

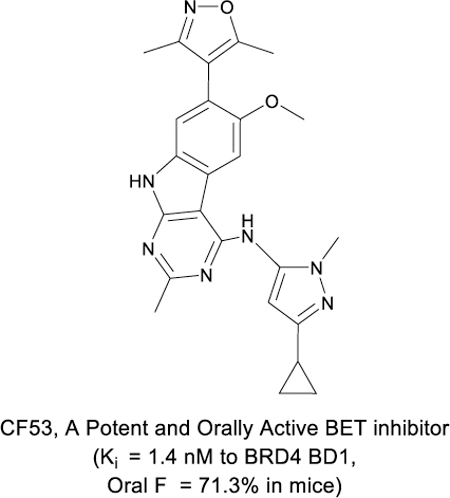
Abstract
We report structure-based discovery of CF53 (28) as a highly potent and orally active inhibitor of bromodomain and extra-terminal (BET) proteins. By incorporation of a NH-pyrazole group into the 9H-pyrimido[4,5-b]indole core, we identified a series of compounds, that bind to BRD4 BD1 protein with Ki values < 1 nM and achieve low nanomolar potencies in cell growth inhibition of leukemia and breast cancer cells. The most promising compound CF53 possesses excellent oral pharmacokinetic properties, and achieves significant antitumor activity in both triple-negative breast cancer and acute leukemia xenograft models in mice. Determination of the co-crystal structure of CF53 with the BRD4 BD1 protein provides a structural basis for its high binding affinity to BET proteins. CF53 is very selective over non-BET bromodomain-containing proteins. These data establish CF53 as a potent, selective, and orally active BET inhibitor, which warrants further evaluation for advanced preclinical development.
Graphical Abstract
INTRODUCTION
Bromodomain and extra-terminal (BET) family proteins include BRD2, BRD3, BRD4, and a testis-specific protein BRDT.1–4 The N-terminal domain of the BET family proteins contains two tandem and characteristic bromodomains (BRD), BD1 and BD2, which share high sequence homology and structural similarities and are a common feature of BET proteins.4,5 The BET BRD domains function as recognition motifs for interaction with acetylated lysine residues (AcK) in histone tails and anchor their associated proteins to the target gene promoter and enhancer sites in chromatins.6–10 BET proteins are thus critical epigenetic “readers” and play a key role in the regulation of gene transcription. They are attractive new therapeutic targets for cancers and a number of other human diseases.1,2,11
In recent years, a number of classes of potent and specific small-molecule inhibitors of BET proteins (hereafter called BET inhibitors) have been developed, with representative compounds shown in Figure 1. JQ-1 (1) was the first reported potent and specific BET inhibitor10 and has been extensively employed to evaluate the therapeutic potential of BET inhibitors in a large number of preclinical human disease models. Several BET inhibitors have subsequently advanced into clinical development.12,13 For examples, compounds 3,14,15 4,16 5,17 6,18 and 7,19,20 are currently being evaluated in Phase I/II clinical trials for treatment of hematological malignancies and solid tumors and compound 821,22 has been tested as a new therapy for the treatment of type II diabetes and chronic kidney failure. Recently reported early clinical data for compounds 314,15 and 517 have also provided clinical evidence that small-molecule BET inhibitors may have therapeutic potential for the treatment of several forms of human cancer.
Figure 1.
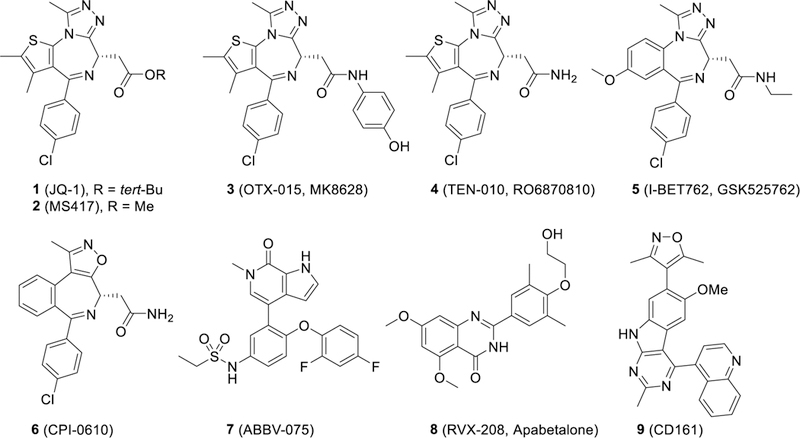
Representative previously reported potent BET bromodomain inhibitors
In our ongoing efforts to identify a potent and selective BET inhibitor for clinical development, we recently reported 9 (4-(6-methoxy-2-methyl-4-(quinolin-4-yl) −9H-pyrimido[4,5-b]indol-7-yl)-3,5-dimethylisoxazole; CD161)23 as a potent and orally bioavailable BET bromodomain inhibitor. Compound 9 binds to BET proteins with low nanomolar affinities and demonstrates high selectivity over 24 non-BET proteins containing bromodomains.23 It shows potent cell growth inhibitor activity in acute leukemia cell lines harboring mixed lineage leukemia 1 (MLL1) fusion protein and in a panel of human breast cancer cell lines.23 Compound 9 has a good pharmacokinetic profile in mice and rats, and demonstrates strong antitumor activity in MV4;11 acute leukemia and MDA-MB-231 breast cancer xenograft models. Overall, compound 9 is a promising lead compound for further optimization toward identifying a suitable clinical candidate.
During the course of our investigation, we found that compound 10 (CD235), a structurally similar analogue of 9, shows restricted rotation of the C-C bond that connects the quinoline and 9H-pyrimido[4,5-b]indole moieties and has a pair of enantiomers in the single crystal structure (Figure 2), which presents a potential manufacturing challenge for further development for this class of compounds. We decided to perform modifications of compound 9 to remove the rotationally restricted C-C bond.
Figure 2:
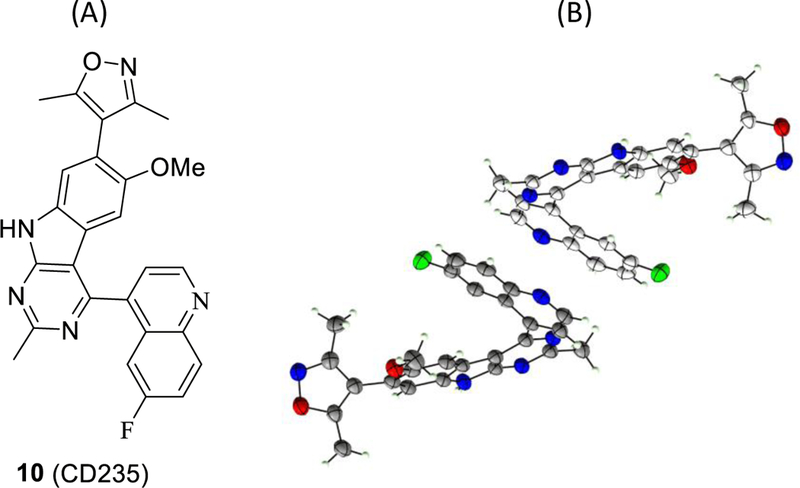
(A). Chemical structure of 10 and (B) single crystal structure of 10.The single crystal structure shows restricted rotation of the C-C bond that connects the quinoline and 9H-pyrimido[4,5-b]indole and contains a pair of enantiomers in the crystal unit.
In the present study, we report the identification of a number of highly potent, orally bioavailable, and achiral small-molecule BET inhibitors. Our effort yielded the discovery of compound 28 (CF53) as a promising BET inhibitor suitable for advanced preclinical development.
RESULTS AND DISCUSSION
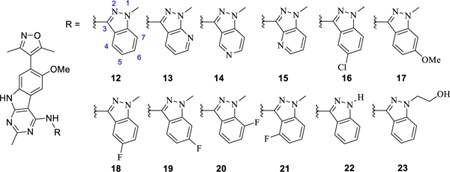
Our optimization effort started with removal of the restricted C-C bond that connects the quinoline and 9H-pyrimido[4,5-b]indolein 9 and 10. We reasoned that addition of a single nitrogen atom between the 9H-pyrimido[4,5-b]indole core and the quinoline moiety will generate achiral compounds which can rotate freely. We synthesized compound 12 (Table 1) bearing a1-methyl-1H-indazol-3-amine moiety based on compound 11 (CD134), because 11 has higher binding affinities to BET proteins than those of compound 9.23,24 Since BRD4 BD1, but not the BD2 domain, was shown to regulate gene transcription,25 we first evaluated the binding affinities of 12 and other synthesized compounds first for their binding affinities to the BRD4 BD1 protein for our structure-activity relationship studies.
Table 1:
Optimization of 9 with NH-indazole type substitutions
 | ||
|---|---|---|
| ID | BRD4 BD1 | |
| IC50 (nM)a | Ki (nM) | |
| 9(CD161) | 28.2±4.4 | 8.2±1.2 |
| 11(CD134) | 6.1±1.4 | 0.8±0.1 |
| 12 | 9.1±0.8 | 0.7±0.3 |
| 13 | 20.8±0.6 | 4.9±0.2 |
| 14 | 228.1±17.3 | 77.9±6.0 |
| 15 | 228.5±25.9 | 78.0±9.0 |
| 16 | >1000 | |
| 17 | 195.9±6.0 | 66.5±2.0 |
| 18 | 54.5±1.8 | 16.7±0.6 |
| 19 | 87.6±3.3 | 28.4±1.1 |
| 20 | 95.2±8.8 | 31.0±3.1 |
| 21 | 68.0±2.0 | 21.5±0.7 |
| 22 | 11.8±0.7 | 1.7±0.2 |
| 23 | 9.8±0.2 | 1.0±0.1 |
Mean of three experiments
Compound 12 binds to BRD4 BD1 with a high affinity (IC50 = 9.1 nM, Ki = 0.7 nM). Based upon the encouraging binding data for 12, subsequent optimization of 1-methyl-1H-indazol-3-amine moiety was carried out and the results are summarized in Table 1. Replacement of one carbon atom at the 7 position of 12 by a nitrogen yielded 13, which binds to BRD4 BD1 with an affinity similar to that of 12. Incorporation of a nitrogen atom at the 5 or 6 position of 12 led to 14 and 15, respectively, which have much weaker binding affinities to BRD4 BD1 than 12. Substitution of the benzene ring of the 1-methyl-1H-indazole moiety of 12 with a halogen atom or a methoxyl group at different positions yielded compounds 16-21, which failed to improve binding affinities to BRD4 BD1 over that of 12. Removal of the methyl group from the 1-methyl-1H-indazole moiety or replacing it with 2-hydroxyethyl yielded compounds 22 and 23, respectively, which bind to BRD4 BD1 with high affinities (Ki values of 1–2 nM).
To facilitate our further optimization effort, we determined the co-crystal structure of 12 complexed with BRD4 BD2 (PDB ID: 6C7Q). Comparison of the co-crystal structures for compounds 9 and 12 complexed with either BRD4 BD1 or BRD4 BD2 shows that the same 3,5-dimethyl isooxazole and 9H-pyrimido[4,5-b]indole moieties in 9 and 12 have very similar binding modes, while the benzene ring of the NH-indazole group in 12 is inserted into the WPF hydrophobic pocket.
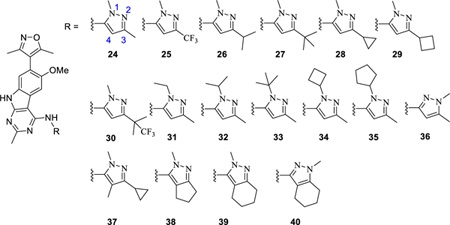
Our co-crystal structure for 12 further shows that while the WPF pocket nicely accommodates the benzene ring of the indazole group in 12, the pocket could fit a bulkier group for enhanced hydrophobic interactions. We therefore decided to replace the NH-indazole group in 12 with a substituted NH-pyrazole and synthesized a series of compounds (24-40) whose binding affinities to BRD4 BD1 are summarized in Table 2.
Table 2:
Optimization of 9 with N-pyrazole type substitutions
 | ||
|---|---|---|
| ID | BRD4 BD1 | |
| IC50 (nM)a | Ki (nM) | |
| 24 | 9.4±0.6 | 0.8±0.2 |
| 25 | 15.8±0.4 | 3.1±0.1 |
| 26 | 6.1±0.1 | <1 |
| 27 | 10.5±1.2 | 1.2±0.4 |
| 28 (CF53) | 2.0±0.3 | <1 |
| 29 | 12.8±2.1 | 2.0±0.7 |
| 30 | 13.8±0.6 | 2.4±0.2 |
| 31 | 16.8±2.5 | 3.4±0.8 |
| 32 | 6.1±0.2 | 0.5±0.1 |
| 33 | 4.4±0.4 | <1 |
| 34 | 14.3±2.5 | 2.6±0.9 |
| 35 | 4.0±0.2 | <1 |
| 36 | 24.9±1.8 | 6.3±0.6 |
| 37 | 51.7±7.9 | 15.8±2.8 |
| 38 | 5.5±1.0 | <1 |
| 39 | 4.9±0.5 | <1 |
| 40 | 5.0±0.3 | <1 |
Mean of three experiments
Compound 24 containing a simple 1,3-dimethyl-1H-pyrazole moiety, binds to BRD4 BD1 with an IC50 value of 9.4 nM (Ki = 1.9 nM). Encouraged by the strong binding affinity of 24, we synthesized 26, 27, and 28 by replacing the methyl group at the 3 position of the pyrazole with an isopropyl, a t-butyl or a cyclopropyl group, respectively. These three compounds bind to BRD4 BD1 with Ki values of <1, 1.2, and <1 nM, respectively. Replacing the 3-methyl group in 24 with CF3 yielded 25, which is slightly less potent than 24 in its binding to BRD4 BD1.Changing the cyclopropyl group in 28 to cyclobutyl resulted in 29, which is less potent than 28. Substituting one of the three methyl groups in the t-butyl group of 27 with a CF3 group generated 30, which has a high affinity to BRD4 BD1, very similar to that of 27.
We next investigated the effect of replacing the 1-methyl group of the pyrazole in 24 with other small hydrophobic groups, and produced 31, 32, 33, 34 and 35. Compounds 32, 33, and 35 have similar binding affinities to BRD4 BD1 to that of 24 but 31 and 34 have higher affinities than 24 for BRD4 BD1.
We synthesized 36 by replacing the 1,3-dimethyl-1H-pyrazole moiety in 24 with 1,5-dimethyl-1H-pyrazole and found that 36 is several times less potent than 24 in binding to BRD4 BD1. We installed a 4-methyl substituent in the 1,3-dimethyl-1H-pyrazole moiety of 24, which led to 37. Compound 37 is >5-times less potent than 24 in binding to BDR4 BD1.
We next synthesized 38 and 39 based upon 24 with a fused five-or six-membered ring, respectively. Both 38 and 39 bind to BRD4 BD1 with high affinities (Ki< 1 nM) and are more potent than 24. We synthesized 40 by moving the 1-methyl group in the pyrazole moiety of 39 to the 2 position. Compound 40 has a high affinity to BRD4 BD1, similar to that of 39.
Thus our further optimization of compound 24 yielded a number of very high affinity BRD4 BD1 inhibitors.
We employed the MOLM-13 acute leukemia and MDA-MB-231breast cancer cell lines to evaluate the cell growth inhibitory activity of these BET inhibitors in Table 2. The data obtained are summarized in Table 3.
Table 3:
Inhibition by BET inhibitors of cell growth in MOLM-13 andMDA-MB-231 cell lines
| ID | MOLM-13 IC50 (nM)a | MDA-MB-231 IC50 (nM) a | ID | MOLM-13 IC50 (nM)a | MDA-MB-231 IC50 (nM)a |
|---|---|---|---|---|---|
| 24 | 10.3 | 55.1 | 33 | 10.6 | 13.9 |
| 25 | 56± 23 | 156 ± 13 | 34 | 38.8 | 26 |
| 26 | 11.7 | 26.0 | 35 | 17.7 | 25.0 |
| 27 | 23.0 | 23.5 | 36 | 30.7 | 81.1 |
| 28 | 7 ± 3 | 85 ± 12 | 37 | 201 | 319 |
| 29 | 26.6 | 63.2 | 38 | 5 ± 1 | 67 ± 9 |
| 30 | 75 | 164 | 39 | 11.3 | 17.5 |
| 31 | 15.4 | 26.3 | 40 | 41.9 | 58.3 |
| 32 | 5.0 | 29.6 |
Mean of three experiments for compounds 25, 28, and 38
Consistent with its high affinity to BRD4 BD1, 24 potently inhibits cell growth in both the MOLM-13 acute leukemia cell line and the MDA-MB-231 breast cancer cell line with IC50 values of 10.3 nM and 55.1 nM, respectively. Several analogues of 24 also have very potent cellular activity with 28, 33, 38 and 39 being the most potent compounds in this series of compounds. Compounds 28, 33, 38 and 39 achieve IC50 values of 11.7 nM, 10.6 nM, 6.0 nM and 11.3 nM, respectively, in the MOLM-13 cell line, and 11.2 nM, 13.9 nM, 7.8 nM and 17.5 nM in the MDA-MB-231 cell line, respectively. In general, compounds with weaker binding affinities than 24 to BRD4 were found to have weaker cellular activity in both cell lines. For example, compounds 30 and 37 have IC50 values of 75 nM and 201 nM in the MOLM-13 cell line and 164 nM and 319 nM in the MDA-MB-231 cell line, respectively.
We next assessed the exposure of 6 representative potent BET inhibitors in plasma and in the xenograft tumor tissue in tumor-bearing mice with 2 time points upon administration for each compound (Table 4). Our data showed that these BET inhibitors all have good oral exposure in the plasma, with 28 being the best. Additionally, several compounds, including 28, 33 and 38, achieve good drug exposure in the tumor tissue, again with 28 being the best.
Table 4:
Oral exposure in plasma and tumor tissues of 6 potent BET inhibitors in mice. Each compound was administered at 50 mg/kg via oral gavage.
| ID | Plasma concentration (ng/mL)a (time-point) |
Tumor concentration (ng/g) a (time-point) |
|---|---|---|
| 26 | 6245 (1h) 1874 (6h) |
736 (1h) 166 (6h) |
| 27 | 923 (1h) 458 (6h) |
305 (1h) 268 (6h) |
| 28(CF53) | 12600 (1h) 7365 (6h) |
7375 (1h) 3671 (6h) |
| 33 | 3675 (0.5h) 127 (6h) |
5231 (0.5h) 621 (6h) |
| 38 | 4880 (1h) 1584 (6h) |
1509 (1h) 600 (6h) |
| 39 | 2983 (1h) 1150 (6h) |
466 (1h) 132 (6h) |
Mean of drug concentrations obtained from two mice/tumors for each time-point
Based upon the initial oral exposure data, 28 was identified as an orally bioavailable, promising BET inhibitor. We next determined the pharmacokinetics of 28 in mice, with the data summarized in Table 5. Compound 28 achieves a cMax of 6.4 µM and an AUC of 32.2 µM*hr with 25 mg/kg oral administration and has an oral bioavailability (F%) of 71.3%. Compound 28 (CF53) has a reasonable aqueous solubility of 19.5±5.5, 72.6 ± 1.3 and 14 ± 0.5µM at pH = 3, 7.4 and 11, respectively.
Table 5.
Pharmacokinetic parameters of 28 (CF53) in mice
| Route /Dose | T1/2 (h) | Cmax (µg/mL) | AUC (µM*h) | Cl (L/h/kg) | Vz (L/kg) | F(%) | |
|---|---|---|---|---|---|---|---|
| 28 | IV/ 5 mg/kg | - | - | 9.05 | 1.25 | 0.83 | |
| PO/ 25 mg/kg | 1.0 | 6.40 | 32.2 | - | - | 71.3 |
To gain structural insights into the high binding affinity of 28 to BRD4 BD1 protein, we determined a high-resolution co-crystal structure at 1.5 Å resolution of 28 in a complex with BRD4 BD1 (Figure 4, PDB ID:6C7R). The co-crystal structure shows that the 3,5-dimethylisooxazole has an H-bond interaction with the conserved water molecules inside the pocket. The 9H-pyrimido[4,5-b]indole moiety of 28 has a binding model similar to that of 9 and 12. The pyrazole projects its 3-cyclopropyl group into the hydrophobic WPF pocket. This high-resolution co-crystal structure thus shows that 28 has optimal interactions with BRD4 BD1, providing a structural basis for the high affinity binding.
Figure 4:
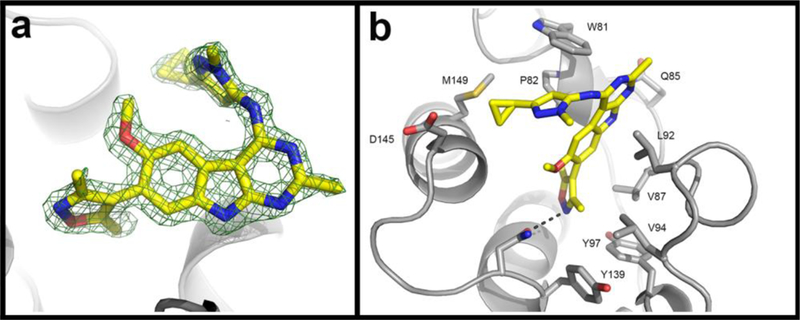
X-ray co-crystal structure of compound 28 (CF53) in complex with BRD4 BD1 protein determined at 1.5 Å resolution (PDB ID:6C7R). (a). The Fo-Fc omit electron density map of CF53 contoured at 3σ. Protein backbone is shown in gray cartoon. (b). Detailed interactions of compound 28 with BRD4 BD1. Protein backbone is shown in gray cartoon with the side chains of residues interacting with compound shown as sticks. For 28, carbons are depicted in yellow, nitrogens in blue, oxygens in red and sulfurs in yellow. Hydrogen bonds are shown as dashed lines.
We evaluated the binding affinities of 28 to the family of BET proteins and its selectivity over bromodomain-containing proteins in the BROMOscan assays by DiscoverX26 and the results are summarized in Table 6. The data showed that 28 binds to both the BD1 and BD2 domains of BRD2, BRD3, BRD4 and BRDT BET proteins with high affinities (Kd = 0.49–2.2 nM). Beyond the BET family proteins, 28 displays good affinities to CREBBP, CRCR2, and EP300 with Kd values of 47, 571, 110 nM, respectively, but shows no appreciable binding affinity to 23 other non-BET family bromodomain containing proteins at concentrations of 3–4 µM. Hence, compound 28 shows >50-fold selectivity for BET bromodomains over CRCR2 and EP300 and >20-fold over CREBBP. We also evaluated the inhibition of CF53 against a panel of 372 kinases by Reaction Biology (Malvern, PA). The kinase activity data showed that CF53 has an IC50 value of 3.9 µM against PLK4 and has no appreciable inhibition against other 371 kinases at 3 µM. Hence, 28 is a very potent BET inhibitor and demonstrates a high selectivity over the other bromodomain-containing proteins and kinases.
Table 6.
Binding affinities of 28 (CF53) to different BET proteins and selectivity over other Bromodomain-containing proteins, measured with the DiscoverX BROMO scan platform.26
| Protein | Kd (nM) | Protein | Kd (nM) | Protein | Kd (nM) |
|---|---|---|---|---|---|
| BRD2 (BD1) | 1.1 | CREBBP | 47 | BRPF3 | >5000 |
| BRD2 (BD2) | 0.6 | CECR2 | 570 | FALZ | >5000 |
| BRD3 (BD1) | 0.52 | EP300 | 110 | GCN5L2 | >5000 |
| BRD3 (BD2) | 0.49 | ATAD2A | >5000 | PBRM1(2) | >3000 |
| BRD4 (BD1) | 2.2 | ATAD2B | >5000 | PBRM1(5) | >5000 |
| BRD4 (BD2) | 0.8 | BAZ2A | >5000 | PCAF | >5000 |
| BRDT (BD1) | 2 | BAZ2B | >5000 | SMARCA2 | >5000 |
| BRDT (BD2) | 2.1 | BRD1 | >5000 | SMARCA4 | >5000 |
| BRD7 | >5000 | TAF1L (BD2) | >5000 | ||
| BRD8 (BD1) | >5000 | TAF1 (BD2) | >5000 | ||
| BRD8 (BD2) | >5000 | TRIM24 | >4000 | ||
| BRD9 | >5000 | TRIM33 (PHD, Bromo.) | >5000 | ||
| BRPF1 | >4000 | WDR9 (BD2) | >5000 |
We evaluated 28 for its in vivo efficacy in the MDA-MB-231 triple-negative breast cancer and RS4;11 acute leukemia xenograft models in mice, with 3 (OTX015) included as the control compound (Figures 5 and 6) because OTX015 has been advanced into phase II clinical trials for the treatment of human cancer. At 25 mg/kg and 50 mg/kg compound 28 was found to be effective in inhibition of tumor growth in both models. In the MDA-MB-231 xenograft tumor model, 28 achieves tumor growth inhibition (TGI) of 67.6% and 77.3% with 25 and 50 mg/kg doses at the end of treatment (day 44) as compared to the control treatment with p value <0.001 for both doses. In comparison, 3 (OTX015) shows only 21.0% TGI at 50 mg/kg (p = 0.056) in the MDA-MB-231 xenograft model at the end of the treatment (day = 44). In the RS4;11 model, 28 achieves TGI of 49.3% and 72.3% with 25 and 50 mg/kg doses at the end of treatment (day 35) as compared to the control treatment with p value <0.005 for both doses. In comparison, 3 (OTX015) at 50 mg/kg shows a TGI of 33.0% at 50 mg/kg (p = 0.036) in the RS4;11 xenograft model at the end of the treatment (day = 35). Significantly, 28 induces no more than 5% of weight loss or does not cause other signs of toxicity in mice at all the three dose-schedules tested.
Figure 5.
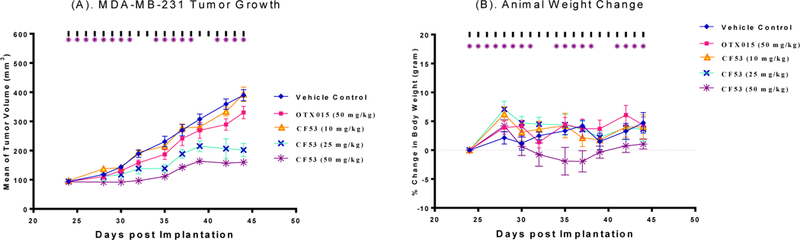
Antitumor efficacy of 28 (CF53) and 3 (OTX015) in the MDA-MB-231 xenograft model in SCID mice. These compounds were administered via oral gavage at indicated dose-schedules. (A). Tumor growth inhibition. (B). Percentage of animal weight change.
Figure 6.
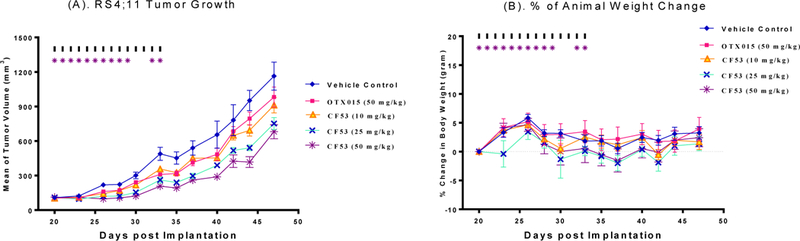
Antitumor efficacy study of 28 (CF53) and 3 (OTX015) in the RS4;11 xenograft model in SCID mice. These compounds were administered via oral gavage at indicated doses. (A). Tumor growth inhibition. (B). Animal weight change.
Our efficacy data showed that 28 (CF53) is effective in inhibition of tumor growth in the MDA-MB-231 triple negative breast cancer and RS4;11 acute leukemia models at well tolerated-dose schedules and is more effective than 3 (OTX015), a BET inhibitor currently in clinical development.
CHEMISTRY
Compounds 12-40 were synthesized from 4-(4-chloro-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-7-yl)-3,5-dimethylisoxazole (S13) and the corresponding 3-amino-1H-indazole or 5-aminopyrazole (S2) as shown in Scheme 1. Generally, the amine (S2) and the key intermediate (S13) were mixed in solvents and this was followed by addition of acid or Pd catalyst. The reaction mixtures were heated and the crude products were further purified by flash chromatography and/or reverse phase HPLC to yield the final products.
Scheme 1:
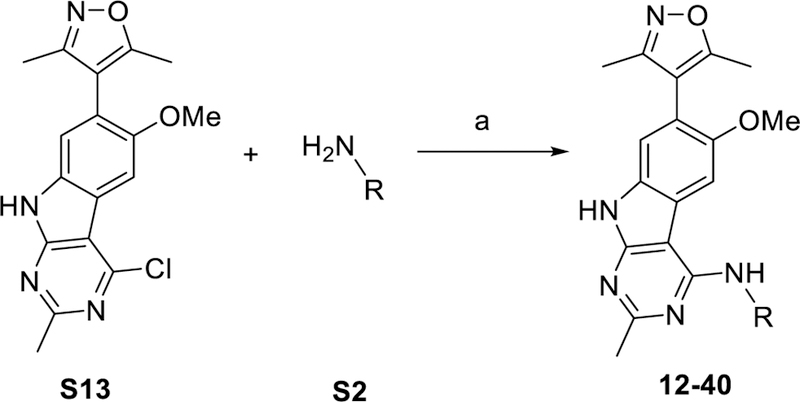
Synthesis of final compounds Reaction Conditions: a. HCl in i-PrOH, reflux, or Pd2(dba)3, Bis(diphenylphosphino)-1,1′-binaphthyl, base, and solvent.
CONCLUSIONS
We have designed and synthesized a series of 9H-pyrimido-[4,5-b]indole-containing compounds by elimination of the restricted rotation around a C-C bond of 9, which yielded achiral, highly potent, and orally bioavailable BET inhibitors. Among them, compound 28 (CF53) binds to BET proteins with Kd values of 0.4–2.2 nM and shows low nanomolar potencies in cell growth inhibition of the MDA-MB-231triple negative breast cell line and the MOLM-13 acute leukemia cell line. It achieves excellent oral pharmacokinetics in mice, and effectively inhibits tumor growth in xenograft models in mice. Determination of the co-crystal structure of 28 with BRD4 BD1 provides a structural basis for its high binding affinity to BET proteins. Testing its binding affinities against other bromodomain-containing proteins shows that 28 is also a highly selective BET inhibitor against bromodomains. Our data show that 28 (CF53) is a potent, selective, and orally active BET inhibitor suitable for preclinical development.
EXPERIMENTAL SECTION
1. General Methods:
All reactions were conducted in a round-bottomed flask equipped with a Teflon-coated magnet stirring bar. Experiments involving moisture and/or air sensitive components were performed under anN2atmosphere. Commercial reagents and anhydrous solvents were used without further purification. The crude reaction products were purified by flash column chromatography using silica gel. Further purification was performed on a preparative HPLC (Waters 2545) with a C18 reverse phase column. The mobile phase used here was a gradient flow of solvent A (water, 0.1% of TFA) and solvent B (CH3CN, 0.1% of TFA) at a flow rate of 40 mL/min. Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectroscopy were performed in Bruker Advance 300/400 NMR spectrometers. Low resolution ESI mass spectrum analysis was performed on a Thermo-Scientific LCQ Fleet mass spectrometer or Advion Expression mass spectrometer. The analytical UPLC model was Waters Acquity H class (UV detection at 230 nm and 254 nm) and the reverse phase column used was the Acquity UPLC® BEH (C18–1.7 µm, 2.1 × 50 mm). Unless otherwise stated, all final compounds were purified to ≽ 95% purity as determined by analytical UPLC analysis.
2. Synthesis of Final Compounds:
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(1-methyl-1H-indazol-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (12) Method A:
S13 (90 mg) and 1-methyl-1H-indazol-3-amine (90 mg) were placed in a round-bottomed flask followed by addition of i-PrOH (30 mL). Four drops of concentrated HCl were added via a glass pipette. The mixture was heated overnight at reflux temperature. The reaction was then concentrated on a rotary evaporator and the residue was purified by HPLC to yield 60 mg of the desired product (12) as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 8.44 (d, J = 7.88 Hz, 1H), 7.84 (s, 1H), 7.68 (d, J = 8.62 Hz, 1H), 7.57 (t, J = 7.63 Hz, 1H), 7.47 (s, 1H), 7.30 (t, J = 7.55 Hz, 1H), 4.16 (s, 3H), 3.86 (s, 3H), 2.73 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H). ESI-MS calculated for C25H24N7O2 [M+H]+ = 454.20; Observed: 454.42
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(1-methyl-1H-pyrazolo[3,4-b]pyridin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (13) Method B:
Pd2(dba)3 (18 mg, 0.02 mmol) and BINAP (26 mg, 0.04 mmol) were mixed in anhydrous toluene (5 mL), and the mixture was heated at reflux for 3–4 min. This mixture was transferred into a round-bottomed flask containing S13 (68 mg, 0.2 mmol), 1-methyl-1H-pyrazolo[3,4-b]pyridin-3-amine (60 mg, 0.4 mmol), K3PO4 (127 mg, 0.6 mmol), and toluene (15 mL). The mixture was heated at reflux overnight before being quenched with MeOH. The reaction mixture was filtered and the filtrate was concentrated and purified by HPLC to yield 32 mg of the desired product (13) as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 8.65 (d, J = 3.99 Hz, 1H), 8.38 (d, J = 8.15 Hz, 1H), 7.89, 7.48, 7.31 (dd, J = 8.08, 4.55 Hz, 1H), 4.17 (s, 3H), 3.86 (s, 3H), 2.71 (s, 3H), 2.32 (s, 3H), 2.15 (s, 3H). ESI-MS calculated for C24H23N8O2 [M+H]+ = 455.19; Observed: 455.50
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(1-methyl-1H-pyrazolo[4,3-c]pyridin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (14) Method B:
S13 (70 mg) and 1-methyl-1H-pyrazolo[4,3-c]pyridin-3-amine (60 mg) were the substrates and upon HPLC purification, 47 mg of the desired product (14) was isolated as the trifluoroacetate. 1H NMR (300 MHz, CDCl3): 9.71 (s, 1H), 8.46 (d, J = 6.97 Hz, 1H), 8.12 (s, 1H), 8.08 (d, J = 7.00 HZ, 1H), 7.38 (s, 1H), 4.23 (s, 3H), 3.98 (s, 3H), 2.65 (s, 3H), 2.34 (s, 3H), 2.18 (s, 3H).
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(1-methyl-1H-pyrazolo[4,3-b]pyridin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (15, 94% purity by UPLC analysis) Method B:
S13 (102 mg, 0.3 mmol) and 1-methyl-1H-pyrazolo[4,3-b]pyridin-3-amine (90 mg, 0.6 mmol) were the substrates and upon HPLC purification, 27.9 mg of the desired product (15) was produced as the trifluoroacetate (94% purity by HPLC).1H NMR (300 MHz, MeOD-d4): 8.69 (d, J = 3.63 Hz, 1H), 8.32 (dd, J = 8.69, 0.79 Hz, 1H), 8.26 (s, 1H), 7.67 (dd, J = 8.74, 4.41 Hz, 1H), 7.49 (s, 1H), 4.22 (s, 3H), 4.01 (s, 3H), 2.84 (s, 3H), 2.35 (s, 3H), 2.18 (s, 3H). ESI-MS calculated for C24H23N8O2 [M+H]+ = 455.19, Observed: 455.42
N-(5-Chloro-1-methyl-1H-indazol-3-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (16) Method A:
S13 (70 mg) and 5-chloro-1-methyl-1H-indazol-3-amine (100 mg) were the substrates and upon HPLC purification, 18 mg of the desired product (16) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.92 (s, 2H), 7.69 (d, J = 9.01 Hz, 1H), 7.52 (d, J = 9.01 Hz, 1H), 7.48 (s, 1H), 4.15 (s, 3H), 3.89 (s, 3H), 2.71 (s, 3H), 2.34 (s, 3H), 2.16 (s, 3H). ESI-MS calculated for C25H2335ClN7O2 [M+H]+ = 488.16; Observed: 488.29
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-N-(6-methoxy-1-methyl-1H-indazol-3-yl)-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (17) Method A:
S13 (68 mg) and 1-methyl-6-methoxy-1H-indazol-3-amine (80 mg) were the substrates and upon HPLC purification, 24 mg of the desired product (17) was isolated as thetrifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.89 (s, 1H), 7.75 (d, J = 8.92 Hz, 1H), 7.47 (s, 1H), 7.07 (d, J = 1.95 Hz, 1H), 6.92 (dd, J = 8.86, 2.13 Hz, 1H), 4.11 (s, 3H), 3.95 (s, 3H), 3.90 (s, 3H), 2.77 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H). ESI-MS calculated for C26H26N7O3 [M+H]+ = 484.21, Observed: 484.37
7-(3,5-Dimethylisoxazol-4-yl)-N-(5-fluoro-1-methyl-1H-indazol-3-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (18) Method A:
5-Fluoro-1-methyl-1H-indazol-3-ylamine (100 mg, 0.6 mol) and S13 (102 mg, 0.3 mmol) were the substrates and upon HPLC purification, 17 mg of the desired product (18) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.93 (s, 1H), 7.71 (dd, J = 9.05, 3.80 Hz, 1H), 7.56 (dd, J = 8.80, 2.24 Hz, 1H), 7.48 (s, 1H), 7.44–7.32 (m, 1H), 4.16 (s, 3H), 3.89 (s, 3H), 2.71 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H). ESI-MS calculated for C25H23FN7O2 [M+H]+ = 472.19, Observed: 472.42
7-(3,5-Dimethylisoxazol-4-yl)-N-(6-fluoro-1-methyl-1H-indazol-3-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (19) Method A:
6-Fluoro-1-methyl-1H-indazol-3-ylamine (100 mg, 0.6 mol) and S13 (102 mg, 0.3 mmol) were the substrates and upon HPLC purification, 77 mg of the desired product (19) was isolated as the trifluoroacetate.1HNMR (300 MHz, MeOD-d4): 8.06 (s, 1H), 7.66 (t, J = 7.97 Hz, 1H), 7.49 (s, 1H), 7.16–7.00 (m, 2H), 3.94 (s, 3H), 3.49 (s, 3H), 2.76 (s, 3H), 2.32 (s, 3H), 2.14 (s, 3H). ESI-MS calculated for C25H23FN7O2 [M+H]+ = 472.19, Observed: 472.36
7-(3,5-Dimethylisoxazol-4-yl)-N-(7-fluoro-1-methyl-1H-indazol-3-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (20) Method A:
7-Fluoro-1-methyl-1H-indazol-3-ylamine (100 mg, 0.6 mol) and S13 (102 mg, 0.3 mmol) were the substrates and upon HPLC purification, 50 mg of the desired product (20)was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.83 (s, 1H), 7.63 (d, J = 7.94 Hz, 1H), 7.48 (s, 1H), 7.30–7.12 (m, 2H), 4.27 (s, 3H), 3.86 (s, 3H), 2.69 (s, 3H), 2.33 (s, 3H), 2.15 (s, 3H). ESI-MS calculated for C25H23FN7O2 [M+H]+ = 472.19, Observed: 472.36
7-(3,5-Dimethylisoxazol-4-yl)-N-(4-fluoro-1-methyl-1H-indazol-3-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (21) Method A:
4-Fluoro-1-methyl-1H-indazol-3-ylamine (102 mg, 0.6 mol) and S13 (102 mg, 0.3 mmol) were the substrates and upon HPLC purification, 75 mg of the desired product (21) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.82 (s, 1H), 7.54–7.45 (m, 2H), 7.49 (s, 1H), 6.98–6.87 (m, 1H), 4.16 (s, 3H), 3.87 (s, 3H), 2.72 (s, 3H), 2.33 (s, 3H), 2.15 (s, 3H). ESI-MS calculated for C25H23FN7O2 [M+H]+ = 472.19, Observed: 472.33
7-(3,5-Dimethylisoxazol-4-yl)-N-(1H-indazol-3-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (22) Method A:
1H-indazol-3-ylamine (84 mg, 0.6 mol) and S13 (102 mg, 0.3 mmol) were the substrates and upon HPLC purification, 27 mg of the desired product (22) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.92 (d, J = 8.29 Hz, 1H), 7.74 (s, 1H), 7.64 (d, J= 8.55 Hz, 1H), 7.57–7.50 (m, 1H), 7.47 (s, 1H), 7.32–7.25 (m, 1H), 3.84 (s, 3H), 2.76 (s, 3H), 2.32 (s, 3H), 2.15 (s, 3H). ESI-MS calculated for C24H22N7O2 [M+H]+ = 440.18, Observed: 440.33
2-(3-((7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-yl)amino)-1H-indazol-1-yl)ethanol (23) Method A:
2-(3-Amino-1H-indazol-1-yl) ethanol (290 mg, 1.6 mol) and S13 (170 mg, 0.5 mmol) were the substrates and upon HPLC purification, 107 mg of the desired product (23) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.91 (d, J = 8.25 Hz, 1H), 7.84 (s, 1H), 7.70 (d, J = 8.65 Hz, 1H), 7.54 (t, J = 7.58 Hz, 1H), 7.47 (s, 1H), 7.27 (t, J = 7.50 Hz, 1H), 4.57 (t, J = 5.10 Hz, 2H), 4.05 (t, J = 5.10 Hz, 2H), 3.83 (s, 3H), 2.71 (s, 3H), 2.32 (s, 3H), 2.15 (s, 3H). ESI-MS calculated for C26H26N7O3 [M+H]+ = 484.21, Observed: 484.25.
N-(1,3-dimethyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (24) Method B:
S13 (68 mg) and 1,3-dimethyl-1H-pyrazol-5-amine (50 mg) were the substrates and upon HPLC purification, 40 mg of the desired product (24) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.46 (s, 1H), 7.43 (s, 1H), 6.25 (s, 1H), 3.87 (s, 3H), 3.76 (s, 3H), 2.70 (s, 3H), 2.31 (s, 3H), 2.30 (s, 3H), 2.14 (s, 3H). ESI-MS calculated for C22H24N7O2 [M+H]+ = 418.20; Observed: 418.92
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-9H-pyrimido[4,5-b]indol-4-amine (25) Method B:
S13 (102 mg) and 2-methyl-5-(trifluoromethyl)pyrazol-3-amine (100 mg) were the substrates and upon HPLC purification, 29 mg of the desired product (25)was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.83 (s, 1H), 7.47 (s, 1H), 6.72 (s, 1H), 3.92 (s, 3H), 3.88 (s, 3H), 2.67 (s, 3H), 2.32 (s, 3H), 2.15 (s, 3H). ESI-MS calculated for C22H21F3N7O2 [M+H]+ = 472.17, Observed: 472.33.
7-(3,5-Dimethylisoxazol-4-yl)-N-(3-isopropyl-1-methyl-1H-pyrazol-5-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (26) Method B:
S13 (102 mg, 0.3 mmol) and 3-isopropyl-1-methyl-1H-pyrazol-5-amine (84 mg, 0.6 mmol) were the substrates and upon HPLC purification, 49 mg of the desired product (26) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.46 (s, 1H), 7.42 (s, 1H), 6.25 (s, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 2.97 (septet, J = 6.92 Hz, 1H), 2.71 (s, 3H), 2.31 (s, 3H), 2.13 (s, 3H), 1.28 (d, J = 6.95 Hz, 6H). ESI-MS calculated for C24H28N7O2 [M+H]+ = 446.23, Observed: 446.42
N-(3-tert-Butyl-1-methyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (27) Method B:
S13 (102 mg, 0.3 mmol) and 3-tert-butyl-1-methyl-1H-pyrazol-5-amine (100 mg, 0.6 mmol) were the substrates and upon HPLC purification, 49 mg of the desired product (27) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.45 (s, 1H), 6.26 (s, 1H), 3.88 (s, 3H), 3.82 (s, 3H), 2.71 (s, 3H), 2.31 (s, 3H), 2.14 (s, 3H), 1.32 (s, 9H). ESI-MS calculated for C25H30N7O2 [M+H]+ = 460.25, Observed: 460.33
N-(3-Cyclopropyl-1-methyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (28) Method B:
S13 (102 mg, 0.3 mmol) and 3-cyclopropyl-1-methyl-1H-pyrazol-5-amine (90 mg, 0.6 mmol) were the substrates and upon HPLC purification, 53 mg of the desired product (28) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.45 (s, 1H), 7.25 (s, 1H), 6.09 (s, 1H), 3.86 (s, 3H), 3.75 (s, 3H), 2.71 (s, 3H), 2.31 (s, 3H), 2.14 (s, 3H), 2.00–1.80 (m, 1H), 1.00–0.90 (m, 2H), 0.76–0.68 (m, 2H). 13C (NMR, 100 MHz): 165.96, 160.91, 159.64, 154.66, 154.53, 153.05, 152.73, 137.03, 131.42, 119.83, 117.44, 114.51, 113.80, 104.65, 99.54, 95.43, 56.56, 35.77, 24.51, 11.73, 10.79, 9.86, 8.11; ESI-MS calculated for C24H26N7O2 [M+H]+ = 444.21, Observed: 444.33
N-(3-Cyclobutyl-1-methyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (29) Method B:
S13 (102 mg, 0.3 mmol) and 1-methyl-3-cyclobutyl-1H-pyrazol-5-amine (90 mg, 0.6 mmol) were the substrates and upon HPLC purification, 49 mg of the desired product (29) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-D4): 7.45 (s, 1H), 7.34 (s, 1H), 6.31 (s, 1H), 3.85 (s, 3H), 3.78 (s, 3H), 3.65–3.50 (m, 1H), 2.71 (s, 3H), 2.50–2.30 (m, 2H), 2.31 (s, 3H), 2.30–2.15 (m, 2H), 2.15–2.00 (m, 1H), 2.14 (s, 3H), 2.00–1.80 (m, 1H). ESI-MS calculated for C25H28N7O2 [M+H]+ = 458.23; Observed: 458.50
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(1-methyl-3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-pyrazol-5-yl)-9H-pyrimido[4,5-b]indol-4-amine (30) Method C:
Pd2(dba)3 (27 mg, 0.03 mmol) and BINAP (37 mg, 0.06 mmol) were mixed in anhydrous toluene (5 mL), and the mixture was heated at reflux for 3–4 min. This mixture was transferred into a round-bottomed flask containing S13 (102 mg, 0.3 mmol), 1-methyl-3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-pyrazol-5-amine (120 mg, 0.6 mmol), t-BuONa (200 mg, 1.2 mmol), and toluene (15 mL). The mixture was heated at reflux overnight before quenching with MeOH. The reaction mixture was filtered and the mother liquid was concentrated and purified by HPLC to yield 48 mg of 30as the trifluoroacetate. 1H NMR (300 MHz, MeOD-D4): 7.67 (s, 1H), 7.46 (s, 1H), 6.41 (s, 1H), 3.90 (s, 1H), 3.83 (s, 3H), 2.69 (s, 3H), 2.32 (s, 3H), 2.15 (s, 3H), 1.55 (s, 6H). ESI-MS calculated for C25H27F3N7O2 [M+H]+ = 514.22; Observed: 514.33
7-(3,5-Dimethylisoxazol-4-yl)-N-(1-ethyl-3-methyl-1H-pyrazol-5-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (31) Method B:
S13 (102 mg, 0.3 mmol) and 1-ethyl-3-methyl-1H-pyrazol-5-amine (75 mg, 0.6 mmol) were the substrates and upon HPLC purification, 19 mg of the desired product (31) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.45 (s, 1H), 7.28 (s, 1H), 6.21 (s, 1H), 4.11 (q, J = 7.22 Hz, 2H), 3.85 (s, 3H), 2.70 (s, 3H), 2.31 (s, 6H), 2.13 (s, 3H), 1.44 (t, J = 7.23 Hz, 3H). ESI-MS calculated for C23H26N7O2 [M+H]+ = 432.21, Observed: 432.92
7-(3,5-Dimethylisoxazol-4-yl)-N-(1-isopropyl-3-methyl-1H-pyrazol-5-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (32) Method B:
S13 (70 mg) and 1-isopropyl-3-methyl-1H-pyrazol-5-amine (640 mg) were the substrates and upon HPLC purification, 26 mg of the desired product (32) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.45 (s, 1H), 7.18 (s, 1H), 6.18 (s, 1H), 4.59 (septet, J = 6.68 Hz, 1H), 3.83 (s, 3H), 2.70 (s, 3H), 2.32 (s, 3H), 2.30 (s, 3H), 2.13 (s, 3H), 1.47 (d, J = 6.66 Hz, 6H). ESI-MS calculated for C24H28N7O2 [M+H]+ = 446.23; Observed: 446.67
N-(1-(tert-Butyl)-3-methyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (33). Method B:
S13 (60 mg) and 1-(tert-butyl)-3-methyl-1H-pyrazol-5-amine (84 mg) were the substrates and upon HPLC purification, 15 mg of the desired product (33) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4) δ 7.44 (s, 1H), 6.59 (s, 1H), 6.24 (s, 1H), 3.80 (s, 3H), 2.74 (s, 3H), 2.31 (s, 3H), 2.30 (s, 3H), 2.14 (s, 3H), 1.72 (s, 9H). ESI-MS calculated for C25H30N7O2 [M+H]+ = 460.24; Observed: 460.55.
N-(1-Cyclobutyl-3-methyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (34) Method B:
S13 (102 mg, 0.3 mmol) and 1-cyclobutyl-3-methyl-1H-pyrazol-5-amine (100 mg, 0.6 mmol) were the substrates and upon HPLC purification, 58 mg of the desired product (34) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.45 (s, 1H), 7.22 (s, 1H), 6.20 (s, 1H), 4.90–4.70 (m, 1H), 3.84 (s, 3H), 2.70–2.50 (m, 2H), 2.69 (s, 3H), 2.40–2.20 (m, 2H), 2.34 (s, 3H), 2.31 (s, 3H), 2.13 (s, 3H), 1.90–1.60 (m, 2H). ESI-MS calculated for C25H28N7O2 [M+H]+ = 458.23, Observed: 548.58
N-(1-Cyclopentyl-3-methyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (35) Method B:
S13 (102 mg, 0.3 mmol) and 1-cyclopentyl-3-methyl-1H-pyrazol-5-amine (100 mg, 0.6 mmol) were the substrates and upon HPLC purification, 60 mg of the desired product (35)was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.44 (s, 1H), 7.19 (s, 1H), 6.18 (s, 1H), 4.69 (quintet, J = 7.95 Hz, 1H), 3.83 (s, 3H), 2.70 (s, 3H), 2.31 (s, 3H), 2.13 (s, 3H), 2.10–2.00 (m, 4H), 2.00–1.80 (m, 2H), 1.70–1.50 (m, 2H). ESI-MS calculated for C26H30N7O2 [M+H]+ = 472.25, Observed: 472.42
N-(1,5-Dimethyl-1H-pyrazol-3-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (36) Method B:
S13 (102 mg, 0.3 mmol) and 1,5-dimethyl-1H-pyrazol-3-amine (70 mg, 0.6 mmol) were the substrates and upon HPLC purification, 31 mg of the desired product (36) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 8.24 (s, 1H), 7.46 (s, 1H), 6.25 (s, 1H), 3.98 (s, 3H), 3.90 (s, 3H), 2.84 (s, 3H), 2.39 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H). ESI-MS calculated for C22H24N7O2 [M+H]+ = 418.20, Observed: 418.50
N-(3-Cyclopropyl-1,4-dimethyl-1H-pyrazol-5-yl)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-2-methyl-9H-pyrimido[4,5-b]indol-4-amine (37) Method C:
S13 (136 mg) and 3-cyclopropyl-1,4-dimethyl-1H-pyrazol-5-amine (120 mg) were the substrates and upon HPLC purification, 16 mg of the desired product (37) was obtained as the trifluoroacetate. 1H NMR (300 MHz, MeOD-D4): 7.45 (s, 1H), 7.30–7.00 (br, 1H), 3.84 (s, 3H), 3.72 (s, 3H), 2.71 (s, 3H), 2.31 (s, 3H), 2.13 (s, 3H), 1.94 (s, 3H), 2.00–1.80 (m, 2H), 1.00–0.75 (m, 4H). ESI-MS calculated for C25H28N7O2 [M+H]+ = 458.23; Observed: 458.50
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(2-methyl-2,4,5,6-tetrahydrocyclopenta-[c]pyrazol-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (38) Method B:
S13 (102 mg, 0.3 mmol) and 2-methyl-2,4,5,6-tetrahydrocyclopenta [c]pyrazol-3-amine (90 mg, 0.6 mmol) were the substrates and upon HPLC purification, 38 mg of the desired product (38) was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.46 (s, 1H), 7.27 (s, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 2.80–2.70 (m, 2H), 2.73 (s, 3H), 2.56–2.34 (m, 4H), 2.31 (s, 3H), 2.14 (s, 3H). ESI-MS calculated for C24H26N7O2 [M+H]+ = 444.21, Observed: 444.42
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(2-methyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (39) Method B:
S13 (136 mg, 0.4 mmol) and 2-methyl-4,5,6,7-tetrahydro-2H-indazol-3-amine (144 mg, 1.0 mmol) were the substrates and upon HPLC purification, 25 mg of the desired product was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.45 (s, 1H), 7.14 (s, 1H), 3.83 (s, 3H), 3.80 (s, 3H), 2.80–2.60 (m, 2H), 2.71 (s, 3H), 2.40–2.20 (m, 2H), 2.31 (s, 3H), 2.13 (s, 3H), 1.90–1.76 (m, 2H), 1.76–1.60 (m, 2H). ESI-MS calculated for C25H28N7O2 [M+H]+ = 458.23, Observed: 458.50
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-2-methyl-N-(1-methyl-4,5,6,7-tetrahydro-1H-indazol-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (40) Method B:
S13 (240 mg, 0.7 mmol) and 1-methyl-4,5,6,7-tetrahydro-1H-indazol-3-amine (220 mg, 1.4 mmol) were the substrates and upon HPLC purification, 100 mg of the desired product was isolated as the trifluoroacetate. 1H NMR (300 MHz, MeOD-d4): 7.71 (s, 1H), 7.45 (s, 1H), 3.90 (s, 3H), 3.81 (s, 3H), 2.77 (s, 3H), 2.71 (t, J = 6.06 Hz, 2H), 2.53 (t, J = 5.95 Hz, 2H), 2.32 (s, 3H), 2.15 (s, 3H), 1.98–1.84 (m, 2H), 1.84–1.70 (m, 2H). ESI-MS calculated for C25H28N7O2 [M+H]+ = 458.23, Observed: 458.75
3. Structure Determination of CD235
Yellow plates of CD235 were grown from a methanol/dichloromethane solution of the compound at 23 oC. A crystal of dimensions 0.17 × 0.07 × 0.02 mm was mounted on a Rigaku AFC10K Saturn 944+ CCD-based X-ray diffractometer equipped with a low temperature device and Micromax-007HF Cu-target micro-focus rotating anode (λ = 1.54187 A) operated at 1.2 kW power (40 kV, 30 mA). The X-ray intensities were measured at 85(1) K with the detector placed at a distance 42.00 mm from the crystal. A total of 2028 images were collected with an oscillation width of 1.0° in ω The exposure times were 1 sec. for the low angle images, 5 sec. for high angle. Rigaku d*trek images27 were exported to CrysAlisPro28 for processing and corrected for absorption. The integration of the data yielded a total of 16081 reflections to a maximum 2θ value of 138.75° of which 3873 were independent and 3470 were greater than 2σ(I). The final cell constants (Table S2) were based on the xyz centroids of 7080 reflections above 10σ(I). Analysis of the data showed negligible decay during data collection. The structure was solved and refined with the Bruker SHELXTL (version 2016/6) software package29, using the space group P1bar with Z = 2 for the formula C26H20N5O2F. All non-hydrogen atoms were refined anisotropically with the hydrogen atoms placed in a combination of idealized and refined positions. Full matrix least-squares refinement based on F2 converged at R1 = 0.0415 and wR2 = 0.1072 [based on I > 2sigma(I)], R1 = 0.0458 and wR2 = 0.1118 for all data. Additional details are presented in SI Table S1 and are given as Supporting Information in a CIF file.
4. Determination of Biochemical Binding Affinities to BET Proteins
Recombinant human proteins corresponding to BRD4 BD1 (residues 44−168) and BRD4 BD2 (residues 333−460) were used in the biochemical binding assays. The binding affinities of BET inhibitors to BRD4 (BD1and BD2 proteins) were determined using our established fluorescence polarization binding assays as described previously.23,24
5. Determination of Co-Crystal Structures for Compounds 12 and 28 complexed with BRD4 BD1 protein
BRD4 BD1 (residues 44–168) was cloned into a N-terminal His-TEV vector and expressed overnight at 20 °C in Rosetta cells. The BRD4 DB1 containing cells were lysed via sonication in 25 mM Tris pH 7.5, 200 mM NaCl and 0.1% β-mercaptoethanol with protease inhibitors. The cellular debris was removed by centrifugation. The protein was purified from the soluble fraction using Ni-NTA resin (Qiagen) followed by tag cleavage with TEV protease. The cleaved protein was further purified by gel filtration on a Superdex75 column (GE Healthcare). The final buffer for BRD4 BD1 was 25mM Tris, pH 8.5, 0.2 M NaCl, and 1 mM TCEP.
For crystallization, BRD4 BD1 was concentrated to 12.1 mg/mL then incubated with a 2-fold molar excess of compound 28 (CF53). Crystals grew from drops containing equal volumes of protein and well solution (20% PEG 3350 and 0.2 M potassium chloride). Prior to data collection, crystals were cryoprotected in well solution containing 25% ethylene glycol. For BRD4 BD2 crystallization, the protein was concentrated to 8.3 mg/mL and incubated with a 3-fold molar excess of 12 followed by a 1 hour incubation with 0.5% β-mercaptoethanol at 20 °C. Crystals grew in drops setup against 50–75% PEG 400 and 0.1 M imidazole, pH 8.0, which proved to be cryoprotective. Data were collected at the Advance Photon Source at Argonne National Lab on the LS-CAT beamlines 21-ID-D (BRD 4 BD1-CF53) and 21-ID-G (BRD4 BD2-12). Diffraction data were processed with HKL200030 and the structures solved by molecular replacement using MOLREP31 for BRD4 BD1 with PDB code 4LYI as a starting model or Phaser32 for BRD4 BD2 using PDB code 2OUO as a starting model. The structures were refined with Buster33 and electron density fit with COOT34. The protein structures were validated using Molprobity35. Ligand structures and restraints were created using Grade33. Refined ligand statistics were obtained from the PDB Validation Server. Data refinement and statistics are given in Supporting Information SI Table I.
6. Cell Growth Inhibition Assay
All human cancer cell lines were purchased from the American Type Culture Collection. Cells were used within 3 months after their thaw and were cultured as recommended. In cell growth assay, cells were seeded in 96-well cell culture plates at 10 000 cells/well for leukemia cells and 3000 cells/well for breast cancer cells in 75 μL of culture medium. Compounds were serially diluted in culture medium, and 75 μL of the diluted compounds was added to the plates. The cells were then cultured for 4 days. Cell growth was evaluated by adehydrogenase-based WST-8 assay (Dojindo Molecular Technologies). The WST-8 reagent was added to the plate at a final concentration of 10% (v/v), incubated for 1−3 h, and read at 450 nm using a Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC). The readings were normalized to the DMSO-treated cells, and the IC50 was calculated by nonlinear regression analysis using GraphPad Prism 6 software.
7. Pharmacokinetics Studies
All animal experiments were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine under the approved protocol (PRO00005315, P.I. Shaomeng Wang).
Limited pharmacokinetics of compounds 26, 27, 28, 33, 38, and 39 were determined in SCID mice bearing MDA-MB-231 tumor and the corresponding drug was orally administrated at 50 mg/kg dose.
Full pharmacokinetics of compounds 28 was determined in Balb/c mice following intravenous (iv) dosing at 5.0 mg/kg or oral (po) dosing at 25 mg/kg. The tested compound was dissolved in a vehicle containing 20% (v/v) PCP and 70% (v/v) PBS. Blood samples (100 μL) were collected from rats with a catheter at 0, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 24 h after administration of the drugs. The blood samples were centrifuged at 15 000 rpm for 10 min, then the supernatant plasma was stored at −80 °C until analysis. Plasma concentrations of the compounds were determined by the LC−MS/MS method developed and validated for this study. The LC−MS/MS method consisted of a Shimadzu HPLC system, and chromatographic separation of tested compounds was achieved using a Waters XBridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB SciexQTrap 4500 mass spectrometer equipped with an electrospray ionization source (Applied Biosystems, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. The mobile phases were 0.1% formic acid in purified water (A) and 0.1% formic acid in CH3CN(B). The gradient (B) was held at 10% (0−0.3 min), increased to 95% at 0.7 min, then stayed at isocratic 95% B for 2.3 min, and then immediately stepped back down to 10% for 2 min of re-equilibration. The flow rate was set at 0.4 mL/min. All pharmacokinetic parameters were calculated by noncompartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
8. Efficacy Studies in the RS4;11 and MDA-MB-231 xenograft Models in Mice
All efficacy experiments were done under the guidelines of the University of Michigan Committee for Use and Care of Animals and using an approved animal protocol (PRO00005315, P.I. Shaomeng Wang).
To develop xenograft tumors, 5 × 106 of RS4;11 or MDA-MB-231 cells with 50% Matrigel were injected subcutaneously on the dorsal side of severe combined immunodeficient (SCID) mice, obtained from Charles River, one tumor per mouse. When tumors reached ca.100 mm3, mice were randomly assigned to treatment and vehicle control groups. Animals were monitored daily for any signs of toxicity and weighed 2−3 times per week during the treatment and weighed at least weekly after the treatment was ended. Tumor size was measured 2−3 times per week by electronic calipers during the treatment period and at least weekly after the treatment was ended. Tumor volume was calculated as V = L × W2/2, where L is the length and W is the width of the tumor. The p value was calculated using unpaired t-test (two tailed) using GraphPad Prism 7 software.
Supplementary Material
Figure 3:
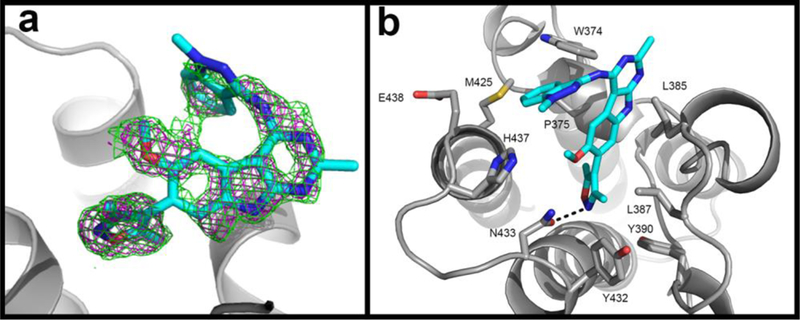
X-ray co-crystal structure of compound 12 in complex with BRD4 BD2 determined at 1.5 Å resolution (PDB ID:6C7Q). (a). The Fo-Fc omit electron density map of 12 contoured at 2σ (magenta) and 1.5σ (green). Protein backbone is shown in gray cartoon. (b). Detailed interactions of compound 12 with BRD4 BD2. Protein backbone is shown in gray cartoon with the side chains of residues interacting with compound shown as sticks. Carbons are depicted in cyan, nitrogens in blue, oxygens in red and sulfurs in yellow. Hydrogen bonds are shown as dashed lines.
ACKNOWLEDGMENTS
This study is supported in part by the Prostate Cancer Foundation, OncoFusion Therapeutics, Inc., the University of Michigan Prostate Cancer SPORE grant (NIH/NCI, Grant P50 CA186786) and the University of Michigan Comprehensive Cancer Core grant (NIH/NCI, Grant P30CA046592). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract DE-AC02–06CH11357. The Life Sciences Collaborative Access Team (LS-CAT) at Sector 21 of the Advanced Photon Source at Argonne National Laboratory was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We thank Dr. David Smith of LS-CAT for his assistance with crystal screening and remote data collection. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other funding agencies.
Notes
The authors declare the following competing financial interest(s): Multiple patents have been filed by University of Michigan on this class of BET bromodomain in inhibitors, which have been licensed by OncoFusion Therapeutics Inc. Shaomeng Wang, Yujun Zhao, Bing Zhou, LiuLiu, Longchuan Bai, Chao-Yie Yang, Bo Wen, Ting Zhao, Duxin Sun, Donna McEachern, and Xiaoqin Li are inventors of the BET bromodomain inhibitors reported in this manuscript and receive royalties from University of Michigan. Shaomeng Wang also owns stock in and serves as a consultant for OncoFusion Therapeutics Inc. The University of Michigan and Shaomeng Wang have also received a research contract from OncoFusion Therapeutics, Inc.
ABBREVIATIONS USED
- BET
Bromodomain and Extra-Terminal
- BD1
the first bromodomain starting from the N-terminal
- BD2
the second bromodomain starting from the N-terminal
- BINAP
(1,1′-Binaphthalene-2,2′-diyl)bis(diphenylphosphine)
- BRD2
Bromodomain -containing protein 2
- BRD3
Bromodomain-containing protein 3
- BRD4
Bromodomain-containing protein 4
- CL
volume of plasma cleared of the drug per unit time
- SCID
severe combined immunodeficient
- TGI
tumor growth inhibition
- Vz
volume of distribution
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI and includes a molecular formula string (CSV) file.
Accession Codes
Atomic coordinates have been deposited in the Protein Data Bank (PDB code: 6C7Qfor compound 12 and 6C7R for compound 28) and will release the atomic coordinates upon publication of this article.
REFERENCES
- (1).Belkina AC; Denis GV BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Filippakopoulos P; Knapp S Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- (3).Smith SG; Zhou MM The bromodomain: A new target in emerging epigenetic medicine. ACS Chem. Biol 2016, 11, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shang E; Salazar G; Crowley TE; Wang X; Lopez RA; Wang X; Wolgemuth DJ Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expression Patterns 2004, 4, 513–519. [DOI] [PubMed] [Google Scholar]
- (5).Florence B; Faller DV You BET-CHA: A novel family of transcriptional regulators. Front Biosci 2001, 6, D1008–D1018. [DOI] [PubMed] [Google Scholar]
- (6).Pivot-Pajot C; Caron C; Govin J; Vion A; Rousseaux S; Khochbin S Acetylation-dependent chromatin reorganization by BRDT, a testis-specific bromodomain-containing protein. Mol. Cell Biol 2003, 23, 5354–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).LeRoy G; Rickards B; Flint SJ The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dey A; Chitsaz F; Abbasi A; Misteli T; Ozato K The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Muller S; Pawson T; Gingras AC; Arrowsmith CH; Knapp S Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Theodoulou NH; Tomkinson NCO; Prinjha RK; Humphreys PG Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr. Opin. Chem. Biol 2016, 33, 58–66. [DOI] [PubMed] [Google Scholar]
- (12).Postel-Vinay S; Herbschleb K; Massard C; Woodcock V; Ocker M; Wilkinson G; Walter A; Ewerton F; Poelman M; Middleton M; Soria JC First-in-human phase I dose escalation study of the bromodomain and extra-terminal motif (BET) inhibitor BAY 1238097 in subjects with advanced malignancies. Eur. J. Cancer 2016, 69, S7–S8. [Google Scholar]
- (13).Liu Z; Wang P; Chen H; Wold EA; Tian B; Brasier AR; Zhou J Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem 2017, 60, 4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Stathis A; Zucca E; Bekradda M; Gomez-Roca C; Delord JP; Rouge TD; Uro-Coste E; de Braud F; Pelosi G; French CA Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov 2016, 6, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Thieblemont C; Stathis A; Inghirami G; Karlin L; Morschhauser F; Gleeson M; Broussais F; Amorim S; Salles G; Facon T; Cunningham D; Vey N; Bourdel F; Herait P; Zucca E A phase 1 study of the BET-bromodomain inhibitor OTX015 in patients with non-leukemic hematologic malignancies. Blood 2014, 124. [Google Scholar]
- (16).Shapiro GI; Dowlati A; LoRusso PM; Eder JP; Anderson A; Do KT; Kagey MH; Sirard C; Bradner JE; Landau SB Abstract A49: Clinically efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma (NMC). Mol. Cancer Ther 2015, 14, A49–A49. [Google Scholar]
- (17).Mirguet O; Gosmini R; Toum J; Clement CA; Barnathan M; Brusq JM; Mordaunt JE; Grimes RM; Crowe M; Pineau O; Ajakane M; Daugan A; Jeffrey P; Cutler L; Haynes AC; Smithers NN; Chung CW; Bamborough P; Uings IJ; Lewis A; Witherington J; Parr N; Prinjha RK; Nicodeme E Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem 2013, 56, 7501–7515. [DOI] [PubMed] [Google Scholar]
- (18).Albrecht BK; Gehling VS; Hewitt MC; Vaswani RG; Cote A; Leblanc Y; Nasveschuk CG; Bellon S; Bergeron L; Campbell R; Cantone N; Cooper MR; Cummings RT; Jayaram H; Joshi S; Mertz JA; Neiss A; Normant E; O’Meara M; Pardo E; Poy F; Sandy P; Supko J; Sims RJ 3rd; Harmange JC; Taylor AM; Audia JE Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J. Med. Chem 2016, 59, 1330–1339. [DOI] [PubMed] [Google Scholar]
- (19).Bui MH; Lin X; Albert DH; Li L; Lam LT; Faivre EJ; Warder SE; Huang X; Wilcox D; Donawho CK; Sheppard GS; Wang L; Fidanze S; Pratt JK; Liu D; Hasvold L; Uziel T; Lu X; Kohlhapp F; Fang G; Elmore SW; Rosenberg SH; McDaniel KF; Kati WM; Shen Y Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res 2017, 77, 2976–2989. [DOI] [PubMed] [Google Scholar]
- (20).McDaniel KF; Wang L; Soltwedel T; Fidanze SD; Hasvold LA; Liu DC; Mantei RA; Pratt JK; Sheppard GS; Bui MH; Faivre EJ; Huang XL; Li LM; Lin XY; Wang RQ; Warder SE; Wilcox D; Albert DH; Magoc TJ; Rajaraman G; Park CH; Hutchins CW; Shen JWJ; Edalji RP; Sun CHC; Martin R; Gao WQ; Wong SM; Fang GW; Elmore SW; Shen Y; Kati WM Discovery of N-(4-(2,4-difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a potent and orally available bromodomain and extraterminal domain (BET) family bromodomain inhibitor. J. Med. Chem 2017, 60, 8369–8384. [DOI] [PubMed] [Google Scholar]
- (21).Siebel AL; Trinh SK; Formosa MF; Mundra PA; Natoli AK; Reddy-Luthmoodo M; Huynh K; Khan AA; Carey AL; van Hall G; Cobelli C; Dalla-Man C; Otvos JD; Rye KA; Johansson J; Gordon A; Wong NCW; Sviridov D; Barter P; Duffy SJ; Meikle PJ; Kingwell BA Effects of the BET-inhibitor, RVX-208 on the HDL lipidome and glucose metabolism in individuals with prediabetes: A randomized controlled trial. Metab., Clin. Exp 2016, 65, 904–914. [DOI] [PubMed] [Google Scholar]
- (22).Nicholls SJ; Puri R; Wolski K; Ballantyne CM; Barter PJ; Brewer HB; Kastelein JJP; Hu B; Uno K; Kataoka Y; Herrman JPR; Merkely B; Borgman M; Nissen SE Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: results of the phase 2b, randomized, double-Blind, multicenter, ASSURE trial. Am. J. Cardiovasc. Drug 2016, 16, 55–65. [DOI] [PubMed] [Google Scholar]
- (23).Zhao Y; Bai L; Liu L; McEachern D; Stuckey JA; Meagher JL; Yang CY; Ran X; Zhou B; Hu Y; Li X; Wen B; Zhao T; Li S; Sun D; Wang S Structure-based discovery of 4-(6-methoxy-2-methyl-4-(quinolin-4-yl)-9H-pyrimido[4,5-b]indol-7-yl)-3,5-dimethy lisoxazole (CD161) as a potent and orally bioavailable BET bromodomain inhibitor. J. Med. Chem 2017, 60, 3887–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ran X; Zhao Y; Liu L; Bai L; Yang CY; Zhou B; Meagher JL; Chinnaswamy K; Stuckey JA; Wang S Structure-based design of gamma-carboline analogues as potent and specific BET bromodomain inhibitors. J. Med. Chem 2015, 58, 4927–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Picaud S; Wells C; Felletar I; Brotherton D; Martin S; Savitsky P; Diez-Dacal B; Philpott M; Bountra C; Lingard H; Fedorov O; Muller S; Brennan PE; Knapp S; Filippakopoulos P RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kruger AW; Rozema MJ; Chu-Kung A; Gandarilla J; Haight AR; Kotecki BJ; Richter SM; Schwartz AM; Wang Z The discovery and development of a safe, practical synthesis of ABT-869. Org. Process Res. Dev 2009, 13, 1419–1425. [Google Scholar]
- (27).CrystalClear Expert 2.0 r16 Rigaku Americas and Rigaku Corporation (2014), Rigaku Americas, 9009, TX, USA 77381–5209, Rigaku Tokyo, 196–8666, Japan. [Google Scholar]
- (28).CrysAlisPRO software system. 1.171.38.41 ed.; Rigaku Oxford Diffraction, Rigaku Corporation, Oxford, U. K. [Google Scholar]
- (29).Sheldrick GM Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (31).Vagin A; Teplyakov A MOLREP: an automated program for molecular replacement. J. Appl. Cryst 1997, 30, 1022–1025. [Google Scholar]
- (32).Mccoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bricogne GB,E; Brandl M; Flensburg C; Keller P; Paciorek W; Roversi P; Sharff A; Smart OS; Vonrhein C; Womack TO Cambridge, United Kingdom: Global Phasing Ltd; BUSTER version X.Y.Z 2011. [Google Scholar]
- (34).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.