

Abstract
Schwannomas are common, highly morbid and medically untreatable tumors that can arise in patients with germ line as well as somatic mutations in neurofibromatosis type 2 (NF2). These mutations most commonly result in the loss of function of the NF2-encoded protein, Merlin. Little is known about how Merlin functions endogenously as a tumor suppressor and how its loss leads to oncogenic transformation in Schwann cells (SCs). Here, we identify nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-inducing kinase (NIK) as a potential drug target driving NF-κB signaling and Merlin-deficient schwannoma genesis. Using a genomic approach to profile aberrant tumor signaling pathways, we describe multiple upregulated NF-κB signaling elements in human and murine schwannomas, leading us to identify a caspase-cleaved, proteasome-resistant NIK kinase domain fragment that amplifies pathogenic NF-κB signaling. Lentiviral-mediated transduction of this NIK fragment into normal SCs promotes proliferation, survival, and adhesion while inducing schwannoma formation in a novel in vivo orthotopic transplant model. Furthermore, we describe an NF-κB-potentiated hepatocyte growth factor (HGF) to MET proto-oncogene receptor tyrosine kinase (c-Met) autocrine feed-forward loop promoting SC proliferation. These innovative studies identify a novel signaling axis underlying schwannoma formation, revealing new and potentially druggable schwannoma vulnerabilities with future therapeutic potential.
Significance Statement
We report the discovery of a novel, proteasome-resistant fragment of NIK that drives schwannoma formation. This NIK/NF-κB signaling axis directs an autocrine HGF–c-Met signaling loop that sustains schwannoma proliferation, identifying vulnerabilities with future therapeutic potential.
Introduction
Neurofibromatosis type 2 (NF2) is an autosomal dominant neurocutaneous syndrome caused by mutations of the NF2 tumor suppressor gene that encodes the protein Merlin. NF2 patients typically present in young adulthood or, less commonly, in childhood, with multiple intracranial and spinal schwannomas, meningiomas and ependymomas, resulting in neurologic disability and early mortality. Tumor-related morbidities range from multiple cranial neuropathies to myelopathy and peripheral neuropathy. Although NF2 is relatively rare (1 in 25 000–33 000 live births), its hallmark tumors [vestibular schwannomas (VSs) and meningiomas] are the world’s most common adult neural tumors (1,2). Similar to NF2 patients, these spontaneously arising tumors demonstrate NF2 mutations. Unfortunately, no medical alternatives to surgery exist for NF2-associated or spontaneously arising NF2-mutated tumors, and surgically related morbidities like permanent loss of nerve function for VS patients are not uncommon.
Despite the NF2 gene’s identification dating back to 1993 (3), an understanding of the gene product’s (Merlin) tumor suppressor function remains incomplete. Merlin-deficient schwannomas have been shown to have hyperactivation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling (4,5), but the precise mechanisms by which Merlin influences NF-κB signaling remain elusive. The NF-κB signaling pathway and its transcription factors (NF-κB1/p105, NF-κB2/p100, RELA, RELB and c-REL) mediate organogenesis, cell proliferation, cell survival, inflammation, and oncogenesis in many cancers, potentially including human nerve sheath and glial tumors (6,7).
To interrogate the signaling pathways critical to schwannoma pathogenesis, we employed transcriptomic profiling of primary human schwannomas. Similar approaches utilizing transcriptomics have yielded unique insights into a related neurocutaneous syndrome, neurofibromatosis type 1 (8,9). Consistent with prior findings, human schwannomas displayed activation of multiple NF-κB-related kinases and increased expression of NF-κB-related genes. Through the use of our recently published NF2 mouse model (10), we confirmed that chronic NF-κB activation was also present in murine Merlin-deficient schwannomas. Biochemical analyses revealed that this activated NF-κB signaling depended on ∼55 kDa NF-κB-inducing kinase (NIK) kinase domain fragment produced by caspase-8-mediated cleavage. Intriguingly, this fragment demonstrated resistance to the proteasome-mediated degradation that normally regulates full-length NIK signaling. Schwann cells (SCs) transduced with this NIK fragment demonstrated increased proliferation, survival and adhesion and formed tumors when transplanted into the sciatic nerve of nude mice. We also describe an NF-κB-driven hepatocyte growth factor (HGF) to c-Met autocrine feed-forward signaling loop that sustains schwannoma cell proliferation. In all, this study reveals that a proteasome-resistant NIK fragment drives oncogenic NF-κB signaling in Merlin-deficient SCs. With multiple NIK small molecules currently in preclinical development (11,12), these studies provide a novel molecular insight into schwannoma genesis with highly relevant therapeutic implications.
Results
The NF-κB pathway is activated in schwannomas
Using microarray data from 31 human VSs in the Gene Expression Omnibus (GEO) (13,14), we identified differentially expressed genes (DEGs) which, when input into Ingenuity Pathway Analysis (IPA; Qiagen, Redwood City, CA, USA) (15), revealed 93 deregulated NF-κB pathway elements as compared to normal nerve samples (Fig. 1A, Supplementary Files 1 and 2), including genes identified as either NF2-regulated or overexpressed in schwannomas (e.g. CCND1 and ERBB2) (16,17). In corroboration, quantitative real time polymerase chain reaction (RT-PCR) of schwannomas from our recently characterized NF2 tumor model (Postn-Cre+; Nf2flox/flox; henceforth NF2-KO) and wild-type (WT) (Postn-Cre−; Nf2flox/flox) nerve tissue confirmed NF-κB target gene (Fig. 1B) and transcription factor (Supplementary Material, Fig. 1A) overexpression. Thus, there appears to be a shared molecular signature between human and murine schwannomas (10). Immunoblotting demonstrated increased levels of p100, RELA, RELB and c-REL, proteins encoded by Nfkb2, Rela, Relb and Rel genes, respectively, and increased levels of p52 (Fig. 1C), a marker of non-canonical NF-κB (18). We also observed an increase in p50 but not the precursor protein p105 (Supplementary Material, Fig. 1B). Immunohistochemistry (IHC) and fractionated immunoblotting similarly demonstrated increased total levels and nuclear translocation of the NF-κB transcriptional activators, RELA and RELB, in human and murine schwannomas (Fig. 1D and E, Supplementary Material, Fig. 1C and D). We examined murine schwannomas in dorsal root ganglia, cranial nerve V (CNV), cranial nerve VIII (CNVIII), sciatic nerves and cutaneous tissues and have found NF-κB activation present in all tumors. In all, these transcriptomic and biochemical analyses reveal a net increase in canonical and non-canonical NF-κB signaling in human and murine schwannomas.
Figure 1.
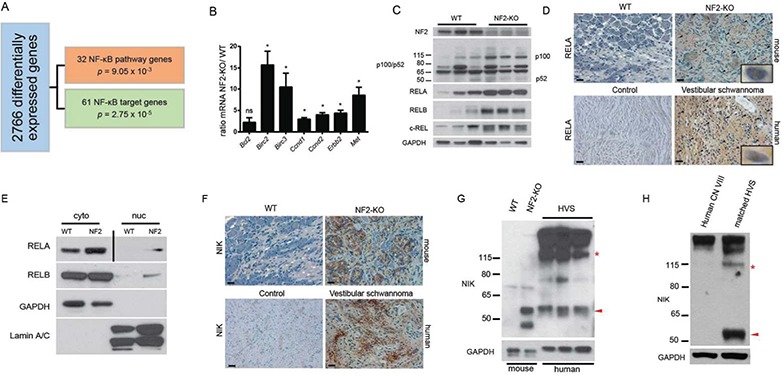
NF-κB pathway activation in schwannomas. (A) IPA of human schwannoma microarray data identifies 2766 differentially regulated genes in the tumor tissue, 93 of which are NF-κB signaling elements and target genes. (B) Validation of deregulated NF-κB genes by qRT-PCR in primary nerve tissues (WT n = 3; NF2-KO n = 3). ns = not significant, *p < 0.05. Unpaired Student’s t-test with Holm–Sidak correction for multiple comparisons, α = 0.05. Error bars represent standard error of the mean (SEM). (C) IB demonstrating increased NF-κB transcription factors and signaling elements in NF2-KO nerve tissues. (D) IHC of RELA in murine (upper) and human (lower) tissues. Scale bar = 20 μm. Original magnification ×400. Arrowheads point to representative nuclei with positive staining for RELA. 1000× insets are examples of positive nuclear staining. (E) IB of fractionated primary nerve tissue from WT and NF2-KO mice demonstrating increased cytoplasmic and nuclear levels of RELA and RELB proteins. Vertical bar indicates different exposures. NF2 = NF2-KO, cyto = cytoplasmic fraction, nuc = nuclear fraction. (F) IHC of NIK in murine (upper) and human (lower) tissues. Scale bar = 20 μm. Original magnification ×400. (G) IB demonstrating the accumulation of fragments of the kinase domain of NIK including a 55 kDa fragment (red arrowhead). Full-length NIK can be identified by the red asterisk at ∼120 kDa. HVS = spontaneous human vestibular schwannoma. (H) IB of primary HVS with matching control CN VIII tissues from the surrounding nerve in the same patient.
We next examined upstream NF-κB regulatory elements in human and murine schwannomas, finding overexpression of NIK (Fig. 1F–H, Supplementary Material, Fig. 2A–C). Intriguingly, immunoblotting of human and murine schwannomas with a kinase domain-specific peptide antibody revealed not only increased total NIK but also accumulation of ∼55 kDa NIK fragment, as compared to non-affected nerve tissue from the same mouse or patient (Fig. 1G and H, Supplementary Material, Fig. 2B and C). The NIK kinase domain is constitutively active (19) and in an in vitro NIK kinase assay using recombinant IKKα as substrate, we observed over a 50% increase in NIK kinase activity in murine schwannomas as compared to normal nerve tissue (Supplementary Material, Fig. 2D). This supports our hypothesis that the increased endogenous levels of NIK in the tumor tissues are sufficient to trigger an increase in substrate phosphorylation and downstream activation NF-κB transcription factors.
p55 NIK is a proteasome-resistant fragment of NIK that drives oncogenic signaling in SCs
Given our findings combined with previous observations that pathologic NIK cleavage drives mucosal associated lymphoid tissue (MALT) lymphoma formation (20) and limited in vitro data showing that caspase-8 can cleave NIK (21), we hypothesized that caspase-8 may produce a kinase-active NIK fragment that underlies schwannoma genesis. An unbiased analysis with Cascleave 2.0 (22) and SitePrediction (23) software predicted species-conserved caspase-8 cleavage clusters in N-terminus sites D291 and D308 and C-terminus sites D801, D807, D808 and D822 capable of producing ∼55 kDa NIK fragments containing the entire kinase domain (Fig. 2A, Supplementary Material, Fig. 3A and B, Supplementary Files 3 and 4). Co-transfection of NIK and caspase-8 into 293 T cells produced a 55 kDa NIK fragment, which was diminished by CrmA, a high-affinity caspase-1 and caspase-8 inhibitor (Fig. 2B). Likewise, incubating purified NIK with caspase-8 in vitro generated a 55 kDa fragment, which could be abrogated by the potent caspase inhibitor Q-VD-Oph (Fig. 2C). Co-immunoprecipitation demonstrated a physical interaction between NIK and caspase-8 (Fig. 2D). Furthermore, peptide-based antibodies specifically targeting murine amino acids 311–328 and a peptide surrounding human residue 659 confirmed that the 55 kDa fragment observed in primary tumor tissues includes the entire kinase domain of NIK (∼390 to 660 amino acids; Supplementary Material, Fig. 4A and B). Finally, mass spectroscopy of the purified 55 kDa fragment independently revealed 12 unique NIK peptides with near exclusive coverage of the NIK kinase domain (Supplementary Material, Fig. 5A–C).
Figure 2.
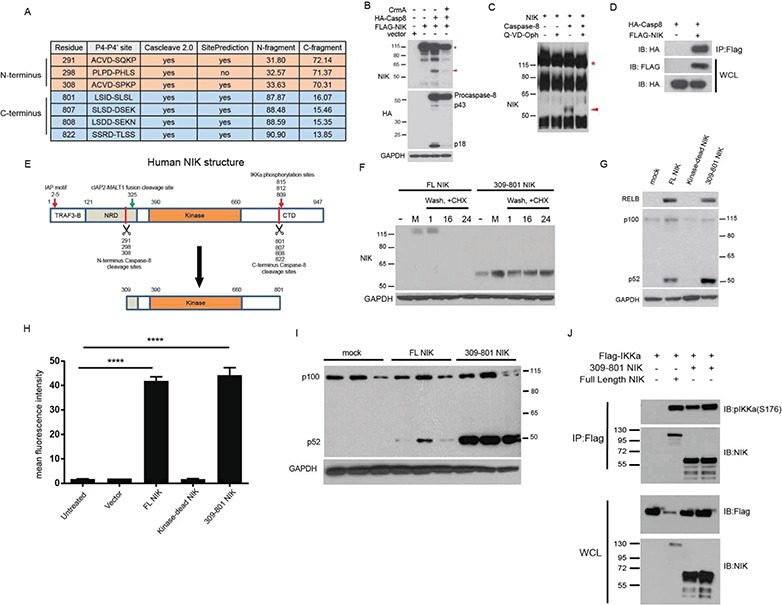
Caspase-8 cleaves NIK, resulting in a 55 kDa fragment of the kinase domain. (A) In silico prediction of human NIK cleavage by caspase-8 using Cascleave 2.0 and SitePrediction software programs. (B) IB of 293 T cells transfection experiment demonstrating that caspase-8 activity is sufficient for the cleavage of NIK to p55 NIK. (C) In vitro cleavage assay with purified NIK and recombinant caspase-8. (D) Co-immunoprecipitation experiment in 293 T cells with IP of FLAG-tagged NIK. (E) Structure of human NIK. TRAF3-B = TRAF3 binding domain, NRD = Negative Regulatory domain, Kinase= Kinase domain and CTD = C-terminal domain. (F) IB of 293 T cells transfected with the indicated plasmids. Cells were pulsed with proteasome inhibitor MG132, washed and incubated with the protein translation inhibitor CHX. Number labels indicate hour time course. (G) IB of 293 T cells transfected with the indicated plasmids. (H) Transfection experiment with Cignal-293 T cells in triplicate possessing an NF-κB responsive promoter upstream of a GFP reporter. ANOVA with Dunnett’s test for post hoc comparisons. ns = not significant, ****p < 0.0001; Error bars represent SEM. (I) Repeat of IB in (G) in triplicate showing increased processing of p100 to p52 in 309-801 NIK-transfected cells. (J) Co-immunoprecipitation experiment in 293 T cells with IP of FLAG-tagged IKKα. The red asterisk in (B) and (C) indicates full-length NIK, while the red arrowhead points to the 55 kDa fragment of the kinase domain. IB = Immunoblot, IP = Immunoprecipitation, WCL = whole cell lysate.
Importantly, this ∼55 kDa NIK fragment (p55 NIK) contains the functional NIK kinase domain but lacks critical NIK negative regulatory elements including the TRAF3 binding domain (TRAF3-B; residues 30–120), the majority of the negative regulatory domain (NRD; 121–317), the inhibitor of apoptosis (N-terminus) binding motif, and the destabilizing IKKα phosphorylation sites at 809, 812, and 815 (19,24–26) (Fig. 2E). We first confirmed that p55 NIK migrated similarly to a cDNA construct encoding NIK residues 309-801, the fragment generated by the innermost N- and C-termini cleavage sites (Supplementary Material, Fig. 5D). Hypothesizing that p55 NIK would demonstrate increased stability, we transfected plasmids encoding full-length NIK or NIK residues 309-801 into 293 T cells, pulsed with the proteasome inhibitor MG132, washed and then incubated for up to 24 h with the translation inhibitor cycloheximide (CHX). While we could not detect full-length NIK protein in the absence of proteasome inhibition or after 16 h CHX treatment, we detected 309-801 NIK at 55 kDa in the same conditions throughout 24 h CHX (Fig. 2F). Both full-length NIK and p55 NIK induced canonical and non-canonical NF-κB signaling, as evidenced by increased RELB levels and proteolytic processing of p100 to p52, respectively (Fig. 2G). Moreover, Cignal-293 T cells possessing an NF-κB responsive element upstream of GFP confirmed the 309-801 NIK fragment induces NF-κB activation (Fig. 2H). Of note, repeated experiments demonstrated that 309-801 NIK more robustly induces p100 processing to p52 as compared to full-length NIK, suggesting p55 NIK preferentially enhances non-canonical NF-kB signaling (Fig. 2G and 2I). Finally, co-immunoprecipitation of p55 NIK with IKKα verified p55 NIK’s ability to both interact with and phosphorylate its putative downstream target (Fig. 2J). Thus, p55 NIK resists proteasome-mediated degradation, retains functional kinase activity, and may preferentially induce non-canonical NF-kB signaling.
We next evaluated the functional effects of NIK signaling on SCs. small hairpin RNA (shRNA)-mediated knockdown of NIK reduced proliferation in an immortalized human schwannoma line and primary murine schwannoma cells by roughly 2-fold with multiple independent shRNAs (Fig. 3A and B, Supplementary Material, Fig. 6A). To examine p55 NIK’s biological impact on non-transformed primary SCs, we used lentiviral-mediated transduction to introduce either internal ribosomal entry site (IRES)-EGFP or 309-801 NIK IRES-EGFP into passage 1 or 2 (P1-P2) murine SCs, which maintained S100-positivity during culture (Supplementary Material, Fig. 6B). 309-801 NIK-transduced SCs demonstrated increased S-phase activity, proliferation, survival and adhesion to poly-D-lysine (PDL)/laminin-coated plates as measured by iCelligence real-time monitoring (Fig. 3C–F, Supplementary Material, Fig. 6C). 309-801 NIK fragment expression also increased Ccnd2, Erbb2 and Met messenger RNA and protein (Fig. 3G, Supplementary Material, Fig. 6D) as well as increased expression of three of five Rel family transcription factors by RT-PCR and immunoblot (IB) (Supplementary Material, Fig. 6E–G). Of note, our in silico analysis and confirmatory studies (Fig. 1) revealed overexpression of many of the same genes in human VS and murine schwannomas.
Figure 3.
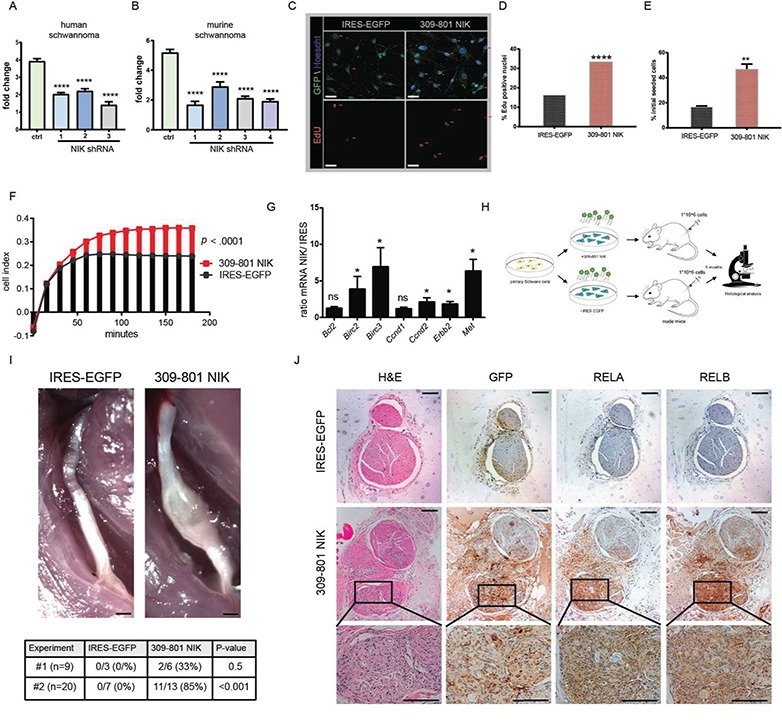
NIK critically regulates SC function. (A) 72 h proliferation study in HEI-193 human schwannoma cell line comparing control and three independent NIK shRNAs. (B) 72 h proliferation study with primary murine schwannoma cells comparing control and four independent NIK shRNAs. In (C)–(J) primary passages 1 and 2 SCs were stably transduced with an IRES-EGFP control or 309-801 NIK IRES-EGFP expressing lentivirus. (C) Representative images of EdU immunofluorescence experiment. Scale bar = 20 μm. Original magnification ×200. (D) Quantitation of (C). Over 400 cells were counted of each genotype in total (n = 3 of each). (E) 48 h serum and growth factor starvation survival assay. (F) iCelligence cell adhesion assay. Adhesion is measured by changes in impedance caused by cells falling out of suspension and depositing onto electrodes, forming focal adhesion complexes. (G) qRT-PCR in transduced primary SCs. (H) Transplant schema of transduced primary SCs into Foxn1nu nude mice. (I, upper) Gross images of dissected sciatic nerves from IRES- and NIK-transduced SC transplants. Scale bar = 500 μm. (I, lower) Tabular results from transplant studies outlined in (H). (J) Serial sections of IRES- and NIK-injected nerves stained with H&E, GFP, RELA and RELB. Original magnification ×100, magnified inset 400×. Scale bar = 100 μm. [ANOVA with Dunnett’s test (α = 0.05) for post hoc comparisons (A) and (B); Fisher’s exact test (D); unpaired Student’s t-test with Holm–Sidak correction for multiple comparisons (α = 0.05, E, F, G, and I). ns = not significant, *P < 0.05, **P < 0.01, ****P < 0.0001. Error bars represent SEM. All experiments with statistical testing were performed in triplicate.]
To test the significance of these findings in vivo, we transduced primary murine SCs with 309-801 NIK IRES-EGFP or IRES-EGFP and injected 1*106 cells with basement membrane matrix into the sciatic nerves of Foxn1-deficient nude mice (Fig. 3H). Five months later, 309-801 NIK-injected nerves showed fusiform tumor nodules (Fig. 3I). Of note, two independent experiments yielded similar results (Fig. 3I, Supplementary Material, Table 1). Haemotoxylin and eosin (H&E) revealed focal SCs dysplasia consistent with schwannoma formation, and IHC demonstrated increased expression of GFP, RELA and RELB (Fig. 3J). Importantly, the SC nucleocentric RELA and RELB staining patterns mimicked those seen in murine model and primary human schwannomas (Fig. 1F, Supplementary Material, Fig. 1C and D). These studies roundly demonstrate that p55 NIK is sufficient to upregulate NF-κB signaling and transform primary SCs in vivo.
NF-κB potentiates an HGF–c-MET activation loop that sustains schwannoma proliferation
c-MET, the receptor for HGF and a known glial cell growth factor, has previously been implicated in the pathogenesis of schwannomas, though the mechanism of how this occurs remains to be identified (27–28). IHC of human vestibular schwannoma revealed marked elevations of c-MET and Phospho-c-MET (Tyr 1234/1235), a marker of c-MET activation (Fig. 4A). Furthermore, kinome profiling (29), a recently developed mass spectrometry (MS)-based assay which utilizes kinase-specific inhibitor beads to measure the activity of up to 275 kinases in a single tissue lysate sample, revealed a nearly 4-fold increase in c-MET receptor activation in schwannoma-bearing murine nerve tissue as compared to control nerve tissues (Fig. 4B). In Figure 3G and Supplementary Material, Figure 6D, we demonstrate 309-801 NIK upregulates c-MET expression in primary SCs. Conditioned media from 309-801 NIK-transduced SCs had increased levels of secreted HGF with no change in neuregulin 1 (NRG1), indicating that NF-kB activation in tumorigenic SCs may also be capable of promoting HGF secretion (Fig. 4C). AliBaba 2.1 software predicted NF-κB binding motifs at −67 (ATTTCCCTCT), −350 (CTTCCCGGGG), −566 (AAACTCCTCT) and −584 (GGAAGTCTCC) and chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) of the MET promoter using anti-RELA antibody in three patient-derived VS amplified a region at −199, corresponding to strong RELA binding at the MET promoter (Fig. 4D), further supporting the notion that NF-kB activation is responsible for the increased expression of c-MET we observe in schwannomas.
Figure 4.
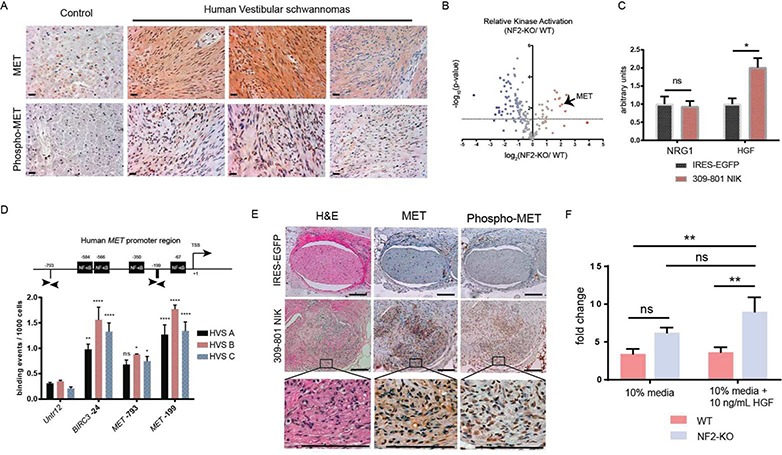
NIK and NF-κB regulate pathologic activation of MET signaling in schwannomas. (A) IHC of MET and Phospho-MET in human nerve tissue and three distinct HVS. Scale bar = 20 μm. Original magnification ×400. Arrowheads point to representative cells with positive staining for Phospho-MET. (B) Kinome analysis in primary murine tissues demonstrating a nearly 4-fold statistically significant increase in MET activation in schwannomas. The dotted line indicates a false discovery rate of 5% (Benjamini–Hochberg correction). (C) ELISA of SC-conditioned medium collected after 96 h of incubation. (D) ChIP-qPCR of human MET promoter region with an antibody targeting RELA. The graph displays the MET promoter region and PCR primers flanking locations at −199 and −793. Untr12 primers serve as a negative control and BIRC3 as a positive control. TSS = transcriptional start site. (E) Serial sections of IRES- and NIK-injected nerves stained with H&E, MET and Phospho-MET from the experiment outlined in Figure 3H. Original magnification ×100, magnified inset 400×. Scale bar = 100 μm. (F) In vitro cell counting assay with primary NF2-KO and WT murine schwannoma/SCs over 10 days. [Unpaired Student’s t-test with Holm–Sidak correction for multiple comparisons (α = 0.05; C). Two-way ANOVA with Dunnett’s test for post hoc comparisons (α = 0.05, D and F). ns = not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars represent SEM. All experiments with statistical testing were performed in triplicate.]
Examination of 309-801 NIK transplanted schwannoma tumors revealed elevations of c-MET and Phospho-c-MET in the murine tumor-bearing tissues, consistent with the hypothesis outlined in the above paragraph (Fig. 4E). Finally, in vitro proliferation studies demonstrated primary NF2-KO SCs were over twice as sensitive to HGF stimulation as WT SCs, indicating that c-Met activation can be strongly mitogenic in tumorigenic SCs (Fig. 4F). These data support c-Met playing a central role in schwannoma development, with NF-κB critically regulating a feed-forward HGF/MET signaling loop that may initiate and sustain NF2-deficient SC tumor formation (30–31).
Discussion
Our data directly implicate NIK/NF-κB as the putative driver of schwannoma genesis, of which there are a number of preclinical drugs in the early stages of development (32). Currently, our understanding of how the loss of NF2 leads to NF-κB is limited. Previously, Merlin has been shown to bind to large tumor suppressor kinase 1/2 (LATS1/2), leading to phosphorylation of LATS1/2 and suppression of the Hippo signaling transcription factors YAP and TAZ, suggesting a critical role for NF2 and the Hippo pathway in branching morphogenesis (33). Prior studies have linked activation of the Hippo pathway to activation of NF-κB (34). Thus, it is possible that Hippo signaling connects loss of functional Merlin to the activation of NF-κB we observe in our tumors. We and others are currently exploring the crosstalk between these pathways in our recently published NF2 mouse model (17).
We further describe an NF-κB-initiated autocrine HGF/c-MET signaling loop that selectively promotes the growth of Merlin-deficient SCs. Activation of c-MET can occur through at least three mechanisms: in response to its ligand HGF, via integrins and via dimerization with ErbB family receptors; the latter two of which are also overexpressed in schwannomas (35–37). Thus, there are multiple avenues leading to the c-MET activation observed in schwannomas, suggesting c-MET may be a critical signaling node in these tumors and, consequently, an excellent therapeutic target. Future genetic and pharmacologic in vitro and in vivo studies are warranted to better evaluate c-MET inhibition as a therapeutic option.
Our studies delineate molecular effectors of schwannoma genesis while elucidating mechanisms pertinent to broader investigations of NF-kB signaling in normal and diseased states. NIK N- and C-termini caspase cleavage sites are conserved across species, suggesting that these sites critically regulate NIK biology (Supplementary Material, Fig. 3). We conclude that the deregulated NIK kinase domain fragment activity explains, at least in part, the functional relevance of caspase-mediated NF-κB signaling, an area that deserves much greater attention (21,38). We additionally provide compelling evidence of a particular signaling pathway’s influence on schwannoma formation, highlighting a downstream NF-κB-mediated c-MET/HGF signaling loop with tremendous therapeutic opportunity given the availability of preclinical and clinically available drugs targeting both NF-κB signaling and c-MET. Furthermore, our orthotopic transplant model provides a reproducible tool for generating schwannomas molecularly and histologically identical to those observed in humans and mice. Of note, the studies presented here principally rest upon observations in primary murine model and human schwannoma tissues, strongly arguing for the broad relevance of our conclusions to NF2, SC and NF-κB biology writ large.
Materials and Methods
Animal and human study approval
Animal studies were conducted in accordance with and approval of Institutional Animal Care and Use Committee of Indiana University Medical School, the US Department of Agriculture’s Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals. Human studies were conducted under IRB approval (IRB# 1107006213, ‘Genotype, Phenotype, and Treatment of Human Vestibular Schwannomas’). Details of our NF2 model can be found in the initial publication (10).
Statistical methods
Statistical analyses were performed with GraphPad Prism 6.0. and 7.0. As noted in the text, analysis of variance (ANOVA) or Student’s t-test was used to test for differences between samples. When applicable, corrections were made for multiple testing. Specific tests and significance levels can be found in the figures and figure legends.
Microarray and IPA
Previously published schwannoma microarray data (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39645, Torres-Martin et al., Int. J. Oncol., 2013) were downloaded from the GEO and loaded into Partek Genomics Suite (Partek Inc., St. Louis, MO, USA). In our analysis, we included 31 schwannoma samples and 8 control samples (the human SC line group was excluded from the controls). The data were then quantile normalized and DEGs were identified by ANOVA as those having P-values <0.05. The Benjamini and Hochberg method was used to correct for multiple testing. Further, filtering of DEGs was completed to remove genes with fold changes between −2 and 2. Network analysis was then completed with this DEG set with IPA (www.qiagen.com/ingenuity). Microarray analysis and IPA were completed by the Indiana University School of Medicine Center for Computational Biology and Bioinformatics.
Preparation of mouse trigeminal nerves for RNA and protein studies
Mice were sacrificed and trigeminal nerve tissue was freshly dissected, minced with a scalpel and washed 3× with cold phosphate-buffered saline (PBS), pelleting the tissue in between washes using a tabletop centrifuge (Eppendorf, Enfield, CT, USA) at 4°C. Tissue was then prepared for protein or RNA studies. For whole lysate protein studies, xTractor Lysis buffer (Takara Bio, Mountain View, CA, USA) with cOmplete Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA) and PhosSTOP Phosphatase Inhibitor Cocktail (Roche, Indianapolis, IN, USA) was incubated with the tissues. This mixture was then sonicated on ice, spun down at max speed on a tabletop centrifuge for 10 min at 4°C and the supernatant was collected and stored at −80°C for future use. Trigeminal nerve tissue was fractionated into cytoplasmic and nuclear protein fractions using the NE-PER subcellular fractionation kit (Thermo Fisher Scientific, Federal Way, WA, USA). Proteins were standardized by bicinchoninic acid assay (BCA) in all immunoblotting studies. For RNA extraction, tissues were suspended in RNALater (Thermo Fisher Scientific, Federal Way, WA, USA) and stored at −20°C and later harvested with Trizol (Thermo Fisher Scientific, Federal Way, WA, USA).
Histology and IHC
Tissues were excised from recently sacrificed animals, fixed with 10% formalin, embedded in paraffin and sectioned. IHC on formalin-fixed, paraffin-embedded mouse and human samples was completed by the Indiana University School of Medicine Immunohistochemistry Core. Schwannoma tissue microarrays (US Biomax, Rockville, MD, USA) were also completed by the Indiana University School of Medicine Immunohistochemistry Core.
Cell culture and transfection
HEK-293 T and HEI-193 cells (American Type Culture Collection, Manassas, VA, USA) were used for transfection experiments with polyethyleneimine (PEI; Millipore-Sigma, St. Louis, MO, USA). Unless otherwise noted, Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Federal Way, WA, USA) containing 10% fetal bovine serum (FBS; Millipore-Sigma, St. Louis, MO, USA), 50 U/ml penicillin, 50 μg/ml streptomycin and 2 mm L-glutamine (Lonza, Allendale, NJ, USA) was used for cell culture. For creation of a stable NF-κB reporter cell line, 293 T cells were transduced with Cignal NF-κB GFP Reporter Lentivirus (Qiagen) and selected with puromycin (Millipore-Sigma, St. Louis, MO, USA). The Cignal NF-κB GFP Reporter Lentivirus stably inserts a modified form of the GFP gene under the control of a minimal CMV promoter with tandem repeats of the canonical NF-κB transcriptional response element. Both 293 T and HEI-193 cell lines were tested for mycoplasma using a PCR kit from Lonza (MycoAlert). Cell lines were used in experimentation prior to 30 passages after thawing.
Primary SC procurement and culture
WT embryonic day 13.5 embryos were harvested from pregnant dams after sacrifice. Embryos were placed in cold PBS + 1% Pen/Strep (Lonza, Allendale, NJ, USA, 10K/10K) and the dorsal root ganglion (DRG) were dissected from each embryo with the aid of a dissecting microscope. All DRGs isolated from an individual were pooled and placed into a tube containing 500 μl of DMEM + 10% FBS and placed on ice. The DRGs were then digested in 0.05% trypsin-EDTA (Thermo Fisher Scientific, Federal Way, WA, USA) and dissociated with syringes. DRGs pooled from a single embryo were then plated on PDL (0.1 mg/ml)/laminin (0.25 mg/ml; Millipore-Sigma, St. Louis, MO, USA)-coated 12 well plates at one embryo per well in SC Media I (SCM-I) comprised of DMEM with 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mm L-glutamine (Lonza, Allendale, NJ, USA), 1X N2 supplement (Thermo Fisher Scientific, Federal Way, WA, USA) and 250 ng/ml Nerve Growth Factor (NGF, Millipore-Sigma, St. Louis, MO, USA). The following day, the medium was changed to SC Media II (SCM-II), which was identical to SCM-I except for the substitution of 2 μm forskolin and 10 ng/ml neuregulin (Millipore-Sigma, St. Louis, MO, USA) for NGF. Embryonic SCs were used for all experiments save for those shown in Figure 4F, where adult primary SCs were used. These were harvested from the trigeminal nerves of adult mice with a 30 min Collagenase A (2 mg/ml; Millipore-Sigma, St. Louis, MO, USA) and 20 min trypsin (0.25%; Thermo Fisher Scientific, Federal Way, WA, USA) digestion. After 7 days in culture in 10% FBS /DMEM medium on PDL/laminin-coated plates, cells were expanded and cultured in 10% FBS/10% human melanocyte growth serum/DMEM solution (10% media; Thermo Fisher Scientific, Federal Way, WA, USA).
Orthotopic transplant studies
One million primary SCs (P1-P2) were transduced with IRES-EGFP or 309-801 NIK lentivirus and transplanted into the sciatic nerves of female B6.Cg-Foxn1/J (nude) mice purchased from Jackson laboratories, Bar Harbor, ME, USA. Mice were roughly 8–12 weeks old at the time of experimentation. Each mouse was anesthetized with isofluorane and buprenorphine and a small incision was made using an 11 blade surgical scalpel. The sciatic nerve was then exposed with blunt dissection using scissors under a dissecting microscope. A 1:1 admixture of suspended SC and Culture® basement membrane extract (Trevigen, Gaithersburg, MD, USA) totaling 20 μl was injected into the right sciatic nerve of each mouse using a 33 gauge syringe (Hamilton, Reno, NV, USA). The wound was then closed with vetbond in experiment #1 and 5-0 prolene suture in experiment #2.
Kinome studies
Isolation and measurement of the activation of endogenous protein kinases from SCs were carried out using multiplexed kinase inhibitor beads and mass spectrometry (MIB/MS). Tissue lysates were sonicated, filtered, pre-cleared with blocked Sepharose beads and then passed over MIB columns. Pelleted protein is digested with trypsin and peptides purified on C18 spin columns. Kinase peptides are labeled with TMT isobaric mass tags, which provide a quantitative method to determine relative differences across samples and between treatments. Labeled kinase peptides are then analyzed using liquid chromatography/mass spectroscopy (LC/MS) on a Q-exactive mass spectrometer and sequence-identified using Proteome Discoverer 1.3 (Thermo Fisher Scientific, Federal Way, WA, USA) using MASCOT 2.2.4 and SEQUEST databases. Statistically significant kinase identifications are declared by adjusting for multiple comparisons using false discovery rate. In general, we are interested in kinases that are either upregulated or downregulated by at least 3-fold in two independent experiments. Examples of such analyses are shown in a recent publication (29). For the study in Figure 4, flash frozen tissue (NF2-KO schwannoma DRG or WT sciatic nerve, n = 3 of each) was processed for MIB/MS analysis. MIB-binding differences were compared (schwannoma versus WT) and the log2 difference in binding (schwannoma versus WT) was plotted versus the –log10 adjusted P-value (Benjamini–Hochberg correction). The dotted line indicates a false discovery rate of 5%.
RNA extraction from trigeminal nerves and primary SCs
RNA extraction, cDNA synthesis, qRT-PCR, data acquisition, analysis and quality control (QC) of mouse trigeminal nerve samples were completed by Genome Explorations USA (Memphis, TN). All other RNA work was completed at Indiana University School of Medicine. RNA was extracted and purified from primary SCs with Trizol (Thermo Fisher Scientific, Federal Way, WA, USA) and the PureLink RNA Mini Kit (Thermo Fisher Scientific, Federal Way, WA, USA). QuantiTect Reverse Transcription Kit (Qiagen) and Fast SYBR Green Real-Time PCR Master Mix (Thermo Fisher Scientific, Federal Way, WA, USA) were then used for two-step qRT-PCR with an ABI 7500 Fast thermal cycler. Expression of GAPDH is used for normalization.
Primary antibody list
The following antibodies were used for western blotting experiments: RELA (Cell Signaling, Danvers, MA, USA, #8242), RELB (Cell Signaling #4954), p100/p52 (Cell Signaling, Danvers, MA, USA, #4882), c-REL (Cell Signaling, Danvers, MA, USA, #12707), GAPDH (Cell Signaling, Danvers, MA, USA, #5174), NIK (Cell Signaling, Danvers, MA, USA, #4994), IKKα (Cell Signaling, Danvers, MA, USA, #2682), IκBα (Cell Signaling #4814), Myc (Cell Signaling, Danvers, MA, USA, #2278), HA (Cell Signaling, Danvers, MA, USA, #3724), FLAG (Cell Signaling, Danvers, MA, USA, #8146), NF2 (Abcam ab30329), ERBB2 (Pierce, MA5-14057), Phospho-MET (Millipore-Sigma, St. Louis, MO, USA, SAB4300182), c-MET (Santa Cruz sc-161), pIKKα (Cell Signaling, Danvers, MA, USA, #2697), p105/p50 (Cell Signaling, Danvers, MA, USA, #13586) and caspase-8 (Santa Cruz sc-6136). The custom polyclonal NIK antibody targeting murine NIK residues 311:328 was generated by Thermo Pierce custom antibody services. In all western blots, GAPDH was used as the loading control. For IHC/immunofluorescence (IF), the following antibodies were used: RELA (Santa Cruz sc-109), RELB (Santa Cruz sc-226), NIK (Santa Cruz sc-7211), GFP (Abcam,Cambridge, MA, USA, ab1218), MET (Santa Cruz sc-8057), Phospho-MET (Cell Signaling, Danvers, MA, USA, #3077) and S100 (Dako, Carpinteria, CA, USA, IS504).
In vitro kinase assay
Freshly dissected trigeminal nerves from four WT and four NF2-KO mice were pooled according to genotype and lysed with immunoprecipitation (IP) lysis buffer (2.5 mm HEPES pH 7.4, 150 mm NaCL, 1 mm EDTA, 1% Triton X-100, 0.02% NaN3) and immunoprecipitated with anti-NIK antibody (Cell Signaling, Danvers, MA, USA, #4994) and agarose-conjugated protein A/G beads (Santa Cruz sc-2003). Recombinant IKKα was purchased from Abcam. The in vitro kinase assay was performed in 30 μl total, comprised of 10× kinase buffer, H20, P32-ATP, substrate and the IP pellet. The reaction mixture was incubated for 30 min, separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and developed by autoradiography.
Western blot densitometry
Western blot film was scanned into a digital format and intensity values were determined using the Image Studio Lite Version 3.1 (LI-COR Biosciences, Lincoln, NE, USA).
Virus generation and titration
The puc2CL6IEGwo lentiviral IRES-EGFP construct was a kind gift of Helmut Hannenberg at Indiana University School of Medicine. C-terminus FLAG-tagged NIK was cloned into the puc2CL6IEGwo construct using the EcoRI and BamHI cloning sites. The puc2CL6IEGwo construct contains an IRES-EGFP cassette and multiple cloning site downstream of the spleen focus-forming virus promoter, which robustly drives transcription in eukaryotic cells. To generate virus, 293 T cells grown to 70% confluence were transfected with 10 μg each of C-FLAG 309-801 NIK puc2CL6IEGwo (Lenti 309-801 NIK) or empty vector puc2CL6IEGwo (Lenti IRES EGFP), 5 μg of gag-pol expressing plasmid and 1 μg of foamyviral envelope containing plasmid (PCOPE01) in 6 ml of DMEM containing 10% FBS and 0.0075 mg/ml PEI (Millipore-Sigma, St. Louis, MO, USA). After roughly 16 h of transfection at 37°C, transfection medium was replaced with 6 ml fresh DMEM containing 10% FBS, 50 U/ml penicillin, 50 μg/ml streptomycin and 2 mm L-glutamine (Millipore-Sigma, St. Louis, MO, USA). After 24 h, cultures were collected and filtered through a 0.22 μm polyethersulfone membrane Stericup unit (Millipore-Sigma, St. Louis, MO, USA) and centrifuged at 30 000 × g at 4°C for 2 h in a polycarbonate Oak Ridge Centrifuge Tube (Thermo Fisher Scientific, Federal Way, WA, USA). Supernatant was then decanted, bleached and placed in biohazard waste. The viral pellet was then resuspended in 1 ml of DMEM containing 10% FBS. Virus was then stored in aliquots at −80°C. Titration of virus to calculate effective infectious particles per unit volume was determined by percent GFP-positivity (as measured by flow cytometry) of serially transduced HT1080 cells plated at 100 000 per well on a 6 well plates in 1 ml of DMEM/10% FBS. Serial dilutions started at 10−3 and ended with 10−8 dilution.
Transduction of primary SCs
Three days after harvesting and plating WT SCs on 12 well plates, cells were then transduced with lentivirus in 1 ml of SCM-II at a multiplicity of infection (MOI) of 20 (based on an approximate cell number of 200 000 cells per well). Cells were incubated in SCM-II (see Primary SC procurement and culture) with virus for 72 h before replating. All experiments utilized P1 SCs.
Fluorescent microscopy
Cells were fixed in 4% paraformaldehyde on coverslips in 12 well plates (BD Falcon). Following fixation, coverslips were washed 2× in PBS and permeabilized in 0.2% Triton X-100 in PBS and blocked in 0.2% Triton X-100, 2% BSA in PBS. Coverslips were then incubated with primary antibody in 1% BSA in PBS overnight at 4°C, washed 3× in PBS and subsequently incubated for 1 h at room temperature with fluorophore-conjugated anti-mouse or anti-rabbit antibodies (Thermo Fisher Scientific, Federal Way, WA, USA). After washing in PBS 3×, they were then incubated in PBS containing Hoechst and to stain the nuclei, respectively. Coverslips were then sealed on microscope slides (Thermo Fisher Scientific, Federal Way, WA, USA) with lacquer and were allowed to dry overnight. Data were acquired on a DeltaVision deconvolution microscopy system (Thermo Fisher Scientific, Federal Way, WA, USA).
ChIP-qPCR on primary tumor tissue
Primary patient-derived human vestibular schwannoma samples were snap frozen and sent to Active Motif (Carlsbad, CA) for completion of the ChIP-qPCR experiment. After ChIP was performed, qPCR reactions were set up in triplicate for each ChIP sample. Each qPCR plate also contained input DNA and a standard curve for normalization. Normalized data are expressed as binding events detected per 1000 Cells. The MET primers used in the experiment are as follows: forward primer MET-199 = GACTCGGTCCCGCTTATCTC, reverse primer MET-199 = GCCCACGACAAGGTGAAAC, forward primer MET-793 = CCTTTGAGTGTTCATTTACTCTTG and reverse primer MET-793 = TTGTGTTGGGATAACTTTGTATTTC. Specific details on the data normalization can be found at https://www.activemotif.com/documents/1837.pdf.
Mass spectroscopy
Excised gel bands were first destained, reduced with 5 mm Tris(2-carboxyethyl)phosphine hydrochloride and then alkylated with10 mm chloroacetamide. Alkylated samples were digested by trypsin (Promega Gold, Sunnyvale, CA, USA) overnight at 37°C. Digested peptides were extracted from the gel spots with (1) 50% acetonitrile/49.9%H2O/0.1% trifluoroacetic acid and (2) 99.9% acetonitrile/0.1% trifluoroacetic acid. The instruments used for analysis were the Thermo Fisher Scientific Orbitrap Velos Pro and Thermo Dionex UltiMate 3000 RSLC nano system. Digested peptides were injected onto the C18 column. Peptides were eluted with a linear gradient from 3 to 40% acetonitrile (in water with 0.1% trifluoroacetic acid) developed over 70 min at room temperature at a flow rate of 300 nL/min, and effluent was electro-sprayed into the mass spectrometer. A blank was run prior to the sample run to make sure there was no significant signal from solvents or the column.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) kits for murine HGF and NRG1 were purchased from R&D Biosystems, Oakville, ON, Canada and USCN Lifescience Inc., Houston, TX, USA, respectively. Experimental design details can be found in Figure 4. Each experiment represents a minimum of three biological replicates.
Plasmids
C-terminal Myc/FLAG-tagged full-length NIK and Caspase-8 cDNAs in the PCMV6-Entry vector were purchased from Origene. The Caspase-8 cDNA was then shuttled into the PCMV6-AC-HA backbone (Origene, Rockville, MD, USA) using the Sgf1 and Mlu1 restriction sites. C-Flag full-length NIK and 309-801 NIK were PCR-amplified (adding epitope tag sequences and cloning sites) using Phusion Hi-Fidelity polymerase (New England Biolabs,Ipswich, MA, USA) from a vector containing full-length NIK, subcloned into a TOPO-TA cloning vector (Thermo Fisher Scientific, Federal Way, WA, USA) and then cloned into the PCMV6-XL5 backbone (Origene, Rockville, MD, USA) using the EcoRI and SalI cloning sites. The pCR-FLAG-IKKalpha-KM, pFlag-CrmA and pcDNA-CrmA plasmids were acquired from Addgene.
SC proliferation assays
Unless otherwise noted, SC-based experiments utilized primary P1-P2 embryonic SCs.
Hemocytometer-based
200000 SCs of each genotype were seeded in triplicate on a PDL/laminin-coated 12 well plates and were allowed to adhere overnight. The following morning, the medium was changed to fresh SCM-II, and 72 h later the cell number in each well was calculated by trypan blue exclusion with a hemocytometer.
EdU with fluorescent microscopy
200 000 SCs of each genotype were plated on coated coverslips in 12 well plates. The following morning, the medium was changed to fresh SCM-II with 10 μm EdU. Cells were pulsed for 8 h and fixed. Life Technologies Alexa 594 EdU Imaging Kit was used to complete the staining of the cells. Coverslips were then imaged on the deconvolution microscope.
SC survival assay
In a PDL/laminin-coated 12 well plates, 200 000 SCs of each genotype were seeded in triplicate and were allowed to adhere overnight. The following day, the medium was removed, cells were washed 1× with PBS and growth-factor free DMEM with 50 U/ml penicillin, 50 μg/ml streptomycin and 2 mm L-glutamine was added. The number of living cells in each well was counted 48 h later by trypan blue exclusion with a hemocytometer.
SC adhesion assay
For measurement of cell adhesion, the ACEA iCelligence system was used. The iCelligence system uses an impedance calculation that allows for real-time monitoring of diverse cellular processes, including cellular adhesion. Adhesion is measured by changes in impedance caused by cells falling out of suspension and depositing onto electrodes, forming focal adhesion complexes. E-plates (ACEA) were coated with PDL/laminin and 250 μl of SCM-II was added to the plates to take an initial impedance reading. Next, IRES-EGFP and 309-801 NIK SCs were added to the wells at 25 000 cells/well with a final volume of 500 μl. Impedance values were measured every second for the following 3 h.
HGF and SC experiments
In Figure 4F, SCs were plated on PDL/laminin-coated wells in 24 well plates and incubated with the indicated medium over the course of 10 days. Cell counts were then determined by hemocytometer counts.
NIK shRNA studies
The HEI-193 human schwannoma cell line was transduced with lentivirus (Sigma control shRNA SHC004 targeting GFP and Sigma NIK shRNAs TRCN0000010542, TRCN0000196415, TRCN0000199183) and selected with puromycin (Millipore-Sigma, St. Louis, MO, USA). Cells were plated in 12 well plates at 100 000 cells per well in triplicate. After 3 days in culture, cells were detached and counted by hemocytometer. Analogous studies were performed in primary NF2-KO schwannoma cells cultured from the peripheral nerve of a NF2-KO mouse. For these studies, Sigma control shRNA SHC007 targeting luciferase and Sigma NIK shRNAs TRCN0000012763, TRCN0000012764, TRCN0000012765 and TRCN0000012767 were used. The pLKO.1-puro lentiviral backbone was used for all shRNA studies, which were performed independently a minimum of three times.
CHX NIK protein stability experiment
293 T cells at 70% confluency in 10 cm dishes (BD Falcon) were transfected with 4 μg/dish of full-length N-Myc NIK or N-Myc 309-801 NIK for 36 h prior to addition of media containing 20 μm of MG132 (Millipore-Sigma, St. Louis, MO, USA) for 5 h. MG132 was then washed out and medium containing 10 μg/ml of CHX (Millipore-Sigma, St. Louis, MO, USA) was added to the cells. Cells were then lysed at the indicated conditions/time points and the proteins visualized by SDS-PAGE and western blot.
Immunoprecipitation experiments in 293 T cells
Immunoprecipitation experiments were completed in 293 T cells transfected with the indicated plasmids/concentrations at 70% confluency in 10 cm dishes. Two days after transfection, cells were lysed with radioimmunoprecipitation assay buffer (RIPA) buffer (50 mm Tris-HCl pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, protease and phosphatase inhibitors), sonicated and samples were standardized by BCA. Samples were then incubated overnight with M2 Flag Affinity Gel (Millipore-Sigma, St. Louis, MO, USA). Immunoprecipitated proteins were then visualized by SDS-PAGE and western blot.
In vitro caspase-8 cleavage assay
Purification of FLAG-NIK
FLAG-NIK was transfected at 20 μg/dish in 70% confluent 15 cm dishes and harvested 48 h later in lysis buffer (25 mm HEPES pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, protease and phosphatase inhibitors) and sonicated. These samples were then incubated overnight with M2 Flag Affinity Gel. Flag NIK was then eluted from the resin with 100 μl of Flag peptide elution solution (200 μg/ml in tris-buffered saline (TBS); Millipore-Sigma, St. Louis, MO, USA).
In vitro Caspase-8 cleavage of NIK
200 ng of activated Caspase-8(BD) was added to 200 ng of purified NIK protein in a 50 μl volume of reaction buffer (20 mm HEPES pH 7.4, 20 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1.5 mm MgCL2, 10 mm DTT) for 1 h at 37°C. Q-VD-Oph, an irreversible pan-caspase inhibitor, was purchased from ApexBio, Boston, MA, USA.
Prediction of caspase-8 cleavage sites and orthologous cleavage site analysis
Caspase-8 cleavage sites were predicted for human NIK using two different applications, Cascleave 2.0 and SitePrediction. Cascleave 2.0 was set to a medium stringency threshold for prediction of cleavage sites. The NCBI homologene application was used to identify orthologous cleavage sites in chimpanzee, macaque, mouse, rat, dog and cow.
Supplementary Material
Funding
Grant support for these studies derived from NF170058, F31 DC016528-02, and the Riley Children's Foundation.
References
- 1. Evans D.G. (1993) In Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., & Amemiya A. (eds), GeneReviews®, [Internet]. Seattle, WA: University of Washington, Seattle, 1993–2018. https://www.ncbi.nlm.nih.gov/books/NBK1201/. [Google Scholar]
- 2. Asthagiri A.R., Parry D.M., Butman J.A., Kim H.J., Tsilou E.T., Zhuang Z. and Lonser R.R. (2009) Neurofibromatosis type 2. Lancet, 373, 1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rouleau G.A., Merel P., Lutchman M., Sanson M., Zucman J., Marineau C., Hoang-Xuan K., Demczuk S., Desmaze C., Plougastel B. et al. (1993) Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature, 363, 515–521. [DOI] [PubMed] [Google Scholar]
- 4. Ammoun S., Provenzano L., Zhou L., Barczyk M., Evans K., Hilton D.A., Hafizi S. and Hanemann C.O. (2014) Axl/Gas6/NFkappaB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene, 33, 336–346. [DOI] [PubMed] [Google Scholar]
- 5. Dilwali S., Briët M.C., Kao S.Y., Fujita T., Landegger L.D., Platt M.P. and Stankovic K.M. (2015) Preclinical validation of anti-nuclear factor-kappa B therapy to inhibit human vestibular schwannoma growth. Mol. Oncol., 9,1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karin M., Cao Y., Greten F.R. and Li Z.W. (2002) NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer, 2, 301–310. [DOI] [PubMed] [Google Scholar]
- 7. Parker M., Mohankumar K.M., Punchihewa C., Weinlich R., Dalton J.D., Li Y., Lee R., Tatevossian R.G., Phoenix T.N., Thiruvenkatam R. et al. (2014) C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature, 506, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Raedt T., Beert E., Pasmant E., Luscan A., Brems H., Ortonne N., Helin K., Hornick J.L., Mautner V., Kehrer-Sawatzki H. et al. (2014) PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature, 514, 247–251. [DOI] [PubMed] [Google Scholar]
- 9. Malone C.F., Fromm J.A., Maertens O., DeRaedt T., Ingraham R. and Cichowski K. (2014) Defining key signaling nodes and therapeutic biomarkers in NF1-mutant cancers. Cancer Discov., 4, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gehlhausen J.R., Park S.J., Hickox A.E., Shew M., Staser K., Rhodes S.D., Menon K., Lajiness J.D., Mwanthi M., Yang X. et al. (2015) A murine model of neurofibromatosis type 2 that accurately phenocopies human schwannoma formation. Hum. Mol. Genet., 24, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castanedo G.M., Blaquiere N., Beresini M., Bravo B., Brightbill H., Chen J., Cui H.F., Eigenbrot C., Everett C., Feng J. et al. (2017) Structure-based design of tricyclic NF-kappaB inducing kinase (NIK) inhibitors that have high selectivity over phosphoinositide-3-kinase (PI3K). J. Med. Chem., 60, 627–640. [DOI] [PubMed] [Google Scholar]
- 12. Ren X., Li X., Jia L., Chen D., Hou H., Rui L., Zhao Y. and Chen Z. (2017) A small-molecule inhibitor of NF-kappaB-inducing kinase (NIK) protects liver from toxin-induced inflammation, oxidative stress, and injury. FASEB J., 31, 711–718. [DOI] [PubMed] [Google Scholar]
- 13. Edgar R., Domrachev M. and Lash A.E. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Torres-Martin M., Lassaletta L., San-Roman-Montero J., De Campos J.M., Isla A., Gavilan J., Melendez B., Pinto G.R., Burbano R.R., Castresana J.S. and Rey J.A. (2013) Microarray analysis of gene expression in vestibular schwannomas reveals SPP1/MET signaling pathway and androgen receptor deregulation. Int. J. Oncol., 42, 848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krämer A., Green J., Pollard J. Jr. and Tugendreich S. (2014) Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics, 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao G.H., Gallagher R., Shetler J., Skele K., Altomare D.A., Pestell R.G., Jhanwar S. and Testa J.R. (2005) The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol. Cell. Biol., 25, 2384–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Houshmandi S.S., Emnett R.J., Giovannini M. and Gutmann D.H. (2009) The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2- and Src-dependent manner. Mol. Cell. Biol., 29, 1472–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karin M. and Greten F.R. (2005) NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol., 5, 749–759. [DOI] [PubMed] [Google Scholar]
- 19. Liu J., Sudom A., Min X., Cao Z., Gao X., Ayres M., Lee F., Cao P., Johnstone S., Plotnikova O. et al. (2012) Structure of the nuclear factor kappaB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem., 287, 27326–27334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosebeck S., Madden L., Jin X., Gu S., Apel I.J., Appert A., Hamoudi R.A., Noels H., Sagaert X., Van Loo P. et al. (2011) Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science, 331, 468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu W.H., Johnson H. and Shu H.B. (2000) Activation of NF-kappa B by FADD, Casper, and caspase-8. J. Biol. Chem., 275, 10838–10844. [DOI] [PubMed] [Google Scholar]
- 22. Wang M., Zhao X.M., Tan H., Akutsu T., Whisstock J.C. and Song J. (2014) Cascleave 2.0, a new approach for predicting caspase and granzyme cleavage targets. Bioinformatics, 30, 71–80. [DOI] [PubMed] [Google Scholar]
- 23. Verspurten J., Gevaert K., Declercq W. and Vandenabeele P. (2009) SitePredicting the cleavage of proteinase substrates. Trends Biochem. Sci., 34, 319–323. [DOI] [PubMed] [Google Scholar]
- 24. Lee S., Challa-Malladi M., Bratton S.B. and Wright C.W. (2014) Nuclear factor-kappaB-inducing kinase (NIK) contains an amino-terminal inhibitor of apoptosis (IAP)-binding motif (IBM) that potentiates NIK degradation by cellular IAP1 (c-IAP1). J. Biol. Chem., 289, 30680–30689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao G., Zhang M., Harhaj E.W. and Sun S.C. (2004) Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem., 279, 26243–26250. [DOI] [PubMed] [Google Scholar]
- 26. Razani B., Zarnegar B., Ytterberg A.J., Shiba T., Dempsey P.W., Ware C.F., Loo J.A. and Cheng G. (2010) Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci. Signal., 3, ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dilwali S., Roberts D. and Stankovic K.M. (2015) Interplay between VEGF-A and cMET signaling in human vestibular schwannomas and schwann cells. Cancer Biol. Ther., 16, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fuse M.A., Plati S.K., Burns S.S., Dinh C.T., Bracho O., Yan D., Mittal R., Shen R., Soulakova J.N., Copik A.J. et al. (2017) Combination therapy with c-Met and Src inhibitors induces caspase-dependent apoptosis of merlin-deficient Schwann cells and suppresses growth of schwannoma cells. Mol. Cancer Ther., 16, 2387–2398. [DOI] [PubMed] [Google Scholar]
- 29. Duncan J.S., Whittle M.C., Nakamura K., Abell A.N., Midland A.A., Zawistowski J.S., Johnson N.L., Granger D.A., Jordan N.V., Darr D.B. et al. (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell, 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krasnoselsky A., Massay M.J., DeFrances M.C., Michalopoulos G., Zarnegar R. and Ratner N. (1994) Hepatocyte growth factor is a mitogen for Schwann cells and is present in neurofibromas. J. Neurosci., 14, 7284–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dai J.Y., DeFrances M.C., Zou C., Johnson C.J. and Zarnegar R. (2009) The Met protooncogene is a transcriptional target of NF kappaB: implications for cell survival. J. Cell. Biochem., 107, 1222–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Demchenko Y.N., Brents L.A., Li Z., Bergsagel L.P., McGee L.R. and Kuehl M.W. (2014) Novel inhibitors are cytotoxic for myeloma cells with NFkB inducing kinase-dependent activation of NFkB. Oncotarget, 5, 4554–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reginensi A., Enderle L., Gregorieff A., Johnson R.L., Wrana J.L. and McNeill H. (2016) A critical role for NF2 and the Hippo pathway in branching morphogenesis. Nat. Commun., 7, 12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu B., Zheng Y., Yin F., Yu J., Silverman N. and Pan D. (2016) Toll receptor-mediated Hippo signaling controls innate immunity in Drosophila. Cell, 164, 406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Engelman J.A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J.O., Lindeman N., Gale C.M., Zhao X., Christensen J. et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science, 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- 36. Mitra A.K., Sawada K., Tiwari P., Mui K., Gwin K. and Lengyel E. (2011) Ligand-independent activation of c-Met by fibronectin and α(5)β(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene, 30, 1566–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Utermark T., Kaempchen K. and Hanemann C.O. (2003) Pathological adhesion of primary human schwannoma cells is dependent on altered expression of integrins. Brain Pathol., 13, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chaudhary P.M., Eby M.T., Jasmin A., Kumar A., Liu L. and Hood L. (2000) Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene, 19, 4451–4460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.