-survival in metastatic tissues (16). Nevertheless, the miRNAs related to the recurrence of OC have not been fully revealed.
Thus, exploring the correlation between miRNAs and the development and recurrence of OC is critical for improving the curative effects and prognosis of OC patients. Based on the miRNA expression profile of OC in the public database, the miRNAs correlated with the recurrence of OC were screened, and then a classifier was constructed to recognize the recurrence of OC. Combined with the prognostic information of the samples, a risk score system was constructed based on the expression levels of significant miRNAs. The present study may provide a theoretical basis for the prognostic prediction and targeted therapy of OC, particularlyrecurrent OC.
Materials and methods
Data source and prescreening of clinical factors
The miRNA expression profile of OC (the training set) was downloaded from The Cancer Genome Atlas (TCGA, http://portal.gdc.cancer.gov/) database (September 10, 2018), based on the Illumina HiSeq 2000 RNA Sequencing platform. The data in the ‘0a07b199-d93d-4202-a63a-b38e39dc5ca4.mirbase21.mirnas.quantification.txt’ file that is level 3 was downloaded and used. Then we used the encoding information to obtain the sample information. There were 415 OC samples with available clinical information in the training set, of which 390 had information regarding recurrence: 170 were non-recurrent OC samples and 220 were recurrent. The human reference genome hg38/GRCh38 was used to annotate the expression information.
Meanwhile, other relevant datasets were searched from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database with the keywords ‘ovarian cancer’ and ‘Homo sapiens’. The inclusive criteria were: i) the samples in the dataset had recurrent information; ii) the samples had prognostic information; iii) the total number of samples was no <50; and iv) the dataset was an miRNA expression profile. Based on these criteria, only two datasets, GSE25204 (17) and GSE27290 (18), were selected and used as validation datasets. The GSE25204 dataset was based on the Illumina Human v2 MicroRNA expression beadchip platform and included 85 OC samples with recurrent information (validation set 1). The GSE27290 dataset was based on the Agilent-015508 Human miRNA Microarray platform (pre-commercial version 6.0) and contained 58 OC samples with recurrent information (the validation set 2).
The clinical information of all the samples in the training set was statistically analyzed. In order to determine the basis for grouping, the univariate and multivariate Cox regression analysis in the R survival package (19) (version 2.41-1, http://bioconductor.org/packages/survivalr/) was used to screen the clinical factors significantly associated with prognosis. P<0.05 was set as the significance threshold.
Data standardization and differential expression analysis
The miRNA expression profile matrixes of the three datasets were stacked, and the matrix for each dataset was scaled according to the expression level. The unit specification was scaled as follows to provide a sample vector:
where is the 2-norm of the vector (l2 norm).
Using the sqrt [sum(data^2)] function in R (20), the square root of the eigenvalue of matrix B = A*AT was obtained. The purpose of this normalization was to obtain the sample values scaled to 1. Using median scaling, the expression level of each miRNA was centralized and normalized according to the median and median absolute deviation (MAD). Specifically, an eigenvector × = (×1,…, xn):
was assigned. The median scale normalization was defined as
For the training set, the samples were grouped according to the recurrence condition of the samples. Using the R package limma (21) (version 3.34.7, http://bioconductor.org/packages/release/bioc/html/limma.html), the differentially expressed miRNAs (DE-miRNAs) between the recurrent and non-recurrent OC samples were selected. The false discovery rate (FDR) <0.05 and |log2fold change (FC)| >0.263 were set as the thresholds for the significant differences. According to the expression levels of the DE-miRNAs in the training datasets, the bidirectional hierarchical clustering for expression levels of these DE-miRNAs was performed based on the centered Pearson correlation algorithm, using the pheatmap package (22) (version 1.0.8, http://cran.r-project.org/web/packages/pheatmap/index.html) in R.
Construction of Support Vector Machine (SVM) classifier
In the training set, the Cox regression analysis in the R survival package (19) was used to select the DE-miRNAs significantly related to prognosis, with the threshold of log-rank P<0.05. The DE-miRNAs significantly related to recurrent prognosis were further selected to perform the follow-up analyses.
Recursive feature elimination (RFE) is an integrated machine learning method, which considers the selection of subset as an optimization problem (23). Using the RFE algorithm in the R package Caret (24) (version 6.0–76, http://cran.r-project.org/web/packages/caret), the optimal miRNA set was filtered from the training dataset. In the 100-fold cross validation, the miRNA with the highest accuracy was selected as the signature miRNA.
SVM is a supervised classification algorithm of machine learning, which discriminates sample types by estimating the probability that a sample belongs to a certain category (25). For the training set, the SVM classifier was constructed based on the optimal miRNA set using the SVM method (Core: Sigmoid Kernel; Cross: 100-fold cross validation) in the R package e1071 (26) (version 1.6–8, http://cran.r-project.org/web/packages/e1071).
The performance of the SVM classifier was separately evaluated in the training set and the validation sets using 4 valuation indicators [Concordance index, C-index; Brier score; Log-rank P-value of Cox-proportional hazard (PH) regression; and area under the receiver operating characteristic (ROC) curve, AUC]. The C-index and Brier score were calculated using the R package survcomp (27) (version 1.30.0, http://www.bioconductor.org/packages/release/bioc/html/survcomp.html). Using the R package survival (19), the Kaplan-Meier (KM) curves for the two groups classified by the SVM classifier were generated, and the log-rank P-value of the difference between the two groups was calculated. Furthermore, the indicators of ROC curves (sensitivity, Sen; specificity, Spe; positive prediction value, PPV; negative prediction value, NPV) were calculated using the R package pROC (28) (version 1.12.1, http://cran.r-project.org/web/packages/pROC/index.html).
Construction of the risk score system
Based on the multivariate Cox regression analysis in the R survival package (19), the prognosis-associated miRNAs were further analyzed to identify the DE-miRNAs independently related to prognosis. The log-rank P<0.05 was set as the threshold.
Based on the regression coefficients of the independent prognostic miRNAs, the risk score system was constructed, and the risk score of each sample was obtained according to the following formula:
where Coef DE-miRNA represents the regression coefficient, and Exp DE-miRNA indicates the expression level of the corresponding miRNA.
For the training set, the samples were divided into high- and low-risk groups with the median of risk scores as the cut-off point. Using the KM curve analysis in the R survival package (19), the correlation between the risk score system and prognosis was evaluated. Meanwhile, the risk score system was confirmed in the validation sets.
Stratified analysis of clinical factors
Using the univariate and multivariate Cox regression analysis in the R survival package (19), the clinical factors independently correlated with prognosis in the training set were screened out. Combined with the high- and low-risk samples determined by the risk score system, stratified analysis was further carried out.
miRNA-target regulatory network analysis and enrichment analysis
The risk scores of the mRNA-sequencing samples matched with the miRNA-sequencing samples were calculated using the risk score system. Based on the risk scores, the samples in the training set were divided into high- and low-risk groups. Using the R package limma (21), the differentially expressed genes (DEGs) between the two groups were selected, with the thresholds of FDR <0.05 and |log2FC| >0.263. Based on the starBase database (29) (version 3.0, http://starbase.sysu.edu.cn/), the miRNA-mRNA regulatory interactions in at least one of the five databases, targetScan, picTar, RNA22, PITA, and miRanda, were selected. Then, the correlation of the expression levels of the miRNAs and target DEGs in the matched samples were calculated, and the interactions with significant negative correlations were selected. Subsequently, the miRNA-target regulatory network was visualized using the Cytoscape software (30) (version 3.6.1, http://www.cytoscape.org/). Using the Database for Annotation, Visualization and Integrated Discovery (DAVID) tool (31) (version 6.8, http://david.ncifcrf.gov/), the functional and pathway enrichment analyses were carried out, with P<0.05 as the screening criterion.
Results
Prescreening of clinical factors and differential expression analysis
The clinical information of the 415 OC samples in the training set was performed with statistical analysis, and then the clinical factors significantly associated with prognosis were screened. The age, tumor recurrence, and neoplasm cancer status were found to be the clinical factors significantly related to prognosis (Table I and Fig. 1). To identify the recurrence prognosis-associated miRNAs, the samples in this study were grouped based on the recurrence information.
Table I.
Clinical information of all the tumor samples in the training set and the prescreening of the clinical factors significantly associated with prognosis.
| Univariables Cox | Multivariables Cox | ||||||
|---|---|---|---|---|---|---|---|
| Clinical characteristics | TCGA (n=415) | HR | 95% CI | P-value | HR | 95% CI | P-value |
| Age (years, mean ± SD) | 59.42±11.41 | 1.018 | 1.006–1.030 | 3.78×١٠−3a | 1.016 | 1.003–1.0301 | 1.73×10−2a |
| Neoplasm histologic grade (G1/G2/G3/G4/-) | 1/52/353/1/8 | 1.385 | 0.959–2.019 | 8.09×10−2 | – | – | – |
| Pathological stage (II/III/IV/-) | 23/327/62/3 | 1.375 | 1.037–1.823 | 2.74×10−2a | 1.001 | 0.730–1.3730 | 9.93×10−1 |
| Tumor recurrence (yes/no/-) | 220/171/24 | 1.316 | 1.057–2.012 | 6.08×10−9a | 1.385 | 1.272–1.544 | 6.76×10−8a |
| Neoplasm cancer status (tumor-free/with tumor/-) | 92/274/49 | 0.070 | 0.026–0.189 | 4.72×10−12a | 0.0481 | 0.017–0.133 | 5.61×10−9a |
| Lymphatic invasion (yes/no/-) | 103/55/257 | 1.144 | 0.704–1.859 | 5.87×10−1 | – | – | – |
| Venous invasion (yes/no/-) | 63/47/305 | 0.788 | 0.437–1.419 | 4.26×10−1 | – | – | – |
| Decreased (deceased/alive) | 121/139 | – | – | – | |||
| Overall survival time (months, mean ± SD) | 34.16±27.67 | – | – | – | |||
TCGA, The Cancer Genome Atlas; HR, hazard ratio; CI, confidence interval.
Significant P-value.
Figure 1.

The Kaplan-Meier (KM) survival curves for the clinical factors significantly associated with prognosis. (A) The KM curve for age (blue and red curves separately represent the tumor samples with age < and >60 years). (B) The KM curve for tumor recurrence (blue and red curves separately represent the tumor samples with information on non-recurrence and recurrence). (C) The KM curve for neoplasm cancer status (blue and red curves separately represent the tumor-free samples and the samples with tumor).
For the training set, a total of 46 DE-miRNAs (18 upregulated and 28 downregulated) were identified between the recurrent and non-recurrent OC samples (Fig. 2A). The clustering heatmap was drawn based on the expression levels of the DE-miRNAs, which indicated that the samples were clearly divided into two types (Fig. 2B).
Figure 2.

Rresults of the differential expression analysis for screening differentially expressed miRNAs (DE-miRNAs). (A) The volcano plot of the DE-miRNAs (blue and red dots represent DE-miRNAs with FDR <0.05 and |log2FC| >0.263). (B) Clustering heatmap of the DE-miRNAs (green and red sample bars separately represent the samples with information on non-recurrence and recurrence).
Construction of the SVM classifier
Using the Cox regression analysis, 24 prognosis-associated miRNAs were selected in the training set. Using RFE algorithm, the optimal miRNA set involving 19 miRNAs (including miR-135b, miR-139, miR-151, miR-187, miR-193b, miR-210, miR-211, miR-218, miR-219, miR-30b, miR-30d, miR-365, miR-505, miR-506, miR-508, miR-509, miR-513c, miR-514 and miR-760) was selected (Fig. 3). Based on the optimal 19-miRNA set, the SVM classifier was constructed. Then, the performance of the SVM classifier in the training set and the validation sets was assessed using the 4 valuation indicators aforementioned. The results showed that the C-index values were >0.80, and Brier score values <0.1 in both the training and validation sets (Table II). As shown in the confusion table diagrams that indicated the sample classification based on the SVM classifier, the 19-miRNA set could distinguish well the recurrent samples from the non-recurrent (Fig. 4). The AUC curves showed that the AUC values of the training set and the validation sets were >0.9 (Fig. 4 and Table II). The KM curves suggested that the predictive results of the SVM classifier were significantly related to prognosis (P<0.05; Fig. 4). These results indicated that the 19-miRNA-based classifier could accurately determine the recurrence type of the OC samples.
Figure 3.
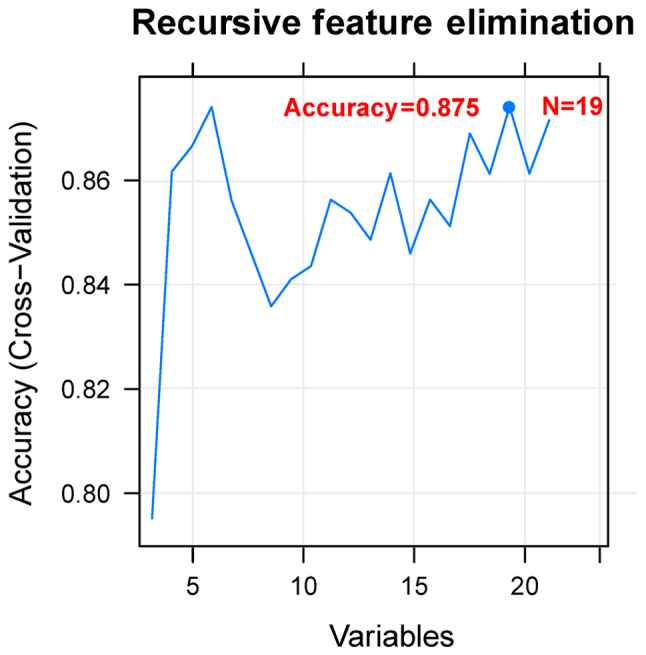
Accuracy curve for screening the optimal miRNA set. The horizontal axis represents the number of miRNA variables, and the vertical axis represents the cross-validation accuracy. The marked content is the number of miRNAs corresponding to the optimal miRNA set.
Table II.
Evaluation indicators for the Support Vector Machine (SVM) classifier in the training set and the validation sets.
| ROC | ||||||||
|---|---|---|---|---|---|---|---|---|
| Datasets | C-index | Brier score | Log rank P-value | AUC | Sensitivity | Specificity | PPV | NPV |
| Training set (TCGA, n=390) | 0.942 | 0.021 | 4.72×10−12a | 0.905 | 0.847 | 0.946 | 0.914 | 0.829 |
| Validation set 1 (GSE25204, n=85) | 0.899 | 0.066 | 7.98×10−7a | 0.941 | 0.783 | 0.903 | 0.75 | 0.918 |
| Validation set 2 (GSE27290, n=58) | 0.842 | 0.063 | 6.39×10−6a | 0.902 | 0.875 | 0.905 | 0.778 | 0.95 |
ROC, receiver operating characteristic; AUC, area under the receiver operating characteristic curve; PPV, positive prediction value; NPV, negative prediction value.
Significant P-value.
Figure 4.

Area under the receiver operating characteristic (AUC) curves, the Kaplan-Meier (KM) survival curves, and the confusion table diagrams based on the Support Vector Machine (SVM) classifier. (A) The AUC curve (left), KM curve (middle) and confusion table diagram (right) for the training dataset. (B) The AUC curve (left), KM curve (middle) and confusion table diagram (right) for the validation set 1 (GSE25204). (C) The AUC curve (left), KM curve (middle) and confusion table diagram (right) for the validation set 2 (GSE27290). In the KM curves, blue and red curves separately represent the non-recurrent and recurrent tumor samples determined by the SVM classifier.
Construction of risk score system
Combining the optimal 19-miRNA set with the recurrence prognosis information of the samples, 6 independent prognosis-related DE-miRNAs (miR-193b, miR-211, miR-218, miR-505, miR-508 and miR-514) were identified (Table III).
Table III.
The 6 differentially expressed miRNAs independently related to prognosis.
| ID | Coef | P-value | Hazard ratio (95% CI) |
|---|---|---|---|
| hsa-mir-193b | 0.2618 | 0.0431a | 1.2993 (1.008–1.674) |
| hsa-mir-211 | 0.1507 | 0.0331a | 1.1627 (1.012–1.336) |
| hsa-mir-218 | 0.1588 | 0.0179a | 1.1721 (1.028–1.337) |
| hsa-mir-505 | −0.1959 | 0.0052a | 0.8221 (0.675–0.902) |
| hsa-mir-508 | −0.1704 | 0.0181a | 0.8433 (0.657–0.983) |
| hsa-mir-514 | 0.2526 | 0.0211a | 1.2874 (1.066–1.913) |
CI, confidence interval.
Significant P-value.
Combined with the regression coefficients of the 6 independent prognostic miRNAs, the risk score system for OC was constructed. The formula for calculating the risk score of each sample was:
Risk score = (0.2618) × Exphsa-mir-193b + (0.1507) × Exp hsa-mir-211 + (0.1588) × Exp hsa-mir-218 + (−0.1959) × Exp hsa-mir-505 + (−0.1704) × Exp hsa-mir-508 + (0.2526) × Exp hsa-mir-514
With the median of risk scores as the cut-off point, the samples were classified into high- and low-risk groups. For the training and the validation sets, the KM curves showed that the high- and low-risk groups determined by the risk score system were significantly associated with the actual recurrence prognosis information (Fig. 5).
Figure 5.

The Kaplan-Meier (KM) survival curves and the area under the receiver operating characteristic (AUC) curves based on the risk score system. (A) The KM curve (left) and AUC curve (right) for the training dataset. (B) The KM curve (left) and AUC curve (right) for the validation set 1 (GSE25204). (C) The KM curve (left) and AUC curve (right) for the validation set 2 (GSE27290). In KM curves, blue and red curves separately represent the low- and high-risk groups.
Stratified analysis of the clinical factors
In the training set, although the age, tumor recurrence, and neoplasm cancer status were all identified as prognosis-associated clinical factors, only the neoplasm cancer status was considered as an independent prognostic factor relating to the recurrence, based on the multivariate Cox regression analysis (Table IV and Fig. 6A). To analyze the correlation between the neoplasm cancer status and recurrence prognosis separately in the high- and low-risk groups, stratified analysis was further performed for the neoplasm cancer status (Fig. 6B and C).
Table IV.
Cox regression analysis for screening the clinical factor independently correlated with prognosis in the training set.
| Uni-variables cox | |||
|---|---|---|---|
| Clinical characteristics | HR | 95% CI | P-value |
| Age (years) | 1.012 | 0.999–1.024 | 6.38×10−2 |
| Neoplasm histologic grade (G1/G2/G3/G4/-) | 1.338 | 0.927–1.932 | 1.19×10−1 |
| Pathological stage (II/III/IV/-) | 1.005 | 0.753–1.342 | 9.73×10−1 |
| Neoplasm cancer status (tumor free/with tumor/-) | 0.279 | 0.129–0.601 | 5.30×10−4a |
| Lymphatic invasion (yes/no/-) | 1.078 | 0.678–1.714 | 7.52×10−1 |
| Venous invasion (yes/no/-) | 0.619 | 0.365–1.049 | 7.21×10−2 |
HR, hazard ratio; CI, confidence interval
indicates significant P-value.
Figure 6.

Kaplan-Meier (KM) survival curves for neoplasm cancer status. (A) The KM curve showing the correlation between neoplasm cancer status and recurrence prognosis in all samples. (B) The KM curve showing the correlation between neoplasm cancer status and recurrence prognosis in the low-risk group. (C) The KM curve showing the correlation between neoplasm cancer status and recurrence prognosis in the high-risk group. Blue and red curves represent the tumor-free samples and the samples with tumor, respectively.
miRNA-target regulatory network analysis and enrichment analysis
In total, we identified 615 DEGs (400 upregulated and 215 downregulated) between the high- and low-risk groups. Based on the StarBase database, the target genes were predicted for the 6 independent prognostic miRNAs. The overlapping genes between the target genes and the DEGs were obtained after comparison, and 601 miRNA-mRNA regulatory interactions were selected. Then, 218 interactions with significant negative correlations were retained for constructing the miRNA-target regulatory network (involving miR-193b, miR-211, miR-505, miR-508, and miR-514) (Fig. 7). In addition, the target genes in the regulatory network were enriched in 25 functional terms (such as blood vessel development and vasculature development) and 6 pathways (such as regulation of actin cytoskeleton and TGF-β signaling pathway) (Table V).
Figure 7.

miRNA-target regulatory network. Squares and circles represent miRNAs and mRNAs, respectively.
Table V.
Gene Ontology (GO) functional terms and pathways enriched in the target genes involved in the regulatory network.
| Category | Term | Count | P-value |
|---|---|---|---|
| GO biology process | GO:0001568~blood vessel development | 16 | 4.26×10−7a |
| GO:0001944~vasculature development | 16 | 5.81×10−7a | |
| GO:0001525~angiogenesis | 10 | 9.95×10−5a | |
| GO:0007155~cell adhesion | 22 | 1.72×10−4a | |
| GO:0022610~biological adhesion | 22 | 1.75×10−4a | |
| GO:0048514~blood vessel morphogenesis | 11 | 3.19×10−4a | |
| GO:0006928~cell motion | 14 | 6.66×10−3a | |
| GO:0051674~localization of cell | 10 | 1.51×10−2a | |
| GO:0048870~cell motility | 10 | 1.51×10−2a | |
| GO:0008219~cell death | 16 | 3.56×10−2a | |
| GO:0016265~death | 16 | 3.75×10−2a | |
| GO:0006915~apoptosis | 14 | 3.87×10−2a | |
| GO:0012501~programmed cell death | 14 | 4.27×10−2a | |
| GO cellular component | GO:0031012~extracellular matrix | 14 | 3.11×10−4a |
| GO:0005578~proteinaceous extracellular matrix | 12 | 1.89×10−3a | |
| GO:0009986~cell surface | 11 | 1.03×10−2a | |
| GO:0044421~extracellular region part | 20 | 2.47×10−2a | |
| GO:0005794~golgi apparatus | 18 | 3.72×10−2a | |
| GO molecular function | GO:0030246~carbohydrate binding | 13 | 1.10×10−3a |
| GO:0005198~structural molecule activity | 17 | 3.75×10−3a | |
| GO:0032555~purine ribonucleotide binding | 32 | 2.72×10−2a | |
| GO:0032553~ribonucleotide binding | 32 | 2.72×10−2a | |
| GO:0017076~purine nucleotide binding | 33 | 2.89×10−2a | |
| GO:0001882~nucleoside binding | 28 | 4.24×10−2a | |
| GO:0005524~ATP binding | 26 | 4.52×10−2a | |
| Pathway | hsa04512:ECM-receptor interaction | 8 | 6.16×10−6a |
| hsa04510:Focal adhesion | 10 | 6.47×10−5a | |
| hsa04810:regulation of actin cytoskeleton | 7 | 4.57×10−3a | |
| hsa03010:ribosome | 4 | 8.91×10−3a | |
| hsa04350:TGF-beta signaling pathway | 3 | 2.87×10−2a | |
| hsa04142:lysosome | 3 | 4.20×10−2a |
Significant P-value.
Discussion
In the present study, we identified 46 DE-miRNAs between the recurrent and non-recurrent ovarian cancer (OC) samples. Nineteen prognosis-associated miRNAs were used to construct an SVM classifier, among which 6 were deregulated and independently related to prognosis. A risk score system based on the 6 miRNAs had a high accuracy for risk prediction in both the training and validation sets. The neoplasm cancer status was a clinical factor independently correlated with recurrence.
miR-193b serves as a tumor suppressor in many cancer types. Its role in OC has recently been investigated. The epigenetic silencing of miR-193a-3p could promote OC progression by targeting the growth factor receptor-bound protein-7 (GRB7) (32). miR-193b-3p has an antitumor effect in OC cells by inhibiting the p21-activated kinase 3 (33). Downregulation of miR-193b could induce OC metastasis (34). These results indicate that miR-193b may be a tumor suppressor in OC. Moreover, low expression of miR-193b is associated with a poor prognosis of OC patients (35). In the present study, miR-193b was one of the 6 miRNA signatures that could predict recurrence of OC, suggesting that its expression also might be linked to recurrence.
Currently, only a few studies have reported the correlations between miR-218 and OC. It was reported that miR-218 prevents the proliferation and invasion in OC by downregulating its target gene, runt-related transcription factor 2 (RUNX2) (36). In colon adenocarcinoma, the long noncoding RNA MNX1-AS1 could promote progression. It acts as a competing endogenous RNA (ceRNA) of miR-218-5p and upregulates SEC61A1, the downstream target gene of miR-218-5p (37). MNX1-AS1 was also found to facilitate the progression of OC (37). However, it is unclear whether MNX1-AS1 also has this competing relationship with miR-218-5p. In the present study, miR-218 was another important miRNA identified to be related to the recurrence of OC, indicating it may be a novel predictive factor for OC recurrence.
It has been found that downregulation of the tumor suppressor miR-211 induces the overexpression of cyclin-dependent kinase 6 (CDK6) and cyclin D1 (CCND1), which contributes to the proliferation of epithelial ovarian cancer (EOC) cells (38). The high expression of PHF19 (PHD finger protein 19) was found to be related to the poor prognosis of OC patients, and it is a target of miR-211. By competing with the lncRNA MALAT1, miR-211 suppresses the expression of PHF19, and thus functions as a suppressor in OC development (39). This suggests that miR-211 is linked to OC prognosis by regulating the downstream targets. However, there are no studies reporting the role of miR-211 in the recurrence of OC. Based on the present study, it was one of the 6 miRNA signatures for OC recurrence. Therefore, miR-211 may be a potential biomarker indicative of the recurrence of OC.
miR-30a, miR-30e and miR-505 exhibit significantly lower expression in ovarian clear cell carcinoma (OCC) compared with those in elderly advanced ovarian papillary serous carcinoma (OPSC) patients, and the activating transcription factor 3 (ATF3) is the primary gene co-targeted by them (40). Overexpression of the tumor suppressors miR-130b-3p, miR-509-3p, miR-509-5p, miR-508-3p and miR-508-5p has been association with the improved survival of OC patients, and these miRNAs may alter the physical properties of OC cells via regulating the actin cytoskeleton (41). Moreover, by downregulating gene expression levels in the MAPK1/ERK signaling pathway, miR-508 acts as an inhibitor for cell proliferation, migration and invasion in OC cells (42). Reduced miR-514 is correlated with adverse prognosis of OC patients, and miR-514 can inhibit cell proliferation and lower cisplatin chemosensitivity in OC by regulating the ATP binding cassette subfamily (43). These findings indicate that the three miRNAs, miR-505, miR-508 and miR-514 may function as suppressors in OC development and their low expression could be associated with poor prognosis. Our results demonstrated that miR-505, miR-508 and miR-514 were three miRNA signatures in recurrent OC, suggesting that they may be the predictive indicators for OC recurrence.
For the target genes in the miRNA-target regulatory network (involving miR-193b, miR-211, miR-505, miR-508 and miR-514), they were significantly enriched in the regulation of the actin cytoskeleton and the TGF-β signaling pathway. The TGF-β signaling pathway functions in various cellular processes correlated with tumorigenesis, and the genetic variants in the pathway are related to OC risk and may help to identify high-risk individuals (44). TGF-β signaling can be suppressed by the accumulation of epigenetic modifications, which contributes to the oncogenesis of OC (45). The dynamic remodeling of the actin cytoskeleton is important for multiple cellular activities, and dysfunction of cytoskeletal proteins can lead to many diseases in humans (46). Therefore, miR-193b, miR-211, miR-505, miR-508 and miR-514 may influence the prognosis of OC via the regulation of the actin cytoskeleton and the TGF-β signaling pathway.
Although we performed comprehensive bioinformatic analyses using the miRNA expression profile of OC and confirmed the classification accuracy by the validation datasets, several limitations remain. First, the sample size with available recurrence information was small. Second, this study lacks validation experiments to validate the expression of these predictive miRNAs and interplayed target genes.. Third, the accuracy of the SVM classifier and clinical value of the 6-miRNA risk score system should be further tested in OC patients. Therefore, further experiments should be prepared and conducted to support our findings.
In conclusion, the SVM classifier may be accurate in determining the recurrence status of OC patients. Moreover, the 6-miRNA risk score system may be effective in predicting the outcome of OC patients. Furthermore, miR-193b, miR-211, miR-505, miR-508 and miR-514 may affect the prognosis of OC via regulation of the actin cytoskeleton and the TGF-β signaling pathway.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets analyzed in the present study are available from the corresponding author on reasonable request.
Authors' contributions
JD performed the data analyses and wrote the manuscript. MX conceived and designed the study. JD and MX read and approved the final manuscript. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
In the original studies that generated the datasets, the trials were approved by the local institutional review boards of all participating centers, and informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Moufarrij S, Dandapani M, Arthofer E, Gomez S, Srivastava A, Lopez-Acevedo M, Villagra A, Chiappinelli KB. Epigenetic therapy for ovarian cancer: Promise and progress. Clin Epigenetics. 2019;11:7. doi: 10.1186/s13148-018-0602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mcguire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for research on cancer, WHO Press, 2015. Adv Nutr. 2016;7:418. doi: 10.3945/an.116.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanley GE, Mcalpine JN, Kwon JS, Mitchell G. Opportunistic salpingectomy for ovarian cancer prevention. Gynecol Oncol Res Pract. 2015;2:5. doi: 10.1186/s40661-015-0014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibson SJ, Fleming GF, Temkin SM, Chase DM. The application and outcome of standard of care treatment in elderly women with ovarian cancer: A literature review over the last 10 years. Front Oncol. 2016;6:63. doi: 10.3389/fonc.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gov E, Kori M, Arga KY. Multiomics analysis of tumor microenvironment reveals Gata2 and miRNA-124-3p as potential novel biomarkers in ovarian cancer. OMICS. 2017;21:603–615. doi: 10.1089/omi.2017.0115. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Ji G, Wu Q, Feng S, Zhao Y, Cao Z, Tao C. Integrated microarray meta-analysis identifies miRNA-27a as an oncogene in ovarian cancer by inhibiting FOXO1. Life Sci. 2018;201:263–270. doi: 10.1016/j.lfs.2018.08.043. [DOI] [PubMed] [Google Scholar]
- 7.Lv T, Song K, Zhang L, Li W, Chen Y, Diao Y, Yao Q, Liu P. MiRNA-34a decreases ovarian cancer cell proliferation and chemoresistance by targeting HDAC1. Biochem Cell Biol. 2018;96:663–671. doi: 10.1139/bcb-2018-0031. [DOI] [PubMed] [Google Scholar]
- 8.Cheng Y, Ban R, Liu W, Wang H, Li S, Yue Z, Zhu G, Zhuan Y, Wang C. MiRNA-409-3p enhances cisplatin-sensitivity of ovarian cancer cells by blocking the autophagy mediated by Fip200. Oncol Res. 2018 Jan 2; doi: 10.3727/096504017X15138991620238. (Epub ahead of print). doi: 10.3727/096504017X15138991620238. [DOI] [PubMed] [Google Scholar]
- 9.Koutsaki M, Libra M, Spandidos DA, Zaravinos A. The miR-200 family in ovarian cancer. Oncotarget. 2017;8:66629–66640. doi: 10.18632/oncotarget.18343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu X, Macdonald DM, Huettner PC, Feng Z, El Naqa IM, Schwarz JK, Mutch DG, Grigsby PW, Powell SN, Wang X. A miR-200 microRNA cluster as prognostic marker in advanced ovarian cancer. Gynecol Oncol. 2009;114:457–464. doi: 10.1016/j.ygyno.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Hong F, Li Y, Xu Y, Zhu L. Prognostic significance of serum microRNA-221 expression in human epithelial ovarian cancer. J Int Med Res. 2013;41:64–71. doi: 10.1177/0300060513475759. [DOI] [PubMed] [Google Scholar]
- 12.Gao YC, Wu J. MicroRNA-200c and microRNA-141 as potential diagnostic and prognostic biomarkers for ovarian cancer. Tumor Biol. 2015;36:4843–4850. doi: 10.1007/s13277-015-3138-3. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Zhao X, Wang J, Wen Y, Zhang L, Wang D, Chen H, Chen Q, Xiang W. Upregulation of microRNA-203 is associated with advanced tumor progression and poor prognosis in epithelial ovarian cancer. Med Oncol. 2013;30:681. doi: 10.1007/s12032-013-0681-x. [DOI] [PubMed] [Google Scholar]
- 14.Xiaohong Z, Lichun F, Na X, Kejian Z, Xiaolan X, Shaosheng W. MiR-203 promotes the growth and migration of ovarian cancer cells by enhancing glycolytic pathway. Tumor Biol. 2016;37:14989–14997. doi: 10.1007/s13277-016-5415-1. [DOI] [PubMed] [Google Scholar]
- 15.Xu YZ, Xi QH, Ge WL, Zhang XQ. Identification of serum microRNA-21 as a biomarker for early detection and prognosis in human epithelial ovarian cancer. Asian Pac J Cancer Prev. 2013;14:1057–1060. doi: 10.7314/APJCP.2013.14.2.1057. [DOI] [PubMed] [Google Scholar]
- 16.Wilczyński M, Żytko E, Danielska J, Szymańska B, Dzieniecka M, Nowak M, Malinowski J, Owczarek D, Wilczyński JR. Clinical significance of miRNA-21, −103, −129, −150 in serous ovarian cancer. Arch Gynecol Obstet. 2018;297:741–748. doi: 10.1007/s00404-018-4660-5. [DOI] [PubMed] [Google Scholar]
- 17.Bagnoli M, De Cecco L, Granata A, Nicoletti R, Marchesi E, Alberti P, Valeri B, Libra M, Barbareschi M, Raspagliesi F, et al. Identification of a chrXq27.3 microRNA cluster associated with early relapse in advanced stage ovarian cancer patients. Oncotarget. 2015;6:9643. doi: 10.18632/oncotarget.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih KK, Qin LX, Tanner EJ, Zhou Q, Bisogna M, Dao F, Olvera N, Viale A, Barakat RR, Levine DA. A microRNA survival signature (MiSS) for advanced ovarian cancer. Gynecol Oncol. 2011;121:444–450. doi: 10.1016/j.ygyno.2011.01.025. [DOI] [PubMed] [Google Scholar]
- 19.Therneau TM. Survival analysis [R package survival version 2.41–3] Technometrics. 2015;46:111–112. [Google Scholar]
- 20.Stadler L, Welc A, Humer C, Jordan M. Optimizing R language execution via aggressive speculation. Symposium on Dynamic Languages. 2016:84–95. [Google Scholar]
- 21.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng Z, Zhu G, Qi J, Ma H, Nian H, et al. RNA-seq analyses of multiple meristems of soybean: Novel and alternative transcripts, evolutionary and functional implications. BMC Plant Biol. 2014;14:169. doi: 10.1186/1471-2229-14-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang X, Zhang L, Wang B, Li F, Zhang Z. Feature clustering based support vector machine recursive feature elimination for gene selection. Appl Intelligence. 2018;48:594–607. doi: 10.1007/s10489-017-0992-2. [DOI] [Google Scholar]
- 24.Kumar A. Pre-processing and modelling using caret package in R. Int J Comput Appl. 2018;181:39–42. [Google Scholar]
- 25.Daliri MR. Feature selection using binary particle swarm optimization and support vector machines for medical diagnosis. Biomed Tech. 2012;57:395–402. doi: 10.1515/bmt-2012-0009. [DOI] [PubMed] [Google Scholar]
- 26.Meyer D. Support vector machines the interface to libsvm in package e1071. R News. 2013;1:1–3. [Google Scholar]
- 27.Schröder MS, Culhane AC, Quackenbush J, Haibe-Kains B. survcomp: An R/BioconductoR package for performance assessment and comparison of survival models. Bioinformatics. 2011;27:3206–3208. doi: 10.1093/bioinformatics/btr511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Müller M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohl M, Wiese S, Warscheid B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 31.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen K, Liu MX, Mak SL, Yung MM, Leung TH, Xu D, Ngu SF, Chan KK, Yang H, Ngan HY, et al. Methylation-associated silencing of miR-193a-3p promotes ovarian cancer aggressiveness by targeting GRB7 and MAPK/ERK pathways. Theranostics. 2018;8:423–436. doi: 10.7150/thno.22377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Qin J, Su Y. miR-193b-3p possesses anti-tumor activity in ovarian carcinoma cells by targeting p21-activated kinase 3. Biomed Pharmacother. 2017;96:1275–1282. doi: 10.1016/j.biopha.2017.11.086. [DOI] [PubMed] [Google Scholar]
- 34.Mitra AK, Chiang CY, Tiwari P, Tomar S, Watters KM, Peter ME, Lengyel E. Microenvironment-induced downregulation of miR-193b drives ovarian cancer metastasis. Oncogene. 2015;34:5923–5932. doi: 10.1038/onc.2015.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Xu Y, Qiu W, Zhao D, Zhang Y. Tissue miR-193b as a novel biomarker for patients with ovarian cancer. Med Sci Monit. 2015;21:3929–3934. doi: 10.12659/MSM.895407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li N, Wang L, Tan G, Guo Z, Liu L, Yang M, He J. MicroRNA-218 inhibits proliferation and invasion in ovarian cancer by targeting Runx2. Oncotarget. 2017;8:91530–91541. doi: 10.18632/oncotarget.21069. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Ye Y, Gu B, Wang Y, Shen S, Huang W. E2F1-mediated MNX1-AS1-miR-218-5p-SEC61A1 feedback loop contributes to the progression of colon adenocarcinoma. J Cell Biochem. 2019;120:6145–6153. doi: 10.1002/jcb.27902. [DOI] [PubMed] [Google Scholar]
- 38.Xia B, Yang S, Liu T, Lou G. miR-211 suppresses epithelial ovarian cancer proliferation and cell-cycle progression by targeting Cyclin D1 and CDK6. Mol Cancer. 2015;14:57. doi: 10.1186/s12943-015-0322-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao F, Tian X, Ruan S, Shen M, Zhang Z. miR-211 sponges lncRNA MALAT1 to suppress tumor growth and progression through inhibiting PHF19 in ovarian carcinoma. FASEB J. 2018:fj201800495RR. doi: 10.1096/fj.201800495RR. [DOI] [PubMed] [Google Scholar]
- 40.Zhao H, Ding Y, Tie B, Sun ZF, Jiang JY, Zhao J, Lin X, Cui S. miRNA expression pattern associated with prognosis in elderly patients with advanced OPSC and OCC. Int J Oncol. 2013;43:839–849. doi: 10.3892/ijo.2013.1988. [DOI] [PubMed] [Google Scholar]
- 41.Chan CK, Pan Y, Nyberg K, Marra MA, Lim EL, Jones SJ, Maar D, Gibb EA, Gunaratne PH, Robertson AG, et al. Tumour-suppressor microRNAs regulate ovarian cancer cell physical properties and invasive behaviour. Open Biol. 2016;6:160275. doi: 10.1098/rsob.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong L, Wang Y, Chen W, Yang S. MicroRNA-508 suppresses epithelial-mesenchymal transition, migration, and invasion of ovarian cancer cells through the MAPK1/ERK signaling pathway. J Cell Biochem. 2011;119:7431–7440. doi: 10.1002/jcb.27052. [DOI] [PubMed] [Google Scholar]
- 43.Xiao S, Zhang M, Liu C, Wang D. MiR-514 attenuates proliferation and increases chemoresistance by targeting ATP binding cassette subfamily in ovarian cancer. Mol Genet Genomics. 2018 May 11; doi: 10.1007/s00438-018-1447-0. (Epub ahead of print). doi: 10.1007/s00438-018-1447-0. [DOI] [PubMed] [Google Scholar]
- 44.Yin J, Lu K, Lin J, Wu L, Hildebrandt MA, Chang DW, Meyer L, Wu X, Liang D. Genetic variants in TGF-β pathway are associated with ovarian cancer Risk. PLoS One. 2011;6:e25559. doi: 10.1371/journal.pone.0025559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsumura N, Huang Z, Mori S, Baba T, Fujii S, Konishi I, Iversen ES, Berchuck A, Murphy SK. Epigenetic suppression of the TGF-beta pathway revealed by transcriptome profiling in ovarian cancer. Genome Res. 2011;21:74–82. doi: 10.1101/gr.108803.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SH, Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Mol Cells. 2010;29:311–325. doi: 10.1007/s10059-010-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed in the present study are available from the corresponding author on reasonable request.