

Abstract
Leber’s hereditary optic neuropathy (LHON) is a classical mitochondrial disease caused by mutations in the mitochondrial DNA encoding complex I subunits. Oxidative stress associated with complex I defect has been implicated in developing LHON phenotype such as retinal ganglion cell (RGC) death and loss of vision. However, the mechanism of LHON pathogenesis is still not very clear and thus no effective therapies are available to date. Using cybrid models for LHON, we show that autophagy is significantly compromised in cells carrying LHON-specific mtDNA mutations, which results in reduced clearance of dysfunctional mitochondria contributing to cell death. We further show that pharmacological activation of autophagy selectively clears the damaged mitochondria and thus repairs mitochondrial defects and improves overall cell survival in LHON cell models. Our results suggest that compromised autophagy is the missing link from oxidative stress to LHON pathogenesis. Activation of mitophagy ameliorates mitochondrial defects and exerts a protective role by improving cell survival in cells carrying LHON mutations that could be utilized as a potential therapeutic target for LHON treatment.
Introduction
Mitochondrial DNA mutations leading to mitochondrial dysfunctions have been linked to a variety of pathological conditions including metabolic syndromes, neurodegenerative diseases, myopathies, cancer and aging (1). Despite the fact that increasing numbers of such mutations have been reported for more than three decades, pathogenesis of such disorders is far from clear. Leber’s hereditary optic neuropathy (LHON) is a maternally inherited neurodegenerative disease that is characterized by selective death of retinal ganglion cells (RGCs) (2). The genetic bases for LHON are the point mutations in mtDNA-encoded subunits of mitochondrial respiratory complex I, particularly those located at the nucleotide positions 3460 (ND1), 11778 (ND4) and 114484 (ND6) in the mitochondrial genome (3–5). Unfortunately, no effective treatment is available for this disease, largely due to the missing link of etiopathogenesis of LHON from mtDNA mutations to degeneration in RGCs. While the primary LHON mutation is ubiquitous, LHON is more likely a non-syndromic disease where these homoplasmic mutations affect primarily RGCs in most of the patients (6). Nevertheless, why these mtDNA mutations trigger the specific death of RGCs only at particular age is still not clear. The transmitochondrial cytoplasmic hybrid (‘cybrid’) model where pathogenic mtDNA carrying mitochondria are transferred to a constant nuclear background has served as a valuable tool to characterize the biochemical and bioenergetics phenotypes of mtDNA mutations. Analysis of cybrids containing LHON-specific mutations have revealed defective complex I respiration, reduced Adenosine triphosphate (ATP) production, loss of mitochondrial membrane potential (MMP; Δψm), increased mitochondrial reactive oxygen species (ROS) production and sensitization to cell death under stress conditions (7–10). Among three primary LHON mutations, ND4 (G11778A) and ND1 (G3460A) mutations cause significant reduction in complex I activity and subsequent biochemical defects, while ND6 (T14484C) mutation only exert mild impact (7,11). In particular, both ND4 and ND1 mutations result in increased ROS levels and decreased antioxidant enzyme activities including glutathione peroxidases and glutathione reductase (7,11). Similarly, investigations carried out with a mouse model resembling LHON, both genetically and phenotypically, indicated oxidative stress as a predominant factor in etiopathogenesis of LHON (12). Autophagy is an important quality control mechanism, which involves lysosomal degradative process for eliminating damaged organelles and protein aggregates. The success of completion of autophagy involves dynamic interactions and integration of multiple pathways. Neuronal cells are particularly vulnerable to disruptions of these interactions, and the risk increases with age (13,14). As such, autophagy has been identified as underlying event involved in pathogenesis and emerging as a potential therapeutic target for several neurodegenerative diseases (15–17). Since autophagy plays a major role in removal of defective and damaged mitochondria (18,19), it becomes imperative to investigate the role of autophagy in pathogenesis of LHON that can further help to identify potential therapeutic target. Thus, to identify additional modifiers for the LHON pathogenesis, in the present study, we explored the quality control mechanism, particularly in the form of autophagy/mitophagy in cells carrying LHON-specific mtDNA mutations.
Results
Impairment in autophagy activation during mitochondrial stress in LHON cybrids
Specific mtDNA mutations in ND4/ND6/ND1 subunits of complex I have been identified in LHON patients and believed to play a causative role in sensitizing RGC to cell death, which is an essential phenotype for LHON (8,20,21). We utilized established cybrids carrying no mutation [wild type (WT); named as KB30 cybrid] or homoplasmic LHON mutations in mtDNA-encoded ND4 (G11778A mutation: named as Le131 cybrid) or ND1 (G3460A mutation: named as RJ206 cybrid), subunit genes of complex I. These mutations have previously been shown to cause significant decrease in complex I activity leading to mitochondrial dysfunctions (7,11). These mitochondrial dysfunctions have been linked to cell stress, thereby leading to cell death (9,11). It has been hypothesized that autophagy, particularly mitophagy, is involved in the removal of damaged and dysfunctional mitochondria, thereby preserving mitochondrial functions and preventing cellular stress. To explore the involvement of autophagy during mitochondrial defects in LHON cybrids, we incubated these cells in galactose media and measured lysosomal numbers that correlates with the level of autophagy, followed by immunoblotting with autophagy marker proteins such as Beclin1 and LC3B-II. In galactose media, cells are forced to rely on mitochondrial OXPHOS for its ATP requirements, and therefore it is a reliable indicator of overall mitochondrial health. Beclin1, is the mammalian orthologous of yeast Atg 6, which has a central role in localizing autophagic proteins to a pre-autophagosomal structure and sorting of the vacuole resident hydrolase carboxypeptidase Y through the Vps pathway (22,23). Similarly, LC3 is a microtubule-associated protein 1 light chain 3, a ubiquitin-like molecule (a mammalian homolog of the Atg8 in yeast). During autophagy, LC3-I is converted to LC3 II (lipidated form), which has been shown to play a role both in selecting cargo for degradation (e.g. interaction of LC3-II with p62/SQTM1 target-associated protein aggregates for turnover) and to promote membrane tethering and fusion in vitro supporting a possible role in fusion of other membrane compartments including mitochondria with autophagosomes (24). However, care should be taken while interpreting the LC3B levels, and it is suggested that LC3-II levels correlates well with the increased levels of autophagic vesicle than the LC3-I/II ratio and should be normalized with loading control for the comparison between samples (25,26).
For measuring lysosomes, cybrids were incubated for 24 h in glucose/galactose media and then stained with Lysotracker Red DND-99 (LTR): a fluorescent probe for lysosomes and Hoechst-33342 (H), a counter stain for nuclei, followed by fixation. As shown in Figure 1A and B, compared with the WT cybrid, the number of lysosomes were significantly less (almost 50%) in LHON cybrids after galactose incubation, suggesting a decrease in autophagic activity. Subsequently, we performed immunoblotting with autophagic marker proteins Beclin1 and LC3B-II at different time points after galactose incubation. As shown in Figure 1C, incubation of WT cybrid in galactose media resulted in an initial decrease in Beclin1 and LC3B-II levels (starting from 30 min to 12 h) indicating a decrease in autophagy upon switch from glucose to galactose media. However, recovery of Beclin1 and LC3B-II at protein levels was observed at later time points (24 and 48 h). In contrast, in LHON cybrids (Le131 and RJ206), Beclin1 and LC3B-II levels declined progressively (except 15 min and 1 h time points for Beclin1 in Le131) and did not show a recovery, suggesting a downregulation of autophagy after galactose incubation. Combining the results of quantitation of lysosomes (Fig. 1A and B) and levels of autophagic marker proteins (Fig. 1C), LHON cybrids exhibited a compromised autophagy upon galactose incubation, which could not be recovered at later stages (24 and 48 h) compared with WT. Since LHON cybrids carry mutations in genes encoding complex I subunits and exhibited reduced complex-I dependent oxygen consumption (27), we then tested how cells modulate autophagy levels during complex I inhibition in WT and LHON cybrids. We utilized pharmacological agents such as rotenone and paraquat, known to produce both complex-I-dependent and -independent mitochondrial stress, respectively (27,28).
Figure 1.
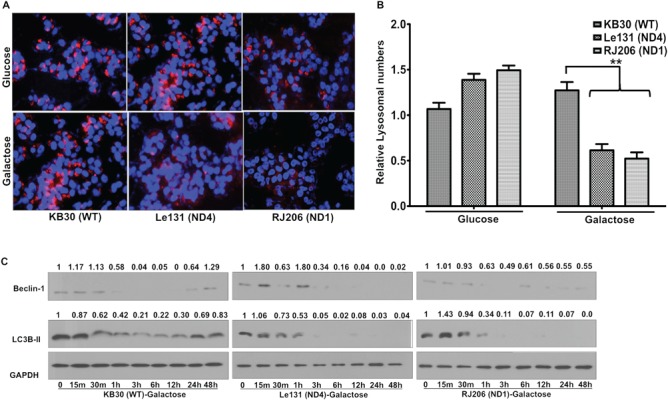
Compromised autophagy activation in LHON cybrids: WT (KB30), ND4 mutant (Le131) and ND1mutant (RJ206) cybrids were incubated for 24 h in glucose/galactose media and analyzed for autophagy activation. (A) Cells were stained with lysotracker red and fixed to visualize lysosomes (indicated as red) under fluorescence microscope (Nikon Eclipse TE2000). In parallel, nuclear stain Hoechst (indicated as blue) was used to normalize the lysosome numbers with total number of nuclei. One of the representative co-localized images for each line is shown. (B) Ten random images for each condition were acquired to quantitate the number of lysosomes by normalizing with number of Hoechst-33258 stained nuclei. Data represents means of three independent experiments. (**P < 0.005). (C) Similarly, cell lysates were used for immunoblotting for measuring autophagy marker proteins Beclin1 and LC3B-II (1:1000 dilutions). GAPDH was used as a loading control (1:2000 dilutions). Band intensities were measured by NIH ImageJ software and fold change to their respective 0 m is presented.
In the presence of rotenone in glucose medium (Fig. 2A), the WT cybrids demonstrated a significant increase in Beclin1 levels at 12 and 24 h time points, while LC3B-II levels initially increased at 15 min and maintained relatively higher levels until 12 h and returned to initial levels later, suggesting that WT cybrids attempt to employ autophagy under stress conditions. However, LHON cybrids failed to maintain the Beclin1 and LC3B-II levels especially at later time points such as 12 and 24 h as determined by immunoblotting. Similarly, for paraquat treatment (Fig. 2B), WT cybrid showed an increase in Beclin1 levels at 6, 12 and 24 hrs and LC3B-II levels increasing at 3, 6 and 12 hrs, and later decreased at 24 hrs time points. Conversely, in LHON cybrids, these marker protein levels were decreased particularly at later time points such as 6, 12 and 24 hrs. These findings further indicated that autophagy is enhanced in response to complex I inhibition in WT cybrids; however, it failed to sustain its level in cells carrying LHON mutation.
Figure 2.

Impairment in autophagy activation in LHON cybrids during complex I stress. Cells were grown in glucose for 24 h and treated with complex I stressors rotenone (1 μM) and paraquat (1 mM) in glucose media for different time points. Cell lysates were used for immunoblotting to measure the autophagy marker proteins (Beclin1 and LC3B-II) level for rotenone (A) and paraquat (B) treatment, respectively. GAPDH was used as a loading control (1:2000 dilution). Relative fold changes in proteins were calculated by measuring band intensities using NIH ImageJ software.
Autophagy impairments lead to accumulation of defective mitochondria
Mitochondrial homeostasis is maintained by balancing mitochondrial biogenesis and degradation (29). Autophagy has been demonstrated to play a protective role in response to cellular stress by clearing out damaged components; therefore, defective autophagy may lead to accumulation of damaged mitochondria (30). To investigate the consequence of compromised autophagy, we first measured the effect on mitochondrial turnover by quantitating mitochondrial mass by staining cells with mitochondrial-specific probe Mito-tracker Red and quantitated using fluorescent measurement (excitation/emission: 579/599 nm). As shown in Figure 3A, in comparison with control cybrids, we observed a significantly higher mitochondrial mass in LHON cybrids. This increase in mitochondrial mass was confirmed by immunoblotting analysis for various mitochondrial markers such as respiratory complexes subunit proteins and voltage-dependent anion channel (VDAC) in cybrids after galactose incubation (Fig. 3B). Overall results of mitochondrial mass measurement suggested an increased accumulation of mitochondria in LHON cybrids.
Figure 3.
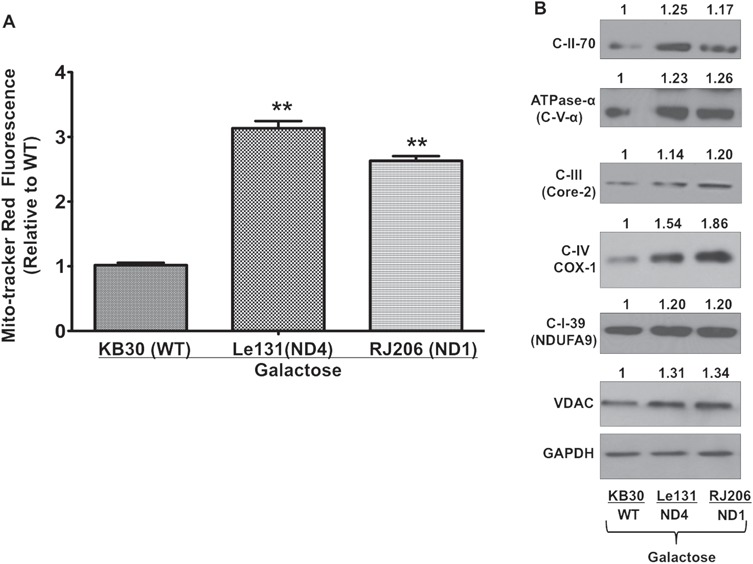
Measurement of mitochondrial mass in LHON cybrids: (A) Cells were grown in galactose media for 24 h in and incubated with MTR (250 nM) and Hoechst (10 μg/ml) and quantitated using fluorescent plate reader. Mitotracker fluorescence was normalizing with Hoechst fluorescence reading and presented as fold change in comparison with WT cybrid. (B) Cell lysates of galactose-incubated cells were used for immunoblotting analysis of different mitochondrial respiratory chain subunits. One of the representative images were used for quantitation of protein bands by ImageJ and normalized with internal control GAPDH. Values were presented as fold change relative to WT cybrid.
To investigate further, if these accumulated mitochondria were functional, we analyzed various mitochondrial parameters such as viability in galactose media, MMP and mitochondrial superoxides and ATP levels. When cybrids were investigated for their viability in galactose media, we found that LHON cybrids exhibited significant reduction in viability in galactose medium, where ATP production relies largely on oxidative phosphorylation (Fig. 4A). To measure the effect on MMP after glucose and galactose incubation, mitochondria were isolated and stained with an MMP indicator, Tetra-methyl-rhodamine methyl ester (TMRM), which is a cell-permeant dye that accumulates in active mitochondria with intact membrane potentials and quantitated using fluorescence measurement (excitation/emission: 548/574 nm). As shown in Figure 4B, in glucose medium, the basal level of MMP in LHON cybrids was lower compared with the WT cybrids, and it was further decreased when incubated in galactose medium (Fig. 4B). We then measured the mitochondrial ROS in the isolated preparations of mitochondria from WT and LHON cybrids after glucose and galactose incubation using Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen) and quantitated the fluorescence at excitation/emission: ∼571/585 nm. We found that LHON cybrids showed higher basal levels of mitochondrial ROS (in glucose media) than WT cybrids (ranging from 1.5- to 2.1-fold) and such overproduction of ROS was further enhanced in galactose medium (ranging from 2.4- to 4.3-fold) when compared in LHON cybrids. We also measured the total ATP content in the cell lysates after glucose and galactose incubation. ATP content was measured using luciferin–luciferase bioluminescence assay-based ATP determination kit (Invitrogen). As shown in Figure 4D, there was no significant difference in the total ATP content at basal levels (in glucose medium) between WT and LHON cybrids. However, upon galactose incubation, LHON cybrids showed a significant decrease in ATP content (ranging from 2.9- to 4.2-fold). Altogether, results on mitochondrial functional parameters clearly indicated functional impairments in mitochondria in LHON cybrids when compared with WT.
Figure 4.
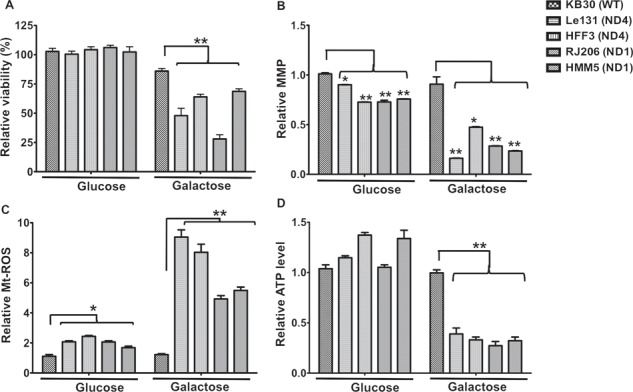
Viability and mitochondrial functional analysis of LHON cybrids after galactose incubation: (A) WT and LHON cybrids were incubated in glucose or galactose media for 48 h, and viability was measured using trypan blue exclusion assay. Relative viability is presented in comparison with WT as 100%. (B) Similarly, different cybrids were grown in glucose or galactose media for 24 h and mitochondria were isolated. MMP levels in these isolated preparations were determined using TMRM staining and fluorescent plate reading as described in Materials and methods. Fluorescence was normalized with total protein content and presented as relative value for comparison between WT and LHON cybrids with glucose or galactose media incubation. (C) Mitochondrial ROS levels were quantitated in the isolated mitochondria using Amplex Red assay kit and compared for their relative ROS levels between WT and LHON cybrids after glucose or galactose incubation for 24 h. (D) Total ATP content was determined in the cell lysates of WT and LHON cybrids using luciferase based ATP detection kit after growing them in glucose or galactose media for 24 h. The data was normalized with their total protein content (mg/ml) and presented as fold change relative to WT. Results are mean ± SEM of three independent experiments.(*P < 0.05; **P < 0.005).
Rapamycin treatment selectively clears the oxidatively damaged mitochondria by mitophagy
To further investigate the effect of compromised autophagy on mitochondrial clearance, we first examined the level of mitophagy during glucose and galactose incubation in WT and one of the representative LHON cybrids (Le131). During mitophagy, the damaged or defective mitochondria are engulfed in autophagosomes and degraded by fusion with lysosomal degradation. Thus, mitophagy can be identified by determining co-localization of mitochondria [by Mitotracker Red (MTR)] with lysosomes [by Lysotracker Green (LTG)] by fluorescence microscopy. We observed that there was no significant difference in the co-localization of mitochondria and lysosomes between LHON and WT cybrids in glucose medium (Fig. 5A, left panel, and 5B: comparison between glucose incubation). However, in galactose media, our results indicated a significant decrease in co-localization of mitochondria with lysosomes for LHON cybrids, suggesting an overall decrease in the clearance of mitochondria through autophagy or mitophagy (Fig. 5A, middle panel, and B: comparison between galactose incubation). Since incubation in galactose media increases mitochondrial ROS levels in LHON cybrids as shown in Figure 4C, we further investigated the fate of these oxidatively stressed mitochondria. We probed the cells with Mitosox Red to detect accumulated free radicals in mitochondria and LTG for lysosomes to see if oxidatively stressed mitochondria are localized to lysosomes for mitophagy. Although there was an increased accumulation of mitochondrial superoxides, indicated by more Mitosox Red staining in LHON cybrids in galactose medium, a significant decrease in the co-localization of these oxidatively stressed mitochondria with lysosomes was observed compared with WT and from glucose media incubation (Fig. 5C, left and middle panel, and D: comparison between glucose and galactose media).
Figure 5.
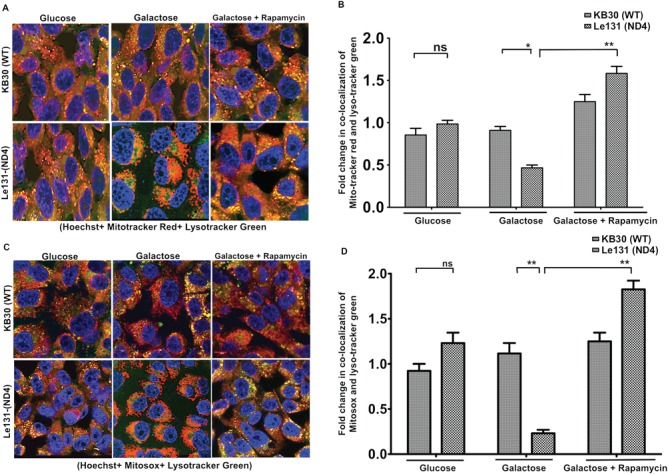
Analysis of mitochondrial autophagy during galactose/galactose + rapamycin incubation in LHON cybrids: cybrids were grown in glucose/galactose/galactose + rapamycin (25 μM) for 24 h and analyzed for mitophagy. (A) Cells were stained with MTR (for mitochondria) and LTG (for lysosomes) together with Hoechst (for nuclei). Cells were fixed and images were acquired. Approximately, 50–100 cells per coverslip were visualized on LSM (Olympus FV1000), and images were digitally captured and scored blindly. Representative images of one of the co-localization in WT (KB30) and ND4 mutant (Le131) are shown in which yellow dots represent co-localization of MTR and LTG reflecting mitophagy. (B) Quantification of co-localized images after normalization with Hoechst stained nuclei using ImageJ software. (C) In a separate experiment, glucose/galactose/galactose + rapamycin incubated cells were labeled with Mitosox (for oxidatively stressed mitochondria) and LTG for 15 min at 37 C in 5% CO2 incubator followed by fixation. One of the representative co-localized images of each line is shown. (D) Quantitation of co-localized images after normalized with total number of nuclei (Hoechst stain). Images (LSM) were taken with a 40× objective, repeated three times with different cultures (ns, non-significant; *P < 0.05; **P < 0.005).
In parallel, we used autophagy inducer rapamycin to further investigate if mitochondrial clearance in LHON cybrids can be activated by autophagy induction. Rapamycin activates autophagy via inhibiting Ser/Thr protein kinase mammalian Target of Rapamycin (mTOR) pathway. As shown in the right panel of Figure 5A, treatment with rapamycin significantly increased the sequestration of mitochondria to lysosomes in LHON cybrid as can be seen by an increased co-localization of MTR and LTG (3.4-fold) compared with their galactose alone incubation (Fig. 5B: comparison between LHON cybrid in galactose and galactose with rapamycin). Further, such increase in co-localization of lysosomes with the mitochondria was associated with increased mitochondrial ROS (almost 8-fold increase; Fig. 5C, middle and right panel, and 5D: comparison between LHON cybrid in galactose and galactose with rapamycin), suggesting selective mitophagy for oxidatively damaged/defective mitochondria that accumulated ROS. These results clearly suggested that due to compromised autophagy, oxidatively damaged mitochondria accumulated during galactose incubation, which resulted in oxidative stress and cell death as reported earlier (8,20,21). In addition, pharmacological activation of autophagy by rapamycin activated the clearance of damaged mitochondria that may contribute in cell survival.
Autophagy activation is crucial for cell survival by improving mitochondrial functions under galactose-induced stress in LHON cybrids
To investigate if the activation of autophagy contributes to the survival of LHON cybrids during galactose stress, we inhibited the autophagy using late-stage pharmacological inhibitor of autophagy, Bafilomycin A-1 (BafA1) that prevents the maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes (31) and measured the cell viability. In parallel, we also induced autophagy by using rapamycin and determined their effects on cell survival. We measured the effect of these agents on cellular viability in multiple cybrids carrying primary LHON mutations at ND1 and ND4. As shown earlier (Fig. 4A), galactose incubation decreases the viability of LHON cybrids. We found that incubation with autophagy inhibitor Baf-A-1 in galactose media further decreased the cell viability (ranging from 1.6- to 3.8-folds) in individual LHON cybrids (Fig. 6A: comparison between galactose and galactose with BafA-1). On the other hand, rapamycin treatment in galactose medium significantly restored the viability of LHON cybrids ranging from 2.6- to 5-fold increases (Fig. 6A: comparison between galactose with BafA-1 and galactose with rapamycin). We confirmed the inhibition of mTOR pathway by rapamycin through measuring phospho-mTOR protein levels by immunoblotting. As shown in Figure 6B, we observed a decreased phosphorylation of p-mTOR proteins upon rapamycin treatment. Similar protective effects were also observed when we utilized an mTOR-independent activator of autophagy, trehalose (32) (Supplementary Material, Fig. S1). To further investigate the consequence of rapamycin induced autophagy on mitochondrial functions; we analyzed the mitochondrial functions in galactose medium with rapamycin treatment. As shown in Figure 7, after rapamycin treatment, MMP levels in LHON cybrids significantly improved (1.6- to 3-fold increase) (Fig. 7A), and the ROS levels in LHON cybrids significantly reduced ranging from 1.3- to 1.9-fold (Fig. 7B). However, we did not observe any significant change in total ATP contents with rapamycin treatment (Fig. 7C).
Figure 6.
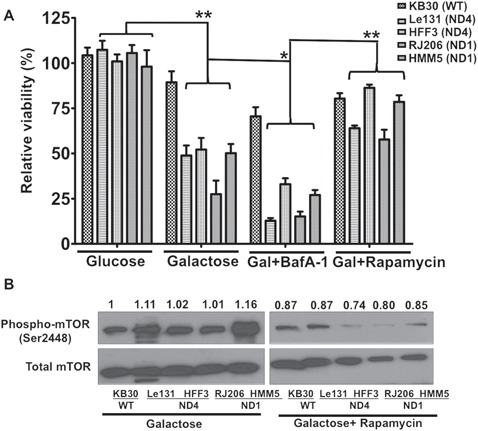
Effect of autophagy inhibition and activation on survival of LHON cybrids: (A) Viability of cybrids were measured by trypan blue exclusion assay after their incubation with autophagy inhibitor (Bafilomycin A1: BafA1: 200 nM) or inducer (Rapamycin: 25 μM) supplemented in galactose media and compared with their glucose/galactose incubation. Viability was compared in individual cybrids for their growth between glucose versus galactose, galactose versus galactose + BafA1 and galactose + BafA1 versus galactose + rapamycin. (B) Immunoblotting with phospho-mTOR and total mTOR proteins from cell lysates of cybrids grown in galactose and galactose + rapamycin for 24 h. Densitometry analysis of band intensities was done using ImageJ software and presented as relative to WT in galactose.
Figure 7.
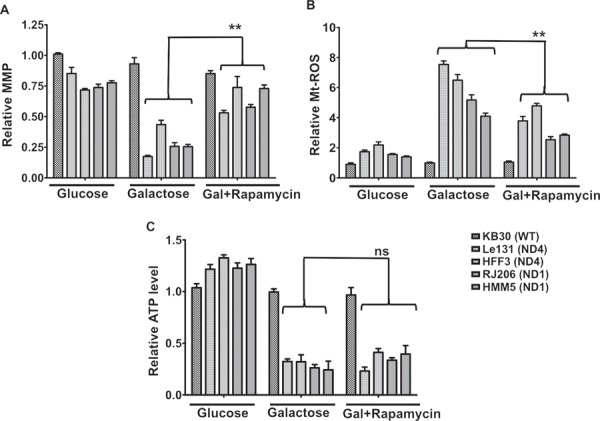
Analysis of mitochondrial functions after rapamycin treatment in LHON cybrids: Cybrids were grown in glucose/galactose/galactose + rapamycin media and measured for different parameters (MMP, ROS and ATP). (A) MMP was measured in the isolated mitochondria from cybrids grown in above conditions and compared individually between galactose and galactose + rapamycin. (B) Similarly, mtROS levels were measured in isolated mitochondria using Amplex Red and compared between galactose and galactose + rapamycin. (C) Total ATP levels were measured in the cell lysates using ATP determination kit. Results are mean ± SEM of three independent experiments (**P < 0.005; ns, non-significant).
Discussion
LHON is a mitochondrial inherited acute or subacute loss of central vision associated with degeneration of RGC layer. On mechanism of LHON pathogenesis, increased oxidative stress and bioenergetics failure have been suggested as major contributors of RGC death (12,33). However, the detail underlying mechanisms connecting the complex I subunit mutations to blindness is still largely unclear, and therefore no effective therapy is available for LHON treatment (34,35). Autophagy is one of the major quality control mechanisms to maintain mitochondrial homeostasis under normal and pathological conditions. Autophagy has been implicated in maintenance of neuronal functions and impairments in autophagy have been associated with a variety of neurodegenerative disorders (36–39). Here, our results demonstrate that autophagy activation is an important protective mechanism, which is significantly compromised in cybrids carrying LHON-specific mtDNA mutations. Such defect in autophagy leads to accumulation of damaged/defective mitochondria due to mtDNA mutations thereby leading to oxidative stress and cell death. Our study suggests that compromised mitophagy could be a missing link between mitochondrial dysfunction and RGC death, and such finding promises novel therapeutic approaches for LHON treatment.
Due to the lack of reliable animal models for LHON with specific mtDNA mutations, cybrid models have served as valuable tools to investigate the effects of LHON mutations on mitochondrial functions and cell death mechanisms (8,11,20,40). Important discoveries through investigations on these cybrids include mitochondrial impairments, oxidative stress and sensitization to cell death, particularly when the cybrids are cultured in galactose media where mitochondrial OXPHOS serves as the major source for ATP production (11,20,40,41). However, the connection among mitochondrial dysfunction, oxidative stress and cell death are not fully established and there are several missing links.
Autophagy has been reported to function as both cell death and cell survival mechanism depending on the context. For example, in the absence of apoptosis, autophagy can serve as an alternative cell death mechanism as shown in knock-out mouse models deficient in pro-apoptotic proteins such as Bax and caspases (42–44). Conversely, beneficial role of autophagy has also been shown to facilitate the clearance of aggregation-prone proteins and promote cell survival in a number of neurodegenerative disease models (45). Cybrid carrying mitochondria from patients with Parkinson’s disease (PD), which exhibited complex I defect, reproduced the alterations in the autophagic system observed in the brain of these patients with PD (46). On the other hand, deletion of autophagy protein (ATG7−/−) disrupts mitochondrial functions results in reduced respiration, increase in glycolysis and increased ROS generation (47). In our study, we found that LHON cybrids failed to sustain the activation of autophagy upon galactose incubation. We determined the levels of LC3B-II, which is continually degraded within autophagolysosomes during autophagy; however, determining autophagic flux by using agents such as pepstatin A and E-64d, or other lysosomal inhibitor, and comparing the levels of LC3B-II could have further demonstrated a better representation of autophagic flux (25), thus further studies are required to investigate autophagic flux under such conditions. In our study, we have shown that inhibitors of autophagy further enhanced cell death, which could be prevented by upregulation of autophagy using rapamycin, suggest that autophagy was compromised in LHON cybrids, corroborative with the evidences of defective autophagy in these cells as compared to the WT. Further, our results suggest that this compromised autophagy is a result of mitochondrial stress more specifically from complex I disturbances.
It has been reported that oxidative stress induced by tert-butyl hydroperoxide (tBHP) treatment in cardiac myoblast H9c2 cells caused significant impairment in autophagy pathway (48). Interestingly, investigations with cybrids harboring the A3243G MELAS mutation exhibited increased autophagy activity, and it can be abolished by treatment with antioxidants, suggesting that oxidative stress participate in this process (49). Further, prevention of autophagy in MELAS fibroblasts resulted in cell death, suggesting a protective role of autophagy. In our study we found that mitochondria are accumulated in LHON cybrids, as suggested by increased mitochondrial numbers and levels of respiratory chain (RC) subunit proteins after galactose incubation (Fig. 3). Further, these accumulated mitochondria are functionally impaired as LHON cybrids showed significant mitochondrial dysfunctions such as reduced viability, increased mt-ROS and lower MMP and ATP levels after galactose incubation (Fig. 4). In addition, co-localization studies between mitochondria and lysosomes (Fig. 5) suggested that mitochondrial accumulation is due to defects in autophagy activation leading to decreased clearance of oxidatively damaged mitochondria. Our results on compromised mitophagy in LHON cybrids are in agreement with a recently published report where a missense mutation in ND5 gene (m.12338 T > C) resulted in complex I dysfunction and decrease in mitophagy linked to increased cell death (50). As a rescue approach for LHON, previously, it was reported that prolong rapamycin treatment in cybrids inhibits mTOR pathway and selectively eliminate mutant mitochondria (with heteroplasmic LHON-ND4 mutation) (51). To further test the selectivity of mitophagy activation with rapamycin treatment, we utilized probes for both functional mitochondria (MTR) and oxidatively stressed mitochondria (MitoSox) separately with lysosomal probe LTG (Fig. 5). Our study utilized homoplasmic mutations for ND4 as well as ND1 and demonstrated further that autophagy activation not only through mTOR inhibition but also mTOR-independent mechanisms could also improve cell survival (Fig. 6 and Supplementary Material, Fig. S1). Together with the measurement of ROS production and membrane potential in LHON cybrids after treatment of rapamycin (Fig. 7A and B), it is quite likely that selective mitophagy contributes to cell survival in LHON cybrids with rapamycin treatment.
Conclusions
Overall, our results revealed novel insights on LHON pathogenesis that autophagy activation could serve as a protective mechanism for LHON survival through clearing damaged mitochondria (Fig. 8). Since LHON shows incomplete penetrance with the primary mtDNA mutations identified, our study also suggests that mitophagy mechanisms could serve as the modifiers for development of LHON phenotypes. Finally, mitophagy activation could serve as a potential therapeutic strategy for LHON treatment.
Figure 8.

Schematic diagram showing the proposed mechanism of LHON pathogenesis including the major conclusions of the current study (highlighted in green color). A potential rescue approach of activating mitophagy leading to clearance of damaged mitochondria and restoring mitochondrial functions and cell survival is also shown.
Materials and Methods
Cell lines and culture conditions
KB30 was used as a WT cybrid, generated after repopulating with healthy mitochondria in an mtDNA-less line ρ°206 derived from 143B cells 143B (ATCC CRL 8303) (52). Similarly, Le131 cybrid was obtained by transferring mitochondria from cultures of skin fibroblast established from a LHON patient carrying homoplasmic ND4 (G11778A) mutation in mtDNA into an mtDNA-less line ρ°206 derived from 143B cell (52). The other ND cybrids for ND4 (G11778A), HFF3, and for ND1 (G3460A) mutations, RJ206 and HMM5, were similar as described previously (9). The WT and cybrid cell lines were maintained in Dulbecco’s Modified Essential medium (DMEM) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin and 0.1 mg/ml bromodeoxyuridine at 37°C and 5% CO2 in humidified incubator. Early passage cybrids were used for all the studies and checked at regular intervals for the presence of homoplasmic ND mutations in different cybrids by sequencing. For the galactose experiments, 4 × 105 cells/cm2 were seeded in DMEM with the above-mentioned supplementation, and, after 24 h, the culture medium was replaced with appropriate media (glucose: DMEM, which contain 4.5 mg glucose/ml and 0.11 mg of pyruvate/l; galactose: DMEM lacking glucose and containing 0.9 mg of galactose/ml and 0.5 mg of pyruvate/l, both supplemented with 10% dialyzed fetal bovine serum: FBS).
Measurement of autophagy by fluorescence imaging
Lysosomal imaging
Control and LHON cybrids (4 × 105 cells/cm2) were seeded separately on glass coverslips in 24-well plates (Goldseal No. 1; Thermo Fisher Scientific, Waltham, MA, USA) for 24–48 h in DMEM containing 10% FBS followed by incubation in appropriate media for different time points. Cells were stained by adding 500 nM of Lysotracker Red DND-99 (Molecular Probes, Inc. Eugene, OR, USA) in the media for 15 min and washed to remove unstained/unbound dye before fixation. Cell permeable nuclear staining dye Hoechst 33258 (10 μg/ml) (Sigma St. Louis, MO, USA) was added at the same time in the cells for staining nuclei. The cells were washed in phosphate buffered saline (PBS) and fixed by the addition of 3.7% paraformaldehyde in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2; pH 7.00) for 20 min at room temperature (RT). Cells were washed with PBS and then with water. The coverslips were mounted with Vectashield mounting medium and sealed. Approximately, 50–100 cells per coverslip were visualized. Cells labeled with LTR and Hoechst 33258 were imaged using standard filter sets in Nikon Eclipse TE2000 fluorescent microscope. LTR dye has excitation/emission maxima of ∼577/590 nm; Hoechst 33342 dye has excitation/emission maxima of ∼350/461 nm. NIH ImageJ software was used to quantitate the total number of lysosomes (represented by red color) normalized by total number of nuclei (represented by blue color) to determine autophagy.
Immunoblotting
Whole-cell protein extracts (protein lysates) were prepared using RIPA lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA and protease). Protein estimation was performed using Bicinchoninic acid (BCA) kit. Equal amounts of protein from the total cell lysates (22–50 μg/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions. The resolved proteins were transferred electrophoretically to polyvinylidene difluoride (PVDF) membrane. The membrane were blocked for non-specific binding by incubating with 5% non-fat dry milk or 2% BSA depending upon antibody in tris-buffered saline-tween 20 (TBST) [150 mM NaCl, 50 mM Tris–HCl (pH 7.4), 0.05% Tween 20] for at least 1 h at RT with gentle shaking. After washing with TBST, the membrane was further incubated with primary antibodies Beclin1, LC3B, β-actin, VDAC, GAPDH, phospho-mTOR and total mTOR, (Cell Signaling Technology, Inc. Danvers, MA) and mitochondrial respiratory chain subunit proteins C-II-70, ATPase-α, C-III core-2, COX-1, NDUFA-9 (Mitoscience, Eugene, Oregon) overnight at 4°C with gentle shaking followed by washing and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000 dilution) for 1 at RT. Protein bands were detected using super signal west pico chemi-luminescent substrate kit (Pierce, Thermo Fisher Scientific, Rockford, IL, USA) and developed on Hyperfilm-ECL (Amersham, GE Healthcare Life Sciences, USA). Band intensity was determined by performing densitometry analysis using ImageJ software.
Mitochondrial mass measurements
Approximately, 10 000 cells/well were seeded in 96-well plates in glucose media for 24 h followed by replacement with glucose/galactose media for the next 24 h. Further, mitochondria were stained with 250 nM MTR for 15 min (Molecular Probes, Inc. Eugene, OR, USA) and Hoechst 33258 (10 μg/ml). Cells were washed and fluorescence was recorded in Synergy™ HT Multi-Detection Microplate Reader (Bio-Tek Instruments, Inc. Winooski, VT, USA). MTR has excitation/emission maxima of ∼577/590 nm; Hoechst 33258 dye has excitation/emission maxima of ∼350/461 nm. Values of MTR TM fluorescence were normalized with Hoechst-33258 fluorescence.
Cell viability analysis
Approximately, 1x105 cells were seeded on 6-well plates in 2 ml of DMEM and cells were cultured at 37°C for 24 h. The medium was changed the next day with either regular growth media or with galactose media (containing 0.9 mg of galactose/ml and 0.5 mg of pyruvate/l, with 10% dialyzed FBS). Cell counting and viability was performed using cell viability analyzer (Beckman Coulter, Inc. Fullerton, CA) based on trypan blue exclusion method. Bright field images were acquired using Nikon Eclipse TE2000 (Nikon Instruments Inc. Melville, NY). Cell death was calculated as a percent of dead cells in the total cell population. Experiments were performed in triplicates and repeated at least three times for reproducible results.
Mitochondrial functional analysis
Mitochondrial isolation
Mitochondria from cultured cells were isolated following previously published protocol (53). Briefly, 2x107 cells were harvested by trypsinization, pelleted at 600xg for 5 min and washed twice with cold isotonic medium. The cell pellets were disrupted by adding one volume of hypotonic homogenization buffer and homogenized on ice with at least 50–100 strokes using a dounce homogenizer. Immediately after, 1/10 of the packed cell volume of hypertonic buffer was added to make the medium isotonic. This homogenate was centrifuged at 1200g for 3 min at 4 C to pellet unbroken cells, debris and nuclei. After the centrifugation, the supernatant was collected and centrifuged again at low speed in the same conditions (1200g for 3 min at 4 C), to ensure a complete elimination of heavy contaminants. The mitochondria contained in the supernatant(s) are pelleted by centrifugation at 15 000g during 2 min at 4 C. The pellets were washed using homogenization buffer A and final mitochondrial pellet was resuspended in the same buffer and kept on ice until used. Mitochondrial protein was estimated using BCA kit (Pierce).
MMP measurements
Basal level of MPP was determined using the cationic fluorescent redistribution dye TMRM (Invitrogen) with lower quenching mode. Freshly isolated mitochondria (up to 10–20 μg in 100 μl volume) were incubated (three replicates for each line) with 200 nM TMRM in homogenization buffer A for 15–20 min in a 37 C CO2 incubator. After incubation, mitochondria were pelleted at 15 000g for 2 min. These mitochondrial pellets were washed three times with homogenization buffer, resuspended in 100 μl of homogenization buffer and transferred to a black 96-well micro-titer plate. Fluorescence was recorded for TMRM (excitation/emission: 540/ 575 nm) in HT-BioTek fluorescence plate reader (BioTek Instrument Inc., Winooski, VT, USA). TMRM fluorescence was normalized with protein concentration (mg/ml), and fold changes were calculated for comparisons.
ROS measurements
ROS measurements in isolated mitochondria were performed using Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen). This kit contains a sensitive, one-step assay that uses the substrate AmplexR Red reagent (10-acetyl-3,7-dihydroxyphenoxazine) to detect hydrogen peroxide (H2O2) or peroxidase activity. In the presence of peroxidase, the Amplex® Red reagent reacts with H2O2 (present in biological sample) in a 1:1 stoichiometry to produce the red-fluorescent oxidation product, resorufin. Resorufin has excitation and emission maxima of ∼571 nm and 585 nm, respectively. The AmplexR Red reagent, in combination with HRP, has been used to detect H2O2 present or released from biological samples including cells or from isolated mitochondria. Mitochondria (up to 10–20 μg in 50 μl volume) were added to 96-well plate in triplicates with a total reaction volume of 100 ml of reaction buffer containing 0.1 U/ml HRP, 50 mM Amplex Red reagents. Fluorescence was measured using fluorescence plate reader with temperature maintained at RT. Relative levels of mitochondrial ROS were plotted after normalization with mitochondrial protein for each sample and presented as fold change to WT.
ATP measurements
Total ATP levels were determined by a bioluminescence assay using an ATP determination kit from Invitrogen/Molecular Probes, according to the instructions of the manufacturer. Briefly, cells were grown in a 100 mm dish to ∼40% confluency and then harvested. About 2 × 106 cells were washed with PBS buffer, resuspended in boiling Buffer (100 mM Tris, 4 mM EDTA) for 5 min, and then centrifuged at 10 000g for 1 min. For luciferase assays, a cell suspension was added to the standard reaction solution and luminescence was measured in a Synergy™ HT Multi-Detection Micro-plate Reader (Bio-Tek Instruments, Inc.). The standard curve was constructed by measuring luminescence derived from ATP solutions. Whole-cell ATP content was determined fluorescence units per milligram protein content and calculated as fold change to WT cybrid.
Mitophagy measurements
For measuring overall mitophagy levels, cells in different media were grown on coverslip and stained with combination of 500 nM LTG (Molecular Probes, Inc. Eugene, OR, USA) for visualizing lysosomes and 250 nM MTR (for visualizing mitochondria) and 10 μg/ml Hoechst (for nuclei) for 15 min at 37 C in 5% CO2 incubator. Following incubations, cells were washed in PBS to remove any dye, fixed and cells were imaged under confocal microscope LSM (Olympus FV1000). For measuring mitophagy of oxidative stress mitochondria, mitochondrial superoxide indicator Mitosox (Molecular Probes, Inc. Eugene, OR, USA) at 5 μM were used in combination with 500 nM LTG (for visualizing lysosomes) and Hoechst-33258 (10 μg/ml) for measuring total number of nuclei. The cells were then fixed and imaged to identify co-localization of ROS accumulating mitochondria with lysosomes to detect mitophagy. Approximately, 50–100 cells per coverslip were visualized, and images were digitally captured and scored blindly. Each experiment was repeated at least three times with similar observations.
Data analysis
Data in graphs are reported as ±S.E. and depict the average of at least three independent experiments. Morphological images are representative of at least three independent experiments with similar results. Statistical analysis was performed using GraphPad software (GraphPad software, La Jolla, CA) with Student’s paired t-test or a one-way analysis of variance followed by a Dunnett’s Multiple Comparison Test when appropriate. (P < 0.05 was considered significant).
Supplementary Material
Acknowledgements
We would like to thank Drs Andrea Martinuzzi and Valerio Carelli for providing LHON cybrids.
Conflict of Interest statement. None declared.
Funding
National Institute of Health (R01 GM109434 and GM130129); William and Ella Owens Medical Research Foundation; Science and Engineering Research Board, Government of India’s scheme for Young Scientist grant (SB/YS/LS-95/2013 to L.K.S.); University Grant Commission Faculty Recharge Programme Start-up grant [N. F.4-5(102-FRP)/2014(BSR) to L.K.S.].
References
- 1. Taylor R.W. and Turnbull D.M. (2005) Mitochondrial DNA mutations in human disease. Nat. Rev. Genet., 6, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koilkonda R.D. and Guy J. (2011) Leber's hereditary optic neuropathy-gene therapy: from benchtop to bedside. J. Ophthalmol., 2011, 179412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huoponen K., Vilkki J., Aula P., Nikoskelainen E.K. and Savontaus M.L. (1991) A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am. J. Hum. Genet., 48, 1147–1153. [PMC free article] [PubMed] [Google Scholar]
- 4. Johns D.R., Neufeld M.J. and Park R.D. (1992) An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun., 187, 1551–1557. [DOI] [PubMed] [Google Scholar]
- 5. Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas L.J. 2nd and Nikoskelainen E.K. (1988) Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science, 242, 1427–1430. [DOI] [PubMed] [Google Scholar]
- 6. Kirches E. (2011) LHON: mitochondrial mutations and more. Curr. Genomics, 12, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown M.D., Trounce I.A., Jun A.S., Allen J.C. and Wallace D.C. (2000) Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber's hereditary optic neuropathy mitochondrial DNA mutation. J. Biol. Chem., 275, 39831–39836. [DOI] [PubMed] [Google Scholar]
- 8. Danielson S.R., Wong A., Carelli V., Martinuzzi A., Schapira A.H. and Cortopassi G.A. (2002) Cells bearing mutations causing Leber's hereditary optic neuropathy are sensitized to Fas-Induced apoptosis. J. Biol. Chem., 277, 5810–5815. [DOI] [PubMed] [Google Scholar]
- 9. Ghelli A., Zanna C., Porcelli A.M., Schapira A.H., Martinuzzi A., Carelli V. and Rugolo M. (2003) Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J. Biol. Chem., 278, 4145–4150. [DOI] [PubMed] [Google Scholar]
- 10. Jun A.S., Trounce I.A., Brown M.D., Shoffner J.M. and Wallace D.C. (1996) Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystonia. Mol. Cell Biol., 16, 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Floreani M., Napoli E., Martinuzzi A., Pantano G., De Riva V., Trevisan R., Bisetto E., Valente L., Carelli V. and Dabbeni-Sala F. (2005) Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber's hereditary optic neuropathy. FEBS J., 272, 1124–1135. [DOI] [PubMed] [Google Scholar]
- 12. Lin C.S., Sharpley M.S., Fan W., Waymire K.G., Sadun A.A., Carelli V., Ross-Cisneros F.N., Baciu P., Sung E., McManus M.J. et al. (2012) Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. U S A, 109, 20065–20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Damme M., Suntio T., Saftig P. and Eskelinen E.L. (2015) Autophagy in neuronal cells: general principles and physiological and pathological functions. Acta Neuropathol., 129, 337–362. [DOI] [PubMed] [Google Scholar]
- 14. Yue Z., Friedman L., Komatsu M. and Tanaka K. (2009) The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim. Biophys. Acta, 1793, 1496–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rubinsztein D.C., Codogno P. and Levine B. (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov., 11, 709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rubinsztein D.C., Bento C.F. and Deretic V. (2015) Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med., 212, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scrivo A., Bourdenx M., Pampliega O. and Cuervo A.M. (2018) Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol., 17, 802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pickles S., Vigie P. and Youle R.J. (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol., 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J., Culp M.L., Craver J.G. and Darley-Usmar V. (2018) Mitochondrial function and autophagy: integrating proteotoxic, redox, and metabolic stress in Parkinson's disease. J. Neurochem., 144, 691–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zanna C., Ghelli A., Porcelli A.M., Carelli V., Martinuzzi A. and Rugolo M. (2003) Apoptotic cell death of cybrid cells bearing Leber's hereditary optic neuropathy mutations is caspase independent. Ann. N. Y. Acad. Sci., 1010, 213–217. [DOI] [PubMed] [Google Scholar]
- 21. Zanna C., Ghelli A., Porcelli A.M., Martinuzzi A., Carelli V. and Rugolo M. (2005) Caspase-independent death of Leber's hereditary optic neuropathy cybrids is driven by energetic failure and mediated by AIF and Endonuclease G. Apoptosis, 10, 997–1007. [DOI] [PubMed] [Google Scholar]
- 22. He C. and Levine B. (2010) The Beclin 1 interactome. Curr. Opin. Cell Biol., 22, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao Y. and Klionsky D.J. (2007) Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res., 17, 839–849. [DOI] [PubMed] [Google Scholar]
- 24. Nakatogawa H., Ichimura Y. and Ohsumi Y. (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell, 130, 165–178. [DOI] [PubMed] [Google Scholar]
- 25. Mizushima N. and Yoshimori T. (2007) How to interpret LC3 immunoblotting. Autophagy, 3, 542–545. [DOI] [PubMed] [Google Scholar]
- 26. Barth S., Glick D. and Macleod K.F. (2010) Autophagy: assays and artifacts. J. Pathol., 221, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baracca A., Solaini G., Sgarbi G., Lenaz G., Baruzzi A., Schapira A.H., Martinuzzi A. and Carelli V. (2005) Severe impairment of complex I-driven adenosine triphosphate synthesis in leber hereditary optic neuropathy cybrids. Arch. Neurol., 62, 730–736. [DOI] [PubMed] [Google Scholar]
- 28. Barrientos A. and Moraes C.T. (1999) Titrating the effects of mitochondrial complex I impairment in the cell physiology. J. Biol. Chem., 274, 16188–16197. [DOI] [PubMed] [Google Scholar]
- 29. Clay Montier L.L., Deng J.J. and Bai Y. (2009) Number matters: control of mammalian mitochondrial DNA copy number. J. Genet. Genomics, 36, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Codogno P. and Meijer A.J. (2005) Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ., 12 (Suppl. 2), 1509–1518. [DOI] [PubMed] [Google Scholar]
- 31. Yamamoto A., Tagawa Y., Yoshimori T., Moriyama Y., Masaki R. and Tashiro Y. (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct., 23, 33–42. [DOI] [PubMed] [Google Scholar]
- 32. Sarkar S., Davies J.E., Huang Z., Tunnacliffe A. and Rubinsztein D.C. (2007) Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem., 282, 5641–5652. [DOI] [PubMed] [Google Scholar]
- 33. Carelli V., Rugolo M., Sgarbi G., Ghelli A., Zanna C., Baracca A., Lenaz G., Napoli E., Martinuzzi A. and Solaini G. (2004) Bioenergetics shapes cellular death pathways in Leber's hereditary optic neuropathy: a model of mitochondrial neurodegeneration. Biochim. Biophys. Acta, 1658, 172–179. [DOI] [PubMed] [Google Scholar]
- 34. Meyerson C., Van Stavern G. and McClelland C. (2015) Leber hereditary optic neuropathy: current perspectives. Clin. Ophthalmol., 9, 1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu-Wai-Man P., Votruba M., Moore A.T. and Chinnery P.F. (2014) Treatment strategies for inherited optic neuropathies: past, present and future. Eye (Lond), 28, 521–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G. et al. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J., 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seppet E., Gruno M., Peetsalu A., Gizatullina Z., Nguyen H.P., Vielhaber S., Wussling M.H., Trumbeckaite S., Arandarcikaite O., Jerzembeck D. et al. (2009) Mitochondria and energetic depression in cell pathophysiology. Int. J. Mol. Sci., 10, 2252–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mandemakers W., Morais V.A. and De Strooper B. (2007) A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J. Cell Sci., 120, 1707–1716. [DOI] [PubMed] [Google Scholar]
- 39. Gellerich F.N., Trumbeckaite S., Muller T., Deschauer M., Chen Y., Gizatullina Z. and Zierz S. (2004) Energetic depression caused by mitochondrial dysfunction. Mol. Cell Biochem., 256–257, 391–405. [DOI] [PubMed] [Google Scholar]
- 40. Beretta S., Mattavelli L., Sala G., Tremolizzo L., Schapira A.H., Martinuzzi A., Carelli V. and Ferrarese C. (2004) Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines. Brain, 127, 2183–2192. [DOI] [PubMed] [Google Scholar]
- 41. Robinson B.H., Petrova-Benedict R., Buncic J.R. and Wallace D.C. (1992) Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol., 48, 122–126. [DOI] [PubMed] [Google Scholar]
- 42. Shimizu S., Kanaseki T., Mizushima N., Mizuta T., Arakawa-Kobayashi S., Thompson C.B. and Tsujimoto Y. (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol., 6, 1221–1228. [DOI] [PubMed] [Google Scholar]
- 43. Tiwari M., Lopez-Cruzan M., Morgan W.W. and Herman B. (2011) Loss of caspase-2-dependent apoptosis induces autophagy after mitochondrial oxidative stress in primary cultures of young adult cortical neurons. J. Biol. Chem., 286, 8493–8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tiwari M., Sharma L.K., Vanegas D., Callaway D.A., Bai Y., Lechleiter J.D. and Herman B. (2014) A nonapoptotic role for CASP2/caspase 2: modulation of autophagy. Autophagy, 10, 1054–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harris H. and Rubinsztein D.C. (2011) Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol., 8, 108–117. [DOI] [PubMed] [Google Scholar]
- 46. Arduino D.M., Esteves A.R., Cortes L., Silva D.F., Patel B., Grazina M., Swerdlow R.H., Oliveira C.R. and Cardoso S.M. (2012) Mitochondrial metabolism in Parkinson's disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum. Mol. Genet., 21, 4680–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu J.J., Quijano C., Chen E., Liu H., Cao L., Fergusson M.M., Rovira I.I., Gutkind S., Daniels M.P., Komatsu M. et al. (2009) Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY), 1, 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luo C., Li Y., Wang H., Feng Z., Li Y., Long J. and Liu J. (2013) Mitochondrial accumulation under oxidative stress is due to defects in autophagy. J. Cell Biochem., 114, 212–219. [DOI] [PubMed] [Google Scholar]
- 49. Cotan D., Cordero M.D., Garrido-Maraver J., Oropesa-Avila M., Rodriguez-Hernandez A., Gomez Izquierdo L., De la Mata M., De Miguel M., Lorite J.B., Infante E.R. et al. (2011) Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J., 25, 2669–2687. [DOI] [PubMed] [Google Scholar]
- 50. Zhang J., Ji Y., Lu Y., Fu R., Xu M., Liu X. and Guan M.X. (2018) Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T > C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum. Mol. Genet., 27, 1999–2011. [DOI] [PubMed] [Google Scholar]
- 51. Dai Y., Zheng K., Clark J., Swerdlow R.H., Pulst S.M., Sutton J.P., Shinobu L.A. and Simon D.K. (2014) Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum. Mol. Genet., 23, 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hofhaus G., Johns D.R., Hurko O., Attardi G. and Chomyn A. (1996) Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber's hereditary optic neuropathy. J. Biol. Chem., 271, 13155–13161. [DOI] [PubMed] [Google Scholar]
- 53. Fernandez-Vizarra E., Lopez-Perez M.J. and Enriquez J.A. (2002) Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods, 26, 292–297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.