

Abstract
Golgi-associated retrograde protein (GARP) and endosome-associated recycling protein (EARP) are related heterotetrameric complexes that associate with the cytosolic face of the trans-Golgi network and recycling endosomes, respectively. At these locations, GARP and EARP function to promote the fusion of endosome-derived transport carriers with their corresponding compartments. GARP and EARP share three subunits, VPS51, VPS52 and VPS53, and each has an additional complex-specific subunit, VPS54 or VPS50, respectively. The role of these complexes in human physiology, however, remains poorly understood. By exome sequencing, we have identified compound heterozygous mutations in the gene encoding the shared GARP/EARP subunit VPS51 in a 6-year-old patient with severe global developmental delay, microcephaly, hypotonia, epilepsy, cortical vision impairment, pontocerebellar abnormalities, failure to thrive, liver dysfunction, lower extremity edema and dysmorphic features. The mutation in one allele causes a frameshift that produces a longer but highly unstable protein that is degraded by the proteasome. In contrast, the other mutant allele produces a protein with a single amino acid substitution that is stable but assembles less efficiently with the other GARP/EARP subunits. Consequently, skin fibroblasts from the patient have reduced levels of fully assembled GARP and EARP complexes. Likely because of this deficiency, the patient's fibroblasts display altered distribution of the cation-independent mannose 6-phosphate receptor, which normally sorts acid hydrolases to lysosomes. Furthermore, a fraction of the patient's fibroblasts exhibits swelling of lysosomes. These findings thus identify a novel genetic locus for a neurodevelopmental disorder and highlight the critical importance of GARP/EARP function in cellular and organismal physiology.
Introduction
The `Golgi-associated retrograde protein' (GARP) and `endosome-associated recycling protein' (EARP) complexes are structurally related, four-subunit complexes that promote SNARE-dependent fusion of endosome-derived transport carriers with the corresponding target organelles in most eukaryotic cells (1,2). GARP and EARP comprise three shared subunits named VPS51, VPS52 and VPS53, and one complex-specific subunit, either VPS54 (GARP) or VPS50 (EARP) (3–8) (Fig. 1A). GARP is mainly associated with the cytosolic face of the trans-Golgi network (TGN), where it participates in the tethering and fusion of tubular–vesicular transport carriers derived from endosomes, as part of a process known as `retrograde transport' (9–11) (Fig. 1B). Examples of mammalian proteins that undergo GARP-dependent retrograde transport are the cation-independent mannose 6-phosphate receptor (CI-MPR) that cycles between the TGN and endosomes in the process of sorting acid hydrolase precursors to lysosomes (12) and the vesicle SNARE VAMP4 that is involved in fusion of endosome-derived carriers with the TGN (11). GARP is also required for retrograde transport of sphingolipids from endosomes to the TGN (13). EARP, on the other hand, is mainly associated with the cytosolic face of endosomes marked by the small GTPase Rab4 and promotes the recycling of proteins such as the transferrin receptor from endosomes to the plasma membrane (7,8) (Fig. 1B). Studies in the nematode Caenorhabditis elegans have additionally implicated EARP in cargo sorting to dense-core vesicles in neurons and neuroendocrine cells (14). Finally, the VPS51 subunit of GARP/EARP was shown to be defective in fat-free mutant zebrafish, which display abnormal Golgi structure and defective lipid transport in the intestinal tract (15).
Figure 1.
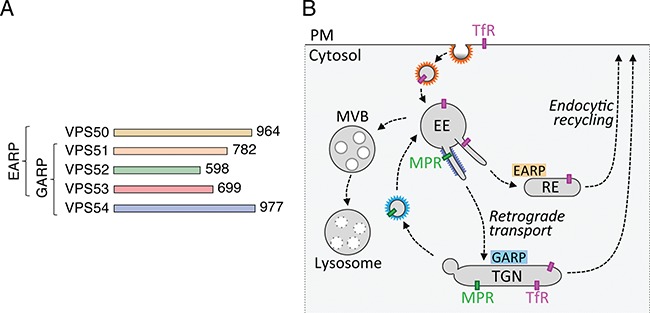
Characteristics of GARP and EARP. (A) The GARP complex is composed of VPS51, VPS52, VPS53 and VPS54 subunits, whereas the EARP complex is composed of VPS50, VPS51, VPS52 and VPS53 subunits (1,2). GARP and EARP were also previously referred to as VFT (VPS fifty three) (3) and GARPII (7), respectively. VPS51 is also known as Ang2 (another new gene 2) (6,38), and VPS50 as syndetin (8) or VPS54L (7). The number of amino acids in each protein is indicated (according to UniProt, http://www.uniprot.org/). The subunits of GARP and EARP are elongated proteins belonging to the family of `complexes associated with tethering containing helical rods' (39). The four subunits of the mammalian complexes are thought to assemble via their N-terminal regions into an X-shaped structure (40). (B) Schematic representation of the localization and function of GARP and EARP (adapted from ref. 8). GARP mediates tethering and fusion of endosome-derived carriers to the TGN, while EARP functions in tethering and fusion of endosome-derived carriers with recycling endosomes. GARP also plays a minor role in this latter pathway. EE, early endosome; MVB, multivesicular body; PM, plasma membrane; RE, recycling endosome; MPR, mannose 6-phosphate receptor; TfR, transferrin receptor.
The functions of GARP and EARP in intracellular transport are essential for the viability of mammalian organisms, as demonstrated by the embryonic lethality of mice with homozygous null mutations in subunits of these complexes (16–19) (www.informatics.jax.org). In addition, a spontaneous, recessive mutation (A>T nucleotide 72 of exon 23) causing substitution of glutamine for leucine-967 (L967Q) in the GARP-specific VPS54 subunit is responsible for the phenotype of the wobbler (wr) mouse (17). This substitution causes structural destabilization and partial degradation of VPS54, resulting in reduced levels of the fully assembled GARP complex (20). Wr mice are viable but develop motor neuron degeneration similar to that of amyotrophic lateral sclerosis (ALS) (21). Despite the similarities between the wr phenotype and ALS, however, mutations in VPS54 have not been shown to cause ALS (22). Recent genome-wide linkage analysis in conjunction with whole exome sequencing identified compound heterozygous mutations (c.2084A>G and c.1556+5G>A) in the gene encoding the shared VPS53 subunit of GARP and EARP in patients with a condition known as progressive cerebello-cerebral atrophy type 2 (PCCA2) or pontocerebellar hypoplasia type 2E (PCH2E) (OMIM no. 615851), a congenital disorder characterized by intellectual disability, progressive microcephaly, spasticity and epilepsy (23). These mutations are predicted to affect the C-terminal region of VPS53 (23), although the effects of these mutations on the properties of the protein were not experimentally addressed. Phenotypic overlap between PCCA2 and an autosomal recessive condition known as progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO) (24) has also been described, as two siblings with a PEHO-like syndrome were recently found to have compound heterozygous mutations in VPS53 (25). This disorder was primarily characterized by severe developmental delay, postnatal microcephaly, progressive cerebellar and cerebral atrophy, seizures, ophthalmologic abnormalities, facial/limb edema and dysmorphic features (25).
Herein we report the identification of compound heterozygous mutations (c.1468C>T and c.2232delC) in the gene encoding the GARP/EARP subunit VPS51 in a 6-year-old patient with severe global developmental delay, pontocerebellar abnormalities, microcephaly, hypotonia, epilepsy, cortical vision impairment, failure to thrive, liver dysfunction, lower extremity edema and dysmorphic features. Biochemical analyses show that a frameshift caused by the c.2232delC mutation (allele 1) produces a longer but highly unstable protein. On the other hand, the c.1468C>T mutation (allele 2) produces a protein with a single amino acid substitution (R490C) that is stable but assembles less efficiently with the other GARP/EARP subunits. As a consequence, skin fibroblasts from the patient have reduced levels of fully assembled GARP and EARP complexes and display altered distribution of the CI-MPR and swelling of lysosomes. These findings thus identify a new genetic locus for a complex neurodevelopmental disorder characterized by pontocerebellar abnormalities and other systemic manifestations and highlight the critical importance of GARP/EARP function in cellular and organismal physiology.
Results
Clinical phenotype
The patient is a 6-year-old Caucasian female who presented in early infancy with cholestatic hepatitis, feeding difficulties and failure to thrive. Her subsequent clinical course has included ongoing liver dysfunction requiring treatment with the bile acid ursodiol, gastrostomy tube dependence, hypotonia, marked microcephaly, severe global developmental delay with an inability to sit and a lack of spoken language, electrical status epilepticus in sleep requiring anticonvulsant therapy, cortical vision impairment with associated strabismus, sleep apnea, a gastric volvulus, constipation, asthma and episodic respiratory infections including respiratory syncytial virus (RSV), adenoviral pneumonia and a pleural effusion. Multiple physical examinations have shown minor dysmorphic features (Fig. 2A), including epicanthal folds, long eyelashes, slightly overfolded ears, an upturned nasal tip, a thin upper lip, a high/narrow anterior palate, full/rounded cheeks, a low posterior hairline, single flexion creases on the fifth fingers, mild clubbing of the thumbnails and other fingernails and increased hair on the upper back. At 6 years of age, she was also noted to have bilateral lower extremity edema. A brain magnetic resonance imaging (MRI) at 4 months of age was normal, while repeat MRI scans at 4 and 6 years of age (Fig. 2B) showed multiple abnormalities, including a small cerebellar vermis, a small pons/brainstem, an enlarged infra-vermian cistern with suspicion for a Dandy–Walker variant, a small hippocampus, a thin corpus callosum and an abnormal white matter signal in the cerebral hemispheres. A more detailed clinical report is included in the Supplementary Material, Supplemental Data.
Figure 2.
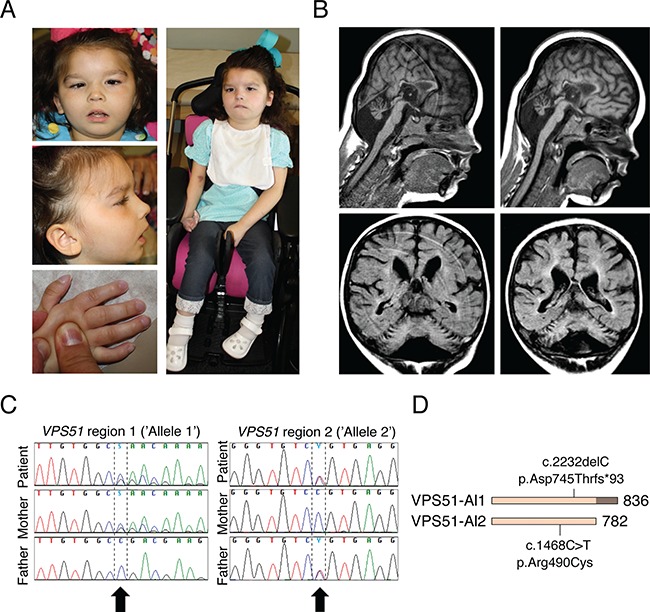
Clinical and sequencing findings in the patient. (A) Photos from ages 4 years and 3 months (top left and middle left) and 6 years and 3 months (right and lower left). Note ptosis, strabismus, epicanthal folds, posteriorly rotated ears, upturned nose, facial hypotonia with open mouth, retrognathia and clubbed fingernails. (B) Selected brain MRI images from ages 4 years and 6 months (left) and 6 years and 3 months (right). Note pontocerebellar hypoplasia, prominent infra-vermian cistern and hypoplastic corpus callosum. (C) Sanger sequencing of DNA samples from the patient and both parents confirming mutations in VPS51 and compound heterozygosity, as originally found by whole exome sequencing (Supplementary Material, Fig. S1). (D) Representation of mutations in VPS51. The mutation in allele 1 causes replacement of threonine for aspartate 745 and a frameshift that adds 54 random amino acids at the C-terminus of the protein. The mutation in allele 2 results in substitution of cysteine for arginine 490.
Biochemical testing has been notable for abnormalities suggesting a possible congenital disorder of glycosylation (CDG), including evidence of hypoglycosylation on serum transferrin isoelectric focusing (manifested by a slightly low percentage of trisialotransferrin and slightly increased percentages of asialo- and monosialo-transferrin) as well as an abnormal pattern of N- and O-glycosylation. Other biochemical tests were normal or non-diagnostic (see Supplementary Material, Supplemental Data).
Identification of compound heterozygous variants in VPS51
Whole exome sequencing was performed through the Molecular Diagnostic Laboratory at the Greenwood Genetic Center using genomic DNA from the patient and both parents and identified compound heterozygous variants in VPS51 (reference sequence NM_013265.3) (Fig. 2C and D; Supplementary Material, Fig. S1), including a maternally inherited frameshift alteration (allele 1: c.2232delC; p.Asp745Thrfs*93) that was considered pathogenic and a paternally inherited missense alteration (allele 2: c.1468C>T; p.Arg490Cys) that was classified as a variant of uncertain significance (VUS) prior to the publication of the American College of Medical Genetics and Genomics (ACMG) variant interpretation guidelines (26). The allele 1 variant results in the substitution of threonine for aspartate at amino acid 745 and a downstream frameshift that causes the normal stop codon to be skipped. The resulting open reading frame is 202 nucleotides longer and encodes a protein with an additional 54 extraneous amino acids C-terminal to aspartate 745 (Fig. 2D). This variant has not been reported in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php). Allele 2 has been reported in the public SNP databases but limited information is available to support it as benign.
Whole exome sequencing also identified heterozygous VUSs in three other genes. These included a paternally inherited missense variant (c.398C>G; p.Pro133Arg) in ALG1, a maternally inherited synonymous variant (c.2022C>G; p.Val674Val) in COG4 and a maternally inherited intronic variant (c.2014-4A>G) in ALG13. Pathogenic alterations in these genes have been associated with autosomal recessive CDG-type Ik (OMIM no. 608540), autosomal recessive CDG-type IIj (OMIM no. 613489) and X-linked recessive CDG-type Is (OMIM no. 300884), respectively. The recessive nature of these disorders, however, made it unlikely that the monoallelic variants in ALG1, COG4 and/or ALG13 were the cause of the disease in our patient.
Reanalysis of the exome data was performed ~18 months after the original analysis and did not identify any other significant variants but did lead to reclassification of the ALG13 variant as benign, based on 339 hemizygotes and 748 heterozygotes having been reported in the gnomAD database (http://gnomad.broadinstitute.org/). According to the ACMG guidelines on variant interpretation, a gene–disease association must be clinically validated prior to using the ACMG criteria; therefore, both of the VPS51 variants would currently be classified as VUSs. Once the gene–disease association for VPS51 has been validated, the use of ACMG criteria would classify the c.2232delC alteration as pathogenic and the c.1468C>T change as likely pathogenic (26).
The allele 1 variant produces an unstable protein that is rapidly degraded by the proteasome
To assess the effects of the mutations on the properties of VPS51, we replicated them in a mammalian expression plasmid encoding human VPS51 tagged with either one copy of the green fluorescent protein (GFP) at the N-terminus or 13 copies of the myc epitope (13myc) at the C-terminus. Wild-type (WT) and mutant forms of tagged VPS51 were expressed by transient transfection in HeLa cells. Immunoprecipitation followed by immunoblotting (IB) of cells expressing GFP-tagged forms of normal VPS51 (GFP-VPS51) or allele 1 mutant VPS51 [GFP-VPS51(Al1)] showed that whereas the normal fusion protein ran at the expected molecular mass of ~113 kDa, the mutant fusion protein was barely detectable (Fig. 3A). By increasing the exposure of the immunoblot, the VPS51 allele 1 GFP fusion can be observed to run at a slightly higher molecular weight when compared to the normal VPS51 (Supplementary Material, Fig. S2). Furthermore, IB of extracts from cells expressing 13myc-tagged normal VPS51 (VPS51-13myc) or allele 1 mutant VPS51 [VPS51(Al1)-13myc] revealed that the mutant protein was expressed at lower levels than the normal protein (Fig. 3B). Incubation with the proteasome inhibitor MG132 did not change the levels of normal VPS51-13myc but progressively increased the levels of mutant VPS51(Al1)-13myc over a 6 h period (Fig. 3B). These observations indicated that the allele 1 mutant protein was unstable and subject to degradation by the proteasome.
Figure 3.
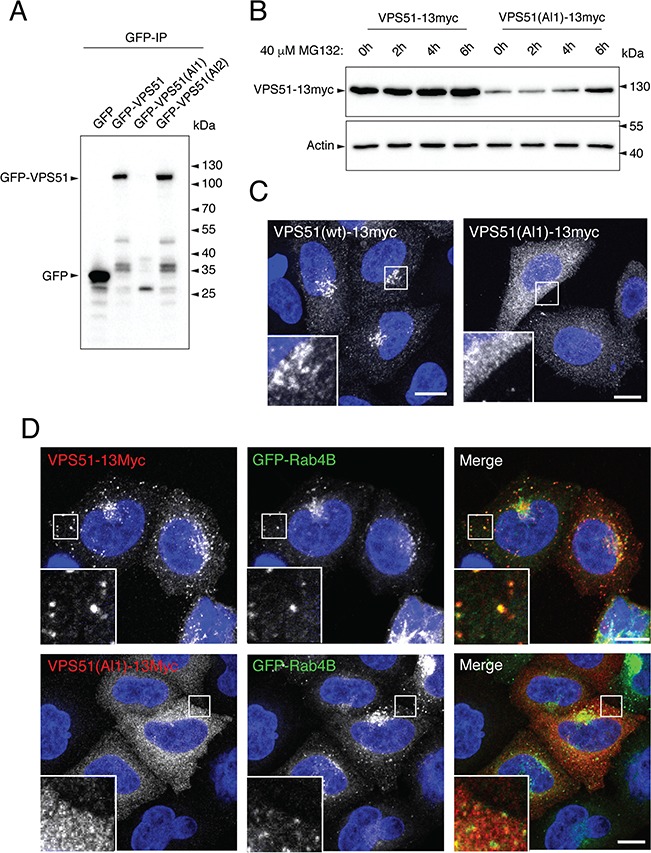
The VPS51(Al1) mutant protein is degraded by the proteasome and fails to associate with the TGN and endosomes. (A) Complementary DNAs encoding allele 1 and allele 2 mutant VPS51 tagged at the N-terminus with GFP [GFP-VPS51(Al1) and GFP-VPS51(Al2)] were expressed by transient transfection in HeLa cells. GFP and GFP-tagged normal VPS51 (GFP-VPS51) were similarly expressed as controls. Cell lysates were subjected to immunoprecipitation with GFP-nanobody-conjugated beads and immunoblot analysis with antibody to GFP. The positions of molecular mass markers are indicated to the right. Notice the undetectable levels of full-length GFP-VPS51(Al1) and the presence of lower molecular mass products of this protein. (B) Complementary DNAs encoding normal and allele 1 mutant VPS51 tagged at the C-terminus with 13myc [VPS51-13myc and VPS51(Al1)-13myc] were expressed by transient transfection in HeLa cells. Cells were incubated in 40 μm of the proteasome inhibitor MG132 for the indicated times, lysed, normalized for protein abundance by the Bradford protein assay and analyzed by IB with an antibody to the myc epitope. The positions of molecular mass markers are indicated to the right. Notice the increase in VPS51(Al1)-13myc levels over the course of the experiment. (C) HeLa cells transiently transfected with plasmids encoding VPS51-13myc and VPS51(Al1)-13myc were immunostained for the myc epitope and imaged by confocal microscopy. Notice that normal VPS51-13myc localizes to the TGN, as previously reported (6). In contrast, VPS51(Al1)-13myc shows cytosolic staining. (D) HeLa cells were transiently co-transfected with plasmids encoding GFP-Rab4B together with VPS51-13myc or VPS51(Al1)-13myc, immunostained for the myc epitope and imaged by confocal microscopy. Notice that GFP-Rab4B promoted recruitment of VPS51-13myc but not VPS51(Al1)-13myc to endosomes. In C and D, nuclei were stained with DAPI (blue). Single channels are shown in grayscale. In the merged images in D, myc staining is shown in red and GFP fluorescence is shown in green. Scale bars, 10 μm; inset scale bars, 2 μm.
We also performed immunofluorescence (IF) microscopy of HeLa cells expressing normal VPS51-13myc or mutant VPS51(Al1)-13myc. We observed that the normal protein mainly localized to the TGN (Fig. 3C), as previously reported (6). This localization is consistent with integration of the normal protein into the endogenous GARP complex. In contrast, expression of the mutant protein was detected in very few cells in comparison to the normal VPS51-13myc. In the few cells with detectable expression, the protein was largely cytosolic (Fig. 3C). The small GTPase Rab4A was previously shown to promote recruitment of the EARP-specific VPS50 subunit (also known as syndetin) to a subpopulation of endosomes (7,8). We observed that co-expression of GFP-Rab4B with VPS51-13myc similarly resulted in co-localization of both proteins to discrete puncta distributed throughout the cytoplasm (Fig. 3D), consistent with the endosomal localization of EARP (7,8). On the other hand, in the few cells that showed expression of VPS51(Al1)-13myc, the protein was cytosolic and did not localize to GFP-Rab4B-positive puncta (Fig. 3D).
Taken together, the above results indicate that VPS51 mutant allele 1 is expressed at very low levels due to its targeting for degradation by the proteasome. Moreover, even in those cells in which some expression is detected, the mutant protein is not recruited to sites of GARP and EARP localization. These defects likely result from misfolding caused by the truncation of the normal sequence and addition of a random sequence C-terminal to amino acid 745.
The allele 2 mutation produces a stable protein that is not efficiently assembled into the GARP and EARP complexes
In contrast to the allele 1 mutation, the substitution of cysteine for arginine 490 in allele 2 did not affect the expression of the GFP-tagged mutant protein [GFP-VPS51(Al2)] relative to the normal protein (GFP-VPS51), as determined by immunoblot analysis of transfected HeLa cells (Figs 3A and 4A). However, mutant GFP-VPS51(Al2) showed reduced co-immunoprecipitation with VPS50 and VPS53 relative to normal GFP-VPS51 (Fig. 4A), consistent with impaired assembly of the mutant protein into the GARP and EARP complexes. Co-immunoprecipitation with VPS54 was not tested because of the lack of a suitable antibody to this subunit. In agreement with the co-immunoprecipitation results, IF microscopy of transfected HeLa cells showed decreased TGN localization of mutant VPS51-Al2-13myc relative to normal VPS51-13myc (Fig. 4B and C). When co-expressed with GFP-Rab4B, mutant VPS51(Al2)-13myc showed co-localization of both proteins to cytoplasmic puncta, but the number of puncta per cell was lower for mutant VPS51(Al2)-13myc relative to normal VPS51-13myc (Fig. 4D and E). From these experiments, we concluded that the allele 2 mutant VPS51 is stable but gets incorporated into the GARP and EARP complexes less efficiently, resulting in reduced levels of the fully assembled complexes.
Figure 4.
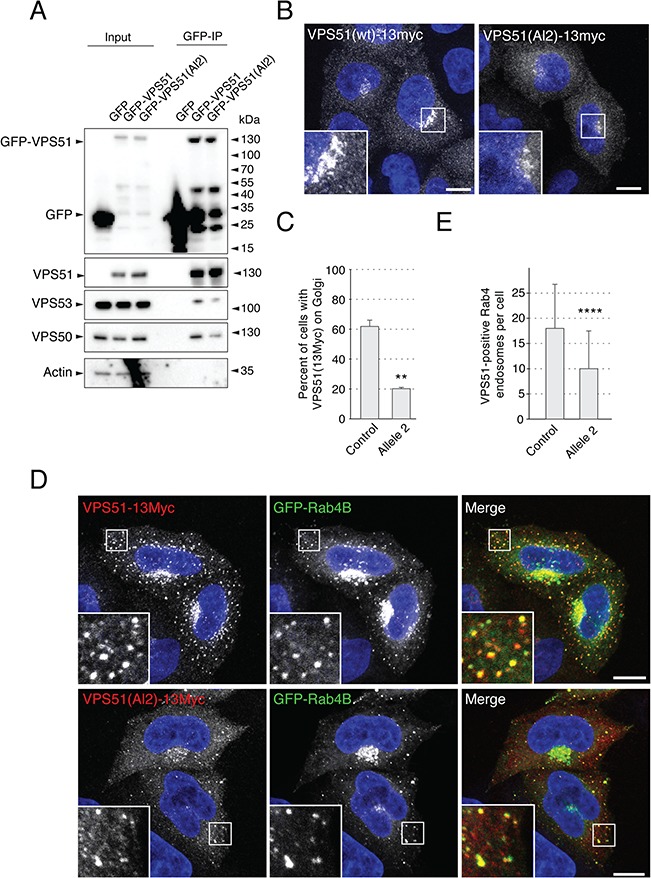
The VPS51(Al2) mutant protein is stable but assembles less efficiently into GARP and EARP. (A) Extracts from HeLa cells transiently transfected with plasmids encoding GFP, GFP-VPS51 or GFP-VPS51(Al2) were analyzed by pull down with a nanobody to GFP conjugated to magnetic beads, followed by SDS-PAGE and IB for GFP, endogenous GARP/EARP subunits and actin (loading and negative control). Positions of molecular mass markers are indicated on the right. Notice the similar levels of GFP-VPS51 and GFP-VPS51(Al2) and the decreased co-immunoprecipitation of VPS53 and VPS50 with GFP-VPS51(Al2). (B) HeLa cells were transiently transfected with plasmids encoding VPS51-13myc, immunostained for the myc epitope and imaged by confocal microscopy. (C) Quantification of the percentage of cells with VPS51-13myc staining at the TGN. Over 300 cells across 3 different independent experiments (over 100 cells per experiment per sample) were classified as having either residual TGN localization or no observable TGN localization. Percentages were calculated and compared with a one-tailed paired t-test, P = 0.0014. VPS51-13myc displayed a perinuclear (TGN) localization in >60% of the transfected cells, whereas VPS51(Al2)-13myc showed a mostly cytosolic localization, with only ~20% of cells having observable residual perinuclear staining. Moreover, VPS51(Al2)-13myc staining at the TGN was less intense than that of VPS51-13myc in comparable cells. (D) HeLa cells were transiently co-transfected with plasmids encoding GFP-Rab4B together with VPS51-13myc or VPS51(Al1)-13myc, immunostained for the myc epitope and imaged by confocal microscopy. (E) Quantification of the number of GFP-Rab4B-positive endosomes that also contained VPS51-13myc or VPS51(Al1)-13myc. VPS51-positive Rab4B endosomes in 62 cells (WT VPS51) and 64 cells (allele 2), from images taken across 3 independent experiments were counted. Data sets were compared with a t-test, P = 5.5 × 10−7. Notice that GFP-Rab4B caused recruitment of VPS51-13myc. GFP-Rab4B also caused VPS51(Al2)-13myc recruitment to endosomes but the number of endosomes was smaller and there was more cytosolic background. In B and D, nuclear staining by DAPI is shown in blue. Single channels are shown in grayscale. In the merged images in D, myc staining is shown in red and GFP fluorescence is shown in green. Scale bars, 10 μm; inset scale bars, 2 μm; error bars = standard deviation.
Decreased levels and membrane association of GARP and EARP in patient's fibroblasts
We next examined the effects of the VPS51 mutations in skin fibroblasts from the patient. Immunoblot analyses of endogenous GARP/EARP subunits showed reduced levels of VPS51 (~25%) as well as VPS50, VPS52 and VPS53 (all ~50%) in fibroblasts from the patient relative to fibroblasts from a control individual (Fig. 5A and B). Because none of the antibodies to GARP/EARP works for IF microscopy of the endogenous proteins, we transfected the fibroblasts with plasmids encoding 13myc-tagged VPS54 (VPS54-13myc), which serves as a surrogate for GARP. We observed that VPS54-13myc exhibited localization to the TGN in control fibroblasts, as previously reported for other cell types (12), and diffuse cytosolic localization in the patient's fibroblasts (Fig. 5C). We also co-expressed 13myc-tagged VPS50 (VPS50-13myc) (to label EARP) with GFP-Rab4B and found that both proteins co-localized on punctate structures distributed throughout the cytoplasm in control fibroblasts (Fig. 5D), consistent with the endosomal localization of EARP (7,8). In contrast, VPS50-13myc failed to localize to GFP-Rab4B puncta and was largely cytosolic in the patient's fibroblasts (Fig. 5D). Both the localization of VPS54-13myc to the TGN and VPS50-13myc to Rab4B-positive endosomes was restored by transfection of the patient's fibroblasts with plasmids expressing normal VPS51 tagged with either GFP (VPS51-GFP) or three copies of the HA epitope (VPS51-3HA), respectively (Fig. 5C and D). These experiments thus demonstrated that the mutations in VPS51 resulted in reduced levels of GARP and EARP and reduced association of both complexes with their corresponding intracellular compartments in the patient's fibroblasts.
Figure 5.
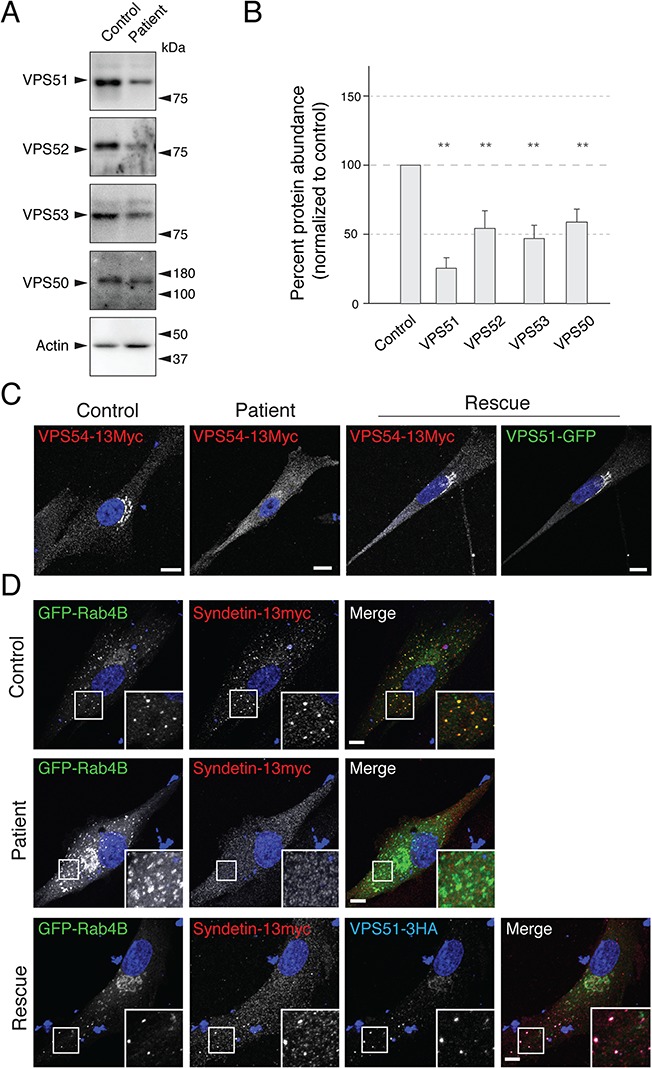
Patient's fibroblasts show defects in GARP/EARP complexes. (A) Patient's and control fibroblasts were lysed, protein abundance was normalized by the Bradford assay and samples were analyzed by SDS-PAGE and IB for endogenous GARP/EARP subunits and actin (negative control). Positions of the molecular mass markers are indicated on the right. (B) Quantification of fold differences in GARP/EARP subunit levels from three independent immunoblot assays. Actin loading control was used to normalize the expression. Samples were compared with a one-tailed paired t-test: VPS51, P = 0.0097; VPS52, P = 0.030; VPS53, P = 0.0470; VPS50, P = 0.0353. Notice the decreased levels of GARP/EARP subunits in the patient's fibroblasts. (C) Control and patient's fibroblasts were transiently transfected with plasmids encoding VPS54-13myc (left two panels), and patient's fibroblasts were transiently co-transfected with plasmids encoding VPS54-13myc and VPS51-GFP (right two panels). Notice that co-expression with VPS51-GFP recovers perinuclear localization of VPS54-13myc in the patient's fibroblasts. (D) Control and patient's cells were co-transfected with DNA plasmids encoding GFP-Rab4B and VPS50-13myc (top two panels), as well as VPS51-3HA (bottom panel). Notice that co-expression with VPS51-3HA recovers the localization of VPS50-13myc to the GFP-Rab4B-positive endosomes. In C and D, nuclear staining by DAPI is shown in blue. Single channels are shown in grayscale. In panel C, GFP signal is shown in green. In the merged images in D, GFP fluorescence is shown in green, myc staining in red and, for the bottom panel, HA staining in blue. Scale bars, 10 μm; inset scale bars, 2 μm; error bars = standard deviation.
Altered distribution of the CI-MPR and abnormal lysosomes in patient's fibroblasts
Previous studies showed that knock-down (KD) of GARP subunits in HeLa cells altered the intracellular distribution of the CI-MPR (6,12,27). In line with these findings, IF microscopy showed that the CI-MPR exhibited bright juxtanuclear staining in control fibroblasts and faint, more dispersed cytoplasmic staining in the patient's fibroblasts (Fig. 6A and G). In addition, we observed that the patient's fibroblasts had lower levels of CI-MPR protein relative to the control fibroblasts, as analyzed by IB (Fig. 6C and F). These results are consistent with reduced retrieval to the TGN and increased turnover of the CI-MPR in the patient's fibroblasts.
Figure 6.
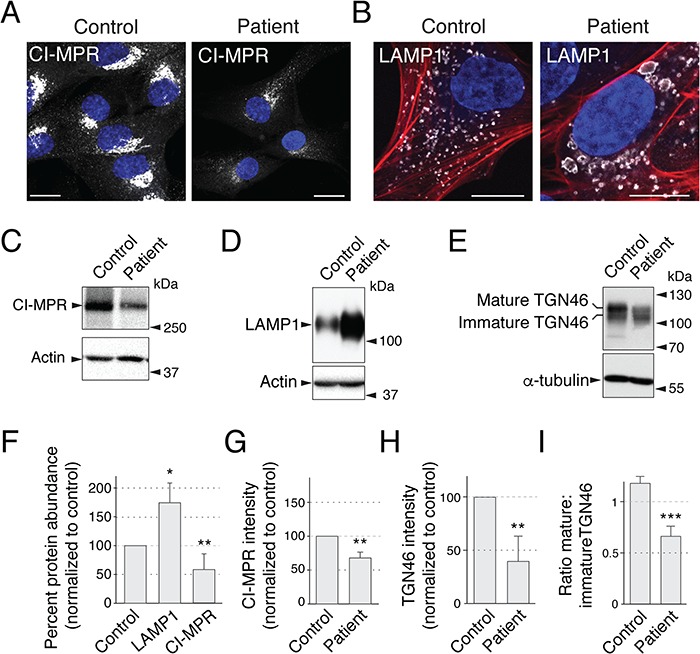
Patient's fibroblasts show defects in GARP/EARP-dependent processes. (A) Control and patient's fibroblasts were immunostained with an antibody to endogenous CI-MPR. (B) Control and patient's fibroblasts were immunostained with antibody to LAMP1 and co-stained with the actin marker phalloidin-555. We observed that ~20% of cells exhibited abnormally large lysosomes, as shown in the figure. (C–E) Patient's and control fibroblasts were lysed, normalized by the Bradford protein assay and analyzed by SDS-PAGE and IB for endogenous CI-MPR (C), LAMP1 (D) and TGN46 (E). (F) Quantification of fold differences in CI-MPR and LAMP1 protein levels in patient's versus normal fibroblasts from a minimum of three independent immunoblot assays per sample. Actin loading control was used to normalize the values. Samples were compared with a one-tailed paired t-test: LAMP1, P = 0.0163; CI-MPR, P = 0.0061. Notice that CI-MPR was less abundant and LAMP1 more abundant in the patient's cells. (G) Quantification of mean CI-MPR intensity of control and patient's fibroblasts was performed by comparing mean intensity from 30 cells per sample across 3 independent experiments (10 cells per sample per experiment). Mean intensity was averaged for each sample in each experiment and the ratio was statistically compared to a value of ‘1’ per experiment for a one-tailed paired t-test, P = 0.008. (H, I) Total intensity of TGN46 normalized to alpha-tubulin (H, P = 0.005) and the mature:immature ratio (I, P = 0.0009) were quantified from three independent experiments. In panels C, D and E, positions of molecular mass markers are indicated to the right. In A and B, nuclear staining by DAPI is shown in blue. CI-MPR and LAMP1 staining is shown in grayscale. In panel B, actin stained with phalloidin-555 is shown in red. Scale bars, 10 μm; error bars = standard deviation.
Another recycled protein is the glycosylated membrane protein TGN46. Accordingly, we observed decreased total levels of TGN46 (Fig. 6E and H), indicative of a recycling defect. Additionally, we observed a decrease in the mature form of TGN46 relative to a lower molecular weight form (Fig. 6E and I), which could be either a partially degraded form or, alternatively, a biosynthetic glycosylation intermediate, in support of the clinical observation of defective N- and O- glycosylation (see clinical report in the Supplementary Material, Supplemental Data).
VPS51-KD and VPS52-KD HeLa cells (6,12), as well as fibroblasts from VPS53-mutant PCCA2/PCH2E patients (23), were previously found to have more numerous and larger lysosomes, as visualized by immunostaining with antibodies to the late endosomal–lysosomal protein CD63. Immunostaining of the VPS51 mutant patient fibroblasts with an antibody to another lysosomal membrane protein, LAMP1, showed enlarged lysosomes in ~20% of the cells (Fig. 6B). In addition, immunoblot analysis showed an increase in the levels of LAMP1 in patient versus control fibroblasts (Fig. 6D and F).
Discussion
This study is the first to demonstrate the involvement of VPS51 in a human disorder and provides further insight into the phenotypic effects resulting from disturbances in the GARP and EARP complexes.
Summary of the patient's phenotype and comparison to pontocerebellar hypoplasia, PCCA2 and PEHO-like syndrome
The clinical phenotype observed in our patient is primarily characterized by neurodevelopmental manifestations, including severe global developmental delay, hypotonia, microcephaly, seizure disorder, cortical vision impairment and abnormal brain imaging with reduced size of the cerebellum and brainstem, hypoplasia of the corpus callosum and cerebral white matter abnormalities. The generally static nature of the patient's condition suggests that these findings may reflect primary developmental abnormalities of the brain. On the other hand, it is possible that the findings could be due to a progressive process, as described in patients with VPS53 mutations (23,25). Our patient's normal early brain MRI scan followed by the discovery of multiple abnormalities on subsequent imaging would support the latter possibility. In either case, the similarities between our patient's neurodevelopmental phenotype and that described in patients with VPS53 mutations suggest that, in general, GARP/EARP dysfunction results in a severe neurodevelopmental disorder. Given that mutations in VPS53 are classified as causing a specific form of pontocerebellar hypoplasia (PCH2E/PCCA2) and have also been associated with a PEHO-like syndrome, we propose that VPS51 mutations may be responsible for a previously unclassified form of these disorders and that perturbation of GARP/EARP function has particularly severe effects on pontocerebellar development.
Our patient's history of poor somatic growth, liver dysfunction, gastric volvulus, other health issues and dysmorphic features is also notable and suggests that GARP/EARP dysfunction has systemic effects beyond the central nervous system. Interestingly, our patient had abnormal glycosylation testing without evidence of a specific CDG, and her phenotype has significant overlap with that seen in CDGs. Moreover, we experimentally observed abnormalities of the N- and O-glycosylated protein TGN46 in the patient's cells. This suggests that GARP/EARP dysfunction may result in a complex phenotype that resembles CDGs both clinically and biochemically. The fact that she developed lower extremity edema at 6 years of age is also noteworthy in light of the previous description of limb edema in the two siblings with a PEHO-like syndrome caused by mutations in VPS53 (25).
Neurological defects in animal models with mutations in GARP/EARP subunits
To date there have been no studies on the loss of VPS51 in a mammalian model. However, homozygous null mutations of the shared GARP/EARP subunits VPS52 or VPS53 in mouse cause embryonic lethality at E6.5 or E10.5–E11.5, respectively (16,18,19). Likewise, homozygous null mutations of the GARP-specific VPS54 subunit in mouse are embryonic lethal at day E12.5 (17), with membrane blebbing into the lumen of the neural tube observed at days E10.5 and E11.5 (19). The earlier lethality resulting from VPS52 and VPS53 mutations relative to VPS54 mutations suggests that combined loss of GARP and EARP has a more deleterious effect than that of GARP alone. Alternatively, EARP may be more important for viability. Distinguishing between these two possibilities will require the study of mice deficient in the EARP-specific VPS50 subunit.
The fact that the VPS51-deficient patient described here survived through embryonic development and infancy is consistent with the presence of residual levels of fully assembled, functional GARP and EARP. This situation is analogous to that of VPS54 mutations in the mouse, for which null mutations are embryonic lethal but the hypomorphic missense mutation in the wr mouse allows viability albeit with neurological defects (17). The L967Q substitution in this mouse results in a protein that is conformationally unstable but partly incorporated into GARP (20). The wr mouse was originally characterized phenotypically by an unsteady gait and head tremor, followed by progressive muscle weakness (28,29). These defects were attributed to motor neuron degeneration similar to that in ALS. As in wr, the motor deficits in the patient described here may derive from loss of GARP function. Other findings in the patient could be due to partial loss of EARP function. In this regard, studies in C. elegans have implicated the EARP-specific VPS50 subunit in the formation of dense-core vesicles, with downstream effects on locomotion (14,30). Additionally, a mutation in TSSC1/EIPR-1, a protein associated with GARP/EARP (14,31), has been shown to cause a similar phenotype in C. elegans (14). These animal studies thus support the conclusion that the neurological deficits in the patient are due to partial loss of GARP and EARP.
Cellular effects of VPS51 mutations
Characterization of skin fibroblasts from the patient revealed several abnormal phenotypes, including reduced GARP/EARP levels, increased levels of LAMP1, swelling of lysosomes and mislocalization of the CI-MPR. These phenotypes are consistent with the reduction in GARP/EARP levels causing lysosomal defects. The CI-MPR and related cation-dependent mannose 6-phosphate receptor mediate sorting of mannose 6-phosphate (M6P)-modified acid hydrolases from the TGN to endosomes (32). The acidic pH of endosomes causes dissociation of the hydrolase-MPR complexes, after which the hydrolases traffic to lysosomes whereas the MPRs return to the TGN for further rounds of sorting. GARP promotes the tethering and fusion of the MPR carriers with the TGN (11). Loss of GARP interrupts this cycle by preventing the retrieval of MPRs to the TGN, with consequent missorting of acid hydrolases toward the extracellular space (6,12). The reduced levels of GARP thus likely explain the mislocalization of the CI-MPR in the patient cells. In this context, the swelling of lysosomes most probably results from accumulation of undegraded materials due to reduction of acid hydrolase levels, as previously observed upon KD of GARP subunits (6,12). The increased levels of LAMP1 could result from decreased turnover of this protein in lysosomes or, alternatively, from increased synthesis due to activation of lysosome biogenesis pathways (33).
The defect in MPR sorting in the patient's cells is also likely to impact the Niemann–Pick C2 protein (NPC2), which, like the lysosomal acid hydrolases, is modified with M6P and sorted by the MPRs (27,34). NPC2, together with NPC1, mediates export of cholesterol from the lumen of late endosomes and lysosomes to the cytosol or other organelles (35). Consistent with this mechanism, cell lines with defects in GARP subunits, as well as the wr mouse, exhibit increased accumulation of cholesterol in lysosomes (20). In addition, GARP mutations in yeast, including one analogous to that in human PCCA2, were shown to alter sphingolipid homeostasis (13). It is thus possible that the patient could also have dysregulation of lipid metabolism. Little is known about other cargos that depend on GARP or EARP for recycling from endosomes. Further studies to identify such cargos will likely shed greater light into the pathogenesis of GARP/EARP deficiency.
Classification of VPS51-related disorders
PCCA2 caused by mutations in VPS53 (23) has been proposed to be part of a more general disease classification with shared pathophysiology, termed the ‘Golgipathies’ (36). Golgipathies are inherited diseases that have a shared set of characteristics including microcephaly, white matter defects and intellectual disability. The causative proteins in this disease class are all functionally associated with the Golgi apparatus. Accordingly, the VPS51 mutations characterized here could be considered in the same class of diseases, occupying a subclass of `GARP/EARP deficiency disorders'. Further understanding of the phenotypic effects of mutations in VPS51 and other genes encoding components of the GARP/EARP complexes will require the identification and characterization of additional cases.
Materials and Methods
Human subjects
The patient was seen for clinical genetic evaluations through the Greenwood Genetic Center. Parental informed consent for human subjects research was obtained through a protocol approved by the institutional review board of Self Regional Healthcare (Greenwood, SC). De-identified skin fibroblasts from the patient and an unrelated control individual were shared with the National Institutes of Health (NIH) laboratory for research purposes. For additional control fibroblasts (Supplementary Material, Fig. S3), unrelated anonymized fibroblasts from an unrelated project were used, as used previously (37).
Whole exome sequencing
DNA libraries were prepared from 3 μg of genomic DNA isolated from the proband and parental peripheral blood samples, using the Agilent SureSelectXT Human All Exon v5 capture kit (Agilent Technologies, Santa Clara, CA). Briefly, DNA was fragmented using the Covaris S220 system (Covaris, Woburn, MA), and fragments of 150–200 bp were selected using AMPure XP beads (Beckman Coulter, Brea, CA). Fragments were subsequently end-repaired, adenylated at the 3′ end, ligated to sequencing adaptors and then polymerase chain reaction (PCR)-amplified using the SureSelectXT Library Preparation kit (Agilent Technologies). DNA was next purified using AMPure XP beads. The concentration of the enriched libraries was determined using a DNA-1000 chip on the 2100 Bioanalyzer (Agilent Technologies). A total of 750 ng of each DNA library was used for hybridization and capture with the SureSelectXT Human All Exon v5 probes (Agilent Technologies). After hybridization, the RNA-bound DNA was retained and unhybridized material was washed away. Captured fragments were amplified by PCR and purified. The quality of the enriched libraries was evaluated using a High Sensitivity DNA chip on the 2100 Bioanalyzer. Libraries were quantified on a Qubit® 2.0 Fluorometer using a Qubit High Sensitivity kit (Life Technologies, Waltham, MA), and separate libraries were pooled and sequenced using an Illumina NextSeq 500® Sequencing System (Illumina Inc., San Diego, CA) per the manufacturer's protocol.
Bioinformatics
Exome sequencing generated a minimum of 97 million reads per patient sample with an average read length of 75 bp. Sequences were processed using NextGENe software (SoftGenetics, LLC, State College, PA) and trimmed for low-quality and duplicate reads. Reads were trimmed or rejected when the median Phred quality score was below 20 and/or had more than 3 bases with a Phred score ≤16. The sequences were then mapped to the February 2009 human reference assembly (GRCh37/hg19). Positions in the coding regions of the reference (with 25 bp flanking) were classified as variants if they had a read depth of more than 3 and a variant allele frequency of ≥12%. The variant reports were then converted into vcf files, which were uploaded to Cartagenia Bench Lab NGS (Agilent Technologies) for filtering and subsequent analysis of variants of interest.
Exome data analysis
Variants were first matched to the HGMD; however, analysis of these variants did not yield promising gene candidates. Therefore, the unmatched variants were further analyzed. Benign variants were separated using the dbSNP, 1000 Genomes, ExAC and ESP6500 databases with an allele frequency cut-off of 0.033. Next, quality and allele frequency filters were used to isolate variants with at least 20× read depth and 22% minor allele frequency. The resulting variants were then further analyzed using additional filters including those for splice site, functional effect prediction and Human Phenotype Ontology (HPO). Reanalysis of the data was performed ~18 months after the initial analysis using the most current databases and literature available (ExAC, gnomAD, HGMD and PubMed).
Antibodies
The following antibodies were used for IB and/or IF: mouse anti-LAMP1 (H4A3; Developmental Studies Hybridoma Bank, Iowa City, IA), used at 1:1000 for IB and 1:500 for IF; mouse anti-CI-MPR (2G11; Abcam, Cambridge, United Kingdom), used at 1:500 for IF; homemade rabbit anti-CI-MPR, used at 1:1000 for IB; rabbit anti-VPS51 (HPA039650; Atlas Antibodies, Bromma, Sweden), used at 1:1000 for IB; homemade rabbit anti-VPS52 (12), used at 1:2000 for IB; rabbit anti-VPS53 (HPA024446; Atlas Antibodies), used at 1:1000 for IB; mouse anti-VPS50 (FLJ20097 monoclonal antibody M01, 2D11; Abnova, Taipei City, Taiwan), used at 1:1000 for IB; mouse anti-myc epitope (9E10; Santa Cruz Biotechnology, Santa Cruz, CA), used at 1:500 for IF; rat anti-HA epitope (3F10; Roche, Basel, Switzerland), used at 1:500 for IF; rabbit anti-GFP (A-11122; Thermo Fisher Scientific, Waltham, MA), used at 1:2000 for IB; and sheep anti-TGN46 (AHP500G; Bio-Rad, Hercules, CA), used at 1:4000 for IB. Horseradish peroxidase (HRP)-conjugated secondary antibodies (Perkin Elmer, Waltham, MA) were used at 1:5000. Alexa Fluor-conjugated secondary antibodies for immunostaining (Thermo Fisher Scientific) were used at 1:1000. Alexa Fluor phalloidin-555 (Thermo Fisher Scientific) was used according to the manufacturer's instructions.
DNA recombinant constructs
Plasmids encoding GFP fused to WT and mutant human VPS51 were constructed by Gibson assembly (New England Biolabs, Ipswich, MA) from two artificially synthesized dsDNA ‘gBlocks’ (Integrated DNA Technologies, Inc., Coralville, IA). The first gBlock (comprising nucleotides 1-1493) was codon optimized for human codon usage to decrease DNA secondary structures. Synthetic gBlocks were assembled into pEGFP-C1 (Clontech, Takara Bio Inc., Kyoto, Japan) by Gibson assembly using a standard procedure (50°C incubation, 1 h). C-terminal 13myc fusions were assembled into a modified pCI-neo (Promega, Madison, WI) by PCR amplification using VPS51-GFP-fusion templates and restriction digestion with EcoRI-SalI sites included in the primers (shared forward primer, 5′ CCTCGAGAATTCGCCACCATGGCCGCAGCCGCAGCGGCAGGG; WT and allele 2 reverse primer, 5′ CCCGGGGTCGACGCCGCGCTCGCAGATGACCTCAAC; allele 1 reverse primer, 5′ CCCGGGGTCGACAAACCCCGCCCCCGGAAAGGCCCG) and ligation into EcoRI-SalI digested vector. A cDNA encoding Canis familiaris Rab4B amplified from MDCK cells and cloned into pEGFP-C1 (Clontech) was a gift of Robert Lodge (National Institute of Child Health and Human Development, NIH). Human VPS54 cDNA was purchased (Origene, Rockville, MD) and subcloned into pCI-neo 13myc vector by Gibson assembly (fragment forward primer, 5′ TAGGCTAGCCCGAGAATTCGCCACCATGGCTTCAAGCCACAGTTC; fragment reverse primer, 5′ TTAATTATCCCGGGGTCGACCCTCTTCTGCTCCCAAATTTC; vector forward primer, 5′ GTCGACCCCGGGATAATTAAC; vector reverse primer, 5′ GAATTCTCGAGGCTAGCCTATAGTGAG). A plasmid encoding VPS50-13myc was previously described (8). p3HA-N1 vector was generated by replacing the EGFP sequence of pEGFP-N1 with a 3HA sequence, followed by sub-cloning of codon-optimized human VPS51 into the HA and EGFP vector (for C-terminally tagged VPS51) using a PCR amplicon (forward primer, 5′ CCTCGAGAATTCGCCACCATGGCCGCAGCCGCAGCGGCAGGG; reverse primer, 5′ CGCGGTACCGTGCCGCGCTCGCAGATGACCT) digested with EcoRI-KpnI and ligated into an EcoRI-KpnI-digested vector backbone.
Cell culture and transfection
HeLa cells (ATCC, Manassas, VA) were cultured in complete Dulbecco's Modified Eagle's Medium [supplemented with 10% fetal bovine serum (FBS), l-glutamine, penicillin-streptomycin (all from Corning, Corning, NY)], at 37°C, 5% CO2 and 95% relative humidity. HeLa cells were transfected with either Lipofectamine 2000 (Thermo Fisher Scientific) or Fugene 6 (Promega) according to the manufacturer's instructions. Skin biopsies obtained at the Greenwood Genetic Center were placed in sterile Petri dishes and minced using scalpels. Minced tissue was incubated for ~1 h in collagenase solution (50 mg collagenase in 50 ml Minimum Essential Medium) to dissociate cells from tissue fragments. The collagenase solution was removed and the remaining enzyme was deactivated using FBS. The cells were plated in complete Chang Medium D (with 1% antibiotic–antimycotic solution) and allowed to grow in an incubator at 37°C, 5% CO2. After shipment to the NIH laboratory, human fibroblasts were cultured in Chang Medium D (Irvine Scientific, Santa Ana, CA) supplemented with MycoZap Plus-CL (Lonza, Walkersville, MD), at 37°C, 5% CO2 and 95% relative humidity. Human fibroblasts were transfected with Fugene HD (Promega) according to the manufacturer's instructions.
Immunoprecipitation and biochemical analysis
Immunoprecipitation of GFP fusions was performed using magnetic GFP-trap beads (ChromoTek, Planegg-Martinsried, Germany) according to the manufacturer's instructions. Briefly, cells transiently transfected on 100 mm cell culture plates were lysed for 30 min on ice in a buffer containing 10 mm Tris-HCl pH 7.4, 150 mm NaCl, 0.5 mm EDTA, 0.1% Nonidet P40 (NP40) and protease inhibitors (Roche; Basel, Switzerland). Lysates were centrifuged at 17 000× g for 15 min at 4°C to remove cell debris. Cell extracts were combined with 25 μl equilibrated GFP-trap magnetic beads; volume was made up to 1 ml with lysis buffer without NP40. Incubation was for 2 h at 4°C under constant rotation. After magnetic separation to remove unbound material, beads were washed three times rotating at room temperature for 5 min with 1 ml of lysis buffer lacking NP40. Protein was eluted from beads by incubation in reducing Laemmli SDS-PAGE sample buffer (Bio-Rad) for 5 min at 98°C. Samples were subsequently analyzed by SDS-PAGE and IB as described below.
For MG132 incubations, 800 000 HeLa cells were plated onto a six-well dish and transfected with plasmids encoding VPS51-13myc or VPS51(Al1)-13myc. The following day, cells were split equally onto 4 wells each of a 12-well plate. The following day, a time course (0, 2, 4 and 6 h) with 40 μm MG132 (700 μl per well) was performed, starting with a medium change at the 6 h time point to allow a simultaneous cell lysis across all samples. At the end of the time course, the cells were transferred to ice, washed in ice-cold phosphate-buffered saline (PBS) and lysed in 250 μl lysis buffer [50 mm Tris pH 7.4, 150 mm NaCl, 1% Triton X-100 with protease inhibitors (Roche)]. Cells were incubated for 30 min on ice with pipetting every 10 min. After lysis, cells were transferred to an ice-cold 1.5 mL microcentrifuge tube, centrifuged at 17 000× g for 15 min at 4°C to pellet cell debris, and the cell lysate was transferred to a new 1.5 ml microcentrifuge tube. Protein concentration was normalized across series by Bradford assay before IB probing.
IB was performed by standard protocols using SDS-PAGE separation and subsequent transfer to polyvinyl difluoride (PVDF) membranes. Membranes were blocked for 1–2 h with 5% non-fat milk (Bio-Rad) in PBS-T [PBS supplemented with 0.05 % Tween 20 (Sigma-Aldrich, St. Louis, MO)], before being incubated overnight in primary antibody diluted in PBS-T with 3% bovine serum albumin (BSA) (Sigma-Aldrich) or PBS-T with 5% non-fat milk. Samples were washed three times for 5 min in PBS-T before being incubated for 1–2 h in HRP-conjugated secondary antibody (1:5000), diluted in PBS-T supplemented with 5% non-fat milk. Membranes were washed three times in PBS-T and once in PBS before being visualized using either Clarity western ECL blotting substrate (Bio-Rad) or SuperSignal West Femto maximum sensitivity substrate (Thermo Fisher Scientific).
Immunofluorescent staining
Cells for IF microscopy were plated onto 12 mm cover glasses (EF15973A; Daigger, Vernon Hills, IL), transfected as described above and fixed using 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS (KD MedicalColumbia, MD). Cells were permeabilized at room temperature for 15 min in IF buffer [0.05% saponin (Sigma-Aldrich), 5% BSA (Sigma-Aldrich) in PBS]. Primary antibodies were diluted in IF buffer and incubated with cells for 1 h at room temperature. Cells were washed three times in IF buffer. Alexa Fluor secondary antibodies (Thermo Fisher Scientific) were diluted to 1:1000 in IF buffer and cells were incubated for 1 h at room temperature. Cells were washed three further times in IF buffer and once in water to remove PBS from the coverslip surface, and mounted to a glass cover slip in Immuno Mount [with or without 4′, 6-diamidino-2-phenylindole (DAPI); Electron Microscopy Sciences].
Microscopy
Imaging was performed on a Zeiss 710 or 880 microscope (Carl Zeiss, Oberkochen, Germany) with an oil-immersion 63×/1.40 NA Plan-Apochromat Oil DIC M27 objective lens (Carl Zeiss). Image settings (i.e. gain, laser power and pinhole) were kept constant for images presented for comparison. ImageJ (http://imagej.nih.gov/ij/) was used for image processing, including cropping, average/maximum intensity projections and contrast and brightness adjustments, with any changes being kept consistent for comparable images. Images were assembled in Adobe Illustrator (Adobe Systems, San Jose, CA).
Quantification
Quantification of percentage of cells with VPS51-13myc at the TGN (Fig. 4C) was performed by counting a total of over 300 cells across 3 different independent experiments (over 100 cells per experiment per sample). Cells in which no TGN localization could be observed were classified in one category, and cells with any residual TGN localization were classified as having ‘VPS51-13myc at the TGN’. Percentages were calculated and compared with a one-tailed paired t-test.
Quantification of VPS51-positive Rab4B endosomes per cell (Fig. 4E) was calculated by manually counting VPS51-positive Rab4B endosomes in 62 cells (WT VPS51) and 64 cells (allele 2) from images taken across 3 independent experiments.
Quantification of fold differences in protein levels (Figs. 5 and 6) was performed in ImageJ. Three independent immunoblot assays (i.e. different cell lysates) were analyzed per sample. Band intensity was calculated as mean intensity in ImageJ, with blot background removed by measuring mean intensity from an area without signal. For each sample, the corresponding actin loading control was used to normalize the expression. Samples were compared with a one-tailed paired t-test.
Quantification of the ratio of mature to immature TGN46 protein levels (Fig. 6) was performed in ImageJ. Three different cell lysates were analyzed. Band intensities of both mature and immature TGN46 were measured and calculated as the ratio using ImageJ gel analysis software. Samples were compared with a one-tailed paired t-test.
Quantification of CI-MPR intensity of control and patient's fibroblasts (Fig. 6) was performed in ImageJ by comparing mean intensity of 30 cells per sample across 3 independent experiments (10 cells per sample per experiment). Mean intensity was averaged for each sample in each experiment and the ratio was statistically compared to a value of ‘1’ per experiment for a one-tailed paired t-test.
Asterisks are indications of calculated statistical significance. ns: P > 0.05, *P ≤ 0.05, ** P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the patient and her parents for their participation in this project and Robert Lodge, PhD, for kind gifts of reagents. We also thank Melanie Jones, PhD, for her initial analysis of the patient's exome data and Gail Stapleton, MS, and Jessica Davis, MS, for their assistance with the patient's clinical evaluations. We would also like to thank Tal Keren-Kaplan, PhD, and Amra Saric, PhD, for their critical reading of this manuscript.
Conflict of Interest statement. The Greenwood Genetic Center receives revenue from diagnostic testing performed in the Greenwood Genetic Center Molecular Diagnostic Laboratory.
Funding
This work was funded by the Intramural Program of the National Institute of Child Health and Human Development (project ZIA HD001607). Support was also provided to the Greenwood Genetic Center through the South Carolina Department of Disabilities and Special Needs.
References
- 1. Bonifacino J.S. and Hierro A. (2011) Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol., 21, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spang A. (2016) Membrane tethering complexes in the endosomal system. Front. Cell Dev. Biol., 4, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siniossoglou S. and Pelham H.R. (2002) Vps51p links the VFT complex to the SNARE Tlg1p. J. Biol. Chem., 277, 48318–48324. [DOI] [PubMed] [Google Scholar]
- 4. Conibear E., Cleck J.N. and Stevens T.H. (2003) Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol. Biol. Cell, 14, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liewen H., Meinhold-Heerlein I., Oliveira V., Schwarzenbacher R., Luo G., Wadle A., Jung M., Pfreundschuh M. and Stenner-Liewen F. (2005) Characterization of the human GARP (Golgi associated retrograde protein) complex. Exp. Cell Res., 306, 24–34. [DOI] [PubMed] [Google Scholar]
- 6. Perez-Victoria F.J., Schindler C., Magadan J.G., Mardones G.A., Delevoye C., Romao M., Raposo G. and Bonifacino J.S. (2010) Ang2/fat-free is a conserved subunit of the Golgi-associated retrograde protein (GARP) complex. Mol. Biol. Cell, 21, 3386–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gillingham A.K., Sinka R., Torres I.L., Lilley K.S. and Munro S. (2014) Toward a comprehensive map of the effectors of rab GTPases. Dev. Cell, 31, 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schindler C., Chen Y., Pu J., Guo X. and Bonifacino J.S. (2015) EARP, a multisubunit tethering complex involved in endocytic recycling. Nat. Cell Biol., 17, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conibear E. and Stevens T.H. (2000) Vps52p, Vps53p, and Vps54p form a novel multisubunit complex required for protein soring at the yeast late Golgi. Mol. Biol. Cell, 11, 305–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siniossoglou S. and Pelham H.R. (2001) An effector of Ypt6p binds the SNARE Tlg1p and mediates selective fusion of vesicles with late Golgi membranes. EMBO J., 20, 5991–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perez-Victoria F.J. and Bonifacino J.S. (2009) Dual roles of the mammalian GARP complex in tethering and SNARE complex assembly at the trans-Golgi network. Mol. Cell. Biol., 29, 5251–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perez-Victoria F.J., Mardones G.A. and Bonifacino J.S. (2008) Requirement of the human GARP complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin D to lysosomes. Mol. Biol. Cell, 19, 2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fröhlich F., Petit C., Kory N., Christiano R., Hannibal-Bach H.K., Graham M., Liu X., Ejsing C.S., Farese R.V. and Walther T.C. (2015) The GARP complex is required for cellular sphingolipid homeostasis. Elife, 4. doi: 10.7554/eLife.08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Topalidou I., Cattin-Ortolá J., Pappas A.L., Cooper K., Merrihew G.E., MacCoss M.J. and Ailion M. (2016) The EARP complex and its interactor EIPR-1 are required for cargo sorting to dense-core vesicles. PLoS Genet., 12, e1006074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ho S.Y., Lorent K., Pack M. and Farber S.A. (2006) Zebrafish fat-free is required for intestinal lipid absorption and Golgi apparatus structure. Cell Metab., 3, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bennett D. and Dunn L.C. (1958) Effects on embryonic development of a group of genetically similar lethal alleles derived from different populations of wild house mice. J. Morphol., 103, 135–157. [Google Scholar]
- 17. Schmitt-John T., Drepper C., Mussmann A., Hahn P., Kuhlmann M., Thiel C., Hafner M., Lengeling A., Heimann P., Jones J.M. et al. (2005) Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet., 37, 1213–1215. [DOI] [PubMed] [Google Scholar]
- 18. Sugimoto M., Kondo M., Hirose M., Suzuki M., Mekada K., Abe T., Kiyonari H., Ogura A., Takagi N., Artzt K. et al. (2012) Molecular identification of t(w5): Vps52 promotes pluripotential cell differentiation through cell–cell interactions. Cell Rep., 2, 1363–1374. [DOI] [PubMed] [Google Scholar]
- 19. Karlsson P., Droce A., Moser J.M., Cuhlmann S., Padilla C.O., Heimann P., Bartsch J.W., Füchtbauer A., Füchtbauer E.M. and Schmitt-John T. (2013) Loss of vps54 function leads to vesicle traffic impairment, protein mis-sorting and embryonic lethality. Int. J.Mol. Sci., 14, 10908–10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez-Victoria F.J., Abascal-Palacios G., Tascon I., Kajava A., Magadan J.G., Pioro E.P., Bonifacino J.S. and Hierro A. (2010) Structural basis for the wobbler mouse neurodegenerative disorder caused by mutation in the Vps54 subunit of the GARP complex. Proc. Natl. Acad. Sci. U. S. A., 107, 12860–12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmitt-John T. (2015) VPS54 and the wobbler mouse. Front. Neurosci., 9, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meisler M.H., Russ C., Montgomery K.T., Greenway M., Ennis S., Hardiman O., Figlewicz D.A., Quenneville N.R., Conibear E. and Brown R.H. (2008) Evaluation of the Golgi trafficking protein VPS54 (wobbler) as a candidate for ALS. Amyotroph. Lateral Scler., 9, 141–148. [DOI] [PubMed] [Google Scholar]
- 23. Feinstein M., Flusser H., Lerman-Sagie T., Ben-Zeev B., Lev D., Agamy O., Cohen I., Kadir R., Sivan S., Leshinsky-Silver E. et al. (2014) VPS53 mutations cause progressive cerebello-cerebral atrophy type 2 (PCCA2). J. Med. Genet., 51, 303–308. [DOI] [PubMed] [Google Scholar]
- 24. Salonen R., Somer M., Haltia M., Lorentz M. and Norio R. (1991) Progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO syndrome). Clin. Genet., 39, 287–293. [DOI] [PubMed] [Google Scholar]
- 25. Hady-Cohen R., Ben-Pazi H., Adir V., Yosovich K., Blumkin L., Lerman-Sagie T. and Lev D. (2018) Progressive cerebello-cerebral atrophy and progressive encephalopathy with edema, hypsarrhythmia and optic atrophy may be allelic syndromes. Eur. J.Paediatr. Neurol., doi: 10.1016/j.ejpn.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 26. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E. and Spector E. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wei J., Zhang Y.Y., Luo J., Wang J.Q., Zhou Y.X., Miao H.H., Shi X.J., Qu Y.X., Xu J., Li B.L. et al. (2017) The GARP complex is involved in intracellular cholesterol transport via targeting NPC2 to lysosomes. Cell Rep., 2835, 19, 2823. [DOI] [PubMed] [Google Scholar]
- 28. Duchen L.W. and Strich S.J. (1968) An hereditary motor neurone disease with progressive denervation of muscle in the mouse: the mutant ‘wobbler’. J. Neurol.Neurosurg. Psychiatry., 31, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moser J.M., Bigini P. and Schmitt-John T. (2013) The wobbler mouse, an ALS animal model. Mol. Genet. Genomics, 288, 207–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paquin N., Murata Y., Froehlich A., Omura D.T., Ailion M., Pender C.L., Constantine-Paton M. and Horvitz H.R. (2016) The conserved VPS-50 protein functions in dense-core vesicle maturation and acidification and controls animal behavior. Curr. Biol., 26, 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gershlick D.C., Schindler C., Chen Y. and Bonifacino J.S. (2016) TSSC1 is novel component of the endosomal retrieval machinery. Mol. Biol. Cell, 27, 2867–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Braulke T. and Bonifacino J.S. (2009) Sorting of lysosomal proteins. Biochim. Biophys. Acta., 1793, 605–614. [DOI] [PubMed] [Google Scholar]
- 33. Raben N. and Puertollano R. (2016) TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol., 32, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Willenborg M., Schmidt C.K., Braun P., Landgrebe J., Figura K., Saftig P. and Eskelinen E.L. (2005) Mannose 6-phosphate receptors, Niemann–Pick C2 protein, and lysosomal cholesterol accumulation. J. Lipid Res., 46, 2559–2569. [DOI] [PubMed] [Google Scholar]
- 35. Luo J., Jiang L., Yang H. and Song B.L. (2017) Routes and mechanisms of post-endosomal cholesterol trafficking: a story that never ends. Traffic, 18, 209–217. [DOI] [PubMed] [Google Scholar]
- 36. Passemard S., Perez F., Colin-Lemesre E., Rasika S., Gressens P. and El Ghouzzi V. (2017) Golgi trafficking defects in postnatal microcephaly: the evidence for `Golgipathies'. Prog. Neurobiol., 153, 46–63. [DOI] [PubMed] [Google Scholar]
- 37. De Pace R., Skirzewski M., Damme M., Mattera R., Mercurio J., Foster A.M., Cuitino L., Jarnik M., Hoffmann V., Morris H.D. et al. (2018) Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet., 14, e1007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lemmens I., Merregaert J., Van de Ven W.J., Kas K., Zhang C.X., Giraud S., Wautot V., Buisson N., De Witte K., Salandre J. et al. (1997) Construction of a 1.2-Mb sequence-ready contig of chromosome 11q13 encompassing the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Genomics, 44, 94–100. [DOI] [PubMed] [Google Scholar]
- 39. Yu I.M. and Hughson F.M. (2010) Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol., 26, 137–156. [DOI] [PubMed] [Google Scholar]
- 40. Chou H.T., Dukovski D., Chambers M.G., Reinisch K.M. and Walz T. (2016) CATCHR, HOPS and CORVET tethering complexes share a similar architecture. Nat. Struct. Mol. Biol., 23, 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.