

Abstract
Mutations in the microtubule-associated protein tau (MAPT) underlie multiple neurodegenerative disorders, yet the pathophysiological mechanisms are unclear. A novel variant in MAPT resulting in an alanine to threonine substitution at position 152 (A152T tau) has recently been described as a significant risk factor for both frontotemporal lobar degeneration and Alzheimer’s disease. Here we use complementary computational, biochemical, molecular, genetic and imaging approaches in Caenorhabditis elegans and mouse models to interrogate the effects of the A152T variant on tau function. In silico analysis suggests that a threonine at position 152 of tau confers a new phosphorylation site. This finding is borne out by mass spectrometric survey of A152T tau phosphorylation in C. elegans and mouse. Optical pulse-chase experiments of Dendra2-tau demonstrate that A152T tau and phosphomimetic A152E tau exhibit increased diffusion kinetics and the ability to traverse across the axon initial segment more efficiently than wild-type (WT) tau. A C. elegans model of tauopathy reveals that A152T and A152E tau confer patterns of developmental toxicity distinct from WT tau, likely due to differential effects on retrograde axonal transport. These data support a role for phosphorylation of the variant threonine in A152T tau toxicity and suggest a mechanism involving impaired retrograde axonal transport contributing to human neurodegenerative disease.
Introduction
The microtubule-associated protein tau (MAPT) gene, located on chromosome 17 (17q21), encodes for the protein tau. Through its microtubule binding, tau impacts multiple cellular functions including microtubule stability, axonal trafficking and development (1–3). Tau is found in the pathological tangles of Alzheimer’s disease (AD), and mutations in MAPT are associated with a variety of neurodegenerative diseases, including frontotemporal lobar degeneration (FTLD), progressive supranuclear palsy and corticobasal syndrome (4). These mutations frequently occur in the carboxy-terminal microtubule-binding domain, leading to reduced microtubule binding and protein aggregation.
Recently, a novel disease-associated variant has been described outside the microtubule-binding domain of tau (5–8). This alanine to threonine variant at position 152 (A152T tau) resides in the projection domain of tau. This is the unstructured, amino-terminal section of the protein (9), not previously implicated in disease. However, human genetic data implicate A152T tau in increased risk of FTLD, AD and Parkinson’s disease, possibly through impaired microtubule seeding (5). This microtubule stabilizing function was not previously thought to require the tau projection domain. However, as tau is a natively unstructured protein, the hinge region of tau containing alanine 152 could regulate protein conformation and/or association. Furthermore, the retention of tau protein within the axon by the axon initial segment (AIS), a specialized cytoskeletal structure, is known to be impaired in an isoform specific manner (10–12). Therefore, it is also possible that the A152T variant could promote AIS leakage of tau into the somatodendritic compartment and disrupt tau localization. In addition, alanine 152 is contained within one of tau’s six canonical phosphodegron sequences, in this case 149-TKIATP-154. Phosphodegrons are short linear amino acid motifs completed through the addition of one or more phosphate groups that robustly control protein stability and half-life through ubiquitination and proteasomal degradation (13). Although better studied in cell cycle regulation (14), phosphorylation of the phosphodegron motifs of tau likely contributes to its turnover (15). Certainly, accumulation of hyperphosphorylated tau is a feature of tau tangles (16) and there are known disease mutations that occur in phosphodegron motifs (17).
A152T tau was shown to bind less tightly to microtubules and is less efficient at promoting microtubule assembly, resembling tau loss-of-function phenotypes (5). However, A152T tau also has a lower tendency to form tau fibrils, and these filaments are more prone to fragmentation (5). As a result, there is significantly more soluble tau oligomers present, and thus these phenotypes suggest an alternative toxic gain-of-function mechanism for A152T tau. In support of this, A152T tau expression in mice results in neuronal loss, cognitive impairments and elevated hippocampal calcium levels without evidence of tau aggregation (18,19), suggesting that these changes are most likely due to soluble oligomeric tau dysfunction. A similar disease mechanism has been implicated in other tauopathies (20). Recent studies expressing A152T tau in Caenorhabditis elegans observed altered distribution of organelles, mitochondria and synaptic vesicles that could be due to defects in intracellular trafficking (21), as well as progressive neuronal loss in the glutamatergic nervous system (22). Despite these recent insights, how the A152T substitution impacts tau molecular function and promotes neurodegeneration remains unclear.
In this study, we take advantage of a diverse array of techniques, including in silico prediction algorithms, mass spectrometry, biochemical, cell biological, genetic, behavioral approaches and high-resolution live imaging in both mouse primary cortical neurons and in C. elegans tauopathy models to investigate the toxicity of A152T tau. We identify 152T as a phosphorylation site and show that expressing A152T as well as a phosphomimetic A152E tau in C. elegans neurons promotes distinct development and post-development toxicities as well as learning and memory deficits compared to wild-type (WT) tau. These phenotypes are consistent with impairment in microtubule binding and retrograde axonal transport. Together, our data suggest that aberrant phosphorylation of A152T tau results in neuronal toxicity via impaired axonal trafficking.
Results
The variant threonine in A152T tau is phosphorylated in C. elegans and mouse
Phosphorylation of serine, threonine and tyrosine residues represents a common form of post-translational modification (23). The substitution of an alanine for a threonine at position 152 generates a potential new phosphorylation site in the tau protein. To investigate whether the threonine variant at position 152 meets consensus criteria for phosphorylation, we utilized the phosphorylation prediction algorithm DISPHOS version 1.3 (DISorder-enhanced PHOSphorylation predictor, http://www.dabi.temple.edu/disphos/) to perform generic phosphorylation site predictions in the A152T tau variant (24). A threonine at position 152 displayed high probability as a phosphorylation site compared to WT tau (Fig. 1A). In addition, by using GPS 3.0 (Group-based Prediction System, version 3.0), we found that the A152T substitution is also predicted to reside within a potential phosphodegron motif, 149-TKIATP-154 (Fig. 1B) (13).
Figure 1.

Threonine 152 is phosphorylated in the A152T tau variant. (A) DISPHOS data: prediction of phosphorylation sites in A152T tau protein by DISPHOS version 1.3. Significant gain of phosphorylation (P = 0.04) was obtained for A152T tau. (B) Amino acid sequence of the 2N4R tau isoform highlighting that the A152T substitution (red) lies within a predicted phosphodegron motif (bold underlined). The full-length 2N4R tau isoform was analyzed to ensure all potential phosphodegron motifs were identified. (C) Annotated tandem mass spectrometry (MS/MS) spectrum obtained from C. elegans from an ion with m/z 691.3517 (z = 3), corresponding to the tryptic tau peptide spanning residues I151 to R170. Two alternative isoforms of the peptide phosphorylated either at residues 152 (green) or 153 (blue) are presented. Detected ions that did not resolve between the two possible sequences are indicated in black. Detection of the y18 + 2 ion (green) indicates phosphorylation of T152 (zoomed inset). (D) Annotated tandem mass spectrometry (MS/MS) spectrum obtained from mouse from an ion with m/z 448.734 (z = 2), corresponding to the tryptic tau peptide spanning residues I149 to R155. Two alternative isoforms of the peptide phosphorylated either at residues 152 (green) or 153 (blue) are presented. Detected ions that did not resolve between the two possible sequences are indicated in black. Detection of the y3 ion (green) indicates phosphorylation of T152.
To directly investigate whether the threonine variant at 152 is phosphorylated, we conducted mass spectrometric survey for phosphorylation of WT and A152T human tau protein expressed in C. elegans and mouse. First, we generated C. elegans lines expressing either the WT or A152T tau variant of the 1N4R tau isoform in all neurons under the aex-3 promoter. The 1N4R tau isoform is the most abundant adult human brain isoform (25,26). We then performed mass spectrometry on tryptic digests of tau protein enriched by immunoprecipitation from lysates from these lines. Tau peptides identified by MS/MS covered >75% of the 1N4R tau isoform. Mass spectrometry detected an MS/MS spectra corresponding to the mono-phosphorylated form of a tau peptide spanning residues 151-ITTPRGAAPPGQKGQANATR-170 in full-length 2N4R tau (Fig. 1C and Supplementary Material, Table S1). Fragment ions in the spectra indicated phosphorylation of the threonine residue at position 152 (we could observe y18+2 corresponding to the mass of the non-phosphorylated sequence, and y19+2 corresponding to the phosphorylated peptide, which would place the phosphorylation at T152). T153 is phosphorylated in WT tau (27–29), and this was observed in our C. elegans line expressing WT tau (Supplementary Material, Table S2), but in A152T tau-expressing animals T152 is phosphorylated instead of T153.
As there are differences between the C. elegans and mammalian kinome (30), we also investigated phosphorylation of WT and A152T human tau expressed in mouse. Tau was enriched by immunoprecipitation from cortex and hippocampus (19). We further confirmed phosphorylation at T152 by mass spectrometry (Supplementary Material, Table S2). Tau peptides identified by MS/MS covered >76% of the 1N4R tau isoform. Two peptides spanning residues 149-TKITTPR-155 and 149-TKITTPRGAAPPGQK-163 were detected, both covering the A152T substitution. Here, the MS/MS spectra show mixed, co-fragmenting positional isomers of the phosphorylation site at T152 and T153 (Fig. 1D). The frequency of phosphorylation at this site increased with age, from one out of six samples in younger mice (hippocampus, 12 months of age) to five out of eight in older mice (cortex and hippocampus, 18–22 months of age, Supplementary Material, Table S2), suggesting an age-dependent increase in phosphorylation at the T152 site.
Strikingly, phosphorylation of the adjacent T153 residue was observed in all A152T mouse samples analyzed, and this was frequently accompanied by phosphorylation at T149 (9 out of 14 samples, Supplementary Material, Table S2). This pattern of phosphorylation was never observed in animals expressing WT tau (Supplementary Material, Table S2). The T153 residue is known to be phosphorylated by cdc2, CDK5 and MAPK (31). In addition, T149 and T153 are also phosphorylated by LRRK2 and found in pathological inclusions of several tauopathies (28,29). Such post-translational modification completes the 149-TKIATP-154 phosphodegron and should lead to the immediate degradation of tau through the ubiquitination and proteasome pathway (32). The fact that both phosphorylation sites are detected only in the A152T tau variant suggests that the variant threonine at 152 interferes with proteasomal recognition and degradation of tau. Taken together, these results suggest that the variant threonine 152 residue undergoes phosphorylation and may promote the accumulation of a pathological tau T149/T153 phosphoepitope.
A152T tau disrupts synaptic transmission in C. elegans
To assess whether the A152T variant has an effect on behavior, we tested a number of phenotypic outputs in our C. elegans tau lines. Others have previously observed that when expressed behind a pan-neuronal promoter, WT tau disrupts a motor circuit required for thrashing (21,33), which we confirmed (Fig. 2A). However, animals expressing A152T tau did not exhibit as marked impairments in thrashing (Fig. 2A). All transgenic tau lines showed a decline in thrashing rates with age (Fig. 2B). We wondered if this finding was specific to an A152T tau, or if another alanine-to-threonine change could cause a similar effect. Therefore, we assessed thrashing in an A178T tau animal, which is also in the hinge domain of tau. In contrast to A152T tau, thrashing in the A178T tau animal was impaired to a similar extent as WT tau (Fig. 2C). To determine if the WT thrashing observed in A152T tau animals is due to phosphorylation of threonine 152, we generated a transgenic C. elegans line expressing a phosphomimetic glutamate at position 152 (A152E tau). Significantly, the A152E tau animals phenocopied A152T tau animals (Fig. 2A and B).
Figure 2.
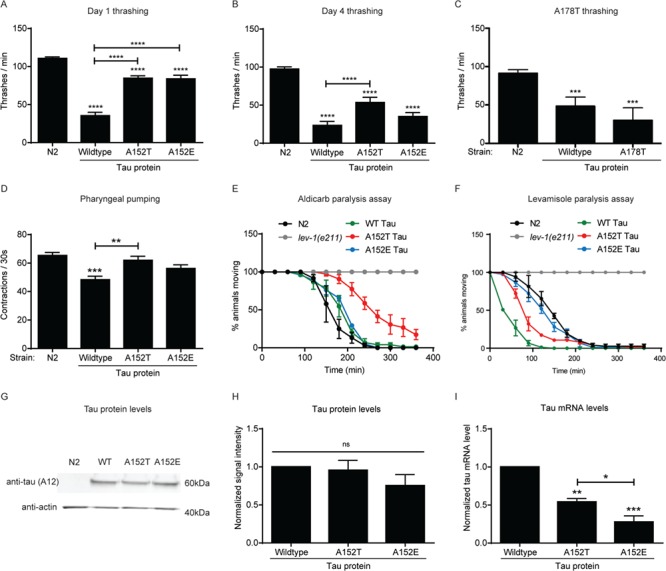
The A152T tau variant impairs synaptic transmission in adult C. elegans. (A) Measurement of the number of thrashes per minute for day 1 adult animals (n = 12, 3 biological replicates). (B) Measurement of the number of thrashes per minute for day 4 adult animals (n = 12, 3 biological replicates). (C) Measurement of the number of thrashes per minute for day 1 adult A178T tau-expressing animals (n = 8, 3 biological replicates). (D) Measurement of pharyngeal pumping per 30 s for day 1 adult animals (n = 10, 3 biological replicates). (E) Time-dependent aldicarb paralysis (n = 25, 3 biological replicates). Two-way ANOVA with Dunnett multiple comparisons test to WT non-transgenic animals. P < 0.05 at time point 150 min for WT tau, P < 0.001 at time point 180 min for WT tau, P < 0.0001 at time points 150–270 min for A152T tau, P < 0.001 at time point 300 min for A152T tau, P < 0.01 at time point 330 min for A152T tau, P < 0.0001 at time point 180 min for A152E tau. (F) Time-dependent levamisole paralysis (n = 25, 3 biological replicates). Two-way ANOVA with Dunnett multiple comparisons test to WT non-transgenic animals. P < 0.0001 at time points 90–150 min for WT and A152T tau, P < 0.01 at time point 180 min for WT tau, P < 0.001 at time point 60 min for A152T tau, P < 0.01 at time point 150 min for A152E tau. Worms lacking a functional nicotinic acetylcholine receptor (lev-1) are resistant to both aldicarb and levamisole and were used as a negative control in paralysis assays. (G) Representative western blot of lysates from N2 control animals and animals expressing WT, A152T or A152E tau. Total tau level was immunoblotted with Tau A12 antibody. (H) Quantification of total tau protein levels from western blot (three biological replicates). (I) Quantification of tau mRNA levels (three biological replicates). Statistical tests for all panels are one-way ANOVA with Tukey’s multiple comparisons test unless otherwise stated. Error bars represent SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001.
To ascertain if A152T expression could impact other neuronal-dependent behaviors we assessed the integrity of pharyngeal pumping in WT, A152T and A152E tau animals, as this behavior is dependent on the proper function of specific neurons (34,35). Similarly, we noted that WT tau animals displayed impaired pumping, while A152T and A152E tau animals were no different than N2 control animals (Fig. 2D).
The observed impairments in thrashing behavior could be due to defective synaptic transmission at the neuromuscular junction. Therefore, we performed pharmacological assays to assess pre- and post-synaptic cholinergic function using aldicarb (an acetylcholinesterase inhibitor) and levamisole (a nicotinic acetylcholine receptor agonist) (36). WT worms exhibit muscle hypercontraction and paralysis in the presence of either chemical. Animals with presynaptic defects or lacking functional acetylcholine receptors are resistant to aldicarb. Only animals lacking functional acetylcholine receptors are resistant to levamisole. A152T tau animals were markedly resistant to aldicarb, with WT and A152E tau animals displaying partial resistance (Fig. 2E). All tau-expressing animals exhibited sensitivity to levamisole (Fig 2F.). On aging to day 4 of adulthood all tau-expressing animals continued to exhibit aldicarb resistance (Supplementary Material, Fig. 1A) and levamisole sensitivity (Supplementary Material, Fig. 1B). These data suggest that transgenic tau expression results in impaired cholinergic presynaptic transmission, being most severe in A152T tau animals. The observed levamisole sensitivity may result from a compensatory upregulation of nicotinic acetylcholine receptors at the neuromuscular junction, as has previously been described for certain synaptic transmission mutants (37).
To ensure that the observed differences in locomotion and pumping were not due to differences in tau expression, we performed anti-tau western blots to determine protein levels and quantitative reverse-transcription PCR (RT-qPCR) to determine mRNA expression levels. WT, A152T and A152E tau protein levels were similar (Fig. 2G and H). Despite comparable protein levels, A152T and A152E tau-expressing animals had significantly lower human tau mRNA levels (Fig. 2I), suggesting that these tau variants may increase the production and/or decrease the clearance of tau protein.
The A152T tau variant impairs hatching, slows development and increases sterility rate in C. elegans
In earlier studies, threonine 153 had been shown to be phosphorylated and integral to the role of tau in development (9,27). Thus, we examined development, hatching and sterility rates in WT, A152T and A152E tau animals and observed that while WT tau minimally affected these parameters, A152T tau impaired hatching and development (Fig. 3). Interestingly, the phosphomimetic A152E tau was even more toxic in this assay than A152T tau (Fig. 3), suggesting that normal cycling of phosphorylation in this region is important for proper development and reproduction.
Figure 3.
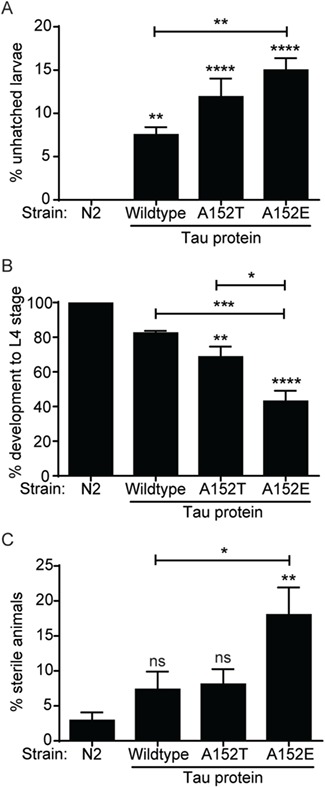
A152T and A152E tau variants impair the hatching, development and reproduction of C. elegans. (A) Quantification of unhatched larvae from animals expressing WT, A152T and A152E tau. One hundred eggs per strain were plated and scored for hatching after 24 h (n = 100, 3 biological replicates). (B) Animals expressing WT, A152T and A152E tau were staged as embryos, and development was scored to L4 stage (n = 50, 3 biological replicates). (C) Quantification of animals expressing WT, A152T and A152E tau that showed sterility. Statistical tests for all panels are one-way ANOVA with Tukey’s multiple comparisons test. Error bars represent SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001.
The A152T tau variant promotes neurodegeneration, impairs short-term associative learning and shortens lifespan in C. elegans
Expression of tau variants in the C. elegans nervous system has been shown to induce progressive axonal degeneration and neuronal loss (21,33). Therefore, to assess the impact of the A152T tau variant on neuronal integrity and morphology, we crossed our tau-expressing animals to marker lines expressing GFP in the γ-aminobutyric acid (GABAergic) motor neurons of the nerve cord (unc-25::gfp) and mechanosensory neurons (mec-4::gfp). Expression of all tau variants induced a significant number of axonal gaps in the ventral nerve cord, with stretches of nerve cord missing and exaggerated varicosities (Fig. 4A and Supplementary Material, Fig. 2A).
Figure 4.

A152T tau variant promotes neurodegeneration, impairs short-term associative learning and shortens lifespan in C. elegans. (A) Quantification of ventral cord gaps in the GABAergic motor neurons of day 1 adult tau-expressing animals (n = 25 animals per strain). One-way ANOVA with Tukey’s multiple comparisons test. Error bars represent SEM. ** = P < 0.01, **** = P < 0.0001. (B) Quantification of ALM mechanosensory neuron deformities (wavy or notched axon and abnormal soma or axon projections) in day 1 and day 4 adult tau-expressing animals (n = 25 animals per strain). One-way ANOVA with Tukey’s multiple comparisons test. Error bars represent SEM. * = P < 0.05, ** = P < 0.01, **** = P < 0.0001. (C) Measurement of short-term associative learning (three biological replicates). The glutamate receptor mutant nmr-1(ak4) was used as a negative control. One-way ANOVA with Bonferroni’s multiple comparisons test. Error bars represent SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001. (D) Representative survival curves of tau-expressing animals with non-transgenic N2 WT as a control. Median survival: N2 non-transgenic = 15 days, WT tau = 13 days, A152T tau = 11 days, A152E tau = 13 days. Mantel–Cox log-rank test was performed to determine statistical significance to WT control, P < 0.001 for WT tau, P < 0.01 for A152T tau, not significant for A152E tau, P < 0.01 for comparison of WT and A152T tau to A152E tau.
We also evaluated the effect of A152T tau expression on several features of neuronal aging in the ALM mechanosensory neuron (notched or wavy axon, axonal or soma projections, posterior projecting neurite) (38). Expression of all tau variants significantly increased the occurrence of these hallmarks of neuronal aging in day 1 adult animals, which was further exacerbated in day 4 adult animals (Fig. 4B and Supplementary Material, Fig. 2B). These results suggest that all three tau variants are effective in inducing neurodegeneration in the C. elegans motor circuit and premature aging phenotypes in the ALM mechanosensory neuron.
As the A152T substitution has been described as a significant risk factor for FTLD and AD, we investigated the effect of each human tau variant on C. elegans learning and memory by performing a short-term associative memory assay on day 1 adult animals. We measured the ability of WT, A152T and A152E tau-expressing animals to learn to associate food with the chemical butanone (39). While WT tau did not affect associative learning, both A152T tau and A152E tau significantly impaired this behavior (Fig. 4C). Again, A152E tau phenocopied A152T tau, although in this assay A152T tau was even more detrimental than the phosphomimetic A152E tau. Animals lacking the glutamatergic NMDA receptor ortholog gene, nmr-1, fail to learn and were used as a negative control in this assay. These data suggest that the A152T and A152E tau variants impair neuronal function in C. elegans learning and memory circuits.
Many studies have highlighted the important role of the nervous system in regulating C. elegans longevity (40,41). Therefore, we assessed the effect of each tau variant on C. elegans lifespan. WT and A152T tau animals displayed a significantly shortened lifespan, with A152T tau expression causing the most severe reduction in survival (Fig. 4D). Altogether, these data suggest that neuronal expression of all tau variants promotes neurodegeneration and accelerated neuronal aging, with A152T tau most negatively impacting short-term associative learning and lifespan.
The phosphorylation status of residues at position 152 and 153 predicts the effect of tau protein expression on C. elegans development and thrashing
The A152T tau variant is adjacent to a threonine residue at position 153 that is also a potential phosphorylation site (9,27). To further investigate the effect of phosphorylation of these two residues, we generated a complete series of C. elegans tau protein variants expressing an alanine, threonine or glutamate at the two positions. We classified these tau protein variants according to whether the residues at position 152 and 153 could be phosphorylated (Fig. 5A). An alanine cannot be phosphorylated, a threonine has the potential to be phosphorylated and a glutamate mimics phosphorylation and does not require the action of a kinase. These designations allow us to order the mutants based on phosphorylation status at each position and provide a conceptual framework to interpret experimental findings relative to phosphorylation (Fig. 5A).
Figure 5.

The phosphorylation status of residues at position 152 and 153 predicts the effect of tau protein expression on C. elegans development and thrashing. (A) The generation of a complete series of C. elegans tau protein variants expressing alanine (A—not phosphorylatable), threonine (T—phosphorylatable) or glutamate (E—phosphomimetic) residues at the 152 and 153 positions. (B) Animals expressing each tau protein variant were staged as embryos, and development was scored to L4 stage (n = 10–20 eggs, 5 biological replicates). One-way ANOVA with Tukey’s multiple comparisons test. Comparisons are to N2 WT animals. Error bars represent SEM. * = P < 0.05, ** = P < 0.01, **** = P < 0.0001. (C) Measurement of the number of thrashes per minute for day 1 adult animals (n ≥ 8, 3 biological replicates). One-way ANOVA with Tukey’s multiple comparisons test. Error bars represent SEM. ** = P < 0.01, **** = P < 0.0001. Comparisons are to N2 WT animals.
We observed that a threonine or glutamate residue at position 152 (TT or ET) delayed development the most severely in our phosphate load series (Fig. 5B). The presence of a phosphomimetic glutamate at the 153 position (AE, TE, EE), which would mimic constitutive phosphorylation at T153, did not have a substantial effect on development (Fig. 5B). An alanine at position 153 displayed a trend to impair development more severely if it was also accompanied by an alanine at position 152 (AA), suggesting that loss of a potential phosphorylation site at both the 152 and 153 positions (AA) may be detrimental (Fig. 5B). Interestingly, and in agreement with our previous data (Fig. 2), the opposite trend was observed for thrashing, with TT and ET tau variants having the least effect (Fig. 5C). In sum, these data suggest that tau variants have the most detrimental impact on development when either a potential or phosphomimetic residue is present at position 152 (TT, ET) or non-phosphorylatable residues are present at both the 152 and 153 positions (AA).
The A152T tau variant confers AIS leakage of tau protein and poor microtubule binding
In healthy and mature neurons the majority of endogenous, WT tau protein is located within the axon. However, during the early stages of AD the protein is observed to redistribute into the somatodendritic compartment (11). Axonal retention of tau has been shown to depend upon the AIS. This specialized cytoskeletal structure is localized between axonal and somatodendritic domains and serves as a physical barrier that helps to maintain neuronal polarity and initiate firing of action potentials (11,42). Tau protein variants that exhibit impaired microtubule binding transit more effectively to the somatodendritic compartment, rendering the AIS barrier less effective (12). Hyperphosphorylation of tau protein in its repeat domain in the context of neurodegenerative disease also prevents microtubule binding and inhibits barrier function, resulting in AIS leakage of the tau protein into somatodendritic compartments (12). Thus tau microtubule binding seems to be crucial for the AIS retention of tau in the axon.
Earlier studies have provided biochemical evidence that A152T tau is less efficient than WT tau at seeding microtubule polymerization and binding microtubules (5). However, no demonstration of this effect has been performed in cellular models. To measure diffusion kinetics of tau variants into the somatodendritic compartment, we utilized tau fusions with a photoconvertible fluorescent protein, Dendra2 (11). Dendra2 constructs were expressed in mouse primary cortical neurons and photoconverted at the proximal axonal region. Photoconversion after expression of Dendra2 alone resulted in the rapid diffusion of this soluble protein out of the axon and into the somatodendritic domain with a half-life of 1.5 min (Fig. 6A). Compared to Dendra2 alone, expression of Dendra2-A152T tau resulted in tau diffusion into the somatodendritic compartment with the next most rapid kinetics (half-life of 14.5 min). Diffusion of A152T tau occurred at a significantly faster rate than that of a Dendra2-WT tau fusion, and the diffusion kinetics were more rapid than a KxGE variant of tau that cannot bind microtubules (43). This KxGE tau variant has all four serine residues of the KxGS motif in the repeat domain mutated to glutamic acid in order to mimic tau phosphorylation, and this promotes detachment from microtubules (43). Here again A152E tau phenocopied A152T tau, displaying a significantly faster diffusion rate than that of WT tau (Fig. 6A). These data suggest that A152T tau may bind microtubules less effectively than WT tau.
Figure 6.

A152T and A152E tau display increased diffusion kinetics. (A) Fluorescence intensity versus time (0 = time of photoconversion) for the diffusion of Dendra2-tau variants out of the photoconverted ROI in the axon; WT tau (green), A152E tau (blue), KxGE tau (black), A152T tau (red), Dendra2 (purple). Error bars represent SEM, n = 2–8 cells. (B) Fluorescence intensity versus time (0 = time of photoconversion) for the diffusion of Dendra2-tau variants into the soma from the photoconverted ROI in the axon. (C) Quantification of Dendra2-tau variant fluorescence in the cell soma 1 h after photoconversion of the ROI in the axon. One-way ANOVA with Dunnett’s multiple comparisons test. Error bars represent SEM. *** = P < 0.001.
We next investigated the ability of tau variants to traverse the AIS by photoconverting Dendra2-tau species in the axon of mouse cortical primary neurons and measuring fluorescence in the soma. Dendra2 alone diffused rapidly from the axon back into the soma with a half-life of 1.5 min for diffusion out of the photoconverted axonal region and displayed the highest fluorescence in the soma 1 h after photoconversion (Fig. 6B–C). WT tau protein displayed the slowest diffusion rate away from the axon and was barely able to penetrate the AIS. The A152T tau species showed faster diffusion away from the photoconverted axonal region and greater AIS leakage than that of WT tau (Fig. 6B–C). Again, these data suggest that A152T and A152E tau species may bind less strongly to microtubules than WT tau, thus allowing their retrograde movement toward the soma.
A152T tau impairs the retrograde axonal transport of synaptic vesicle precursors
Previous studies showed that overexpression of tau protein in cell culture systems resulted in mislocalization of overexpressed as well as of endogenous tau to somatodendritic compartments, and in disruption of anterograde and retrograde axonal transport (44). We also observed that A152T and A152E tau mislocalized to the soma to a greater extent than WT tau (Fig. 6), suggesting that these tau variants bind less strongly to microtubules and this could affect the transport of cargo that routinely move on microtubules. The C. elegans RAB-3 protein is involved in synaptic transmission (45), and movement of RAB-3 vesicles along axons has been shown to be dependent on Kinesin-3 (UNC-104) function (46). To image live RAB-3 vesicle transport, we used a fluorescently tagged GFP::RAB-3 fusion expressed uniquely in mechanosensory neurons, including PLM, under the mec-7 promoter. To test whether expression of WT tau, A152T tau or A152E tau disrupts the axonal transport of vesicular cargo in neurons, we imaged the transport of GFP::RAB-3 synaptic vesicles in PLM, a touch receptor neuron, in day 1 old adult animals (Fig. 7A–B). A152T tau-expressing animals displayed a selective impairment in the retrograde transport of synaptic vesicles (Fig. 7). In all PLM neurons imaged, vesicles moved in anterograde and retrograde directions, reversed directions during the imaging period, and a large percentage were stationary (Fig. 7B and C). Expression of all tau variants resulted in a significant increase in the percentage, as well as in the overall density, of stationary vesicles, at the expense of those vesicles moving in the anterograde direction (toward the axonal tip; Fig. 7B, C and D), suggesting that neuronal overexpression of tau impairs the delivery of Golgi-derived RAB-3::GFP vesicles into the axon. Moreover, expression of WT, A152T and A152E tau resulted in lower GFP::RAB-3 vesicle anterograde velocities (Fig. 7E). However, only axons of animals expressing A152T tau displayed lower retrograde velocities compared to non-tau control animals (Fig. 7F). Expression of all tau sequences resulted in shorter anterograde GFP::RAB-3 vesicle run lengths compared to non-tau control axons (Fig. 7G), but only GFP::RAB-3 vesicles from animals expressing A152T tau underwent shorter retrograde runs (Fig. 7H). Thus, expression of WT tau, A152T tau and the phosphomimetic A152E tau impaired the anterograde transport of pre-synaptic vesicles, while only expression of A152T tau resulted in the impairment of bidirectional—including retrograde—axonal transport of GFP::RAB-3 pre-synaptic vesicles. Overall, these observations suggest that changes in the phosphorylation/dephosphorylation state of the T152 site, rather than abrogation of phosphorylation/dephosphorylation cycles at this position (as in the case for A152E tau), specifically impaired the retrograde movement of pre-synaptic vesicles.
Figure 7.
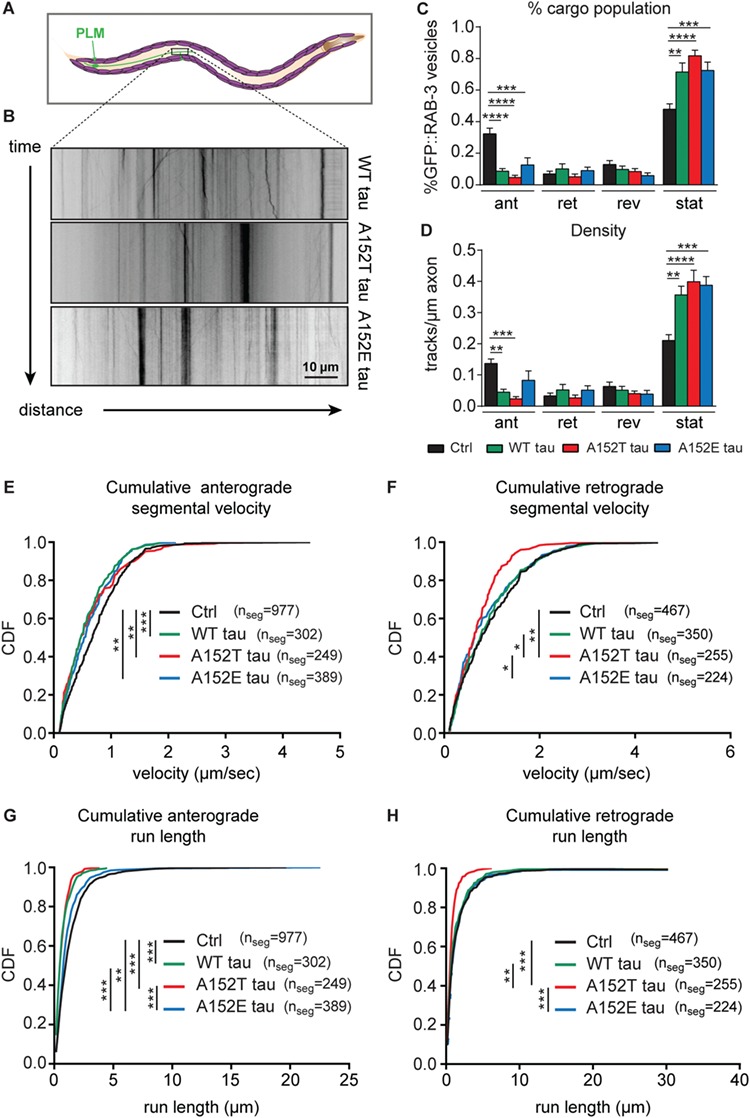
A152T tau impairs both anterograde and retrograde axonal transport of synaptic vesicles. (A) Schematic of PLM neuron trajectory, highlighting the region of axon for synaptic vesicle imaging (brown box). (B) Representative kymographs for movement of GFP::RAB-3 puncta along PLM axons in animals expressing WT tau, A152T tau and A152E tau. (C) Percentage of GFP::RAB-3 puncta that display anterograde, retrograde, reversing or stationary behavior. Mean ± SEM, one-way ANOVA, Tukey’s multiple comparisons test. (D) Number of tracks per μm displaying anterograde, retrograde, reversing or stationary behavior. Mean ± SEM, one-way ANOVA, Tukey’s multiple comparisons test. (E–F) Cumulative distribution function (CDF) of segmental velocities for E, anterograde and F, retrograde moving GFP::RAB-3 puncta. (G–H) CDF of G, anterograde and H, retrograde run lengths. Distribution differences in E–H determined by non-parametric Kolmogorov–Smirnov test. Multiple comparisons Bonferroni correction. Number of animals imaged per strain: control GFP::RAB-3 n = 15, WT tau n = 9, A152T tau n = 10, A152E tau n = 11. * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001.
Discussion
Tau is a complex protein with a multitude of regulatory mechanisms, including phosphorylation and dephosphorylation at multiple sites by multiple enzymes, acetylation and O-GlcNAc modification (9,47). Of the identified tau mutations, A152T is the only variant of known pathogenicity that may potentially disrupt a phosphodegron through the creation of a new phosphorylation site. Hypothetically, nearly 20% of tau amino acids can undergo phosphorylation and hyperphosphorylated tau is found in tau tangles, suggesting that either increased kinase activity, decreased phosphatase activity or impaired clearance of phosphorylated tau contributes to neurodegenerative disease pathogenesis.
The MS/MS spectra showed mixed, co-fragmenting positional isomers of phosphorylation at the T152 and T153 residues. Such isomer-specific phosphorylation has also been observed for protein kinase AKT1/2 activation (48). Phosphorylation of the variant threonine in A152T tau was frequently accompanied by phosphorylation at T149 and T153. There are several potential mechanisms through which the A152T substitution could alter kinase interaction and phosphorylation of tau, including through a change in conformation or accessibility to kinases (28,29). In addition, it is possible that disruption of the 149-TKIATP-154 phosphodegron motif could disrupt the clearance of A152T tau. Indeed, it has recently been shown that mice expressing the A152T tau variant have a higher tau protein: mRNA ratio in the cortex and hippocampus (19) that could indicate impaired tau clearance. Our data for C. elegans A152T tau protein and mRNA expression levels agree with this (Fig. 2). In addition, pharmacological and genetic upregulation of autophagy and proteasome function has recently been shown to ameliorate pathology in an A152T tau-expressing zebrafish model (49). Phosphorylation at T153 likely creates a consensus priming site for the serine/threonine kinase glycogen synthase kinase 3 (GSK3), which phosphorylates residues four places upstream (i.e. T149) (50–52). Phosphorylation sites spaced four residues apart create a favorable interaction site for the SCF ubiquitin ligase Cdc4/Fbw7 (53,54). Such post-translational modification completes a phosphodegron and should lead to the immediate degradation of tau through the ubiquitination and proteasome pathway (32). The fact that we were able to detect both phosphorylation sites, only in the A152T tau variant, suggests that the variant threonine at 152 may interfere with proteasomal recognition and degradation of tau protein. However, we did not detect an increase or modification in ubiquitin patterns around the A152T variant in mouse cortex or hippocampus by mass spectrometry analysis (Supplementary Material, Table S2). Despite this, these data suggest that the variant threonine residue at 152 undergoes phosphorylation and that this variant promotes the presence and accumulation of the pathogenic T149/T153 phosphoepitope (27–29).
We observe that the A152T substitution has a deleterious effect on developmental phenotypes in our transgenic animals (impaired hatching, slowed development and increased sterility, Fig. 3), with a lesser effect on adult locomotion behaviors such as thrashing and pharyngeal pumping (Fig. 2). Tau is highly phosphorylated during embryonic and early postnatal development (55,56), and this correlates with the time of intense neurite outgrowth (57). Interestingly, several of these phosphorylation sites are observed in AD brain (55,58), suggesting some similarity between tau phosphorylation in dividing and degenerating neurons. However, although hyperphosphorylated embryonic and fetal tau is functional under these conditions, the hyperphosphorylated tau from neurodegenerative brain inhibits microtubule assembly and aggregates (57). Furthermore, changes in tau phosphorylation state have been linked to progression through the cell cycle (31). We speculate that phosphorylation of the variant threonine at residue 152 and the subsequent disruption of normal phosphorylation signatures of T149/T153 may impede proper tau function during development. This could potentially result in abnormal microtubule dynamics during neurite outgrowth and maturation and contribute to the developmental phenotypes that we observe. If the A152E variant effectively prevents normal phosphorylation cycling at these residues, it may explain why developmental phenotypes were more severely affected in this strain.
In agreement with the first biochemical reports that A152T tau binds less tightly to microtubules, we have demonstrated in cellular models that Dendra2-A152T tau displays enhanced diffusion kinetics and greater AIS leakage into the soma compared to WT tau (Fig. 6). Indeed, the A152T tau variant diffuses faster than the KxGE tau variant, which cannot bind to microtubules (Fig. 6A). The microtubule surface has an overall negative charge, and interactions with tau are favorable because the microtubule-binding region of tau is predominantly positively charged (59). One potential explanation for these data is that the A152T variant induces aberrant tau phosphorylation, and these post-translational modifications would increase the negative charge of tau protein, making microtubule interaction less favorable. If this were to disrupt microtubule-tau electrostatic interactions more than the KxGE variant, it may explain the faster rate of diffusion for A152T tau.
Expression of A152T tau in neurons in C. elegans decreased the overall number of GFP::RAB-3 pre-synaptic vesicles moving inside axons—in both anterograde and retrograde directions—and impaired the transport dynamics of the remaining retrograde-moving vesicles (Fig. 7). These data are consistent with the hypothesis that phosphorylation of a variant threonine at position 152 either retains the pre-synaptic vesicles destined for the axon in the cell body or might drive them into somatodendritic domains. This latter possibility needs to be further addressed in the case of GFP::RAB-3 vesicles. Retrograde axonal transport allows for synaptic renewal (60), and impairments in this process could contribute to the defective neurotransmission and short-term learning observed in our A152T tau animals. These data, as well as our observation that A152T tau more easily diffuses into somatodendritic compartments (Fig. 6), are consistent with the possibility that neuronal domain polarity shifts could occur in A152T-expressing axons, whereby the identities of axonal versus somatodendritic compartments are no longer maintained. Reversals of neuronal polarity could thus explain the significant impaired translocation of axonal GFP::RAB-3 vesicles into the axon and could be relevant to pathology, as polarity shifts have been implicated in AD, although the mechanisms remain unclear (61).
It remains unclear why retrograde transport might be more notably affected. However, we also observed anterograde disruptions in transport of GFP::RAB-3 vesicles (Fig. 7). As impairing the activity of an anterograde motor has been shown to affect the transport of both anterograde and retrograde-moving cargoes (62–69), it is possible that the impairments observed here for GFP::RAB-3 vesicles could be the result of A152T tau impairments to either kinesins and/or dyneins, which modulate the anterograde and retrograde transport of cargoes, respectively (70). Indeed, tau has been implicated in regulating the transport of both kinesin and dynein along microtubules, as the presence of endogenous WT tau on microtubules in vitro resulted in the reversal of dynein and in the detachment of kinesin from microtubules (71).
These data also showed significant impairments in the anterograde transport of GFP::RAB-3 vesicles in C. elegans mechanosensory axons from animals expressing all tau variants. Previous studies have shown that overexpression of WT tau perturbs anterograde axonal transport and induces axonal collapse by binding microtubules and preferentially impairing plus-end-directed transport mediated by kinesin-like motor protein (72–74). As neurodegenerative phenotypes were observed in all transgenic tau-expressing animals, it is possible that the A152T and A152E tau variants may be acting in a similar manner. Expression of the phosphomimetic A152E mutation affected anterograde velocities and run lengths of GFP::RAB-3 vesicles, but not retrograde transport parameters, as was the case of the A152T mutation (Fig. 7). We hypothesize that the constitutive phosphorylation of the 152E site might putatively abrogate phosphorylation/dephosphorylation cycles at this position that could have a measured effect on the transport of otherwise axonally destined cargoes by strongly retaining pre-synaptic vesicles at the cell body and preventing them from reaching the axon. Thus, primarily movement in the anterograde direction could be significantly affected, while the observed retrograde impairments might be a reflection of changes in the phosphorylation/dephosphorylation state of the T152 site.
Overall, the data showing impaired axonal transport of pre-synaptic vesicles are consistent with previous in vitro studies showing that decorating microtubules with 4RL and 3RS tau isoforms resulted in differential effects on the anterograde or retrograde transport of cargoes along microtubules, including in shorter run lengths and altered velocities (75,76). The role of phosphorylation of tau was not tested in all of these in vitro studies, so it remains unclear to what extent tau phosphorylation affects transport by affecting the number of motors on cargo and thus affecting motility parameters. However, it is clear that tau phosphorylation states influence microtubule-based cargo transport, and our studies here point towards a biased directional impairment of retrograde movement of pre-synaptic cargo in in vivo studies of axons expressing A152T. A recent study expressing A152T tau in C. elegans neurons reported defects in the distribution of mitochondria and synaptic vesicles along sensory neurons, including the accumulation of RAB-3 puncta in the distal axon and minor neurite (21), suggestive of transport defects. Furthermore, a transgenic A152T mouse model observed electron dense degenerating presynaptic terminals filled with swollen synaptic vesicles (18), suggestive of defects in the transport and localization of these vesicles upon active phosphorylation of the 152 tau site. Specifically, the altered distribution of organelles and synaptic vesicles at distal axonal termini observed in these studies could be due to the impaired retrograde movement of these organelles and vesicles that remained sequestered at distal sites due to impaired retrograde motility. In contrast, expression of the anti-aggregant tau, ∆K280-PP, has been shown to increase the retrograde transport velocity of mitochondria when measured in organotypic transgenic hippocampal slices (42). These results suggest a possible disease mechanism, whereby lack of microtubule binding by A152T tau causes tau to relocate from axonal to somadendritic domains, but which also could be responsible for the dysfunctional retrograde transport of the decreased percentage of cargo that do mobilize to distal axonal areas. This could be specifically detrimental to the retrograde movement of cargoes that need to be taken back to proximal sites near the neuronal soma, where lysosomal-based degradation has been shown to occur (77). Indeed, many studies have linked impaired retrograde transport directly with neurodegeneration (78) and this could contribute to the age-dependent neuronal loss observed in transgenic A152T tau mouse models (18,19).
Defective axonal transport could also result in the impaired movement of other cargo critical for neuronal function, for example of nerve growth factor (NGF) (79) that could result in developmental abnormalities and explain the phenotypes observed here in our C. elegans tau models. The current study is consistent with previous work that showed phosphorylated tau immunoreactivity in the soma of in vivo aged rat brains, while young animals showed primarily neurite staining (80). This redistribution of phosphorylated tau to the soma during aging correlated with reduced trafficking of NGF in cholinergic basal forebrain neurons (80). It is unclear how expression of A152T tau could orchestrate the directional impairment of retrograde transport of pre-synaptic vesicles. However, the N-terminal domain of tau where A152T resides interacts with p150/Dynactin (81), the microtubule binding subunit of Dynactin that forms a large complex with Dynein and aids its processivity. Therefore, it is possible that A152T tau has reduced interactions with microtubules but also impaired interactions with the dynein/dynactin complex, being able to impair both anterograde and retrograde mobilization. Alternatively, the retrograde movement of A152T tau towards the soma could be driven by the binding of A152T tau to dynein, thus depleting dynein from axonal domains. The lack of dynein in axons could then in turn result in impairments in the retrograde transport of other axonal cargoes by virtue of the depletion of dynein motors from this domain. Taken together, these data suggest a possible disease mechanism whereby phosphorylation of the variant threonine in A152T tau impairs microtubule binding and leads to abnormal cargo transport within neurons, contributing to human neurodegenerative disease.
Materials and Methods
In silico analysis of WT versus A152T tau phosphorylation sites
We generated two sequences in FASTA format, one for WT tau and the other for A152T tau. DISPHOS (version 1.3; http://www.dabi.temple.edu/disphos/pred.html) was applied to each sequence with default predictor. On each amino acid position, we calculated the probability of gain of phosphorylation as follows:
P(gain) = (1 − P(wild)) * P(mutant),
where P(wild) and P(mutant) are DISPHOS scores from WT and mutant sequence, respectively. A152T was predicted to most likely gain a phosphorylation site across the whole tau protein sequence with a probability of 0.59.
In silico analysis of WT tau phosphodegron motifs
We used GPS 3.0 (Group-based Prediction System, version 3.0) to find potential Cdk1 and GSK3 phosphorylation sites within the 2N4R tau protein sequence that display the preferred residue spacing for binding by SCF ubiquitin ligase Cdc4/Fbw7 (50–54).
Tau immunoprecipitation and mass spectrometry
C. elegans expressing WT or A152T tau variants were grown on high-growth nematode growth medium (NGM) plates (10 × 15 cm plates per strain). Animals were collected and washed in M9 buffer and then re-suspended in lysis buffer (20 mM HEPES, 150 mM NaCl, 2 mM EDTA, 0.1% Triton-X, 10% glycerol, 40 mM NaF) containing protease and phosphatase inhibitors (Roche cOmplete Protease Inhibitor Cocktail Tablets #04693124001 and Roche PhosSTOP Phosphatase Inhibitor Cocktail Tablets #04906845001). Animals were drop-frozen into liquid nitrogen, and the resulting beads were manually homogenized using a pre-chilled mortar and pestle. The paste was re-suspended in lysis buffer (recipe above). 10 μg of HT7 human tau antibody (Thermo Scientific MN1000) was DSS cross-linked to immunoprecipitation columns using the Pierce Crosslink Immunoprecipitation Kit (#26147). 600 μl of 1 mg/ml total protein pre-cleared worm lysate was added to each column and incubated for 90 min at 4°C. The column was washed with kit-provided IP/lysis wash buffer, and bound tau protein was recovered using Eluting buffer. Eluted protein was resolved on a Novex NuPAGE 4–12% Bis Tris SDS-PAGE gel (NP0321BOX) and stained with Colloidal Coomassie Stain (Bio-Rad #161–0803). Bands at 30–50 kDa corresponding to tau protein were cut and digested in-gel with trypsin as described previously (82).
Murine lines expressing WT and A152T tau were reported previously (19). Mouse cortex and hippocampus from 12-, 18- and 22-month-old animals were dissected from hemi-brains and manually homogenized with a tissue homogenizer (Thermo Fisher #K7496250010) for 1 min in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1% NP-40, pH 7.4) containing protease and phosphatase inhibitors (Roche cOmplete Protease Inhibitor Cocktail Tablets #04693124001 and Roche PhosSTOP Phosphatase Inhibitor Cocktail Tablets #04906845001). Samples were sonicated (three rounds of 1 min on, 10 s off) and then centrifuged at 10 000 rpm for 10 min at 4°C. Tau protein was immunoprecipitated from the supernatant using 10 μg of HT7 anti-human tau antibody (Thermo Scientific MN1000) and Pierce Protein A/G magnetic beads (Thermo Fisher #88802). Eluted protein was resolved on a Novex NuPAGE 4–12% Bis Tris SDS-PAGE gel (NP0321BOX) and stained with SilverQuest Silver Staining Kit (Thermo Fisher #LC6070). Bands at 50–60 kDa corresponding to tau protein were cut and digested in-gel with trypsin as described previously (82).
Peptides recovered were analyzed by reversed-phase liquid chromatography-electrospray tandem mass spectrometry (LC–MS/MS) essentially as described (83). Briefly, peptides were separated by nano-flow chromatography in C18 column, and the eluate was coupled to a hybrid linear ion trap-Orbitrap mass spectrometer (LTQ-OrbitrapVelos, ThermoScientific, San Jose, CA) equipped with a nanoelectrospray ion source. Following LC–MS/MS analysis using different energy deposition methods (CID, HCD, ETD), peak lists generated from spectra were searched against the human and C. elegans subset of the Uniprot database plus the A125T_Tau mutant sequence, using in-house ProteinProspector (84).
C. elegans strains
The Bristol strain was used as the WT strain. All strains were maintained at 20°C according to standard methods (85). We used site-directed mutagenesis to introduce the A152T and A152E variations into the cDNA of 4R1N WT human tau, expressed under the pan-neuronal aex-3 promoter (plasmid pPD49.26, a generous gift from Dr Brian Kraemer, University of Washington, Seattle) (45). Extrachromosomal arrays were generated by injection of tau transgenes (10 ng/μl) into day 1 adult C. elegans, along with Pmyo-3::RFP as a co-injection marker (50 ng/μl). Extrachromosomal arrays were integrated by UV irradiation (86) and outcrossed 5× to our lab’s WT N2E animals.
For behavioral assays, the following transgenic strains were used: CF3810 N2E; muIs216 [aex-3p::huMAPT 4RIN + myo-3p::rfp], CF3827 N2E; muIs217 [aex-3p:: huMAPT 4RIN A152T + myo-3p::rfp], AWK63 N2E; rocEx27 [aex-3p:: huMAPT 4R1N A152E + myo-3p::rfp], AWK114 N2E; rocEx36 [aex-3p:: huMAPT 4R1N T153A + myo-3p::rfp], AWK223 N2E; rocEx47 [aex-3p:: huMAPT 4R1N A152T, T153A + myo-3p::rfp], AWK121 N2E; rocEx37 [aex-3p:: huMAPT 4R1N A152E, T153A + myo-3p::rfp], AWK70 N2E; rocEx34 [aex-3p:: huMAPT 4R1N T153E + myo-3p::rfp], AWK136 N2E; rocEx38 [aex-3p:: huMAPT 4R1N A152T, T153E + myo-3p::rfp], AWK169 N2E; rocEx39 [aex-3p:: huMAPT 4R1N A152E, T153E + myo-3p::mCherry], AWK56 N2E; rocEx20 [aex-3p:: huMAPT 4R1N + myo-3p::rfp], AWK58 N2E; rocEx22 [aex-3p:: huMAPT 4R1N A152T + myo-3p::rfp], CZ1200 juIs76 [unc-25p::GFP + lin-15(+)] II; lin-15B(n765) X, AWK530 juIs76 [unc-25p::GFP + lin-15(+)] II; lin-15B(n765) X; muIs216 [aex-3p::huMAPT 4RIN + myo-3p::rfp], AWK531 juIs76 [unc-25p::GFP + lin-15(+)] II; lin-15B(n765) X; rocEx22 [aex-3p:: huMAPT 4R1N A152T + myo-3p::rfp], AWK532 juIs76 [unc-25p::GFP + lin-15(+)] II; lin-15B(n765) X; rocEx27 [aex-3p:: huMAPT 4R1N A152E + myo-3p::rfp], AWK533 juIs76 [unc-25p::GFP + lin-15(+)] II; lin-15B(n765) X; rocEx36 [aex-3p:: huMAPT 4R1N T153A + myo-3p::rfp], AWK534 juIs76 [unc-25p::GFP + lin-15(+)] II; lin-15B(n765) X; rocEx47 [aex-3p:: huMAPT 4R1N A152T, T153A + myo-3p::rfp], ZB154 zdIs5 [mec-4p::gfp], AWK535 zdIs5 [mec-4p::gfp]; muIs216 [aex-3p::huMAPT 4RIN + myo-3p::rfp], AWK536 zdIs5 [mec-4p::gfp]; muIs217 [aex-3p::huMAPT 4RIN A152T + myo-3p::rfp], AWK537 zdIs5 [mec-4p::gfp]; rocEx27 [aex-3p::huMAPT 4R1N A152E + myo-3p::rfp], AWK538 zdIs5 [mec-4p::gfp]; rocEx36 [aex-3p::huMAPT 4R1N T153A + myo-3p::rfp], AWK539 zdIs5 [mec-4p::gfp]; rocEx47 [aex-3p::huMAPT 4R1N A152T, T153A + myo-3p::rfp], CB211 lev-1(e211).
The jsIs821[mec-7p::gfp::rab-3 unc-119(+)] strain was provided by M. Nonet (Washington University in St Louis) and was outcrossed to the WT strain in the Encalada lab four times. To image axonal transport of GFP::RAB-3 vesicles in PLM touch receptor neurons of WT tau, A152T tau and A152E tau transgenic animals, the following strains were constructed:
SEE175 N2E; muIs217[aex-3p::huMAPT 4R1N A152T + myo-3p::rfp]; mec-7p::gfp::rab-3, SEE174 N2E; muIs216[aex-3p::huMAPT 4R1N + myo-3p::rfp]; mec-7p::gfp::rab-3
and AWK288 N2E; rocEx27 [aex-3p::huMAPT 4R1N A152E + myo-3p::rfp]; mec-7p::gfp::rab-3.
Behavioral assays
All behavioral assays were performed at 20°C.
Thrashing assay
Day 1 and day 4 adult animals were placed in a drop of M9 buffer and allowed to equilibrate for at least 1 min. The number of thrashes (defined as a movement of the worm that swings its head to the same side) was counted for 1 min.
Pumping assay
Pumping rates were quantified by measuring the number of contractions of the terminal bulb over 30 s.
Pharmacological paralysis assays
Aldicarb (acetylcholine esterase inhibitor) and levamisole (acetylcholine receptor agonist) sensitivity assays were performed on day 1 and day 4 adulthood worms by transferring animals to plates containing 1 mM aldicarb (Sigma #33386) and 0.2 mM levamisole (Fisher Scientific #AC187870100), respectively. Twenty-five animals per strain were scored for paralysis every 30 min for 6 h, and each experiment was repeated three times.
Hatching assay
Embryos were collected at the comma stage (390–420 min after first cleavage), and the fraction of unhatched larvae remaining contained within their eggshell was quantified 24 h later.
Development assay
Embryos were collected at the comma stage and scored for development to either larval stage L4 or adulthood after 72 h.
Sterility assay
L4 animals were singled onto individual plates, and plates were scored 48 h later for progeny. Animals with no viable progeny (no embryos or no embryos that hatched) were scored as sterile.
Learning and memory assay
Short-term associative memory assays were performed as described previously (39). Briefly, 100–200 synchronized, ad libitum–fed day 1 adult animals were starved in S-basal for 1 h and then conditioned with food and 2 ul of 10% butanone (in ethanol) for 1 h. Animals were then used in a chemotaxis assay where they were allowed to choose between a source of 10% butanone and a source of ethanol. After 1 h a chemotaxis index was calculated as the number of animals at the butanone source minus those at the ethanol source divided by the total number of animals. The learning index was calculated as the chemotaxis index of conditioned animals minus the chemotaxis index of ad libitum–fed animals that had not been previously starved or exposed to butanone.
Lifespan assay
One hundred forty L4 stage animals were transferred to regular NGM plates seeded with Escherichia coli OP50 and scored every day for survival by prodding gently on the nose with a platinum wire covered with bacteria. Worms that failed to respond were scored as dead. Animals were transferred to new plates every day for the first 5 days and then every 2–3 days after the worms had stopped laying eggs. Animals that crawled off the plate, bagged or died from vulval bursting were censored from the analysis.
Neuronal imaging
The ALM mechanosensory neuron and the GABAergic motor neurons were imaged by mounting immobilized young adult animals in 2.5 mM levamisole (Fisher Scientific #AC187870100) on glass slides with 2% agarose pads and imaged using a 40× and 63× objective on a Zeiss M2 AxioImager.
Immunoblotting
Synchronized day 1 adult animals were collected from plates with ice cold M9 and washed twice to remove OP50 food. The worm pellet was resuspended 1:1 in freshly made ice cold RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% SDS, 0.5% SDO, 1% NP-40, 1 mM PMSF, cOmplete protease inhibitor (Roche, #04693124001) and PhosSTOP phosphatase inhibitor (Roche, #04906837001)). Worms were transferred to Eppendorf tubes and sonicated for four cycles of 1 min on and 2 min off (BioRuptor, Diagenode). Lysates were centrifuge for 5 min at 13 000 rpm at 4°C. Supernatant was transferred to a fresh Eppendorf tube, and samples were boiled at 95°C (with 4× LDS, 10% reducing agent) for 5 min and analyzed by SDS PAGE. A total of 50 μg total protein was resolved on 4–12% gradient SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membrane. Total tau level was immunoblotted with Tau A12 antibody (Santa Cruz Biotechnology #sc-166062) and normalized to actin (Millipore #MAB1501).
RT-qPCR
Total RNA was harvested from synchronized day 1 adult animals. Animals were collected from plates with ice cold M9 and washed three times to remove OP50 food. After harvesting, the animals were resuspended in QIAzol (Qiagen #79306) and flash frozen in liquid nitrogen. RNA was extracted and purified using a Qiagen miRNeasy kit (Qiagen #217004) and subjected to an additional DNA digestion step (Thermo Fisher #18068015). Reverse transcription (1 μg RNA per sample) was performed using SuperScript III Reverse Transcriptase (Thermo Fisher #18080044), random primers (Thermo Fisher #48190011) and dNTPs (Sigma #11969064001). RT-qPCR was performed using TaqMan Fast Advanced Master Mix (Thermo Fisher #4444557) in a ABI Prism 7900HT Sequence Detection System (Applied Biosystems), with TaqMan FAM probes for human MAPT (Thermo Fisher #Hs00902194_m1) and the C. elegans house-keeping gene pmp-3 (putative ABC transporter) (Thermo Fisher #Ce02485188_m1). A standard curve was obtained for each primer set by serially diluting a mixture of different complementary DNAs, and the standard curves were used to convert the observed CT values to relative values. Three biological samples were analysed, each with three technical replicates. mRNA levels of target genes were normalized to the mean of the housekeeping gene pmp-3. Data are displayed as relative values compared to WT tau.
Dendra2 photoconversion assay
A total of 7 to 10 DIV mouse cortical primary neurons were transfected using Lipofectamine 2000 (Thermo Fisher) with the respective Dendra2 constructs. We previously showed that Tau-Dendra2 is very stable when transfected into primary neurons and shows no signs of degradation or truncation (11,12,43). Transfected cells were analyzed 48 h post-transfection. The axon of healthy transfected cells was found based on morphology. A reference image was taken (green channel, unconverted Dendra2), and a rectangular region of interest (ROI) was drawn for photoconversion. This ROI was drawn as such to have an exposure time of 1 s (405 nm laser, power = 15%) for photoconversion. After photoconversion, 120 images were taken (red channel, converted Dendra2) with intervals of 30 s (total 1 h) to monitor the fluorescence in the axon and the soma simultaneously.
In vivo imaging and analyses of C. elegans axonal transport
In vivo live imaging
L4 larvae of each of the three strains, SEE175 N2E; muIs217[aex-3p::huMAPT 4R1N A152T + myo-3p::rfp]; mec-7p::gfp::rab-3 unc-119(+), SEE174 N2E; muIs216[aex-3p::huMAPT 4R1N + myo-3p::rfp]; mec-7p::gfp::rab-3 unc-119(+) and AWK288 N2E; rocEx27 [aex-3p::huMAPT 4R1N A152E + myo-3p::rfp]; mec-7p::gfp::rab-3, were picked onto 6 cm plates and placed at 22°C overnight. Day 1 animals were immobilized inside microfluidic devices (Alex Groisman, Edgar Gutierrez, Sandra Encalada, unpublished data) in M9 buffer (22 mM KH2PO4; 42 mM Na2PO4; 86 mM NaCl; 1 μM MgSO4). The movement of GFP::RAB-3 vesicles in either of the PLM neurons was imaged live with a Nikon Ti-E Perfect Focus inverted microscope equipped with a iXon+ DU897 EM Camera, using a 100X/1.49 NA oil objective, and a 488 nm laser with a pseudo-total internal reflection fluorescent angle setting. PLM neurons were identified by their position toward the posterior of the animal, near the tail (http://www.wormatlas.org/neurons/Individual%20Neurons/PLMframeset.html). Movies were 15 s long and collected at 10 frames per second at 100 ms exposure (10 Hz) using the Nikon Elements acquisition software. The pixel size was 0.126 μm.
Transport data analyses
We used a custom ImageJ software called ‘KymoAnalyzer’ with a series of macros (87) to trace the trajectories of each GFP::RAB-3 particle at a high temporal and spatial resolution of 10 Hz and 0.126 μm, respectively. We assigned tracks (trajectories) and segments within tracks for each particle for each animal of each strain analyzed. The cumulative segmental velocities and cumulative run lengths were calculated on ‘combined segments’, which are defined as uninterrupted periods of anterograde or retrograde movement within a track. Once transport parameters were obtained from KymoAnalyzer, figures were plotted in Excel spreadsheets.
Statistical analyses for axonal transport parameters
All transport parameters were first tested for normality using the Lilliefors test implemented in the nortest package of R. The cargo population and density parameters were normally distributed so the data plotted were for the mean percentage of anterograde, retrograde, reversing and stationary vesicles ± SEM and were analyzed using parametric one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons tests. The cumulative segmental velocities and cumulative run lengths data were analyzed using the non-parametric Kolmogorov–Smirnov test and multiple comparisons Bonferroni’s correction. Statistical analyses were performed using Matlab, R and PRISM GraphPad, and results were considered significant when the P-value was <0.05.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We thank Michael Nonet and Lenny Guarente for providing C. elegans strains. We thank Lennart Mucke and Melanie Das for provision of brain tissue from hTau-A152T and hTau-WT transgenic mice. We thank Bikash Choudhary and Jeelani Pir for helpful discussion. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Conflict of Interest statement. None declared.
Funding
Rainwater Tau Consortium, National Institutes of Health/National Institute on Aging (R01NS095257) and the Paul G. Allen Family Foundation (to A.W.K.); National Institutes of Health/National Institute on Aging (R01AG046400, R01AG011816 to K.A.); a Glenn/American Federation for Aging Research (AFAR) Aging Research Scholarship and Hillblom Foundation Graduate Student Fellowship (to M.V.); The Ellison Medical Foundation, The Baxter Foundation, The Glenn Foundation for Medical Research, Arlene and Arnold Goldstein and National Institutes of Health/National Institute on Aging (R01AG049483 to S.E.E.); FCT fellowship (SFRH/BD/101352/2014 to M.A.F). Mass spectrometry was provided by the Bio-Organic Biomedical Mass Spectrometry Resource at University of California San Francisco (A.L. Burlingame, Director) supported by the Biomedical Technology Research Centers program of the National Institutes of Health/National Institute of General Medical Sciences (8P41GM103481) and Howard Hughes Medical Institute.
References
- 1. Andreadis A., Brown W.M. and Kosik K.S. (1992) Structure and novel exons of the human tau gene. Biochemistry, 31, 10626–10633. [DOI] [PubMed] [Google Scholar]
- 2. Weingarten M.D., Lockwood A.H., Hwo S.Y. and Kirschner M.W. (1975) A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U. S. A., 72, 1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Witman G.B., Cleveland D.W., Weingarten M.D. and Kirschner M.W. (1976) Tubulin requires tau for growth onto microtubule initiating sites. Proc. Natl. Acad. Sci. U. S. A., 73, 4070–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ballatore C., Lee V.M. and Trojanowski J.Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci., 8, 663–672. [DOI] [PubMed] [Google Scholar]
- 5. Coppola G., Chinnathambi S., Lee J.J., Dombroski B.A., Baker M.C., Soto-Ortolaza A.I., Lee S.E., Klein E., Huang A.Y., Sears R. et al. (2012) Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum. Mol. Genet., 21, 3500–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kara E., Ling H., Pittman A.M., Shaw K., Silva R., Simone R., Holton J.L., Warren J.D., Rohrer J.D., Xiromerisiou G. et al. (2012) The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol. Aging, 33, 2231 e2237–2231 e2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kovacs G.G., Wohrer A., Strobel T., Botond G., Attems J. and Budka H. (2011) Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin. Neuropathol., 30, 3–10. [DOI] [PubMed] [Google Scholar]
- 8. Labbe C., Ogaki K., Lorenzo-Betancor O., Soto-Ortolaza A.I., Walton R.L., Rayaprolu S., Fujioka S., Murray M.E., Heckman M.G., Puschmann A. et al. (2015) Role for the microtubule-associated protein tau variant p.A152T in risk of alpha-synucleinopathies. Neurology, 85, 1680–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y. and Mandelkow E. (2016) Tau in physiology and pathology. Nat. Rev. Neurosci., 17, 5–21. [DOI] [PubMed] [Google Scholar]
- 10. Leterrier C. (2018) The axon initial segment: an updated viewpoint. J. Neurosci., 38, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X., Kumar Y., Zempel H., Mandelkow E.M., Biernat J. and Mandelkow E. (2011) Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J., 30, 4825–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zempel H., Dennissen F.J.A., Kumar Y., Luedtke J., Biernat J., Mandelkow E.M. and Mandelkow E. (2017) Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J. Biol. Chem., 292, 12192–12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holt L.J. (2012) Regulatory modules: coupling protein stability to phopshoregulation during cell division. FEBS Lett., 586, 2773–2777. [DOI] [PubMed] [Google Scholar]
- 14. Davey N.E. and Morgan D.O. (2016) Building a regulatory network with short linear sequence motifs: lessons from the degrons of the anaphase-promoting complex. Mol. Cell, 64, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poppek D., Keck S., Ermak G., Jung T., Stolzing A., Ullrich O., Davies K.J. and Grune T. (2006) Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem. J., 400, 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alonso A.C., Grundke-Iqbal I. and Iqbal K. (1996) Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med., 2, 783–787. [DOI] [PubMed] [Google Scholar]
- 17. Uyar B., Weatheritt R.J., Dinkel H., Davey N.E. and Gibson T.J. (2014) Proteome-wide analysis of human disease mutations in short linear motifs: neglected players in cancer? Mol. BioSyst., 10, 2626–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Decker J.M., Kruger L., Sydow A., Dennissen F.J., Siskova Z., Mandelkow E. and Mandelkow E.M. (2016) The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep., 17, 552–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maeda S., Djukic B., Taneja P., Yu G.Q., Lo I., Davis A., Craft R., Guo W., Wang X., Kim D. et al. (2016) Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep., 17, 530–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shafiei S.S., Guerrero-Munoz M.J. and Castillo-Carranza D.L. (2017) Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci., 9, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pir G.J., Choudhary B., Mandelkow E. and Mandelkow E.M. (2016) Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener., 11, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choudhary B., Mandelkow E., Mandelkow E.M. and Pir G.J. (2018) Glutamatergic nervous system degeneration in a C. elegans Tau(A152T) tauopathy model involves pathways of excitotoxicity and Ca(2+) dysregulation. Neurobiol. Dis., 117, 189–202. [DOI] [PubMed] [Google Scholar]
- 23. Krebs E.G. (1983) Historical perspectives on protein phosphorylation and a classification system for protein kinases. Philos. Trans. R. Soc. Lond. B Biol. Sci., 302, 3–11. [DOI] [PubMed] [Google Scholar]
- 24. Iakoucheva L.M., Radivojac P., Brown C.J., O'Connor T.R., Sikes J.G., Obradovic Z. and Dunker A.K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res., 32, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goedert M. and Jakes R. (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J., 9, 4225–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hong M., Zhukareva V., Vogelsberg-Ragaglia V., Wszolek Z., Reed L., Miller B.I., Geschwind D.H., Bird T.D., McKeel D., Goate A. et al. (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science, 282, 1914–1917. [DOI] [PubMed] [Google Scholar]
- 27. Augustinack J.C., Schneider A., Mandelkow E.M. and Hyman B.T. (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol., 103, 26–35. [DOI] [PubMed] [Google Scholar]
- 28. Bailey R.M., Covy J.P., Melrose H.L., Rousseau L., Watkinson R., Knight J., Miles S., Farrer M.J., Dickson D.W., Giasson B.I. et al. (2013) LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol., 126, 809–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamm M., Bailey R., Shaw G., Yen S.H., Lewis J. and Giasson B.I. (2015) Physiologically relevant factors influence tau phosphorylation by leucine-rich repeat kinase 2. J. Neurosci. Res., 93, 1567–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manning G., Plowman G.D., Hunter T. and Sudarsanam S. (2002) Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci., 27, 514–520. [DOI] [PubMed] [Google Scholar]
- 31. Illenberger S., Zheng-Fischhofer Q., Preuss U., Stamer K., Baumann K., Trinczek B., Biernat J., Godemann R., Mandelkow E.M. and Mandelkow E. (1998) The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer's disease. Mol. Biol. Cell, 9, 1495–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ravid T. and Hochstrasser M. (2008) Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol., 9, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kraemer B.C., Zhang B., Leverenz J.B., Thomas J.H., Trojanowski J.Q. and Schellenberg G.D. (2003) Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. U. S. A., 100, 9980–9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Avery L. and Horvitz H.R. (1987) A cell that dies during wild-type C. elegans development can function as a neuron in a ced-3 mutant. Cell, 51, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Avery L. and Horvitz H.R. (1989) Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron, 3, 473–485. [DOI] [PubMed] [Google Scholar]
- 36. Mahoney T.R., Luo S. and Nonet M.L. (2006) Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat. Protoc., 1, 1772–1777. [DOI] [PubMed] [Google Scholar]
- 37. Miller K.G., Alfonso A., Nguyen M., Crowell J.A., Johnson C.D. and Rand J.B. (1996) A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. U. S. A., 93, 12593–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pan C.L., Peng C.Y., Chen C.H. and McIntire S. (2011) Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. U. S. A., 108, 9274–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kauffman A.L., Ashraf J.M., Corces-Zimmerman M.R., Landis J.N. and Murphy C.T. (2010) Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol., 8, e1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alcedo J. and Kenyon C. (2004) Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron, 41, 45–55. [DOI] [PubMed] [Google Scholar]
- 41. Taylor R.C. and Dillin A. (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell, 153, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones S.L. and Svitkina T.M. (2016) Axon initial segment cytoskeleton: architecture, development, and role in neuron polarity. Neural Plast., 2016, 6808293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Konzack S., Thies E., Marx A., Mandelkow E.M. and Mandelkow E. (2007) Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J. Neurosci., 27, 9916–9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zempel H. and Mandelkow E. (2014) Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci., 37, 721–732. [DOI] [PubMed] [Google Scholar]
- 45. Iwasaki K. and Toyonaga R. (2000) The Rab3 GDP/GTP exchange factor homolog AEX-3 has a dual function in synaptic transmission. EMBO J., 19, 4806–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kumar J., Choudhary B.C., Metpally R., Zheng Q., Nonet M.L., Ramanathan S., Klopfenstein D.R. and Koushika S.P. (2010) The Caenorhabditis elegans Kinesin-3 motor UNC-104/KIF1A is degraded upon loss of specific binding to cargo. PLoS Genet., 6, e1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iqbal K., Liu F. and Gong C.X. (2016) Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol., 12, 15–27. [DOI] [PubMed] [Google Scholar]
- 48. Lawrence R.T., Searle B.C., Llovet A. and Villen J. (2016) Plug-and-play analysis of the human phosphoproteome by targeted high-resolution mass spectrometry. Nat. Methods, 13, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lopez A., Lee S.E., Wojta K., Ramos E.M., Klein E., Chen J., Boxer A.L., Gorno-Tempini M.L., Geschwind D.H., Schlotawa L. et al. (2017) A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain, 140, 1128–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Doble B.W. and Woodgett J.R. (2003) GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci., 116, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fiol C.J., Mahrenholz A.M., Wang Y., Roeske R.W. and Roach P.J. (1987) Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J. Biol. Chem., 262, 14042–14048. [PubMed] [Google Scholar]
- 52. Xu C., Kim N.G. and Gumbiner B.M. (2009) Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle, 8, 4032–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lyons N.A., Fonslow B.R., Diedrich J.K., Yates J.R. 3rd and Morgan D.O. (2013) Sequential primed kinases create a damage-responsive phosphodegron on Eco1. Nat. Struct. Mol. Biol., 20, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Welcker M. and Clurman B.E. (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer, 8, 83–93. [DOI] [PubMed] [Google Scholar]
- 55. Goedert M., Jakes R., Crowther R.A., Six J., Lubke U., Vandermeeren M., Cras P., Trojanowski J.Q. and Lee V.M. (1993) The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. USA, 90, 5066–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J.Z. and Liu F. (2008) Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol., 85, 148–175. [DOI] [PubMed] [Google Scholar]
- 57. Yu Y., Run X., Liang Z., Li Y., Liu F., Liu Y., Iqbal K., Grundke-Iqbal I. and Gong C.X. (2009) Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem., 108, 1480–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kenessey A. and Yen S.H. (1993) The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res., 629, 40–46. [DOI] [PubMed] [Google Scholar]
- 59. Serrano L., Montejo de Garcini E., Hernandez M.A. and Avila J. (1985) Localization of the tubulin binding site for tau protein. Eur. J. Biochem., 153, 595–600. [DOI] [PubMed] [Google Scholar]
- 60. Yagensky O., Kalantary Dehaghi T. and Chua J.J. (2016) The roles of microtubule-based transport at presynaptic nerve terminals. Front. Synaptic Neurosci., 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tienari P.J., De Strooper B., Ikonen E., Simons M., Weidemann A., Czech C., Hartmann T., Ida N., Multhaup G., Masters C.L. et al. (1996) The beta-amyloid domain is essential for axonal sorting of amyloid precursor protein. EMBO J., 15, 5218–5229. [PMC free article] [PubMed] [Google Scholar]
- 62. Encalada S.E., Szpankowski L., Xia C.H. and Goldstein L.S. (2011) Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell, 144, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gross S.P., Welte M.A., Block S.M. and Wieschaus E.F. (2002) Coordination of opposite-polarity microtubule motors. J. Cell Biol., 156, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kural C., Kim H., Syed S., Goshima G., Gelfand V.I. and Selvin P.R. (2005) Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science, 308, 1469–1472. [DOI] [PubMed] [Google Scholar]
- 65. Lyman M.G. and Enquist L.W. (2009) Herpesvirus interactions with the host cytoskeleton. J. Virol., 83, 2058–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Plitz T. and Pfeffer K. (2001) Intact lysosome transport and phagosome function despite kinectin deficiency. Mol. Cell. Biol., 21, 6044–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shubeita G.T., Tran S.L., Xu J., Vershinin M., Cermelli S., Cotton S.L., Welte M.A. and Gross S.P. (2008) Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell, 135, 1098–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Soppina V., Rai A.K., Ramaiya A.J., Barak P. and Mallik R. (2009) Tug-of-war between dissimilar teams of microtubule motors regulates transport and fission of endosomes. Proc. Natl. Acad. Sci. U. S. A., 106, 19381–19386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Welte M.A. (2004) Bidirectional transport along microtubules. Curr. Biol., 14, R525–R537. [DOI] [PubMed] [Google Scholar]
- 70. Hancock W.O. (2014) Bidirectional cargo transport: moving beyond tug of war. Nat. Rev. Mol. Cell Biol., 15, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dixit R., Ross J.L., Goldman Y.E. and Holzbaur E.L. (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science, 319, 1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ebneth A., Godemann R., Stamer K., Illenberger S., Trinczek B. and Mandelkow E. (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol., 143, 777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mandelkow E.M., Stamer K., Vogel R., Thies E. and Mandelkow E. (2003) Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging, 24, 1079–1085. [DOI] [PubMed] [Google Scholar]
- 74. Stamer K., Vogel R., Thies E., Mandelkow E. and Mandelkow E.M. (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol., 156, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hoeprich G.J., Thompson A.R., McVicker D.P., Hancock W.O. and Berger C.L. (2014) Kinesin's neck-linker determines its ability to navigate obstacles on the microtubule surface. Biophys. J., 106, 1691–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vershinin M., Carter B.C., Razafsky D.S., King S.J. and Gross S.P. (2007) Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. U. S. A., 104, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maday S. and Holzbaur E.L. (2014) Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell, 30, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Perlson E., Maday S., Fu M.M., Moughamian A.J. and Holzbaur E.L. (2010) Retrograde axonal transport: pathways to cell death? Trends Neurosci., 33, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hendry I.A., Stockel K., Thoenen H. and Iversen L.L. (1974) The retrograde axonal transport of nerve growth factor. Brain Res., 68, 103–121. [DOI] [PubMed] [Google Scholar]
- 80. Niewiadomska G., Baksalerska-Pazera M. and Riedel G. (2005) Altered cellular distribution of phospho-tau proteins coincides with impaired retrograde axonal transport in neurons of aged rats. Ann. N. Y. Acad. Sci., 1048, 287–295. [DOI] [PubMed] [Google Scholar]
- 81. Magnani E., Fan J., Gasparini L., Golding M., Williams M., Schiavo G., Goedert M., Amos L.A. and Spillantini M.G. (2007) Interaction of tau protein with the dynactin complex. EMBO J., 26, 4546–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rosenfeld J., Capdevielle J., Guillemot J.C. and Ferrara P. (1992) In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem., 203, 173–179. [DOI] [PubMed] [Google Scholar]
- 83. Duong B.H., Onizawa M., Oses-Prieto J.A., Advincula R., Burlingame A., Malynn B.A. and Ma A. (2015) A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity, 42, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Clauser K.R., Baker P. and Burlingame A.L. (1999) Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal. Chem., 71, 2871–2882. [DOI] [PubMed] [Google Scholar]
- 85. Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Frank C.A., Baum P.D. and Garriga G. (2003) HLH-14 is a C. elegans achaete-scute protein that promotes neurogenesis through asymmetric cell division. Development, 130, 6507–6518. [DOI] [PubMed] [Google Scholar]
- 87. Neumann S., Chassefeyre R., Campbell G.E. and Encalada S.E. (2017) KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic, 18, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.