

Abstract
The 16p11.2 BP4-BP5 deletion and duplication syndromes are associated with a complex spectrum of neurodevelopmental phenotypes that includes developmental delay and autism spectrum disorder, with a reciprocal effect on head circumference, brain structure and body mass index. Mouse models of the 16p11.2 copy number variant have recapitulated some of the patient phenotypes, while studies in flies and zebrafish have uncovered several candidate contributory genes within the region, as well as complex genetic interactions. We evaluated one of these loci, KCTD13, by modeling haploinsufficiency and complete knockout in mice. In contrast to the zebrafish model, and in agreement with recent data, we found normal brain structure in heterozygous and homozygous mutants. However, recapitulating previously observed genetic interactions, we discovered sex-specific brain volumetric alterations in double heterozygous Kctd13xMvp and Kctd13xLat mice. Behavioral testing revealed a significant deficit in novel object recognition, novel location recognition and social transmission of food preference in Kctd13 mutants. These phenotypes were concomitant with a reduction in density of mature spines in the hippocampus, but potentially independent of RhoA abundance, which was unperturbed postnatally in our mutants. Furthermore, transcriptome analyses from cortex and hippocampus highlighted the dysregulation of pathways important in neurodevelopment, the most significant of which was synaptic formation. Together, these data suggest that KCTD13 contributes to the neurocognitive aspects of patients with the BP4-BP5 deletion, likely through genetic interactions with other loci.
Introduction
During the past decade, several neurodevelopmental disorders (NDDs) have been associated with copy number variants (CNVs) (1,2). One of the most common of these lies on 16p11.2 and comprises a ~600 kb genomic segment flanked by two low-copy repeats that also define its most common breakpoints (BP4-BP5) (3). The BP4-BP5 deletion and duplication have a combined population prevalence of ~1/1000 (4) and contribute to as much as ~1% of the genetic burden of intellectual disability (1) and autism spectrum disorder (ASD) (5–9). The 16p11.2 BP4-BP5 dosage imbalances have also been associated with epilepsy (10–13), schizophrenia (SZ), bipolar disorder and depression (14–16). Additionally, 16p11.2 CNVs have been reported to produce reciprocal changes in body mass index (BMI) and head size (4,11,12,17,18). Gray matter volume and specific cortico-subcortical regions implicated in reward processes, language and social cognition are similarly correlated with the number of copies of the 600 kb region (19).
The BP4-BP5 region contains –32 known and predicted protein-coding genes (20). The high gene density of the region and the phenotypic heterogeneity of human patients (7,11,21) have rendered the study of this CNV challenging. To study the contribution of dosage-sensitive loci within the CNV and to probe the mechanistic basis of the human genetic data, animal models have been generated and characterized. In mice, the BP4-BP5 syntenic region lies on chromosome 7F3 and contains 31 protein-coding genes (based on GRCm38.83). To date, three 16p11.2 deletion (Del/+) and two duplication (Dup/+) mouse models have been generated (22–24). Del/+ mice display hyperactivity, repetitive behaviors and recognition memory deficits, while Dup/+ mice show hypoactivity and superior short-term memory. We showed previously a genetic background effect on the social interaction phenotypes of the 16p11.2 CNV mouse models, wherein on a C57BL/6NxC3B hybrid background, both Del/+ and Dup/+ mice display social interaction deficits (22), reminiscent of the ASD phenotypes of patients.
Using the zebrafish embryo, we have also reported that overexpression of human KCTD13 (Potassium Channel Tetramerization Domain containing 13) can induce microcephaly, whereas suppression of the zebrafish ortholog yields a macrocephalic phenotype, potentially mimicking the phenotypes seen in 16p11.2 CNV carriers (25). More recently, transcription-wide association studies suggested that the expression quantitative trait loci in MAPK3 might be a driver of that signal, in part through the control of KCTD13 expression (26). KCTD13 encodes a protein that act as an adaptor for interactions between the Cul3 ubiquitin ligase and its substrates (27,28). KCTD13 has been shown to regulate ubiquitination and degradation of the Ras homolog gene family member A (RhoA) which may, in turn, lead to disrupted cell migration and influence brain size during prenatal development (29). Recently, Escamilla et al. (30) reported that gene editing of Kctd13 does not increase brain size or neurogenesis in mice or zebrafish, respectively. However, Kctd13 deletion in the mouse induced an increased RhoA levels, deficits in spine densities on CA1 pyramidal neurons and reduced synaptic transmission in the hippocampus (30).
To evaluate further the potential contributory role of KCTD13 in the 16p11.2 CNV syndromes, we generated a global deletion of murine Kctd13. In agreement with published results, we did not observe brain structure anomalies in Kctd13 mutants. However, testing for epistasis with two previously proposed interaction candidates, Mvp and Lat, showed double mutants to exhibit sex-dependent volumetric brain changes that might be relevant to the gender bias observed in the phenotype of the human lesion (31,32). Moreover, we found a deficiency of spine maturation in the dendrites of pyramidal neurons of the CA1 hippocampus in both Kctd13 heterozygous (Het) and homozygous (Hom) mutants, as well as an impairment in short-term memory, a phenotype that was not seen in the Escamilla et al (30) mutant but was reported in mouse models for the 16p11.2 BP4-BP5 deletion (22). To probe the possible mechanisms underscoring these phenotypes, we performed transcriptome profiling using RNA sequencing (RNAseq). We found that loss of Kctd13 induced significant expression changes of genes involved in synaptic transmission. Finally, we evaluated RhoA protein levels in cortex and hippocampus. We found no evidence for dysregulation at 3 or 15 weeks of age, which suggests that the spine maturation deficit in Kctd13 mutants might be independent of RhoA/ROCK signaling. Together, our observations suggest that KCTD13 is a potential contributor to the complex neurodevelopmental phenotypes associated with 16p11.2 BP4-BP5 deletion, likely through genetic interaction with other transcripts both within and outside the BP4-BP5 region.
Results
Generation of Kctd13 Het and Hom mice
To generate Kctd13-deficient mice, we introduced LoxP sites flanking exon 2 (Fig. 1A), predicted to induce a frameshift and a premature stop codon in exon 4. We crossed Kctd13-floxed mice with mice expressing Cre recombinase under the cytomegalovirus (CMV) ubiquitous promoter. Compared to wild-type (wt) littermates, the segregation of the Kctd13 mutant allele was not distorted, suggesting no prenatal lethality (Fig. 1B; Supplementary Material, Table S1). We confirmed the excision of exon 2 by polymerase chain reaction (PCR) (Fig. 1C) and we also examined Kctd13 protein levels by western blot at postnatal day 1 (P1; Fig. 1D). Compared to wt littermates, Kctd13 protein levels were decreased approximately by 40% in Het and by 95% in Hom animals, indicative of efficient excision and loss of functional transcript. We also recorded body weights of mice between weeks 5–14; Het and Hom animals did not show any alterations compared to wt (Fig. 1E), suggesting that Kctd13 deficiency does not affect gross postnatal growth and viability.
Figure 1.
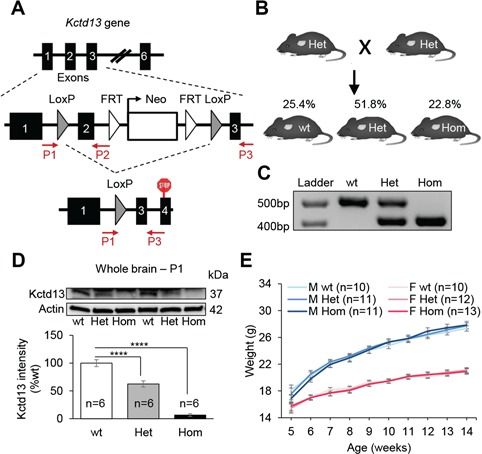
Kctd13 deficiency does not affect postnatal growth and viability. (A)Kctd13 deletion targeting strategy. We introduced LoxP sites flanking exon 2 of Kctd13 gene. Animals haploinsufficient for Kctd13 (Het) were obtained by crossing floxed mice with animals expressing the Cre under the CMV promoter, allowing the deletion of targeted exon in all tissues. (B) Mouse mating strategy used to generate mouse cohorts and ratios per genotype observed. (C) Molecular validation by PCR. (D) Western blots against Kctd13 and β-actin. Whole brains from P1 mice were used as samples. (E) Evolution of the body weight (g). Data are represented as the mean ± s.e.m., ****P < 0.0001 versus wt. Tukey’s test applied following a significant one-way ANOVA.
Kctd13 mutant mice are deficient in short-term recognition memory
As a first analysis for possible consequences of Kctd13 loss in neuronal function, we performed a battery of behavioral studies. In a first cohort of wt, Het and Hom male mice (9–10 mice per genotype), all three genotypes engaged in similar levels of exploratory activity, circadian activity, anxiety- and repetitive-like responses, locomotor coordination, social behaviors and long-term learning and memory (Supplementary Material, Table S2). In contrast, we found significant short-term memory deficits in both Het and Hom animals in the novel object recognition memory task (one-way ANOVA for genotype: F(2,26) = 9.37, P = 0.0009; Tukey’s post hoc tests: Het versus wt: P = 0.006, Hom versus wt: P = 0.001) and in Hom animals in the novel location recognition memory task (F(2,20) = 4.55, P = 0.02; Het versus wt: P = 0.1, Hom versus wt: P = 0.03) with a 1 h delay. The specificity of these memory deficits was confirmed, as no genotype distinctions could be detected in the total times the three genotypes mice spent interacting with objects during training or test sessions (Supplementary Material, Table S2).
To replicate our observations, we tested a second cohort, while at the same time increasing the numbers of male and female mice (n = 20–24 animals per genotype, 50:50 sex ratio). We first evaluated the performances of males and females separately. As no significant sex differences were noted, we collapsed these data across both sexes. In the open field test, we once again observed no significant differences between genotypes in locomotor activity; rearing behavior or time spent in the center of the arena were observed (Supplementary Material, Table S3). To test whether social behavior was perturbated, we evaluated responses in the social affiliation (33) and social preference tests (22). All three genotypes responded similarly in the social tests where they preferred the social over the non-social stimulus in the social affiliation and the novel social over the familiar social stimulus in the social preference test (Supplementary Material, Table S3). Aside from the social affiliation test, we also examined social behaviors by placing two unfamiliar animals of similar genotypes into the open field and did not observe a difference in interaction time between genotypes (Supplementary Material, Table S3).
Next, we evaluated mice in the novel object recognition memory test, a common assay for assessing short-term memory of rodents (34). We first investigated whether Het and Hom mice could discriminate a novel object from a previously explored object after a retention delay of 1 h (Fig. 2A). During both acquisition session or session 1, and test session or session 2, mice of all genotypes spent an equal amount of time exploring the sample object (Fig. 2B). In the test session, Het and Hom mice displayed a significant memory impairment compared with wt (F(2,59) = 14.35, P < 0.0001; Het versus wt: P < 0.0001, Hom versus wt: P < 0.0001; Fig. 2C). We also compared the discrimination index of animals with the level of chance (50%), whereas wt animals differentiated objects (one-sample t-test: t(1,15) = 8.29, P < 0.0001), Het (t(1,21) = 0.69, P = 0.5) and Hom (t(1,23) = 0.45, P = 0.6) mice were not able to discriminate the novel and the familiar objects.
Figure 2.
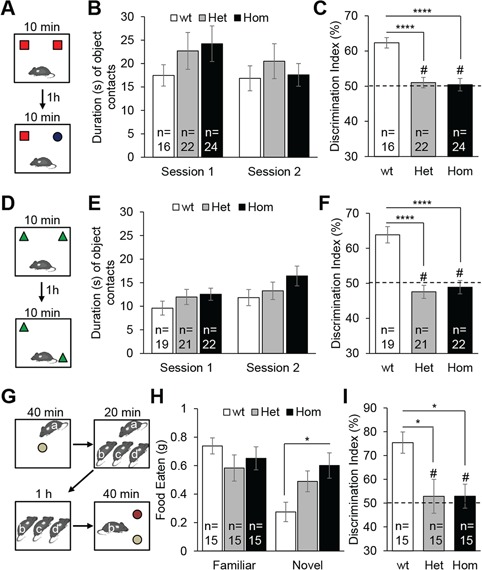
Kctd13 mutant mice show deficiency in short-term memory. (A) Experiment design of novel object recognition test consisting in two 10 min sessions with a delay of 1 h between sessions 1 and 2. (B) Exploration time (s) of the two familiar objects in session 1 and the familiar and novel objects in session 2. (C) Discrimination index (%) reflects the ability of mice to distinguish the novel object from the familiar object after a 1 h retention delay. The dashed line denotes a chance level of 50%. (D) Experiment design of novel location recognition test. (E) Exploration time (s) of the two familiar objects in session 1 and the unmoved and moved objects in session 2. (F) Discrimination index (%). (G) Experiment design of social transmission of food preference test. Mouse a: demonstrator; mice b, c, d: observers. (H) Quantity of food eaten (g) by the observer mice for the familiar and novel flavored pellets. (I) Discrimination index (%) reflects the ability of the observer mice to distinguish the familiar flavored food from the novel flavored food after a 1 h retention delay. Data are represented as the mean ± s.e.m., # not significant compared to 50%, *P < 0.05 versus wt, ****P < 0.0001 versus wt. Tukey’s test applied following a significant one-way ANOVA.
Given these observations, we next evaluated the memory of animals in the novel location recognition test, in which animals are challenged to discriminate a moved object from an unmoved object (Fig. 2D). All mice spent an equal amount of time exploring the sample object (Fig. 2E). After a retention delay of 1 h, we found that Het and Hom mice displayed a significant memory impairment compared with wt (F(2,59) = 19.07, P < 0.0001; Het versus wt: P < 0.0001, Hom versus wt: P < 0.0001; Fig. 2F). Again, although wt differentiated objects (t(1,18) = 5.97, P < 0.0001), Het (t(1,20) = 1.29, P = 0.2) and Hom (t(1,21) = 0.58, P = 0.6) mice were not able to discriminate the moved and the unmoved objects.
Finally, we evaluated the memory of animals with the social transmission of food preference test (Fig. 2G). In this test, used to assess olfactory memory, the observer mice interact with a demonstrator that has eaten a flavored food. When the observer mice have a choice between the food eaten by the demonstrator and a novel flavored food, they prefer the food eaten by the demonstrator (35). After a delay of 1 h, we observed that Hom animals ate significantly more novel flavored flood than wt animals (F(2,42) = 4.59, P = 0.016; Hom versus wt: P = 0.013; Fig. 2H). The ratio of familiar versus novel flavored food eaten by Het and Hom was significantly lower compared with wt (F(2,42) = 5.28, P = 0.009; Het versus wt: P = 0.02, Hom versus wt: P = 0.02; Fig. 2I). Whereas wt animals showed a significant preference for the familiar flavored food (t(1,14) = 5.71, P < 0.0001), Het (t(1,14) = 0.40, P = 0.69) and Hom (t(1,14) = 0.58, P = 0.57) mice displayed a memory deficit as they were not able to discriminate the familiar and the novel flavored foods. The phenotype was not likely to be driven by an olfactory deficit; we verified the olfactory discrimination of animals after the exposure of non-social odors and found no effect in mutants (Supplementary Material, Fig. S1).
Kctd13 deficit is associated with spine maturation deficit in the hippocampal CA1 region
The hippocampus is a brain structure essential for memory formation. We focused on the dorsal CA1 region of the hippocampus, in which neuronal density renders the assessment of synaptic connectivity experimentally tractable (Fig. 3A). We asked whether there were any genotype-driven changes in spine density and maturation (Fig. 3B) in the basal dendrites of pyramidal neurons using Golgi-Cox stained brain sections (n = 6 animals per genotype, 50:50 sex ratio). We found a significant reduction in the density of dendritic spines in Het and Hom animals compared with wt (F(2,207) = 6.47, P = 0.002; Het versus wt: P = 0.009, Hom versus wt: P = 0.004; Fig. 3C). This reduction in spine density was driven exclusively by a loss of mature mushroom and branched spines (F(2,207) = 29.56, P < 0.0001; Het versus wt: P < 0.0001, Hom versus wt: P < 0.0001; Fig. 3D). In the same coronal sections used to perform hippocampal analysis, we also evaluated spine density and maturation in the retrosplenial cortex (Supplementary Material, Fig. S2A), implicated in visuospatial integration and spatial learning (36). We did not observe aberrant spine density (Supplementary Material, Fig. S2B) or morphology (Supplementary Material, Fig. S2C) in our mutants, indicating that the hippocampal phenotype is region specific; although, this does not preclude potential spine maturation alterations in other brain regions.
Figure 3.
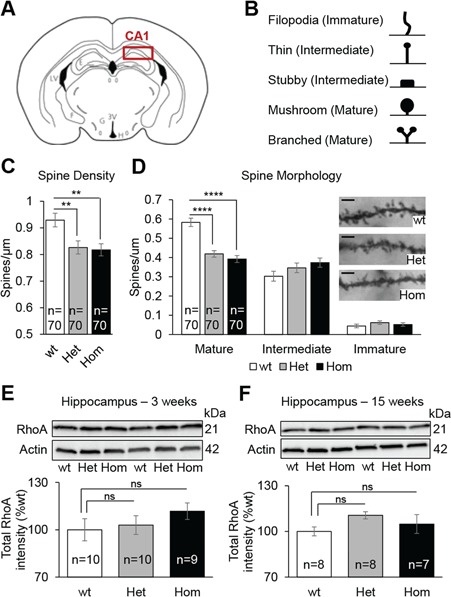
Kctd13 deficiency induce spine maturation deficit in the CA1 region of the hippocampus. (A) Coronal brain diagram indicating the region of interest. (B) Categorization of Golgi-Cox-stained dendritic spine types. Ten micrometer stretches of dendrites were analyzed. (C) Quantification of dendritic spine density of CA1 pyramidal neurons. (D) Representative dendrite stretches (scale bar: 2 μm) and spine morphology of dendritic stretches analyzed. (E and F) Western blots against RhoA and β-actin. Hippocampi from 3- and 15-week-old mice were used as samples. Data are represented as the mean ± s.e.m.; ns, not significant; ****P < 0.0001 versus wt. Tukey’s test applied following a significant one-way ANOVA.
Previous studies have intimated a role for RhoA dysregulation to the observed spine density/morphology phenotype (29). We therefore evaluated the level of RhoA at three different ages: day 1 or P1 (whole brain), 3 weeks (hippocampus) and 15 weeks (hippocampus and cortex). Consistent with prior observations (30), we observed a significant increase of the total RhoA levels at P1, but only in Hom animals (F(2,27) = 4.55, P = 0.02; Het versus wt: P = 0.3, Hom versus wt: P = 0.015; Supplementary Material, Fig. S2D). However, we did not find any differences in RhoA levels during the synaptogenic period at 3 weeks of age in the hippocampus (Fig. 3E) or at 15 weeks of age in either hippocampus (Fig. 3F) or cortex (Supplementary Material, Fig. S2E). These data raise the possibility that spine maturation deficits in Het and Hom animals is independent of RhoA/ROCK. We also tested RhoA protein levels at 15 weeks of age in both the hippocampus and the cortex of mouse models of the 16p11.2 deletion and duplication (23). After confirming a Kctd13 gene dosage in both brain regions (Supplementary Material, Fig. S3A and C), we found that RhoA levels were not perturbed in either region of 16p11.2 deletion models, but were decreased in both regions of 16p11.2 duplication models (Supplementary Material, Fig. S3B and D).
Transcriptome analyses of Kctd13 mutant mice reveals dysregulation of Dctn5, Mapk3 and genes associated with NDDs
Next, to gain insight into some of the pathways that might underscore the behavioral and neuroanatomical deficits of our mutants, we performed transcriptome analyses. We compared Kctd13 Het and Hom mice to matching wt littermates for both cortex and hippocampus brain samples. We first confirmed the ~50% reduction in Kctd13 expression for Het mice and near complete loss of Kctd13 in Hom mice in both tissues (Supplementary Material, Fig. S4). We then performed analyses using DESeq2 (Wald test) with surrogate variables (~SVs) to account for unknown factors. These models fit the expected distribution for Hom mice in cortex and both genotypes in hippocampus. We found significant inflation in Het mice in the cortex data set, and we thus restricted all interpretations of differentially expressed genes (DEGs) to Benjamini Hochberg (BH) adjusted thresholds or DEGs that overlapped between Het and Hom mice.
In cortex, we detected 419 DEGs (adjusted P < 0.05) in Het mice and 116 DEGs in Hom mice, with significant overlap between genotypes (59% of Hom DEGs, Fig. 4A, Supplementary Material, Table S4 and Fig. S5). In the hippocampus, 587 Het DEGs and 113 Hom DEGs were significant (adj. P < 0.05), with 24% overlap between Homs and Hets (Fig. 4B, Supplementary Material, Table S4 and Fig. S6). Further analyses revealed that differences in the number of Het and Hom DEGs became comparable in both tissues at larger fold changes (Supplementary Material, Fig. 7).
Figure 4.
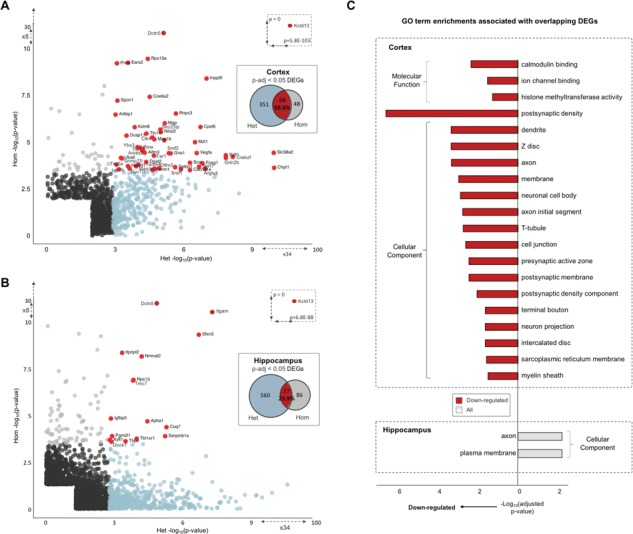
DEGs and associated enriched GO pathways. Cortex (A) and hippocampus (B) significant DEGs. The −log10P-values for significant (P-nominal < 0.05) protein-coding Het DEGs (x-axis) are plotted against Hom DEGs (y-axis). Het DEGs that reach significance at adjusted thresholds (P-adj < 0.05) are colored in blue, and Hom DEGs in gray. Overlapping adjusted significant DEGs across genotype comparisons are colored in red and gene names given. (C) Significant GO term enrichments (P-adj < 0.05) for overlapping Het and Hom DEGs (P-value < 0.05) for cortex and hippocampus. Red shading indicates terms associated with downregulated DEGs, and blue for upregulated terms. GO term categories are given outside of brackets.
As shown in Figure 4, the most significant DEG across tissues and genotypes was Kctd13, while the Gdpd3 alterations are a known consequence of the parental strain background (23,37). Two additional genes in the 16p11.2 CNV region were also significant in cortex of Het mice, upregulation of Fam57b (adj. P = 0.027) and downregulation of Mapk3 (adj. P = 0.019), indicating a potential feedback relationship between the two loci (26). Among the remaining DEGs, the most significant was a downregulation of Dctn5 in both Hom (cortex adj. P = 2.75E−26; hippocampus adj. P = 1.94E−24) and Het mice (cortex adj. P = 1.11E−03; hippocampus adj. P = 1.09E−03). Dctn5 (p25) encodes a subunit of the dynactin/dynein motor complex important for retrograde dendritic transport in neurons and has been associated with bipolar disorder (38–41). Notably, Kctd13 DEGs were also enriched for gene-sets previously associated with ASD and other NDDs such as the Deciphering Developmental Disorders (DDD) study (42) (e.g. DDD gene enrichment cortex Het adj. P = 4.58E−05, nominal P = 5.81E−07, Supplementary Material, Table S5).
Gene ontology (GO) analysis from the restricted set of genes that achieved nominal significance in cortex of both Het and Hom animals (n = 789 genes) showed enrichment for terms corresponding to postsynaptic density, neuronal cell body and projection and calmodulin binding (Supplementary Material, Table S6; Fig. 4C). Overlapping hippocampus Het and Hom DEGs (n = 299 genes) showed enrichment for neuron projection and plasma membrane (Supplementary Material, Table S6; Fig. 4C). When we assessed GO enrichments for each genotype comparison individually (Supplementary Material, Tables S7 and S8), as well as enrichments that were shared across genotypes (Supplementary Material, Tables S9 and S10), we observed a greater number of enriched terms associated with neurodevelopment for hippocampus than cortex. These included myelin sheath, neuronal cell body, axon, dendrite, postsynaptic and presynaptic membrane [e.g. myelin sheath (GO:0043209): hippocampus adj. P = 8.52E−17; cortex adj. P = 5.82E−05; Supplementary Material, Fig. S8].
Finally, we sought evidence for overlap between proximal expression changes associated with reduction of Kctd13 and those altered by deletion of the full 16p11.2 segment. To accomplish this, we compared previously published RNAseq from a 16p11.2 Het deletion cortex mouse model (43), which was re-analyzed here using methods identical to those for the data Kctd13 Het deletion analyses (Methods, Wald Test under design ~SVs + genotype). DEGs identified for 16p11.2 genetic lesion mice (adj. P < 0.05: 629, P < 0.05: 2628 DEGs), overlapped with 54 Kctd13 human ortholog DEGs at adjusted thresholds (15.3%) and 527 genes at nominal levels (25.6%), with a Spearman rank correlation of 0.101 for common DEGs (P < 2.2e-16). We looked for evidence of enrichment over expectations between the two data sets using Fisher’s exact test and calculating Cohen’s kappa coefficient but found no significant differences (overlapping directional DEGs at adjusted P, Cohen's kappa = 0.077; at nominal P, Cohen's kappa = 0.14; Supplementary Material, Table S11). Nominal DEGs common to both data sets (P < 0.05) were enriched for GO terms corresponding to the nucleus synaptobrevin 2-SNAP-25-syntaxin-1a complex and axonal growth cone (adj. P < 0.05) with nervous system-specific genes contributing significantly to the latter two neurodevelopment terms (Supplementary Material, Tables S12 and S13). Phenotypes associated with these common DEGs included long term potentiation (adj. P = 1.8E−05), abnormal nervous system physiology (2.85E−05), abnormal cognition (1.19E−04), abnormal learning, memory and conditioning (1.19E−04) and abnormal CNS synaptic transmission (5.27E−04; Supplementary Material, Table S14).
Double heterozygous Kctd13xMvp and Kctd13xLat mutant mice show sex-specific brain structure alterations
To evaluate the candidacy of KCTD13 in the gross neuroanatomical phenotypes in patients carrying the 16p11.2 BP4-BP5 CNVs, we analyzed brain structures of Kctd13 mutants using magnetic resonance imaging (MRI). Compared with wt littermates, we did not observe any changes in total brain volume or in any of 182 brain regions in Kctd13 Het and Hom animals (Supplementary Material, Table S15 and Fig. S9A). In previous work in zebrafish, we reported a genetic interaction between KCTD13 and MVP (25), and KCTD13 and LAT (44). We wondered whether, in the mouse, such interactions might underscore brain volume regulation that are either too subtle to be recorded in single mutants or require the perturbation of more than one gene. To test this idea, we generated double heterozygous (DblHet) Kctd13xMvp and Kctd13xLat mice. Given the known differences reported in the hippocampus (45) and striatum (24) in 16p11.2 deletion mice, we focused on these two regions. Our high-resolution analysis revealed sex-specific brain structure anomalies in both strains. Specifically, we found smaller hippocampus (4.65% Diff, P = 0.03) and striatum (4.88% Diff, P = 0.02) in DblHet Kctd13xMvp males (Fig. 5A; Supplementary Material, Table S16). Strikingly, the volumes of these regions in mutant females were normal. For the second genetic interaction experiment, we observed the opposite sex-dependent effect: when we evaluated DblHet Kctd13xLat animals, we found a reduction in whole brain and striatum volumes of similar magnitude in females (whole brain: 4.04% Diff, P = 0.02; striatum: 3.50% Diff, P = 0.03; Fig. 5B; Supplementary Material, Table S17 and Fig. S10), while males were normal. Importantly neither Mvp nor Lat single mutants had appreciable brain volumetric changes (Supplementary Material, Fig. S9B and C and Tables S18 and S19). These data support previously observed genetic interactions between Kctd13 and Mvp, and Kctd13 and Lat during zebrafish development (25,44) and intimate possible sex-specific neuroanatomical phenotypes associated with the 16p11.2 BP4-BP5 CNVs.
Figure 5.
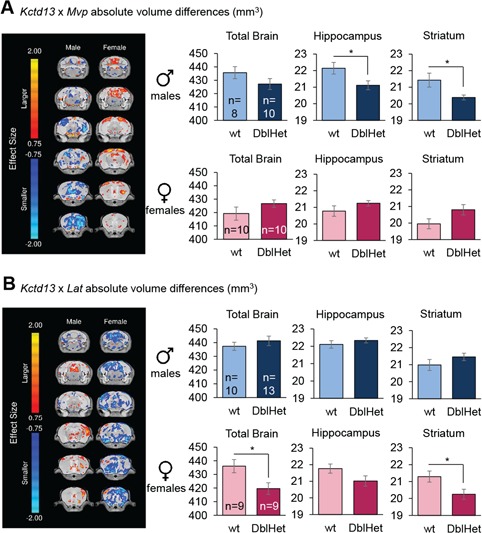
Kctd13xMvp and Kctd13xLat DblHet mutant mice show sex-specific brain structure alterations. (A)Kctd13xMvp Het mice. (B)Kctd13xLat mice. Highlighted are the trends, shown with effect size differences, in absolute volume throughout the brain on six different coronal sections. Bar graphs represent absolute volume on the total brain size, hippocampus and striatum. DblHet, double heterozygous. Data are represented as the mean ± s.e.m., *P < 0.05 versus wt. Student’s t-test.
Discussion
The 16p11.2 BP4-BP5 CNVs are associated with reciprocal effects on head circumference, brain structure and BMI phenotypes. In 2012, we proposed KCTD13 as a significant contributor to mirrored neuroanatomical phenotypes observed in zebrafish for 16p11.2 BP4-BP5 CNVs (25). The injection of human KCTD13 RNA was associated with a 20% decrease of the interocular distance of 4.5 day post fertilization zebrafish larvae whereas the injection of morpholino against the kctd13 zebrafish ortholog was associated with a 20% increase of the same distance. In 2015, similar injections induced a 10% change, showing variability in the phenotypes (20). More recently, Escamilla et al. (30) found that gene editing of kctd13 does not lead to head size alterations in zebrafish. Those discordant observations prompted us to generate and characterize Kctd13 mutant mice. Kctd13 deficiency did not have an impact on postnatal development and weight gain of animals. We also found no detectable brain volume or brain structure anomalies in 10-week-old mutants suggesting that, at least in this model, Kctd13 deficiency is not sufficient to induce appreciable gross neuroanatomical pathologies. This is consistent with human genetic studies, in which KCTD13 has not emerged as significant in cohorts with NDDs. However, studies in zebrafish and Drosophila suggested that genetic interactions between dosage-sensitive genes within the 16p11.2 BP4-BP5 region (25,46–48) might underscore some of the anatomical and behavioral defects of patients and also help explain discordant data. To test this idea, we crossed Kctd13 mutants with genetic interactors established in zebrafish that lie both within the BP4-BP5 region (Mvp) (25) and in the neighboring BP2-BP3 region (Lat) (44). Neither of these mutants, when examined on their own, showed any brain phenotypes at 10 weeks. However, DblHet for either Kctd13 and Mvp, or Kctd13 and Lat, exhibited significant changes that were only detectable when considering sex as a covariate. Specifically, we found bilaterally smaller hippocampus and striatum in Kctd13xMvp DblHet males and a reduction of the total brain volume and bilaterally smaller striatum in Kctd13xLat DblHet females. Future studies will be necessary to dissect the behavioral consequences of these phenotypes. Importantly, male-specific behavioral phenotypes (50,51) and brain structure alterations (52) have also been described in mice carrying a deletion for the whole 16p11.2 BP4-BP5 synthetic region. These data suggest that some of these murine models might represent useful proxies to understand the sex bias reported in patients carrying 16p11.2 BP4-BP5 CNVs (31,32), as well as, more broadly, in NDDs (53–55). We also note that work in human genetics identified in a three-generation pedigree an atypical ~118 kb 16p11.2 deletion encompassing five genes, MVP, CDIPT1, SEZ6L2, ASPHD1 and KCTD13, segregating with ASD (49). We do not know whether patients with the atypical deletion exhibit volumetric brain defects and the family is too small to investigate any sex bias. Nonetheless, based on our present data, as well as prior evidence for interaction between kctd13 and mvp in zebrafish, it will be interesting to evaluate social behaviors of Kctd13xMvp DblHet animals, especially males.
Our studies showed a robust recognition memory deficit phenotype in both Het and Hom mice that we reported previously in mice carrying a deletion for the 16p11.2 BP4-BP5 syntenic region (22) (Supplementary Material, Table S20). Of note, this phenotype was not observed in the recently reported Kctd13 mutant mouse model (30). As the genetic background is similar in both models, we speculate that the different protocols used to assess recognition memory might be responsible for this discrepancy, or that genetic drift has introduced additional variation between the strains. In that regard, it would be useful to understand whether such differences are genetic, as they might illuminate genomic sites that also contribute to phenotypic variability in humans. Finally, given that Escamilla et al. deleted the entire Kctd13 locus whereas we only excised exon 2, it is possible that their larger genetic lesion affects regulatory elements that might be responsible for some of the divergent phenotypes.
Recognition memory is the capacity to judge a previously encountered item as familiar and depends on the integrity of the hippocampus, the adjacent perirhinal cortex and the medial temporal lobe structures (56). We examined basal dendrites in the CA1 where the CA3 Schaeffer collateral axons establish excitatory synaptic connections that are required for learning and memory (57). We found a significant reduction in the density of dendritic spines in the Het and Hom animals driven by a loss of mature mushroom and branched spines. Those data suggest that there are fewer synaptic connections and that the synaptic connections that exist are less mature and weaker, potentially explaining the recognition memory deficit of Kctd13 mutants. Dendritic spine maturation is a dynamic process that requires feedback between the presynaptic and postsynaptic terminals. Given the enrichment of genes associated with axons and presynaptic function emergent from the GO analysis of the hippocampal transcriptome, it is possible that the postsynaptic changes of CA1 dendrites may derive from dysfunction in the CA3 Schaffer collateral axons.
The RhoA/ROCK pathway regulates cytoskeletal reorganization and is associated with various neuronal functions such as migration and neurite development (58). A protein–protein interaction study implicated the physical KCTD13–Cul3 interaction within the inner cortical plate layer in regulating RhoA levels by ubiquitination and potentially influencing brain size and connectivity (29). Consequently, a KCTD13 deficiency should induce an increase of RhoA levels, leading to disrupted cell migration, neurite retraction and spine shrinkage. Escamilla et al. found an 1.5–2-fold upregulation of RhoA in cortex and hippocampus of P18 and 5-week-old Kctd13 Het and Hom mice. We evaluated the level of RhoA at three different ages: P1 (whole brain), during synaptogenesis at 3 weeks (hippocampus) and at 15 weeks of age (hippocampus and cortex). We found a 1.17-fold upregulation of RhoA protein level in the whole brain of Hom mice only at P1 but no dysregulation at 3 and 15 weeks of age in the hippocampus and cortex of Het and Hom animals. As Het and Hom mice show similar behavioral and spine phenotypes, our results suggest that the spine maturation deficit of Kctd13 mutants may be independent of RhoA/ROCK. Although less likely, it does not preclude the possibility that a RhoA dysregulation at an earlier age might introduce developmental defects that persist. We corroborated these findings in a mouse model of the 16p11.2 BP4-BP5 deletion in which we found that the level of RhoA was not affected in the hippocampus and in the cortex. Interestingly, however, we did see a decrease of RhoA protein levels in both hippocampus and cortex of the 16p11.2 duplication mouse model, suggesting that the brain structure and behavioral phenotypes of those mice (23) might be related to the RhoA/ROCK pathway.
Finally, to glean insight into the mechanisms by which Kctd13 haploinsufficiency might contribute to observed pathologies, we performed transcriptional profiling of mutants. We found significant enrichment for altered expression of genes that map to pathways involved in cognitive development. Consistent with this observation, we also found enrichment for genes associated with NDDs, as well as for genes whose products map to GO terms relating to postsynaptic density, neuronal cell body and projection and calmodulin binding in the cortex, as well as neuron projection in the hippocampus. Critically, when evaluating DEGs that are dysregulated in both our deletion models and the cortex of 16p11.2 BP4-BP5 deletion mice, we found significant enrichment for transcripts associated with abnormal cognition, abnormal learning, memory and conditioning, as well as abnormal synaptic transmission. Taken together, these data support a contributory role of this locus to the neurodevelopmental phenotypes of patients carrying 16p11.2 BP4-BP5 CNVs, and begin to dissect its most potent contributions to the network. There are additional analyses that could be performed in larger cohorts and that may yield intriguing insights, such as allele-specific expression patterns and extensive tissue-specific expression studies between KCTD13 and 16p11.2 BP4-BP CNVs. It will be interesting to assemble transcriptomic data from other loci, as well as from Kctd13-interactor double mutants to ask how such data sets overlap with the overall deletion. This may illuminate some of the genome-wide expression differences and mechanisms that underlie the dosage-specific phenotypic effects observed.
16p11.2 BP4-BP5 CNVs are associated with phenotypic heterogeneity and incomplete penetrance of phenotypes (7,11,21). Furthermore, cellular studies and animal modeling in mouse, zebrafish and Drosophila have implicated several genes from the 16p11.2 BP4-BP5 region as contributing to head size, brain structure and neuropsychiatric phenotypes (25,30,46–48,59–62). A recent study in Drosophila showed that cellular and developmental phenotypes are modulated by pervasive genetic interactions between 16p11.2 BP4-BP5 orthologs and neurodevelopmental genes (48). Our findings support this model and are also consistent with the fact that no single gene has been found to bear excessive mutations in AS/ASD or SZ patient cohorts. As such, the 16p11.2 CNVs syndromes are likely a contiguous syndrome not only architecturally but also functionally, in which genes inside (but also outside the BP4-BP5 region) interact to modulate phenotypic expression.
Materials and Methods
Mouse lines, genotyping and ethical statement
To generate a global deletion of Kctd13 in the mouse, we introduced LoxP sites flanking exon 2 of the gene. The targeting vector was generated using BAC Recombineering (63). BAC clone bmq384b14 was used as template. A LoxP site was placed upstream of exon2 while a Frt-flanked Neo cassette and LoxP site were placed downstream of exon2. The final targeting vector contained 8 kb of 5′ homology sequence and 2 kb of 3′ homology sequence. The HSV-TK cassette at the 3′ end of the targeting vector was used for negative selection in ES cell targeting. G4 ES cells (64) (from male blastocyst derived from the natural mating of 129S6/SvEvTac female with C57BL/6Ncr male) were injected into C57BL/6 N blastocysts. Animals haploinsufficient for Kctd13 (Het) were obtained by crossing floxed mice with B6.C-Tg(CMV-cre)1Cgn/J strain (Jackson Laboratory, Bar Harbor, ME, allowing the deletion of targeted exon in all tissues. After segregation of the Cre, Kctd13 Het mice were crossed together in order to generate wt, Het and mice deficient for Kctd13 (Hom). In total, six backcrosses on a C57BL/6 J background were done from the initial floxed mice. The wt allele and the deletion of the exon 2 of Kctd13 gene were identified by using primers Kctd13F (5′-GGGGAAGGGCTTAACATAGA-3′), Kctd13wtR (5′-AGATCCCTCCCAAATCTGCT-3′) and Kctd13koR (5′-GGCCAAACTTGATGACCAGT-3′). PCR reactions amplified specific products from deletion, and wt alleles of 521 and 414 bp, respectively, by using the following program: 95°C /5 min; 35× (94°C/30 s, 60°C/40 s, 72°C/30 s), 72°C/5 min. Colony founders from the 16p11.2 deletion and duplication line generated by the laboratory of Dr Alea Mills on a C57BL/6 N and 129Sv F1 background were obtained from Jackson Laboratories. Animals were backcrossed at least 10 times on a C57BL/6 J background. All experiments were conducted in accordance with National Institutes of Health guidelines for the care and use of laboratory animals and with an approved protocol from the Duke University Institutional Animal Care and Use Committee.
Behavioral analysis
Behavioral studies were conducted in 12–20-week-old animals. All assessments were scored blind to the genotype as recommended by the ARRIVE guidelines (65,66). After weaning, animals were sorted by genotype and sex where they had free access to water and food (Richmond Diet 5001, Lab Diet Inc., Richmond, VA). Animals were housed at 4–5 animals per cage in a humidity- (45%) and temperature-controlled (22°C) room. A 14:10 h light:dark cycle (lights on at 08:00 h) was used, and all behavioral testing was conducted between 08:00 and 15:00 h. Mice were transferred from the animal housing facility to the phenotyping area at the age of 9 weeks. Animals were transferred to the experimental room 1 day before the testing day. A resting period of 2 days to 1 week was used between two consecutive tests.
To study the behavioral of Kctd13 mutant mice, two cohorts were generated. The first cohort comprised 10 wt, 9 Het and 10 Hom male animals. Tests were administered in the following order: elevated zero maze (12 weeks), open field (12 weeks), novel object recognition (13 weeks), hole board (13 weeks), social affiliation (14 weeks), spray test (15 weeks), novel location recognition (15 weeks), rotarod (16 weeks), Morris water maze (17–18 weeks) and circadian activity (19 weeks). To confirm behavior phenotypes observed and to study potential differences related to the gender, we generated a second cohort including males (10 wt, 11 Het and 11 Hom) and females (10 wt, 12 Het and 13 Hom). Tests were administered in the following order: open field (12 weeks), novel object recognition (13 weeks), social affiliation (14 weeks), social interaction (15 weeks), novel location recognition (16 weeks), social transmission of food preference (16 weeks) non-social odor test (17 weeks) and fear conditioning (17 weeks). Experimental procedures for behavioral assessments other than those described below are detailed in the supplementary information.
Novel object recognition memory test
The test was carried out in the open field arenas. On the first day, mice were habituated to the arena for 30 min at 60 lux. On the following day, animals were submitted to the first 10 min acquisition trial during which they were individually placed in the presence of two similar objects (green Lego® or white dice) placed 10 cm away from one of the box corners (the distance between the two objects was ~20 cm). The exploration time of familiar objects (when the animal’s snout was directed toward the object at a distance ≤1 cm) was recorded. A 10 min retention trial was conducted 1 h later. One of the familiar objects was randomly replaced by a novel object (white dice or green Lego®) and the exploration time of these two objects was recorded. Object explorations were hand scored by visualizing videos with a stopwatch. A discrimination index was defined as (t novel object /(t novel object + t familiar object)) × 100. All mice that did not explore each object for more than 2 s during the acquisition trial were excluded from the analysis.
Novel location recognition memory test
In the first 10 min acquisition trial, animals were exposed to two similar objects consisting in blue marbles. One h later, one of the familiar objects was displaced to a novel location and the exploration time of the two objects was recorded for 10 min. Tests were recorded and scored as for the novel object recognition test.
Social transmission of food preference test
48h prior to testing, group-housed mice were placed into clean cages with full access to food and water. On the day prior to the start of testing, the food was removed from the cage ~3–4 h before the onset of the dark cycle. The following morning, a mouse in each cage was designated as the ‘demonstrator’ test animal. Importantly, mice were housed per genotype. Mouse chow was ground to a fine consistency and mixed with equal parts water in order to create a mouse mash. The mash was divided into two equal portions. One portion was flavored with maple extract and the remaining portion with banana extract (J.R. Watkins, Winona, MN). Both flavors were added so the final concentration was 1% of the mash weight. Demonstrator mice were randomized into two groups equally distributed across genotypes, one group which received the maple-demonstrator diet and the other group which received the banana-flavored diet. The demonstrator animal was removed from the cage and placed in a clean empty mouse cage with a single bowl that contained ~10 g of flavored diet. Mice were allowed to freely eat for 40 min, after which the mouse was removed from the cage and the bowl weighed to determine the amount of diet the demonstrator mouse consumed. Demonstrator mice were immediately returned to the home cages and allowed to interact freely with their cage mates for 20 min, at which time the demonstrator mice were removed from the home cage and individually housed for the rest of testing. After removal of the demonstrating, the remaining cage mates, or observer animals, were left alone for 1 h, at which time the short-term recall of the demonstrator diet was examined. Preference for the demonstrator diet versus an unfamiliar diet was assessed by placing an observer mouse into an empty clean mouse cage that had two small bowls, one which contained the familiar demonstrator diet and the other which contained the novel diet. For mice that received the maple demonstrator diet, banana was the novel diet, and for those which received the banana demonstrator diet, maple was the novel diet. The observer mice were allowed to freely eat from the bowls and explore the cage. After 40 min the mice were removed from the test cage and the bowls weighed to determine how much of each flavored diet each tester mouse consumed. A discrimination index was defined as (g familiar food /(g familiar food + g novel food)) × 100. A score above 50% indicated a preference for the familiar demonstrator diet, whereas a score approaching 50% indicates no preference for either diet.
Non-social odor discrimination test
Animals were first habituated to the clean cage for 3 min without bedding. For odor discrimination, scents were presented on a piece of Whatman paper placed in a perforated tube (5 × 3 × 3 cm). Mice were given five consecutive 2 min test sessions, with an 8 min ITI. For the test sessions 1–4, the scent was orange flower water. On the last session, orange flower scent was replaced with a new paper soaked with vanilla extract. Tests were recorded and odor-sniffing duration (when the animal’s snout was directed toward the perforated tube at a distance ≤1 cm) were hand scored by visualizing videos with a stopwatch.
Golgi-Cox staining and dendritic spine analysis
Golgi-Cox staining was performed on Kctd13 Het, Hom and wt gender-matched littermates (n = 6 animals per genotype, 50:50 sex ratio) using the FD Rapid GolgiStain Kit (FD NeuroTechnologies, Columbia, MD). Dye-impregnated brains were embedded in Tissue Freezing Medium (TBS) and were rapidly frozen on ethanol pre-chilled with dry ice. Brains were cryosectioned coronally at 100 μm thickness and were mounted on gelatin-coated microscope slides (FD NeuroTechnologies). Sections were stained according to the directions provided by the manufacturer. Sections that contain the CA1 region of the hippocampus were imaged at 60× magnification on a Nikon Eclipse 90i. To analyze spine density, we chose 10 μm length of secondary basal dendrites. A total of 10–12 secondary basal dendrites were analyzed per animal. Spines were measured and categorized as previously described (67).
Western blot
Whole brain (P1) and fresh hippocampal and cortex tissue (3 weeks and 15 weeks of age) were isolated by decapitation/dissection of males and females and lysed in ice-cold RIPA buffer (50 mm NaCl, 1% NP40, 50 mm Tris–HCl pH 7.5, 0.1% SDS, 0.5% Na deoxycholate, 1 mm Na3VO4 and 1 mm NaF). Total protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA) and 50 μg lysate per condition was subjected to 4–15% SDS-PAGE (Bio-Rad, Hercules, CA) and transferred to a PVDF membrane. Immunoblots were blocked in 3% BSA in PBS containing 0.1% Tween20 and probed with KCTD13 (1:250, HPA043524, Atlas Antibodies, Bromma, Sweden), RhoA (1:1000, 2117, Cell Signaling Technology, Danvers, MA) and Actin (1:2000, #MABT825, Sigma-Aldrich, Saint-Louis, MO). Blots were developed using an enhanced chemiluminescence system, Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), visualized on a ChemiDoc (Bio-Rad) and quantified with ImageJ.
RNA sequencing
A total of 50 RNA sequencing libraries were generated for cortex and hippocampus tissues extracted from 8 Kctd13 Hom, 8 Kctd13 Het and 9 control mice, using Illumina’s TruSeq stranded mRNA protocol. One microliter of a 1:10 dilution of External RNA Controls Consortium spike-ins (Thermo Fisher Scientific, Waltham, MA) containing 92 synthetic RNA standards of known concentrations and sequence was added to each RNA-sequencing library to assess library quality. Libraries were multiplexed, pooled and sequenced on Illumina HiSeq 2500, generating an average of 43.7 million paired-end reads of 75 bp.
RNA sequencing analysis
Quality of sequence reads was assessed by fastQC (version 0.10.1) (Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/ projects/fastqc/, doi: citeulike-article-id:11583827). Gene-based counts for all libraries were generated relying on the Ensembl mouse gene annotation, GRCm38 (v83), while sequence reads were aligned to the mouse reference genome GRCm38 (v83) using STAR (version 2.4.2a; 68) with parameters ‘--outSAMunmapped Within --outFilterMultimapNmax 1 --outFilterMismatchNoverLmax 0.1 --alignIntronMin 21 --alignIntronMax 0 --alignEndsType Local --quantMode GeneCounts --twopassMode Basic’. Quality of alignments was assessed by custom scripts utilizing Picard Tools and SamTools. Further differential expression (DE) and exploratory analyses were implemented in R (version 3.4). Previous QC analyses and exploratory analyses performed using a DE analysis package in R/Bioconductor, DESeq2 (69; version 1.18.1) and custom scripts identified one control mouse library (2_11_1) and one Hom mouse (5_1_5) from cortex tissue, and one control mouse library (2_11_1) from hippocampus tissue as outliers, which were excluded from further analyses. DE analyses of genes in Het versus wt and Hom versus wt sample comparisons separately for both hippocampus and cortex tissues were performed using DESeq2 with surrogate variable analysis package in R/Bioconductor, sva (70; version 3.26.0) to account for unknown factors. DEGs were identified at the nominal level (P < 0.05) or at a more stringent BH adjusted P < 0.05 level, using Wald Test under design ~SVs + genotype. In these analyses, genes with less than or equal to four counts in at least NLG samples, where NLG is the number of samples in the largest group from the comparison, were filtered out, while DESeq2’s independent filtering and cooksCutoff options were turned off. For GO enrichment analyses, DEGs were grouped based on direction of fold change with respect to wt, ‘up-regulated’, ‘down regulated’ or ‘all’, the latter of which no direction criterion was applied. Enriched DEG GO categories were identified using an enrichment analysis package in R/Bioconductor for GO, topGO (version 2.28.0) with nodeSize = 5 option and significant terms at BH P-adjusted < 0.05 reported. To assess enrichment of NDD-associated and other enrichment lists of interest (Supplementary Material, Table S7), as well as Kctd13 and 16p11.2 DEG overlap, we applied one-tailed Fisher’s exact test to the resultant overlap contingency tables. Our analyses were restricted to mouse genes with single human orthologs, which were obtained from the Mouse Genomics Informatics database (04/2018, http://www.informatics.jax.org/homology.shtml).
Data repository
The RNAseq raw data are available under ArrayExpress accession number E-MTAB-7398.
MRI and anatomical scan
MRI was used to study brain region structures in 10-week-old Kctd13 mutant mice (males: 11 wt, 11 Het, 10 Hom; females: 8 wt, 10 Het, 9 Hom), Lat mutant mice (females: 10 wt, 11 Het, 9 Hom), Mvp mutant mice (males: 8 wt, 11 Het; females: 10 wt, 12Het), Kctd13xLat mutant mice (males: 10 wt, 13 Het/Het; females: 9 wt, 9 Het/Het) and Kctd13xMvp mutant mice (males: 8 wt, 9 Het/Het; females: 10 wt, 13 Het/Het). A multichannel 7.0 Tesla MRI scanner (Agilent Inc., Palo Alto, CA) was used to image the brains within their skulls. Sixteen custom-built solenoid coils were used to image the brains in parallel (71,72). In order to detect volumetric changes, we used the following parameters for the MRI scan: T2- weighted, 3-D fast spin-echo sequence, with a cylindrical acquisition of k-space, a TR of 350 ms and TEs of 12 ms per echo for six echoes, field-of-view equaled to 20 × 20 × 25 mm3 and matrix size equaled to 504 × 504 × 630. Our parameters output an image with 0.040 mm isotropic voxels. The total imaging time was 14 h (73).
MRI registration and analysis
To visualize and compare any changes in the mouse brains the images are linearly (6 followed by 12 parameter) and non-linearly registered together. Registrations were performed with a combination of mni_autoreg tools (74) and ANTS (advanced normalization tools) (75,76). All scans are then resampled with the appropriate transform and averaged to create a population atlas representing the average anatomy of the study sample. The result of the registration is to have all images deformed into alignment with each other in an unbiased fashion. For the volume measurements, this allows for the analysis of the deformations needed to take each individual mouse’s anatomy into this final atlas space, the goal being to model how the deformation fields relate to genotype (77,78). The jacobian determinants of the deformation fields are then calculated as measures of volume at each voxel. Significant volume differences can then be calculated by warping a pre-existing classified MRI atlas onto the population atlas, which allows for the volume of 182 different segmented structures encompassing cortical lobes, large white matter structures (i.e. corpus callosum), ventricles, cerebellum, brain stem and olfactory bulbs (79–82) to be assessed in all brains. Further, these measurements can be examined on a voxel-wise basis in order to localize the differences found within regions or across the brain. Multiple comparisons in this study were controlled for using the False Discovery Rate (83).
Statistical analyses
Results were processed for statistical analysis using the GraphPad Prism 7 software. All acquired behavioral data were analyzed using a one-way ANOVA analysis with a post hoc Tukey’s test. The Pearson’s chi-squared test was used to evaluate mutant allele transmission. Data are represented as the mean ± standard error of the mean (s.e.m.) and difference were considered to be significant if P < 0.05.
Supplementary Material
Acknowledgements
We are grateful to Dr Weiguo Zhang who provided the Lat mutant mice and to Dr Binnaz Yalcin who provided the Mvp mutant mice. We also thank the members of the Center for Human Disease Modeling for their thoughtful comments.
Conflict of Interest statement. None declared.
Funding
Conte Center for Schizophrenia (P50 MH094268); National Institutes of Health (R01HD096326 and R01NS093200); Simons Foundation for Autism Research Initiative (#308955).
References
- 1. Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. et al. (2011) A copy number variation morbidity map of developmental delay. Nat. Genet., 43, 838–U844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grayton H.M., Fernandes C., Rujescu D. and Collier D.A. (2012) Copy number variations in neurodevelopmental disorders. Prog. Neurobiol., 99, 81–91. [DOI] [PubMed] [Google Scholar]
- 3. Moreno-De-Luca D., Sanders S.J., Willsey A.J., Mulle J.G., Lowe J.K., Geschwind D.H., State M.W., Martin C.L. and Ledbetter D.H. (2013) Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry, 18, 1090–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jacquemont S., Reymond A., Zufferey F., Harewood L., Walters R.G., Kutalik Z., Martinet D., Shen Y.P., Valsesia A., Beckmann N.D. et al. (2011) Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature, 478, 97–U111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weiss L.A., Shen Y.P., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A.R., Green T. et al. (2008) Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med., 358, 667–675. [DOI] [PubMed] [Google Scholar]
- 6. Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet., 82, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fernandez B.A., Roberts W., Chung B., Weksberg R., Meyn S., Szatmari P., Joseph-George A.M., MacKay S., Whitten K., Noble B. et al. (2010) Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet., 47, 195–203. [DOI] [PubMed] [Google Scholar]
- 8. Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. et al. (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron, 70, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanson E., Bernier R., Porche K., Jackson F.I., Goin-Kochel R.P., Snyder L.G., Snow A.V., Wallace A.S., Campe K.L., Zhang Y. et al. (2015) The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol. Psychiatry, 77, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghebranious N., Giampietro P.F., Wesbrook F.P. and Rezkana S.H. (2007) A novel microdeletion at 16p11.2 harbors candidate genes for aortic valve development, seizure disorder, and mild mental retardation. Am. J. Med. Genet. A, 143A, 1462–1471. [DOI] [PubMed] [Google Scholar]
- 11. Shinawi M., Liu P.F., Kang S.H.L., Shen J., Belmont J.W., Scott D.A., Probst F.J., Craigen W.J., Graham B.H., Pursley A. et al. (2010) Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet., 47, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zufferey F., Sherr E.H., Beckmann N.D., Hanson E., Maillard A.M., Hippolyte L., Mace A., Ferrari C., Kutalik Z., Andrieux J. et al. (2012) A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J. Med. Genet., 49, 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reinthaler E.M., Lal D., Lebon S., Hildebrand M.S., Dahl H.H., Regan B.M., Feucht M., Steinböck H., Neophytou B., Ronen G.M.. et al. (2014) 16p11.2 600 kb Duplications confer risk for typical and atypical Rolandic epilepsy. Hum. Mol. Genet., in press. [DOI] [PubMed] [Google Scholar]
- 14. McCarthy S.E., Makarov V., Kirov G., Addington A.M., McClellan J., Yoon S., Perkins D.O., Dickel D.E., Kusenda M., Krastoshevsky O. et al. (2009) Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet., 41, U1223–U1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bergen S.E., O'Dushlaine C.T., Ripke S., Lee P.H., Ruderfer D.M., Akterin S., Moran J.L., Chambert K.D., Handsaker R.E., Backlund L. et al. (2012) Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry, 17, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steinberg S., Jong S., Mattheisen M., Costas J., Demontis D., Jamain S., Pietilainen O.P.H., Lin K., Papiol S., Huttenlocher J. et al. (2014) Common variant at 16p11.2 conferring risk of psychosis. Mol. Psychiatry, 19, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walters R.G., Jacquemont S., Valsesia A., Smith A.J., Martinet D., Andersson J., Falchi M., Chen F., Andrieux J., Lobbens S. et al. (2010) A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature, 463, 671–U104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bochukova E.G., Huang N., Keogh J., Henning E., Purmann C., Blaszczyk K., Saeed S., Hamilton-Shield J., Clayton-Smith J., O'Rahilly S. et al. (2010) Large, rare chromosomal deletions associated with severe early-onset obesity. Nature, 463, 666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maillard A.M., Ruef A., Pizzagalli F., Migliavacca E., Hippolyte L., Adaszewski S., Dukart J., Ferrari C., Conus P., Männik K. et al. (2015) The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol. Psychiatry, 20, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Migliavacca E., Golzio C., Mannik K., Blumenthal I., Oh E.C., Harewood L., Kosmicki J.A., Loviglio M.N., Giannuzzi G., Hippolyte L. et al. (2015) A potential contributory role for ciliary dysfunction in the 16p11.2 600 kb BP4–BP5 pathology. Am. J. Hum. Genet., 96, 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenfeld J.A., Coppinger J., Bejjani B.A., Girirajan S., Eichler E.E., Shaffer L.G. and Ballif B.C. (2010) Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J. Neurodev. Disord., 2, 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arbogast T., Ouagazzal A.M., Chevalier C., Kopanitsa M., Afinowi N., Migliavacca E., Cowling B.S., Birling M.C., Champy M.F., Reymond A. et al. (2016) Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genet., 12, e1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horev G., Ellegood J., Lerch J.P., Son Y.E., Muthuswamy L., Vogel H., Krieger A.M., Buja A., Henkelman R.M., Wigler M. et al. (2011) Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. USA, 108, 17076–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Portmann T., Yang M., Mao R., Panagiotakos G., Ellegood J., Dolen G., Bader P.L., Grueter B.A., Goold C., Fisher E. et al. (2014) Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep., 7, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Golzio C., Willer J., Talkowski M.E., Oh E.C., Taniguchi Y., Jacquemont S., Reymond A., Sun M., Sawa A., Gusella J.F. et al. (2012) KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature, 485, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gusev A., Mancuso N., Won H., Kousi M., Finucane H.K., Reshef Y., Song L., Safi A., Schizophrenia Working Group of the Psychiatric Genomics Consortium, McCarroll S. et al. (2018) Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet., 50, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Z., Xiang Y. and Sun G. (2013) The KCTD family of proteins: structure, function, disease relevance. Cell Biosci., 3, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pinkas D.M., Sanvitale C.E., Bufton J.C., Sorrell F.J., Solcan N., Chalk R., Doutch J. and Bullock A.N. (2017) Structural complexity in the KCTD family of Cullin3-dependent E3 ubiquitin ligases. Biochem. J., 474, 3747–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin G.N., Corominas R., Lemmens I., Yang X., Tavernier J., Hill D.E., Vidal M., Sebat J. and Iakoucheva L.M. (2015) Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron, 85, 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Escamilla C.O., Filonova I., Walker A.K., Xuan Z.X., Holehonnur R., Espinosa F., Liu S., Thyme S.B., Lopez-Garcia I.A., Mendoza D.B. et al. (2017) Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature, 551, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polyak A., Rosenfeld J.A. and Girirajan S. (2015) An assessment of sex bias in neurodevelopmental disorders. Genome Med., 7, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu Y., Zhu H., Miller D.T., Gusella J.F., Platt O.S., Wu B.L., Shen Y. and Children's Hospital Boston Genotype Phenotype Study Group (2011) Age- and gender-dependent obesity in individuals with 16p11.2 deletion. J. Genet. Genomics, 38, 403–409. [DOI] [PubMed] [Google Scholar]
- 33. Rodriguiz R.M., Colvin J.S. and Wetsel W.C. (2011) Neurophenotyping genetically modified mice for social behavior. Methods Mol. Biol., 768, 343–363. [DOI] [PubMed] [Google Scholar]
- 34. Broadbent N.J., Gaskin S., Squire L.R. and Clark R.E. (2010) Object recognition memory and the rodent hippocampus. Learn. Mem., 17, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wrenn C.C. (2004) Social transmission of food preference in mice. Curr. Protoc. Neurosci., Chapter 8, Unit 8 5G. [DOI] [PubMed] [Google Scholar]
- 36. Mao D., Neumann A.R., Sun J.J., Bonin V., Mohajerani M.H. and McNaughton B.L. (2018) Hippocampus-dependent emergence of spatial sequence coding in retrosplenial cortex. Proc. Natl. Acad. Sci. U. S. A., 115, 8015–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luo R., Sanders S.J., Tian Y., Voineagu I., Huang N., Chu S.H., Klei L., Cai C., Ou J., Lowe J.K. et al. (2012) Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am. J. Hum. Genet., 91, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wellcome Trust Case Control C. (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature, 447, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeGiorgis J.A., Galbraith J.A., Dosemeci A., Chen X. and Reese T.S. (2006) Distribution of the scaffolding proteins PSD-95, PSD-93, and SAP97 in isolated PSDs. Brain Cell Biol., 35, 239–250. [DOI] [PubMed] [Google Scholar]
- 40. Kirkpatrick B., Xu L.Y., Cascella N., Ozeki Y., Sawa A. and Roberts R.C. (2006) DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J. Comp. Neurol., 497, 436–450. [DOI] [PubMed] [Google Scholar]
- 41. Kwinter D.M., Lo K., Mafi P. and Silverman M.A. (2009) Dynactin regulates bidirectional transport of dense-core vesicles in the axon and dendrites of cultured hippocampal neurons. Neuroscience, 162, 1001–1010. [DOI] [PubMed] [Google Scholar]
- 42. Mcrae J.F., Clayton S., Fitzgerald T.W., Kaplanis J., Prigmore E., Rajan D., Sifrim A., Aitken S., Akawi N., Alvi M. et al. (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blumenthal I., Ragavendran A., Erdin S., Klei L., Sugathan A., Guide J.R., Manavalan P., Zhou J.Q., Wheeler V.C., Levin J.Z. et al. (2014) Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet., 94, 870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Loviglio M.N., Arbogast T., Jonch A.E., Collins S.C., Popadin K., Bonnet C.S., Giannuzzi G., Maillard A.M., Jacquemont S., p, C. et al. (2017) The immune signaling adaptor LAT contributes to the neuroanatomical phenotype of 16p11.2 BP2-BP3 CNVs. Am. J. Hum. Genet., 101, 564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pucilowska J., Vithayathil J., Pagani M., Kelly C., Karlo J.C., Robol C., Morella I., Gozzi A., Brambilla R. and Landreth G.E. (2018) Pharmacological inhibition of ERK signaling rescues pathophysiology and behavioral phenotype associated with 16p11.2 chromosomal deletion in mice. J. Neurosci., 38, 6640–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blaker-Lee A., Gupta S., McCammon J.M., De Rienzo G. and Sive H. (2012) Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis. Model. Mech., 5, 834–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCammon J.M., Blaker-Lee A., Chen X. and Sive H. (2017) The 16p11.2 homologs fam57ba and doc2a generate certain brain and body phenotypes. Hum. Mol. Genet., 26, 3699–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iyer J., Singh M.D., Jensen M., Patel P., Pizzo L., Huber E., Koerselman H., Weiner A.T., Lepanto P., Vadodaria K. et al. (2018) Pervasive genetic interactions modulate neurodevelopmental defects of the autism-associated 16p11.2 deletion in Drosophila melanogaster. Nat. Commun., 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crepel A., Steyaert J., De la Marche W., De Wolf V., Fryns J.P., Noens I., Devriendt K. and Peeters H. (2011) Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am. J. Med. Genet. B, 156b, 243–245. [DOI] [PubMed] [Google Scholar]
- 50. Angelakos C.C., Watson A.J., O'Brien W.T., Krainock K.S., Nickl-Jockschat T. and Abel T. (2017) Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res., 10, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grissom N.M., McKee S.E., Schoch H., Bowman N., Havekes R., O'Brien W.T., Mahrt E., Siegel S., Commons K., Portfors C. et al. (2018) Male-specific deficits in natural reward learning in a mouse model of neurodevelopmental disorders. Mol. Psychiatry, 23, 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kumar V.J., Grissom N.M., McKee S.E., Schoch H., Bowman N., Havekes R., Kumar M., Pickup S., Poptani H., Reyes T.M. et al. (2018) Linking spatial gene expression patterns to sex-specific brain structural changes on a mouse model of 16p11.2 hemideletion. Transl. Psychiatry, 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Werling D.M. and Geschwind D.H. (2013) Sex differences in autism spectrum disorders. Curr. Opin. Neurol., 26, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jacquemont S., Coe B.P., Hersch M., Duyzend M.H., Krumm N., Bergmann S., Beckmann J.S., Rosenfeld J.A. and Eichler E.E. (2014) A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet., 94, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martin J., Walters R.K., Demontis D., Mattheisen M., Lee S.H., Robinson E., Brikell I., Ghirardi L., Larsson H., Lichtenstein P. et al. (2018) A genetic investigation of sex bias in the prevalence of attention-deficit/hyperactivity disorder. Biol. Psychiatry, 83, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Squire L.R., Wixted J.T. and Clark R.E. (2007) Recognition memory and the medial temporal lobe: a new perspective. Nat. Rev. Neurosci., 8, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lovett-Barron M., Kaifosh P., Kheirbek M.A., Danielson N., Zaremba J.D., Reardon T.R., Turi G.F., Hen R., Zemelman B.V. and Losonczy A. (2014) Dendritic inhibition in the hippocampus supports fear learning. Science, 343, 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lehmann M., Fournier A., Selles-Navarro I., Dergham P., Sebok A., Leclerc N., Tigyi G. and McKerracher L. (1999) Inactivation of Rho signaling pathway promotes CNS axon regeneration. J. Neurosci., 19, 7537–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Richter M., Murtaza N., Scharrenberg R., White S.H., Johanns O., Walker S., Yuen R.K.C., Schwanke B., Bedurftig B., Henis M. et al. (2018) Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yadav S., Oses-Prieto J.A., Peters C.J., Zhou J., Pleasure S.J., Burlingame A.L., Jan L.Y. and Jan Y.N. (2017) TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through septin7 phosphorylation. Neuron, 93, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Anda F.C., Rosario A.L., Durak O., Tran T., Graff J., Meletis K., Rei D., Soda T., Madabhushi R., Ginty D.D. et al. (2012) Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci., 15, 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pucilowska J., Vithayathil J., Tavares E.J., Kelly C., Karlo J.C. and Landreth G.E. (2015) The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci., 35, 3190–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu P., Jenkins N.A. and Copeland N.G. (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res., 13, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. George S.H., Gertsenstein M., Vintersten K., Korets-Smith E., Murphy J., Stevens M.E., Haigh J.J. and Nagy A. (2007) Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc. Natl. Acad. Sci. USA, 104, 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Karp N.A., Meehan T.F., Morgan H., Mason J.C., Blake A., Kurbatova N., Smedley D., Jacobsen J., Mott R.F., Iyer V. et al. (2015) Applying the ARRIVE guidelines to an in vivo database. PLoS Biol., 13, e1002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kilkenny C., Browne W.J., Cuthill I.C., Emerson M. and Altman D.G. (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol., 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McKinstry S.U., Karadeniz Y.B., Worthington A.K., Hayrapetyan V.Y., Ozlu M.I., Serafin-Molina K., Risher W.C., Ustunkaya T., Dragatsis I., Zeitlin S. et al. (2014) Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci., 34, 9455–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Love M.I., Huber W. and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Leek J.T. and Storey J.D. (2007) Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet., 3, 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bock N.A., Nieman B.J., Bishop J.B. and Henkelman R.M. (2005) In vivo multiple-mouse MRI at 7 Tesla. Magn. Reson. Med., 54, 1311–1316. [DOI] [PubMed] [Google Scholar]
- 72. Lerch J.R., Sled J.G. and Henkelman R.M. (2011) MRI phenotyping of genetically altered mice. Methods Mol. Biol., 711, 349–361. [DOI] [PubMed] [Google Scholar]
- 73. Noakes T.L.S., Henkelman R.M. and Nieman B.J. (2017) Partitioning k-space for cylindrical three-dimensional rapid acquisition with relaxation enhancement imaging in the mouse brain. NMR Biomed., 30. [DOI] [PubMed] [Google Scholar]
- 74. Collins D.L., Neelin P., Peters T.M. and Evans A.C. (1994) Automatic 3d intersubject registration of mr volumetric data in standardized talairach space. J. Comput. Assist. Tomogr., 18, 192–205. [PubMed] [Google Scholar]
- 75. Avants B.B., Epstein C.L., Grossman M. and Gee J.C. (2008) Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med. Image Anal., 12, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Avants B.B., Tustison N.J., Song G., Cook P.A., Klein A. and Gee J.C. (2011) A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage, 54, 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lerch J.P., Carroll J.B., Spring S., Bertram L.N., Schwab C., Hayden M.R. and Henkelman R.M. (2008) Automated deformation analysis in the YAC128 Huntington disease mouse model. Neuroimage, 39, 32–39. [DOI] [PubMed] [Google Scholar]
- 78. Nieman B.J., Flenniken A.M., Adamson S.L., Henkelman R.M. and Sled J.G. (2006) Anatomical phenotyping in the brain and skull of a mutant mouse by magnetic resonance imaging and computed tomography. Physiol. Genomics, 24, 154–162. [DOI] [PubMed] [Google Scholar]
- 79. Dorr A.E., Lerch J.P., Spring S., Kabani N. and Henkelman R.M. (2008) High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. Neuroimage, 42, 60–69. [DOI] [PubMed] [Google Scholar]
- 80. Steadman P.E., Ellegood J., Szulc K.U., Turnbull D.H., Joyner A.L., Henkelman R.M. and Lerch J.P. (2014) Genetic effects on cerebellar structure across mouse models of autism using a magnetic resonance imaging atlas. Autism Res., 7, 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ullmann J.F.P., Watson C., Janke A.L., Kurniawan N.D. and Reutens D.C. (2013) A segmentation protocol and MRI atlas of the C57BL/6J mouse neocortex. Neuroimage, 78, 196–203. [DOI] [PubMed] [Google Scholar]
- 82. Richards K., Watson C., Buckley R.F., Kurniawan N.D., Yang Z.Y., Keller M.D., Beare R., Bartlett P.F., Egan G.F., Galloway G.J. et al. (2011) Segmentation of the mouse hippocampal formation in magnetic resonance images. Neuroimage, 58, 732–740. [DOI] [PubMed] [Google Scholar]
- 83. Genovese C.R., Lazar N.A. and Nichols T. (2002) Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage, 15, 870–878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.