

Abstract
Background
Clonidine is a presynaptic alpha‐2‐adrenergic receptor agonist used for many years to treat hypertension and other conditions, including chronic pain. Adverse events associated with systemic use of the drug have limited its application. Topical use of drugs is currently gaining interest, as it may limit adverse events without loss of analgesic efficacy. Topical clonidine (TC) formulations have been investigated recently in clinical trials.
Objectives
The objectives of this review were to assess the analgesic efficacy of TC for chronic neuropathic pain in adults and to assess the frequency of adverse events associated with clinical use of TC for chronic neuropathic pain.
Search methods
We searched the Cochrane Register of Studies (CRS) Online (Cochrane Central Register of Controlled Trials (CENTRAL)), MEDLINE and EMBASE databases, reference lists of retrieved papers and trial registries, and we contacted experts in the field. We performed the most recent search on 17 September 2014.
Selection criteria
We included randomised, double‐blind studies of at least two weeks' duration comparing TC versus placebo or other active treatment in patients with chronic neuropathic pain.
Data collection and analysis
Two review authors extracted data from the studies and assessed bias. We planned three tiers of evidence analysis. The first tier was designed to analyse data meeting current best standards, by which studies reported the outcome of at least 50% pain intensity reduction over baseline (or its equivalent) without use of the last observation carried forward or other imputation method for dropouts, reported an intention‐to‐treat (ITT) analysis, lasted eight weeks or longer, had a parallel‐group design and included at least 200 participants (preferably at least 400) in the comparison. The second tier was designed to use data from at least 200 participants but in cases in which one of the above conditions was not met. The third tier of evidence was assumed in other situations.
Main results
We included two studies in the review, with a total of 344 participants. Studies lasted 8 weeks and 12 weeks and compared TC versus placebo. 0.1%. TC was applied in gel form to the painful area two to three times daily.
Studies included in this review were subject to potential bias and were classified as of moderate or low quality. One drug manufacturer supported both studies.
We found no top‐tier evidence for TC in neuropathic pain. Second‐tier evidence indicated slight improvement after the drug was used in study participants with painful diabetic neuropathy (PDN). A greater number of participants in the TC group had at least 30% reduction in pain compared with placebo (risk ratio (RR) 1.35, 95% confidence interval (CI) 1.03 to 1.77; number needed to treat for an additional beneficial outcome (NNTB) 8.33, 95% CI 4.3 to 50). Third‐tier evidence indicated that TC was no better than placebo for achieving at least 50% reduction in pain intensity and on the Patient Global Impression of Change Scale. The two included studies could be subject to significant bias. We found no studies that reported other neuropathic pain conditions.
The rate of adverse events did not differ between groups, with the exception of a higher incidence of mild skin reactions in the placebo group, which should have no clinical significance.
Authors' conclusions
Limited evidence from a small number of studies of moderate to low quality suggests that TC may provide some benefit in peripheral diabetic neuropathy. The drug may be useful in situations for which no better treatment options are available because of lack of efficacy, contraindications or adverse events. Additional trials are needed to assess TC in other neuropathic pain conditions and to determine how patients who have a chance to respond to the drug should be selected for treatment.
Plain language summary
Clonidine applied to the skin for neuropathic pain
The aim of this review was to examine how clonidine applied to the skin works in people with neuropathic pain. To answer this question, we searched medical databases up to 17 September 2014. We found only two studies that provided information. They lasted 8 weeks and 12 weeks and included a total of 344 participants with painful diabetic neuropathy (PDN). One drug manufacturer supported both studies, which were of low quality.
From these studies, we know that clonidine applied to the skin probably gives little benefit to patients with PDN, but we cannot be sure of this. Clonidine may provide partial pain relief to one out of eight people treated with it. We do not know how clonidine applied to the skin works in other neuropathic pain conditions. Treatment with the drug for short periods probably will not cause side effects, but we do not know from the studies if clonidine is safe for long‐term use. Researchers have reported no differences in the total numbers of side effects among people taking TC or placebo.
The most important message from this review is that clonidine applied to the skin may give partial pain relief for only some people with PDN.
Summary of findings
for the main comparison.
| TC for PDN | |||||||
|
Patient or population: adult patients with PDN Settings: primary care, outpatient Intervention: 0.1% or 0.2% clonidine gel applied to both feet 2‐3 times daily Comparison: placebo | |||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number needed to treat (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | ||||||
| Placebo | TC | ||||||
| Pain intensity reduction ≥ 50% (follow‐up 12 weeks) |
29 per 100 | 35 per 100 (23‐54) | RR 1.21 (0.78‐1.86) | Not calculated | 179 (1) |
Low1, a, c | Results not statistically significant |
| Pain intensity reduction ≥ 30% (follow‐up 8‐12 weeks) |
36 per 100 | 49 per 100 (37‐64) |
RR 1.35 (1.03‐1.77) | NNTB 8.3 (4.3‐50) | 344 (2) |
Low2, b, c | Results statistically significant |
| PGIC much or very much improved (follow‐up 12 weeks) |
42 per 100 | 45 per 100 (32‐63) |
RR 1.06 (0.76‐1.49) | Not calculated | 179 (1) |
Low1, a, c | Results not statistically significant |
| PGIC very much improved (follow‐up 12 weeks) |
11 per 100 | 20 per 100 (10‐41) | RR 1.82 (0.89‐3.72) | Not calculated | 179 (1) | Very low1, a, c, d | Results not statistically significant |
| Withdrawals due to adverse events (follow‐up 12 weeks) |
3 per 100 | 1 per 100 (0‐10) |
RR 0.33 (0.03‐3.23) | Not calculated | 179 (1) |
Very low1, a, c, d | Results not statistically significant |
| Participants with ≥ 1
adverse event (follow‐up 8‐12 weeks) |
13 per 100 | 10 per 100 (6‐19) |
RR 0.65 (0.14‐3.05) | Not calculated | 344 (2) |
Very low2, a, b, c, d | Results not statistically significant |
| Participants experiencing any serious adverse event | No clear data; probably no serious adverse events occurred. Campbell 2009 reports 1 severe adverse event in the placebo group. It is not specified in the study, however, what adverse event it was, and whether it met criteria for serious adverse events specified in this review | ||||||
| Rate of withdrawal due to lack of efficacy | 1 per 100 | 1 per 100 (0‐16) | RR 1.01 (0.06‐16.42) | Not calculated | 179 (1) |
Very low1, a, c, d | Results not statistically significant |
| *Mean baseline risk was chosen to determine the assumed risk in the control group. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; NNTB: Number needed to treat for an additional beneficial outcome; RR: Risk ratio. | |||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||||
1Evidence from 1 moderate‐quality randomised controlled trial.
2Evidence from 1 moderate‐quality randomised controlled trial and 1 low‐quality randomised controlled trial.
aDowngraded 1 level because of imprecision (< 400 participants total).
bDowngraded 1 level because of inconsistency (I2 > 50%).
cDowngraded 1 level because of risk of bias.
dDowngraded 1 level because of imprecision of results.
Background
This review is based on a template for reviews of drugs used to relieve neuropathic pain. The aim is for all reviews to use the same methods, based on new criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Appendix 1).
Description of the condition
Neuropathic pain comprises a wide range of pain conditions. It is defined by the International Association of the Study of Pain as "pain caused by lesion or disease of the somatosensory nervous system" (IASP Taxonomy Working Group 2011; Jensen 2011), based on an earlier consensus meeting (Treede 2008). Neuropathic pain may be caused by nerve damage but is often followed by changes in the central nervous system (Moisset 2007). It tends to be chronic and may be present for months or years. It is complex (Apkarian 2011; Tracey 2011), and neuropathic pain features can be found in patients with joint pain (Soni 2013). The pathomechanism of neuropathic pain differs significantly from that of nociceptive pain. Nociceptive pain is a consequence of tissue damage, whereas neuropathic pain results from maladaptive changes that can occur in injured sensory neurons and along the entire nociceptive pathway within the central nervous system (CNS), possibly leading to spontaneous pain or pain hypersensitivity. The most characteristic clinical symptoms of neuropathic pain are spontaneous pain, hyperalgesia and allodynia; this has been easily demonstrated in various animal models (Hurley 2013; Woolf 1999).
In primary care in the UK, the incidence per 100,000 person‐years of observation has been reported as 28 (95% confidence interval (CI) 27 to 30) for postherpetic neuralgia, 27 (95% CI 26 to 29) for trigeminal neuralgia, 0.8 (95% CI 0.6 to 1.1) for phantom limb pain and 21 (95% CI 20 to 22) for painful diabetic neuropathy (PDN) (Hall 2008). Estimates vary between studies, often because of small sample sizes. The incidence of trigeminal neuralgia has been estimated at 4 in 100,000 per year (Katusic 1991; Rappaport 1994), and more recently, a study of facial pain in the Netherlands found an incidence per 100,000 person‐years of 12.6 for trigeminal neuralgia and 3.9 for postherpetic neuralgia (Koopman 2009). A systematic review of chronic pain indicated that some neuropathic pain conditions, such as PDN, can be more common than others, with prevalence rates up to 400 per 100,000 person‐years (McQuay 2007), illustrating how common the condition is as well as its chronicity. The prevalence of neuropathic pain was reported as 3.3% in Austria (Gustorff 2008), 6.9% in France (Bouhassira 2008), as high as 8% in the UK (Torrance 2006) and about 7% in a systematic review of studies published since 2000 (Andrew 2014). The incidence of some forms of neuropathic pain, such as diabetic neuropathy and postsurgical chronic pain (often neuropathic in origin), is increasing (Hall 2008).
Neuropathic pain is known to be difficult to treat effectively; only a minority of individuals experience clinically relevant benefit from any one intervention. A multi‐disciplinary approach is now advocated, with pharmacological interventions combined with physical and/or cognitive interventions. Conventional analgesics usually are not effective. Some patients may benefit from a topical lidocaine patch or low‐concentration topical capsaicin, although evidence showing benefits is uncertain (Anitescu 2013; Derry 2012; Khaliq 2007). High‐concentration topical capsaicin may prove helpful for some patients with postherpetic neuralgia (Derry 2013). Treatment more usually consists of so‐called unconventional analgesics such as antidepressants (e.g. duloxetine, amitriptyline) or antiepileptics (e.g. gabapentin, pregabalin) (Lunn 2009; Moore 2009; Moore 2011a; Moore 2012a; Sultan 2008). An overview of treatment guidelines points out general similarities, as well as differences, in treatment approaches (Hurley 2013; O'Connor 2009; Smith 2013). The proportion of patients who achieve worthwhile pain relief (typically ≥ 50% reduction in pain intensity) is small, generally 10% to 25% greater than with placebo, and numbers needed to treat for additional benefit (NNTBs) are usually between four and 10 (Moore 2013).
Chronic painful conditions accounted for five of the 11 top‐ranking conditions for years lived with disability in 2010 (Vos 2012), and they are responsible for considerable loss of quality of life and employment, and for increased healthcare costs (Andrew 2014).
Description of the intervention
Clonidine is a presynaptic alpha‐2‐adrenergic receptor agonist and an agonist of imidazoline receptors (Eisenach 1996). It has been in clinical use for over 40 years. It was first registered for treatment of hypertension but later proved effective for treatment of acute and chronic pain (Neil 2011). Clonidine is an extremely potent antinociceptive agent with potency equal to or greater than that reported for morphine (Gentili 1997; Samso 1996). Clonidine has been used to treat acute and chronic pain and may be effective when applied intravenously, epidurally and intrathecally (Asano 2000; Eisenach 1995; Hassenbusch 2002; Sierralta 1996). However, systemic and central use of clonidine is limited by undesirable adverse events including sedation, dry mouth, hypotension and rebound hypertension (Dias 1999; Puskas 2003). Recently topical forms of administration have been developed with intention to limit centrally mediated adverse events without reduction in analgesic efficacy (Sawynok 2003). Clonidine is lipophilic and easily penetrates skin to reach the local antinociceptive pathways. The half‐life of clonidine is about eight hours; thus it should be applied three times daily. Clonidine can be prepared in various concentrations by compounding pharmacies (Flores 2012).
Topical clonidine (TC) proved to be an effective analgesic in several animal studies. Dogrul 2004 demonstrated that topical administration of clonidine increased the pain threshold to radiant heat stimuli (measured by tail‐flick test) in mice. Antinociceptive activity was limited to the portion of the tail exposed to drug solution. Systemic administration of the alpha‐2‐receptor antagonist, yohimbine, before immersion of the tail blocked the antinociceptive activity of TC (Dogrul 2004). Chi 2007 studied the efficacy of topically applied clonidine in an animal model of neuropathic, postoperative and inflammatory pain. Clonidine turned out to be effective in neuropathic pain, only partially effective in postoperative pain and not effective in inflammatory pain. The analgesic efficacy of clonidine in postoperative pain manifested on the sixth day of application and reduction in thermal hyperalgesia ‐ not mechanical allodynia ‐ were observed (Chi 2007).
How the intervention might work
Target receptors for clonidine ‐ alpha‐2 receptors ‐ are located in the brain, spinal cord and dorsal root ganglia and on sensory neurons (Kawaski 2003; Ongioco 2000; Riedl 2009). Activation of alpha‐2 receptors leads to release of an inhibitory G‐protein, which down‐regulates adenylate cyclase and other second messengers responsible for initiating and maintaining the abnormal excitability of nociceptors (Lavand'homme 2002). Antinociceptive effects of clonidine are mediated via spinal and supraspinal sites of action (Asano 2000; Bernard 1994; Buerkle 1998). However, investigators in previous studies proved that peripheral administration of alpha‐2‐receptor agonists also induces antinociception (Aley 1997; Buerkle 1998; Buerkle 2000; Gentili 1996). The mechanism of action of clonidine is similar to that of opioids. Antinociceptive effects of topically administered opioids have been reported previously (Kolesnikov 1999; Kolesnikov 2000); however, tolerance to antinociceptive action was observed after repeated administration (Kolesnikov 1999). Tolerance to the antinociceptive action of clonidine was observed in animal studies and was not attenuated by N‐Methyl‐D‐aspartate (NMDA)‐receptor antagonists such as ketamine (Dogrul 2004).
Clonidine is also an imidazoline‐receptor agonist. Stimulation of the I2‐imidazoline subclass of receptors causes analgesia. I2‐imidazoline receptors are located centrally in the brain and spinal cord and peripherally on peripheral nerve endings. Activation of peripheral imidazoline receptors may be responsible for additional mechanisms of analgesic activity of TC (Khan 1999).
Why it is important to do this review
Practitioners have attempted to use TC in the past to treat neuropathic pain; however, no clear evidence is available to support this clinical practice. Recently, new randomised clinical trials investigating this topic have been published. The aim of this review is to determine whether TC is effective in neuropathic pain and to specify in which particular neuropathic pain conditions it is effective. No Cochrane review authors have examined this topic.
Standards used to assess evidence in chronic pain trials have changed substantially, with particular attention paid to trial duration, withdrawals and statistical imputation following withdrawal ‐ all of which can substantially alter estimates of efficacy. The most important change is the move from use of average pain scores, or average change in pain scores, to the numbers of study participants who report a large decrease in pain (≥ 50%); this level of pain relief has been shown to correlate with improvement in co‐morbid symptoms, function and quality of life. These standards are set out in the reference guide for pain studies (Cochrane PaPaS Group 2011).
This Cochrane review was designed to assess evidence in ways that make both statistical and clinical sense, and to use developing criteria for what constitutes reliable evidence in chronic pain (Moore 2010a). Trials included and analysed had to meet minimum criteria for reporting quality (blinding, randomisation), validity (duration, dose and timing, diagnosis, outcomes, etc.) and size (ideally ≥ 500 participants in a comparison in which the number needed to treat for an additional beneficial outcome (NNTB) is ≥ 4) (Moore 1998). This approach imposes high standards and marks a departure from the way previous reviews were conducted.
Objectives
The objectives of this review were to assess the analgesic efficacy of TC for chronic neuropathic pain in adults and to assess the frequency of adverse events associated with clinical use of TC for chronic neuropathic pain.
Methods
Criteria for considering studies for this review
Types of studies
Studies meeting the inclusion criteria were randomised controlled trials (RCTs) with double‐blind assessment of participant outcomes following two weeks of treatment or longer. Cross‐over studies could also be included, provided results for the first phase were reported clearly. We required full journal publications, with the exception of online clinical trial results summaries of otherwise unpublished clinical trials and abstracts, with sufficient data for analysis. We did not include short abstracts (usually meeting reports). We excluded studies that were non‐randomised studies of experimental pain, case reports and clinical observations. We applied no language restrictions.
Types of participants
We included adult participants 18 years of age and older. Participants had to have one or more of a wide range of chronic neuropathic pain conditions including the following.
PDN.
Postherpetic neuralgia.
Trigeminal neuralgia.
Phantom limb pain.
Postoperative or traumatic neuropathic pain.
Complex regional pain syndrome.
Cancer‐related neuropathy.
Human immunodeficiency virus (HIV) neuropathy.
Spinal cord injury.
Types of interventions
TC had to be administered to a painful area for relief of neuropathic pain in a form of cream, ointment, gel, patch or plaster and compared with placebo or any active comparator. We included studies in which the active comparator was administered via any route: topically, orally, intravenously, subcutaneously, etc. We did not include studies clonidine was applied transdermally with intention to produce a systemic effect ‐ not a local effect.
Types of outcome measures
We anticipated that studies would have used a variety of outcome measures, with most studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS)) for pain intensity or pain relief, or both. We were particularly interested in Initiative on Methods, Measurement and Pain Assessment in Clinical Trials (IMMPACT) definitions of moderate and substantial benefit in chronic pain studies (Dworkin 2008). Benefit is defined as at least 30% pain relief over baseline (moderate), at least 50% pain relief over baseline (substantial), much or very much improved on the Patient Global Impression of Change (PGIC) (moderate) and very much improved on PGIC (substantial). These outcomes are different from those used in most earlier reviews, concentrating as they do on dichotomous outcomes, when pain responses do not follow a normal (Gaussian) distribution. People with chronic pain desire high levels of pain relief, ideally more than 50%, and pain not worse than mild (O'Brien 2010).
We included a 'Summary of findings' table, which contains outcomes of at least 50% and at least 30% reduction in pain intensity, PGIC, adverse event withdrawals and occurrence at least one adverse event.
Primary outcomes
Participant‐reported pain relief of 30% or greater.
Participant‐reported pain relief of 50% or greater.
Much or very much improved on patient global impression of change scale (PGIC).
Very much improved on patient global impression of change scale (PGIC) .
Secondary outcomes
Any pain‐related outcome indicating some improvement.
Withdrawals due to lack of efficacy.
Participants experiencing at least one adverse event.
Participants experiencing any serious adverse event. Serious adverse events typically include any untoward medical occurrence or effect that at any dose results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect or is an ‘important medical event’ that may jeopardise the participant or may require an intervention to prevent one of the above characteristics/consequences.
Withdrawals due to adverse events.
Specific adverse events, particularly somnolence and dizziness.
Skin biopsy results.
Search methods for identification of studies
Electronic searches
We searched the following databases.
Cochrane Register of Studies (CRS) Online (Cochrane Central Register of Controlled Trials (CENTRAL)), 17 September 2014.
MEDLINE and MEDLINE in Process (Ovid), 1946 to 16 September 2014.
EMBASE (Ovid), 1974 to 16 September 2014.
We used medical subject headings (MeSH) or equivalent and text word terms and applied no language restrictions. We tailored searches to individual databases and have provided our search strategies in Appendix 2, Appendix 3 and Appendix 4. The most recent search was performed on 17 September 2014.
Searching other resources
On 30 September 2014, we searched the metaRegister of controlled trials (mRCT) (http://www.controlled‐trials.com/mrct/), ClinicalTrials.gov (http://clinicaltrials.gov/) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) search portal (http://apps.who.int/trialsearch/) for ongoing trials. In addition, we checked the reference lists of reviews and retrieved articles for additional studies and searched citations on key articles. We contacted experts in the field to ask about unpublished and ongoing trials and contacted investigators or study sponsors when necessary.
Data collection and analysis
We intended to perform separate analyses according to particular neuropathic pain conditions; however, we found only studies on PDN.
Selection of studies
We determined eligibility by reading the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy our inclusion criteria, and we obtained full copies of remaining studies; three review authors (A. Wrzosek, J. Jakowicka, J. Woron) made these decisions. We obtained full texts of studies identified by at least one review author. Two review authors (A. Wrzosek, J. Jakowicka) independently read the full texts of these studies and decided whether they met the inclusion criteria. In cases of disagreement, review authors reached conclusions by discussion or, if not possible, by seeking the opinion of a third review author (J. Wordliczek or J. Dobrogowski) to resolve the disagreement. We did not anonymise the studies in any way before assessment. We created a Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) flow diagram to document the screening process (Liberati 2009), as recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Data extraction and management
Two review authors (A. Wrzosek, J. Jakowicka) independently extracted data from the studies using a standard data extraction form (Appendix 5). We resolved disagreements by consultation and discussion with a third review author (J. Wordliczek). One review author (A. Wrzosek) entered data into the statistical software of The Cochrane Collaboration ‐ Review Manager 2014 ‐ and another review author (J. Jakowicka) checked data for correctness. We included the following data when available.
Study design.
Pain condition.
Inclusion criteria.
Exclusion criteria.
Number of participants screened/enrolled/randomly assigned to each treatment arm.
Mean age.
Number of males.
Duration of pain condition.
Mean baseline pain intensity.
Intervention: form of application, place of application, concentration, dose, dosing regimen.
Control: form of application, place of application, concentration, dose, dosing regimen.
Outcomes.
Other important information.
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for inclusion (Jadad 1996), limiting inclusion to studies that were randomised and double‐blind at a minimum.
Two review authors (A. Wrzosek, J. Wordliczek) independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and adapted from those used by the Cochrane Pregnancy and Childbirth Group, resolved disagreements by discussion.
We assessed the following for each study.
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as follows: low risk of bias (any truly random process, e.g. random number table, computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process (e.g. odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions before assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or could have changed after assignment. We assessed methods as follows: low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (method not clearly stated). We excluded studies that did not conceal allocation (e.g. open list).
Blinding of participants and personnel (checking for possible performance bias). We assessed methods used to blind study participants and personnel from knowledge of which intervention a participant received. We assessed these methods as follows: low risk of bias (study states that it was blinded and describes the method used to achieve blinding, e.g. identical tablets; matched in appearance and smell); unclear risk of bias (study states that it was blinded but does not provide an adequate description of how this was achieved). We excluded studies that were not double‐blind.
Blinding of outcome assessment (checking for possible detection bias). We assessed methods used to blind outcome assessors from knowledge of which intervention a participant received. We assessed these methods as follows: low risk of bias (study states that outcome assessor was blinded and describes the method used to achieve blinding); unclear risk of bias (information indicates that the assessor was blinded; however, it is not clear whether the method used for assessor blinding was suitable); high risk of bias (lack of blinding).
Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data). We assessed methods used to deal with incomplete data as follows: low risk (< 10% of participants did not complete the study and/or used ‘baseline observation carried forward’ analysis); unclear risk of bias (used 'last observation carried forward' analysis); high risk of bias (used 'completer' analysis).
Size of the study (checking for possible biases confounded by small size). We assessed studies as having low risk of bias (≥ 200 participants per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); high risk of bias (< 50 participants per treatment arm).
Measures of treatment effect
We used dichotomous data to calculate relative risk (or 'risk ratio' (RR)) and number needed to treat for an additional beneficial outcome (NNTB) with 95% confidence intervals to establish statistical differences. We calculated NNTBs as the reciprocal of absolute risk reduction (ARR). For unwanted effects, the NNTB becomes the number needed to treat for an additional harmful outcome (NNTH), which is calculated in the same manner. We used a fixed‐effect model unless significant statistical heterogeneity was found (Data synthesis). Because the amount of evidence was small, we decided to include continuous outcomes for illustrative purposes only.
Unit of analysis issues
We accepted randomisation by individual participant only.
We accepted cross‐over studies only if clear reporting for the first cross‐over phase was available.
We planned to split the control treatment arm between active treatment arms in a single study in a situation in which active treatment arms were not combined for analysis; however, this was not the case in this review.
Dealing with missing data
We used intention‐to‐treat (ITT) analysis when the ITT population consisted of participants who were randomly assigned, took at least one dose of assigned study medication and provided at least one postbaseline assessment. Missing participants were assigned zero improvement.
Assessment of heterogeneity
We dealt with clinical heterogeneity by combining studies that examined similar conditions. We assessed statistical heterogeneity by using the I2 statistic (Higgins 2011). When I2 was greater than 50%, we considered possible reasons.
Assessment of reporting biases
The aim of this review was to use dichotomous data of known utility (Moore 2010c). The review does not depend on what authors of the original studies chose to report, although clearly difficulties could arise in studies that failed to report dichotomous results. For illustrative purposes, we used one continuous outcome ‐ change in average pain severity as reported by participants using the Numerical Pain Rating Scale (NPRS), which, however, poorly reflects efficacy and utility.
We did not undertake a specific analysis to detect publication bias due to the small number of included studies, although we had planned to assess publication bias using a method designed to detect the quantity of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean an NNTB ≥ 10) (Moore 2008).
Data synthesis
We used a fixed‐effect model for meta‐analysis when we found no significant heterogeneity among studies, and in cases of significant heterogeneity, we used a random‐effects model.
We analysed data for each painful condition in three tiers, according to outcome and freedom from known sources of bias.
The first tier uses data meeting current best standards, when studies report the outcome of at least 50% reduction in pain intensity over baseline (or its equivalent), without use of last observation carried forward (LOCF) or other imputation methods for dropouts; report an ITT analysis; last eight weeks or longer; have a parallel‐group design and include at least 200 participants (preferably ≥ 400) in the comparison (Moore 2010a, Moore 2012b). These top‐tier results are reported first.
The second tier uses data from at least 200 participants in cases where one or more of the above conditions are not met (e.g. reporting ≥ 30% pain intensity reduction, using LOCF or a completer analysis, or lasting four to eight weeks).
The third tier of evidence relates to data from fewer than 200 participants, or when significant problems are expected because, for example, studies are of very short duration (< 4 weeks); major heterogeneity is noted between studies; shortcomings are evident in allocation concealment; or attrition or incomplete outcome data are reported. For this third tier of evidence, no data synthesis is reasonable, and this may be misleading, but an indication of beneficial effects might be possible.
Subgroup analysis and investigation of heterogeneity
We planned all analyses according to individual painful conditions because placebo response rates with the same outcome can vary between conditions, as can drug‐specific effects (Moore 2009). However, data were insufficient for performance of any meaningful subgroup analysis.
Sensitivity analysis
We did not conduct sensitivity analyses because available evidence was too limited to allow reliable analysis. Also, sensitivity analyses based on different concentrations of drug were not possible.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
Our search strategy identified 2234 studies (see Figure 1), and we identified 25 additional studies by searching reference lists of reviews and retrieved articles. Elimination of duplicates resulted in 1666 references. Three independent review authors (A. Wrzosek, J. Jakowicka, J. Woron) assessed abstracts and titles to eliminate all studies clearly outside the scope of this review. We identified seven full texts as potentially meeting our inclusion criteria. We excluded five studies and listed reasons for exclusion in the Characteristics of excluded studies table (Byas‐Smith 1995; Davis 1991; Lauretti 2009; Meno 2001; Zeigler 1992). Two studies met our inclusion criteria (Campbell 2009; Campbell 2012). The study of Campbell 2009 was reported only in abstract form. We contacted study authors and obtained additional unpublished information.
1.
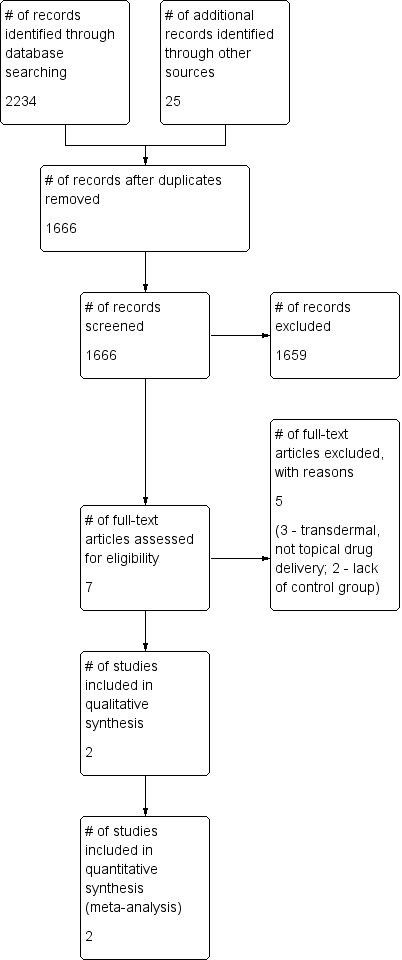
Study flow diagram.
We found one ongoing study (NCT02068027), which should have started in March 2014. This multi‐centre, double blind, randomised, placebo‐controlled trial has been designed to assess the effectiveness of 0.1% TC gel in PDN. Study duration is 12 weeks, and planned outcome measures include change in pain intensity, mean pain intensity and mean daily worst pain. Study authors plan to enrol 140 participants. No results of this study are available at this time.
We found one registered, randomised study (NCT00661063) on efficacy and safety of TC 1% gel compared with placebo or ketamine. Planned study duration was 12 weeks. The study was to start in 2008, but we have found no study results. We tried to contact study authors by phone and by email (as provided in the study description on clinicaltrials.gov), but they have not responded.
Included studies
Both included studies are published in English and were conducted in the USA by the same first author. Both studies assessed efficacy and safety of topically applied clonidine gel in adult patients with PDN. The total number of participants in both studies was 344.
In Campbell 2009, investigators applied gel to both feet twice daily for two weeks, then three times daily for eight weeks total. A total of 54 participants received 650 µL of 0.1% TC per foot, and another 54 participants received 500 µL of 0.2% clonidine per foot. The control group (57 participants) was given placebo.
In Campbell 2012, 91 participants were allocated to the clonidine group and received 650 µg of 0.1% clonidine gel three times daily for 12 weeks. The control group (91 participants) received matching placebo. One participant in each group did not receive allocation intervention because these participants were found to be ineligible after randomisation. One participant in the clonidine group was excluded from the ITT population because no baseline NPRS score was obtained. During the screening phase of the study, researchers assessed nociceptor function by determining pain response to 0.1% topical capsaicin applied to the pretibial area for 30 minutes. Capsaicin responders were defined as participants with pain intensity of 2 or more points on the NPRS during capsaicin stimulation; investigators identified 33 such individuals in the clonidine group and 30 in the placebo group.
Baseline participant characteristics did not differ significantly between groups in both studies. More than 80% of participants had type 2 diabetes. In Campbell 2012, mean duration of diabetes was approximately 10 years, and mean duration of pain was approximately three years. Mean baseline pain intensity was about 6.5 points on the NPRS scale. Campbell 2009 did not provide such information.
The drug manufacturer Arcion Therapeutics supported both studies.
Excluded studies
We excluded five potentially relevant studies from the analysis. Reasons for exclusion included lack of a control group in two studies (Davis 1991; Meno 2001) and transdermal ‐ not topical ‐ drug delivery in three studies (Byas‐Smith 1995; Lauretti 2009; Zeigler 1992). Transdermal application is intended to exert predominantly systemic effects, and skin is only a vehicle for administration. This form of application allows slow and gradual release of medication into the bloodstream with relatively constant blood levels. Topical administration exerts mainly peripheral effects at the site of application.
Risk of bias in included studies
We used the domain‐based evaluation table of The Cochrane Collaboration, which is provided in Review Manager 2014, to assess the validity and quality of included trials. Details of the assessment are specified in Characteristics of included studies tables, and summaries of assessments are given in Figure 2 and Figure 3. Campbell 2009 was determined to be at high risk of bias, and Campbell 2012 at moderate risk.
2.
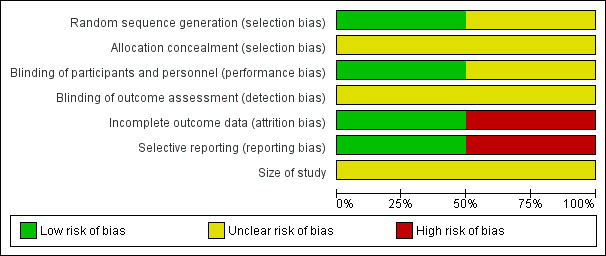
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
In Campbell 2009, information about selection bias is unclear. Study authors stated only that the study was randomised and provided no information on methods of randomisation and allocation concealment. In Campbell 2012, randomisation was adequate, and we have no information on allocation concealment. We considered this study to be at moderate risk of selection bias.
Blinding
Campbell 2009 provides no information on blinding methods, and we considered this study to have unclear risk of bias. Campbell 2012 stated that the placebo formulation was identical in appearance, consistency, packaging and labelling, and we considered this study to have low risks of performance and detection bias.
Incomplete outcome data
Campbell 2009 was defined as having high risk of attrition bias because the number of participants randomly assigned is not equal to the number described in the demographics table (one participant is missing). Some results are missing, and researchers offer no information about how they dealt with missing data in this study. Campbell 2012 was defined as having low risk of bias as baseline observation carried forward was used as an imputation method, and clear information about the number of participants lost from observation was given.
Selective reporting
Campbell 2009 was defined as having high risk of bias because results as presented were unclear. Campbell 2012 was defined as having low risk of reporting bias, as all results were stated clearly and were consistent with the Methods section of the study.
Other potential sources of bias
Both studies included fewer than 200 participants; thus we identified a potentially moderate risk of bias.
The same company supported both studies, which were conducted by the same main author; this may contribute to potential sources of bias.
Effects of interventions
See: Table 1
First‐tier evidence
We found no first‐tier evidence on the efficacy and safety of TC in neuropathic pain.
Second‐tier evidence
The two included studies provided some evidence that TC may be effective in the treatment of PDN. We found no studies on the efficacy of TC in other pain conditions.
Primary outcome measures
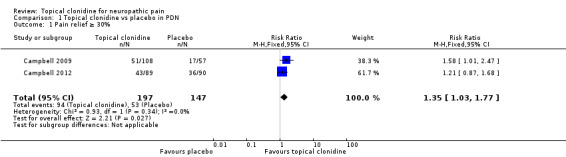
Meta‐analysis of results of included studies (Campbell 2009; Campbell 2012) showed that TC might be effective for treatment of adults with PDN. More participants in the clonidine group experienced at least 30% pain reduction compared with those given placebo (RR 1.35, 95% CI 1.03 to 1.77) during a an eight‐ to 12‐week treatment period (Figure 4). The number needed to treat to achieve this endpoint was 8.33, with 95% CI 4.3 to 50.
4.
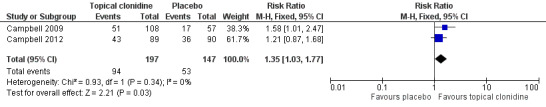
Forest plot of comparison: 1 Topical clonidine vs placebo in PDN, outcome: 1.1 Pain relief ≥ 30%.
Secondary outcome measures
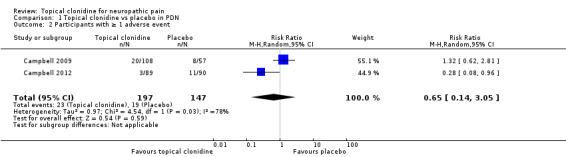
Results of the included studies showed no statistically significant differences in the numbers of participants with at least one adverse event (11.7% vs 12.9% in TC and placebo groups, respectively; RR 0.65, 95% CI 0.14 to 3.05; Figure 5).
5.
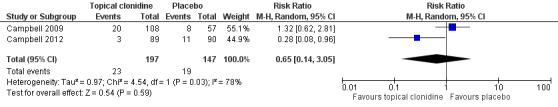
Forest plot of comparison: 1 Topical clonidine vs placebo in PDN, outcome: 1.2 Participants with ≥ 1 adverse event.
Campbell 2012 reported no serious adverse events, and Campbell 2009 reported one severe adverse event in the placebo group. However, the study did not specify what adverse event occurred and whether it met the criteria for serious adverse events as specified in this review.
Third‐tier evidence
Third‐tier evidence is based on the results of one study ‐ Campbell 2012 or Campbell 2009.
Primary outcome measures
Results from Campbell 2012 suggest no statistically significant differences with regard to pain relief of 50% or greater (RR 1.21, 95% CI 0.78 to 1.86) or PGIC scale (very much improved: RR 1.82, 95% CI 0.89 to 3.72; much or very much improved: RR 1.06, 95% CI 0.76 to 1.49).
Secondary outcome measures
In Campbell 2012, a statistically significant difference between changes was noted in average pain severity from baseline to week 12 (reported by participants in their diaries using the NPRS scale). Patients in the TC group had a 2.3‐point reduction compared with a 1.7‐point reduction in the placebo group.
Campbell 2012 reported no statistically significant differences between groups with regard to rate of withdrawal due to adverse events (RR 0.3, 95% CI 0.03 to 3.23) or rate of withdrawal due to lack of efficacy (RR 1.01, 95% CI 0.06 to 16.42). Investigators reported no statistically significant differences with regard to scales assessing quality of life, such as the Brief Pain inventory, the Chronic Pain Sleep Inventory and the Hospital and Depression Scale.
Campbell 2012 reported the same number of participants with adverse events associated with the nervous system (two participants per group), which included burning sensation, dizziness and headache. Campbell 2009 described 11 adverse events associated with the nervous system in the TC group and eight in the placebo group. Study authors did not specify these adverse events.
No studies analysed difference in skin biopsy results between TC and placebo.
'Summary of findings'
Available evidence suggests that TC might be effective for treatment of PDN. This must be interpreted with caution because of the limited number of available studies, the relatively low number of participants included and the moderate to high risk of bias assigned to studies. Efficacy of TC in other neuropathic pain conditions remains unclear because evidence is insufficient.
Discussion
The major finding of this review is that very little evidence is available on the efficacy of topical clonidine (TC) in neuropathic pain. The only two randomised controlled trials included individuals suffering from painful diabetic neuropathy (PDN). We found no studies on other neuropathic pain conditions. Neither trial meets current standards of evidence for chronic pain as described in Initiative on Methods, Measurement and Pain Assessment in Clinical Trials (IMMPACT) recommendations (Dworkin 2008). The two included studies provide only second‐ and third‐tier evidence and might be associated with meaningful bias regarding study design, conduct and reporting, which can lead to favouring of active treatment. Studies lasted eight to 12 weeks, which is a short period for assessment of efficacy of drugs in chronic pain, and the observation period is definitely too short to permit judgements about drug safety.
Available evidence indicates that we can suspect little benefit derived from TC treatment of patients with PDN. We found no evidence on TC given in other neuropathic pain conditions.
Summary of main results
Limited evidence based on a small number of middle‐ to low‐quality studies suggests that TC may provide some benefit for individuals with PDN. We found no evidence on use of TC in other neuropathic pain conditions. Two studies met the inclusion criteria of the review and provided second‐ and third‐tier evidence. For PDN, TC was no better than placebo for at least 50% reduction in pain intensity and on the Patient Global Impression of Change (PGIC) scale. TC was significantly better than placebo with regard to at least 30% reduction in pain intensity (risk ratio (RR) 1.35, 95% confidence interval (CI) 1.03 to 1.77). The number needed to treat for an additional beneficial outcome for this endpoint was relatively high and equalled 8.33 with a wide 95% confidence interval (4.3 to 50), which brings additional uncertainty to the results. Moreover, many participants (36%) in the placebo group reached 30% pain reduction. For the TC group, this value was 49%. This means that only 13% of patients would benefit from the drug. It has to be pointed out that in Campbell 2012, mean pain duration was around three years, and individuals were unsuccessfully treated with other drugs, including antidepressants, anticonvulsants and opioids. Mean pain intensity at inclusion was approximately 6.5 on the Numerical Pain Rating Scale (NPRS). No such information is given in Campbell 2009. Use of active treatment rather than placebo could reveal more information about the comparative efficacy of TC versus other drugs; however, such studies were not done.
In a study of Campbell 2012, participants experienced stimulation with 0.1% capsaicin during the screening phase. Even though topical capsaicin in a concentration of 8% may produce long‐lasting pain relief (Derry 2013), we believe that 0.1% capsaicin should not influence response to clonidine; however, such a situation cannot be ruled out completely. Even though authors of the study claim better results among capsaicin responders, these findings are not statistically significant (change in PGIC scale), and results for at least 50% and 30% pain relief are not presented for this selected group of patients.
Investigators found no differences in the numbers of participants with adverse events, the numbers of withdrawals due to adverse events, lack of efficacy and overall withdrawal rate; however, differences could not be detected because relatively few participants were included and trial duration was short. As only a very small concentration of clonidine is reached in plasma during topical application (Campbell 2012), it can be assumed that topical use will be associated with few important adverse events.
One ongoing trial may bring new evidence (NCT02068027). This trial, which was supposed to start in March 2014, will include individuals with PDN, and about 140 participants are expected to enrol. The other registered trial (NCT00661063) is not likely to bring any evidence, as it should have started in 2008, and we do have had no results until now. We tried to contact the authors of this study by email and by phone, but they have not responded.
Overall completeness and applicability of evidence
It is likely that all reliable trials have been found during the search; however, they might be subject to significant bias favouring active treatment. The number of identified trials is very small (two trials), and we have assessed them as having moderate and high risk of bias. The assessment of Campbell 2009 was based only on the abstract publication and on unpublished data provided by study authors. This limits to a great extent the applicability of the evidence.
Other drugs are available for treatment of individuals with PDN with lower numbers needed to treat for an additional beneficial outcome (NNTBs). Duloxetine has an NNTB of five for at least 50% and 30% pain reduction (Lunn 2014). Gabapentin, pregabalin and oxcarbazepine have NNTBs around six (Moore 2014; Wiffen 2013; Zhou 2013). Carbamazepine has an NNTB of 1.9 for PDN, trigeminal neuralgia and chronic poststroke pain (Wiffen 2014).
Thus, we can conclude that TC should not be used as first‐line treatment for chronic neuropathic pain. TC probably could be used in individuals with PDN when no other treatment options are available. These results cannot be extrapolated to other neuropathic pain conditions.
New clinical trials are needed to establish the role of TC in other neuropathic pain conditions. Studies are also needed to assess use of the drug in PDN for patients with recently diagnosed pain.
Quality of the evidence
All studies included in this review had to meet basic requirements such as randomisation and blinding; however, none of the studies met current standards of efficacy trials on chronic pain, as described in the IMMPACT recommendations (Dworkin 2008). One study was classified as low quality, and the other as moderate quality.
The drug manufacturer Arcion Therapeutics supported both studies.
Campbell 2009 was based on the published abstract and on unpublished data, so the study has high potential of reporting bias.
Potential biases in the review process
We used a comprehensive search strategy based on previous Cochrane reviews for randomised controlled trials on neuropathic pain. We did not restrict our search to topical application of the drug, so we could identify all relevant studies. Additionally, we searched reference lists of potentially relevant studies and reviews, searched trial registries and contacted experts in the field. Two independent review authors read abstracts identified by the search. The probability that any important studies were omitted in the search process is low, as is the possibility of bias in this review process.
Agreements and disagreements with other studies or reviews
Recently, a systematic review with meta‐analysis that focused on pharmacotherapy for neuropathic pain in adults was published (Finnerup 2015). Review authors included studies on topical clonidine for neuropathic pain treatment and state that Grades of Recommendation, Assessment, Development and Evaluation (GRADE) recommendations are inconclusive because of discrepant findings.
Authors' conclusions
Implications for practice.
Limited evidence based on a small number of studies of moderate to low quality indicates that TC may provide some benefit for individuals with peripheral diabetic neuropathy.
Implications for research.
Good quality randomised controlled trials are needed to establish the role of TC in various neuropathic pain conditions such as postherpetic neuralgia, trigeminal neuralgia, phantom limb pain, complex regional pain syndrome and other peripheral neuropathies. Comparative efficacy of TC versus other drugs and efficacy of clonidine in patients with recently diagnosed PDN should be investigated. Researchers should look for ways to identify patients who may respond to TC treatment.
What's new
| Date | Event | Description |
|---|---|---|
| 4 April 2017 | Review declared as stable | See Published notes. |
Notes
No potentially relevant new studies have been published. Therefore, this review has now been stabilised following discussion with the authors and editors. If appropriate, we will update the review if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
We would like to thank Anna Hobson for help with writing this review and Joanne Abbott for helping with searches.
Cochrane Review Group funding acknowledgement: The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane PaPaS Group.
Disclaimer: The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the NIHR, the National Health Service (NHS) or the Department of Health.
Appendices
Appendix 1. Methodological considerations for chronic pain
There have been several recent changes in how efficacy of conventional and unconventional treatments is assessed in chronic painful conditions. The outcomes are now better defined, particularly with new criteria of what constitutes moderate or substantial benefit (Dworkin 2008); older trials may only report participants with "any improvement". Newer trials tend to be larger, avoiding problems from the random play of chance. Newer trials also tend to be longer, up to 12 weeks, and longer trials provide a more rigorous and valid assessment of efficacy in chronic conditions. New standards have evolved for assessing efficacy in neuropathic pain, and we are now applying stricter criteria for inclusion of trials and assessment of outcomes, and are more aware of problems that may affect our overall assessment. To summarise some of the recent insights that must be considered in this new review:
Pain results tend to have a U‐shaped distribution rather than a bell‐shaped distribution. This is true in acute pain (Moore 2011b; Moore 2011c), back pain (Moore 2010c), arthritis (Moore 2010b), as well as in fibromyalgia (Straube 2010); in all cases average results usually describe the experience of almost no‐one in the trial. Data expressed as averages are potentially misleading, unless they can be proven to be suitable. As a consequence, we have to depend on dichotomous results (the individual either has or does not have the outcome) usually from pain changes or patient global assessments. The IMMPACT group has helped with their definitions of minimal, moderate, and substantial improvement (Dworkin 2008). In arthritis, trials shorter than 12 weeks, and especially those shorter than eight weeks, overestimate the effect of treatment (Moore 2009); the effect is particularly strong for less effective analgesics, and this may also be relevant in neuropathic‐type pain.The proportion of patients with at least moderate benefit can be small, even with an effective medicine, falling from 60% with an effective medicine in arthritis, to 30% in fibromyalgia (Moore 2009; Moore 2010b; Straube 2008; Sultan 2008). A Cochrane review of pregabalin in neuropathic pain and fibromyalgia demonstrated different response rates for different types of chronic pain (higher in diabetic neuropathy and postherpetic neuralgia and lower in central pain and fibromyalgia) (Moore 2009). This indicates that different neuropathic pain conditions should be treated separately from one another, and that pooling should not be done unless there are good grounds for doing so. Finally, presently unpublished individual patient analyses indicate that patients who get good pain relief (moderate or better) have major benefits in many other outcomes, affecting quality of life in a significant way (Moore 2010d).
Appendix 2. CRS Online (CENTRAL) search strategy
#1 MESH DESCRIPTOR PAIN EXPLODE ALL TREES
#2 MESH DESCRIPTOR PERIPHERAL NERVOUS SYSTEM DISEASES EXPLODE ALL TREES
#3 MESH DESCRIPTOR SOMATOSENSORY DISORDERS
#4 MESH DESCRIPTOR MYOFASCIAL PAIN SYNDROMES EXPLODE ALL TREES
#5 MESH DESCRIPTOR POLYMYALGIA RHEUMATICA
#6 ((pain* or discomfort*) near10 (central or complex or rheumat* or muscl* or muscul* or myofasci* or nerv* or neuralg* or neuropath*)):TI,AB,KY
#7 ((fibromyalgi* or fibrost* or FM or FMS)):TI,AB,KY
#8 ((neur* or nerv*) near6 (compress* or damag*)):TI,AB,KY
#9 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8
#10 MESH DESCRIPTOR Clonidine EXPLODE ALL TREES
#11 ((Clonidin* or clofelin or klofelin or m5041t or catapres* or clopheline or m‐5041t or st‐155 or klofenil or isoglaucon or clofenil or hemiton or st155 or catapresan or chlophazolin or gemiton or dixarit)):TI,AB,KY
#12 #10 OR #11
#13 #9 AND #12
Appendix 3. MEDLINE and MEDLINE In‐Process (via Ovid) search strategy
1. exp PAIN/
2. exp PERIPHERAL NERVOUS SYSTEM DISEASES/
3. SOMATOSENSORY DISORDERS/
4. FIBROMYALGIA/ or exp MYOFASCIAL PAIN SYNDROMES/ or POLYMYALGIA RHEUMATICA/
5. ((pain* or discomfort*) adj10 (central or complex or rheumat* or muscl* or muscul* or myofasci* or nerv* or neuralg* or neuropath*)).mp.
6. (fibromyalgi* or fibrost* or FM or FMS).mp.
7. ((neur* or nerv*) adj6 (compress* or damag*)).mp.
8. or/1‐7
9. Clonidine/
10. (Clonidin* or clofelin or klofelin or m5041t or catapres* or clopheline or m‐5041t or st‐155 or klofenil or isoglaucon or clofenil or hemiton or st155 or catapresan or chlophazolin or gemiton or dixarit).mp.
11. or/9‐10
12. 8 and 11
13. randomized controlled trial.pt.
14. controlled clinical trial.pt.
15. randomized.ab.
16. placebo.ab.
17. drug therapy.fs.
18. randomly.ab.
19. trial.ab.
20. or/13‐19
21. exp animals/ not humans.sh.
22. 20 not 21
23. 12 and 22
Appendix 4. EMBASE search strategy (via Ovid)
1. exp PAIN/
2. exp PERIPHERAL NERVOUS SYSTEM DISEASES/
3. SOMATOSENSORY DISORDERS/
4. FIBROMYALGIA/ or exp MYOFASCIAL PAIN SYNDROMES/ or POLYMYALGIA RHEUMATICA/
5. ((pain* or discomfort*) adj10 (central or complex or rheumat* or muscl* or muscul* or myofasci* or nerv* or neuralg* or neuropath*)).mp.
6. (fibromyalgi* or fibrost* or FM or FMS).mp.
7. ((neur* or nerv*) adj6 (compress* or damag*)).mp.
8. or/1‐7
9. Clonidine/
10. (Clonidin* or clofelin or klofelin or m5041t or catapres* or clopheline or m‐5041t or st‐155 or klofenil or isoglaucon or clofenil or hemiton or st155 or catapresan or chlophazolin or gemiton or dixarit).mp.
11. or/9‐10
12. 8 and 11
13. random$.tw.
14. factorial$.tw.
15. crossover$.tw.
16. cross over$.tw.
17. cross‐over$.tw.
18. placebo$.tw.
19. (doubl$ adj blind$).tw.
20. (singl$ adj blind$).tw.
21. assign$.tw.
22. allocat$.tw.
23. volunteer$.tw.
24. Crossover Procedure/
25. double‐blind procedure.tw.
26. Randomized Controlled Trial/
27. Single Blind Procedure/
28. or/13‐27
29. (animal/ or nonhuman/) not human/
30. 28 not 29
31. 12 and 30
Appendix 5. Data extraction form
Study ID
| Methods | |
| Participants | Inclusion criteria: Exclusion criteria: Number of participants screened/enrolled: Number of randomly assigned participants (C/P): Number of participants who received allocated intervention (C/P): Mean age (C/P): Number of males (C/P): Duration of pain condition (years ± SD; C/P): Mean baseline pain intensity (NPRS; mean ± SD; C/P): |
| Interventions | Intervention group: Control group: |
| Outcomes | |
| Notes |
C: clonidine group; P: placebo group.
Risk of bias assessment
| Domain | Risk of bias |
Support for judgement |
||
| Low risk | High risk | Unclear | ||
|
Random sequence generation (selection bias) |
||||
|
Allocation concealment (selection bias) |
||||
|
Blinding of participants and personnel (performance bias) |
|
|||
|
Blinding of outcome assessment (detection bias) |
||||
|
Incomplete outcome data (attrition bias) |
||||
|
Selective outcome reporting? (reporting bias) |
||||
| Size of study | [suggest Size of study is added as an additional row] | |||
| Notes: | ||||
Data and analyses
Comparison 1. Topical clonidine vs placebo in PDN.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain relief ≥ 30% | 2 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [1.03, 1.77] |
| 2 Participants with ≥ 1 adverse event | 2 | 344 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.14, 3.05] |
1.1. Analysis.
Comparison 1 Topical clonidine vs placebo in PDN, Outcome 1 Pain relief ≥ 30%.
1.2. Analysis.
Comparison 1 Topical clonidine vs placebo in PDN, Outcome 2 Participants with ≥ 1 adverse event.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Campbell 2009.
| Methods | Randomised, double‐blind, placebo‐controlled study with 8‐week treatment period | |
| Participants | Adult patients with PDN Number of randomly assigned participants (C 0.1%/C 0.2%/P): 166 (54/54/57) Study authors declare that 166 participants were randomly assigned; however, only 165 are described in the demographics table |
|
| Interventions | Intervention group: 0.1% clonidine gel, 2 times daily, 650 µL per foot for the first 2 weeks and 3 times daily thereafter (n = 54) 0.2% clonidine gel, 2 times daily, 500 µL per foot for the first 2 weeks and 3 times daily thereafter (n = 54) Control group: placebo applied in the same way (n = 57) |
|
| Outcomes | Numerical pain rating scale (NPRS) Participants with > 30% pain reduction Adverse events |
|
| Notes | Unpublished data acquired from study authors Participants were allowed to remain on concomitant medications Drug manufacturer Archion Therapeutics was involved in the study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of randomly assigned participants not equal to the number described in the demographics table (1 participant missing). Some results are missing |
| Selective reporting (reporting bias) | High risk | No clear presentation of results |
| Size of study | Unclear risk | Size of study: more than 50 and fewer than 199 participants per treatment arm |
Campbell 2012.
| Methods | Randomised, double‐blind, multi‐centre (USA), parallel‐group study, 12‐week treatment period Study consists of a screening phase (28 ± 7 days), a baseline phase (7 days), a treatment phase and a follow‐up period During screening phase, nociceptor function was tested by determining pain response to 0.1% topical capsaicin applied to the pretibial area of each participant for 30 minutes |
|
| Participants | Inclusion criteria
Exclusion criteria
Number of participants screened: 464 Number of randomly assigned participants (C/P): 182 (91/91) Number of participants who received allocated intervention (C/P): 180 (90/90) Mean age (C/P): 59.4/57.6 years Number of males (C/P): 44/42 Duration of foot pain (years ± SD; C/P): 3.0 ± 1.3/2.9 ± 1.3 Mean baseline pain (0 to 10 NPRS ± SD): 6.4 ± 1.4/6.5 ± 1.5 |
|
| Interventions | Intervention group: clonidine gel 650 µg per foot, 3 times daily, concentration 0.1% self administered on both feet (n = 91) Control group: matching placebo (n = 91) 464 participants were screened, 182 were randomly assigned (91/91), 90 participants in both groups received allocated intervention (1 participant in each group was found to be ineligible after randomisation, 1 participant in the clonidine group was excluded from analysis because no baseline NPRS score was available Intention‐to‐treat population: clonidine 89/placebo 90 Discontinuation: participants lost to follow‐up: C‐3/ P‐4; withdrawal of participant consent: C‐1/P‐1; protocol violation: C‐2/P‐4; adverse events: C‐1/P‐3; lack of efficacy: C‐1/P‐1 Clonidine gel was self administered by participants at home |
|
| Outcomes | Participants with > 30% pain reduction Participants with > 50% pain reduction Avarage pain severity Brief Pain Inventory: severity scale, average pain, functional interference scale Chronic Pain Sleep Inventory: overall seep quality Clinician and Patient Global Impressions of Change: overall change in pain status Hospital Anxiety and Depression Scale: anxiety scale, depression scale Adverse events |
|
| Notes | Participants discontinued use of "as needed" pain medications other than acetaminophen (paracetamol), Daily pain medications were continued on stable daily dosing 97 participants underwent a 3‐mm skin punch biopsy performed to quantify intraepidermal nerve fiber density Study was supported by the drug manufacturer Arcion Therapeutics |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation in blocks with stratifications with regard to baseline pain severity |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo formulation was identical in appearance, consistency, packaging and labelling |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Baseline observation carried forward in cases of missing results |
| Selective reporting (reporting bias) | Low risk | Clear presentation of results |
| Size of study | Unclear risk | Size of study: more than 50 and fewer than 199 participants per treatment arm |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Byas‐Smith 1995 | Transdermal, not topical drug delivery |
| Davis 1991 | Lack of control group |
| Lauretti 2009 | Transdermal, not topical drug delivery |
| Meno 2001 | Lack of control group |
| Zeigler 1992 | Transdermal, not topical drug delivery |
Characteristics of ongoing studies [ordered by study ID]
NCT00661063.
| Trial name or title | Diabetic Neuropathy Topical Treatment |
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study Study duration: 12 weeks |
| Participants | Patients with a diagnosis of diabetes mellitus type I or II and mononeuropathy or polyneuropathy. Patients had to be treated with tricyclic antidepressants or carbamazepine ≥ 3 weeks Age ≥ 18 years |
| Interventions | Participants were to receive clonidine 1% gel or ketamine 150 mcg/g bid or both, or placebo |
| Outcomes | Pain evaluation by visual analogue scale Pain evaluation by amount of rescue medication required |
| Starting date | Study should have started in 2008; however, no results were available until now. We tried to contact the study author but without success |
| Contact information | Judymara L Gozzani; gozzani@osite.com.br |
| Notes |
NCT02068027.
| Trial name or title | Safety and Efficacy Study of Clonidine Hydrochloride Topical Gel, 0.1%, in the Treatment of Pain Associated With Diabetic Neuropathy |
| Methods | Randomised, multi‐centre, double blind, placebo‐controlled, parallel‐group study Study duration: 12 weeks |
| Participants | Established diagnosis of diabetes (type I or II) with pain attributable to a symmetrical stocking distribution neuropathy in lower extremities lasting ≥ 3 months Age 18 to 85 years Estimated enrolment: 140 participants |
| Interventions | Intervention group: 0.1% clonidine gel Control group: identical placebo |
| Outcomes | Change in average pain score (NPRS) Mean daily average pain score (NPRS) Mean daily worst pain intensity (NPRS) |
| Starting date | March 2014 |
| Contact information | Tim M Warneke; MS twarneke@bdsi.com |
| Notes |
Differences between protocol and review
The protocol and the review are different in several ways.
In the protocol, we planned to analyse evidence in two tiers. The Cochrane PaPaS Group recommendations have changed since that time and advise three tiers of evidence, so in this final review, we have included three tiers of evidence.
As evidence was limited, we decided to include continuous outcomes in the review for illustrative purposes only.
We decided to include an additional outcome in the 'Summary of findings' table to better illustrate results of the review reporting participants with at least one adverse event.
Contributions of authors
Conceiving of the review: A. Wrzosek. Co‐ordinating the review: A. Wrzosek. Undertaking manual searches: A. Wrzosek, J. Jakowicka, J. Woron, J. Wordliczek. Screening search results: A. Wrzosek, J. Jakowicka, J. Woron. Organizing retrieval of papers: A. Wrzosek, J. Woron. Screening retrieved papers against inclusion criteria: A. Wrzosek, J. Jakowicka, J. Woron, J. Wordliczek. Appraising quality of papers: A. Wrzosek, J. Jakowicka, J. Dobrogowski, J. Wordliczek. Abstracting data from papers: A. Wrzosek, J. Jakowicka, J. Wordliczek. Writing to authors of papers for additional information: A. Wrzosek. Obtaining and screening data on unpublished studies: A. Wrzosek. Managing data for the review: A. Wrzosek. Entering data into Review Manager (RevMan 5.3): A. Wrzosek. Analysing RevMan statistical data: A. Wrzosek. Performing other statistical analysis not using RevMan 5.3: none. Double‐checking data entered by person one: A. Wrzosek; data checked by person two: J. Jakowicka. Writing the review: A. Wrzosek, J. Jakowicka, J. Woron, J. Dobrogowski, J. Wordliczek. Providing general advice on the conduct of this review: J. Wordliczek, J. Dobrogowski. Taking responsibility for reading and checking the review before submission: A. Wrzosek, J. Wordliczek.
All authors will be responsible for updating the review.
Declarations of interest
Anna Wrzosek has no relevant conflicts of interest to declare.
Jaroslaw Woron has no relevant conflicts of interest to declare.
Jan Dobrogowski has no relevant conflicts of interest to declare.
Joanna Jakowicka‐Wordliczek has no relevant conflicts of interest to declare.
Jerzy Wordliczek has no relevant conflicts of interest to declare.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Campbell 2009 {published and unpublished data}
- Campbell C, Campbell J, Schmidt W, Brady K, Stouch B. Topical clonidine gel reduces pain caused by diabetic neuropathy: results of a multicenter, placebo‐controlled clinical trial. The Journal of Pain 2009;4:S55. [Google Scholar]
Campbell 2012 {published data only}
- Campbell CM, Kipnes MS, Stouch BC, Brady KL, Kelly M, Schmidt WK, et al. Randomized control trial of topical clonidine for treatment of painful diabetic neuropathy. Pain 2012;153(9):1815‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Byas‐Smith 1995 {published data only}
- Byas‐Smith MG, Max MB, Muir J, Kingman ACN, CN‐00115607. Transdermal clonidine compared to placebo in painful diabetic neuropathy using a two‐stage 'enriched enrollment' design. Pain 1995;60(3):267‐74. [DOI] [PubMed] [Google Scholar]
Davis 1991 {published data only}
- Davis KD, Treede RD, Raja SN, Meyer RA, Campbell JN. Topical application of clonidine relieves hyperalgesia in patients with sympathetically maintained pain. Pain 1991;47(3):309‐17. [DOI] [PubMed] [Google Scholar]
Lauretti 2009 {published data only}
- Lauretti GR, Mattos AL, Lima ICPR, Matsumoto M, Resende CS. The clinical and laboratorial evaluation of transdermal ketamine, fentanyl, clonidine or their combination in chronic low back pain. European Journal of Pain 2009;13:S126. [Google Scholar]
Meno 2001 {published data only}
- Meno A, Arita H, Hanaoka K. [Preliminary report: the efficacy of clonidine hydrochloride ointment for postherpetic neuralgia]. Masui ‐ Japanese Journal of Anesthesiology 2001;50(2):160‐3. [PubMed] [Google Scholar]
Zeigler 1992 {published data only}
- Zeigler D, Lynch SA, Muir J, Benjamin J, Max MBCN, CN‐00084468. Transdermal clonidine versus placebo in painful diabetic neuropathy. Pain 1992;48(3):403‐8. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT00661063 {published data only}
- Gozzani JL. Diabetic Neuropathy Topical Treatment. www.clinicaltrials.gov 2008.
NCT02068027 {published data only}
- Shaibani A. Safety and efficacy study of clonidine hydrochloride topical gel, 0.1% in the treatment of pain associated with diabetic neuropathy. www.clinicaltrials.gov 2014.
Additional references
Aley 1997
- Aley KO, Levine JD. Multiple receptors involved in peripheral alpha 2, mu, and A1 antinociception, tolerance, and withdrawal. Journal of Neuroscience 1997;17(2):735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andrew 2014
- Andrew R, Derry S, Taylor RS, Straube S, Phillips CJ. The costs and consequences of adequately managed chronic non‐cancer pain and chronic neuropathic pain. Pain Practice 2014;14(1):79‐94. [DOI] [PubMed] [Google Scholar]
Anitescu 2013
Apkarian 2011
- Apkarian AV, Hashmi JA, Baliki MN. Pain and the brain: specificity and plasticity of the brain in clinical chronic pain. Pain 2011;152(3 Suppl):S49‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Asano 2000
- Asano T, Dohi S, Ohta S, Shimonaka H, Lida H. Antinociception by epidural and systemic alpha(2)‐adrenoceptor agonists and their binding affinity in rat spinal cord and brain. Anesthesia & Analgesia 2000;90(2):400–7. [DOI] [PubMed] [Google Scholar]
Bernard 1994
- Bernard JM, Kick O, Bonnet F. Which way for the administration of alpha2‐adrenergic agents to obtain the best analgesia?. Cahiers d Anesthésiologie 1994;42(2):223–8. [PubMed] [Google Scholar]
Bouhassira 2008
- Bouhassira D, Lanteri‐Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008;136:380–7. [DOI] [PubMed] [Google Scholar]
Buerkle 1998
- Buerkle H, Yaksh TL. Pharmacological evidence for different alpha2‐adrenergic receptor sites mediating analgesia and sedation in the rat. British Journal of Anaesthesia 1998;81(2):208–15. [DOI] [PubMed] [Google Scholar]
Buerkle 2000
- Buerkle H. Peripheral antinociceptive action of alpha2‐adrenoceptoragonist. Baillière's Clinical Anaesthesiology 2000;2:411–8. [Google Scholar]
Chi 2007
- Chi L, Sekiyama H, Hayashida M, Takeda K, Sumida T, Sawamura S, et al. Effects of topical application of clonidine cream on pain behaviors and spinal Fos protein expression in rat models of neuropathic pain, postoperative pain, and inflammatory pain. Anesthesiology 2007;107(3):486‐94. [DOI] [PubMed] [Google Scholar]
Cochrane PaPaS Group 2011
- Cochrane Pain, Palliative and Supportive Care Group. PaPaS author and referee guidance. http://papas.cochrane.org/sites/papas.cochrane.org/files/uploads/L%20‐%20PaPaSAuthor%26RefereeGuidance.pdf (accessed 11 December 2013).
Derry 2012
- Derry S, Moore RA. Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD010111] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2013
- Derry S, Sven‐Rice A, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2013, Issue 2. [DOI: 10.1002/14651858.CD007393.pub3] [DOI] [PubMed] [Google Scholar]
Dias 1999
- Dias VC, Tendler B, Oparil S, Reilly PA, Snar P, White WB. Clinical experience with transdermal clonidine in African‐American and Hispanic‐American patients with hypertension: evaluation from a 12‐week prospective, open‐label clinical trial in community‐based clinics. American Journal of Therapeutics 1999;6(1):19–24. [DOI] [PubMed] [Google Scholar]
Dogrul 2004
- Dogrul A, Uzbay IT. Topical clonidine antinociception. Pain 2004;111(3):385‐91. [DOI] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI] [PubMed] [Google Scholar]
Eisenach 1995
- Eisenach JC, DuPen S, Dubois M, Miguel R, Allin D, Epidural Clonidine Study Group 2. Epidural clonidine analgesia for intractable cancer pain. Pain 1995;61(3):391–9. [DOI] [PubMed] [Google Scholar]
Eisenach 1996
- Eisenach JC, Kock M, Klimscha W. Alpha2‐adrenergic agonists for regional anesthesia: a clinical review of clonidine (1984–1995). Anesthesiology 1996;85:655–74. [DOI] [PubMed] [Google Scholar]
Finnerup 2015
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurology 2015;14(2):162‐73. [PUBMED: 25575710.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Flores 2012
- Flores MP, Castro AP, Nascimento Jdos S. Topical analgesics. Revista Brasileira de Anestesiologia 2012;62(2):244‐52. [DOI] [PubMed] [Google Scholar]
Gentili 1996
- Gentili M, Juhel A, Bonnet F. Peripheral analgesic effect of intra‐articular clonidine. Pain 1996;64(3):593–6. [DOI] [PubMed] [Google Scholar]
Gentili 1997
- Gentili M, Houssel P, Osman M, Henel D, Juhel A, Bonnet F. Intra‐articularmorphine and clonidine produce comparable analgesia but the combination is not more effective. British Journal of Anaesthesia 1997;79(5):660–1. [DOI] [PubMed] [Google Scholar]
Gustorff 2008
- Gustorff B, Dorner T, Likar R, Grisold W, Lawrence K, Schwarz F, et al. Prevalence of self‐reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiologica Scandinavica 2008;52(1):132‐6. [DOI] [PubMed] [Google Scholar]
Hall 2008
- Hall GC, Carroll D, McQuay HJ. Primary care incidence and treatment of four neuropathic pain conditions: a descriptive study, 2002‐2005. BMC Family Practice 2008;9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hassenbusch 2002
- Hassenbusch SJ, Gunes S, Wachsman S, Willis KD. Intrathecal clonidine in the treatment of intractable pain: a phase I/II study. Pain Medicine 2002;3:85–91. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
Hurley 2013
IASP Taxonomy Working Group 2011
- IASP Taxonomy Working Group. Classification of Chronic Pain. Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. Second Edition (Revised). http://www.iasp‐pain.org/AM/Template.cfm?Section=Classification_of_Chronic_Pain&Template=/CM/HTMLDisplay.cfm&ContentID=2687 (accessed 11 December 2014).
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17(1):1‐12. [DOI] [PubMed] [Google Scholar]
Jensen 2011
Katusic 1991
- Katusic S, Williams DB, Beard CM, Bergstralh EJ, Kurland LT. Epidemiology and clinical features of idiopathic trigeminal neuralgia and glossopharyngeal neuralgia: similarities and differences, Rochester, Minnesota,1945‐1984. Neuroepidemiology 1991;10:276‐81. [DOI] [PubMed] [Google Scholar]
Kawaski 2003
- Kawasaki Y, Kumamoto E, Furue H, Yoshimura M. Alpha 2 adrenoceptor mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology 2003;98(3):682–9. [DOI] [PubMed] [Google Scholar]
Khaliq 2007
- Khaliq W, Alam S, Puri N. Topical lidocaine for the treatment of postherpetic neuralgia. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI: 10.1002/14651858.CD004846.pub2] [DOI] [PubMed] [Google Scholar]
Khan 1999
- Khan ZP, Ferguson CN, Jones RM. Alpha‐2 and imidazoline receptor agonists. Their pharmacology and therapeutic role. Anaesthesia 1999;54(2):146‐65. [DOI] [PubMed] [Google Scholar]
Kolesnikov 1999
- Kolesnikov YA, Pasternak GW. Topical opioid in mice: analgesia and reversal of tolerance by a topical N‐methyl‐D‐aspartate antagonist. Journal of Pharmacology and Experimental Therapeutics 1999;290:247–52. [PubMed] [Google Scholar]
Kolesnikov 2000
- Kolesnikov YA, Chereshnev I, Pasternak GW. Analgesic synergy between topical lidocaine and topical opioids. Journal of Pharmacology and Experimental Therapeutics 2000;295:546‐51. [PubMed] [Google Scholar]
Koopman 2009
- Koopman JS, Dieleman JP, Huygen FJ, Mos M, Martin CG, Sturkenboom MC. Incidence of facial pain in the general population. Pain 2009;147(1‐3):122‐7. [DOI] [PubMed] [Google Scholar]
Lavand'homme 2002
- Lavand‘homme PM, Ma W, De KM, Eisenach JC. Perineural alpha(2A)‐adrenoceptor activation inhibits spinal cord neuroplasticity and tactile allodynia after nerve injury. Anesthesiology 2002;97:972–80. [DOI] [PubMed] [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ 2009;339:b2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lunn 2009
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy or chronic pain. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD007115.pub2] [DOI] [PubMed] [Google Scholar]
Lunn 2014
- Lunn Michael PT, Hughes Richard AC, Wiffen Philip J. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd, 2014, issue 1. [DOI: 10.1002/14651858.CD007115.pub3; CD007115] [DOI] [PMC free article] [PubMed]
McQuay 2007
- McQuay HJ, Smith LA, Moore RA. Chronic pain. In: Stevens A, Raftery J, Mant J, Simpson S editor(s). Health Care Needs Assessment. 3rd Edition. Oxford: Radcliffe Publishing, 2007. [Google Scholar]
Moisset 2007
- Moisset X, Bouhassira D. Brain imaging of neuropathic pain. Neuroimaging 2007;37(Suppl 1):S80‐8. [DOI] [PubMed] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything ‐ large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. McQuay HJ, Kalso E, Moore RA, editors(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle: IASP Press, 2008:15‐24. [ 978–0–931092–69–5] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010a
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. "Evidence" in chronic pain ‐ establishing best practice in the reporting of systematic reviews. Pain 2010;150:386‐9. [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(2):360‐4. [DOI] [PubMed] [Google Scholar]
Moore 2010d
- Moore RA, Smugar SS, Wang H, Peloso PM, Gammaitoni A. Numbers‐needed‐to‐treat analyses ‐ do timing, dropouts, and outcome matter? Pooled analysis of two randomized, placebo‐controlled chronic low back pain trials. Pain 2010;151(3):592‐7. [DOI] [PubMed] [Google Scholar]
Moore 2011a
- Moore RA, Wiffen PJ, Derry S, McQuay HJ. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2011, Issue 3. [DOI: 10.1002/14651858.CD007938.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2011b
- Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta‐analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain 2011;152(5):982‐9. [DOI] [PubMed] [Google Scholar]
Moore 2011c
- Moore RA, Mhuircheartaigh RJ, Derry S, McQuay HJ. Mean analgesic consumption is inappropriate for testing analgesic efficacy in post‐operative pain: analysis and alternative suggestion. European Journal of Anaesthesiology 2011;28(6):427‐32. [DOI] [PubMed] [Google Scholar]
Moore 2012a
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD008242.pub2] [DOI] [PubMed] [Google Scholar]
Moore 2012b
- Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265‐8. [DOI] [PubMed] [Google Scholar]
Moore 2013
- Moore RA, Straube S, Aldington D. Pain measures and cut‐offs ‐ 'no worse than mild pain' as a simple, universal outcome. Anaesthesia 2013;68(4):400‐12. [DOI] [PubMed] [Google Scholar]
Moore 2014
- Moore RA, Wiffen Philip J, Derry S, Toelle T, Rice Andrew SC. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd, 2014, issue 4. [DOI: 10.1002/14651858.CD007938.pub3; CD007938] [DOI] [PMC free article] [PubMed]
Neil 2011
- Neil MJ. Clonidine: clinical pharmacology and therapeutic use in pain management. Current Clinical Pharmacology 2011;6:280‐7. [DOI] [PubMed] [Google Scholar]
O'Brien 2010
- O'Brien EM, Staud RM, Hassinger AD, McCulloch RC, Craggs JG, Atchison JW, et al. Patient‐centered perspective on treatment outcomes in chronic pain. Pain Medicine 2010;11(1):6‐15. [DOI] [PubMed] [Google Scholar]
O'Connor 2009
- O'Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. American Journal of Medicine 2009;122(10 Suppl):S22‐32. [DOI] [PubMed] [Google Scholar]
Ongioco 2000
- Ongioco RR, Richardson CD, Rudner XL, Stafford‐Smith M, Schwinn DA. Alpha2‐adrenergic receptors in human dorsal root ganglia: predominance of alpha2b and alpha2c subtype mRNSs. Anesthesiology 2000;92(4):968–76. [DOI] [PubMed] [Google Scholar]
Puskas 2003
- Puskas F, Camporesi EM, O'Leary CE, Hauser M, Nasrallah FV. Intrathecal clonidine and severe hypotension after cardiopulmonary bypass. Anesthesia & Analgesia 2003;97(5):1251–3. [DOI] [PubMed] [Google Scholar]
Rappaport 1994
- Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self‐sustaining discharge in the trigeminal ganglion. Pain 1994;56:127‐38. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Riedl 2009
- Riedl MS, Schnell SA, Overland AC, Chabot‐Doré AJ, Taylor AM, Ribeiro‐da‐Silva A, et al. Coexpression of alpha 2A‐adrenergic and delta‐opioid receptors in substance P‐containing terminals in rat dorsal horn. Journal of Comparative Neurology 2009;513:385–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Samso 1996
- Samso E, Valles J, Pol O, Gallart L, Puig MM. Comparative assessment of the anaesthetic and analgesic effects of intramuscular and epidural clonidine in humans. Canadian Journal of Anesthesia 1996;43(12):1195–202. [DOI] [PubMed] [Google Scholar]
Sawynok 2003
- Sawynok J. Topical and peripherally acting analgesics. Pharmacological Reviews 2003;55(1):1–20. [DOI] [PubMed] [Google Scholar]
Sierralta 1996
- Sierralta F, Naquira D, Pinardi G, Miranda HF. Alpha‐adrenoceptor and opioid receptor modulation of clonidine‐induced antinociception. British Journal of Pharmacology 1996;119(3):551–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 2013
- Smith BH, Lee J, Price C, Baranowski AP. Neuropathic pain: a pathway for care developed by the British Pain Society. British Journal of Anaesthesia 2013;111:73‐9. [DOI] [PubMed] [Google Scholar]
Soni 2013
- Soni A, Batra R, Gwilym S, Spector T, Hart D, Arden N, et al. Neuropathic features of joint pain: a community‐based study. Arthritis & Rheumatism 2013;65(7):1942‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2008
- Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrolment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal of Clinical Pharmacology 2008;66(2):266‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2010
- Straube S, Derry S, Moore RA, Paine J, McQuay HJ. Pregabalin in fibromyalgia ‐ responder analysis from individual patient data. BMC Musculoskeletal Disorders 2010;11:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sultan 2008
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurology 2008;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Torrance 2006
- Torrance N, Smith BH, Bennett MI, Lee AJ. The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. Journal of Pain 2006;7(4):281–9. [DOI] [PubMed] [Google Scholar]
Tracey 2011
- Tracey I. Can neuroimaging studies identify pain endophenotypes in humans?. Nature Reviews Neurology 2011;7(3):173‐81. [DOI] [PubMed] [Google Scholar]
Treede 2008
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008;70(18):1630‐5. [DOI] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2163‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2013
- Wiffen Philip J, Derry S, Moore RA, Aldington D, Cole P, Rice Andrew SC, et al. Antiepileptic drugs for neuropathic pain and fibromyalgia ‐ an overview of Cochrane reviews. Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd, 2013, issue 11. [DOI: 10.1002/14651858.CD010567.pub2; CD010567] [DOI] [PMC free article] [PubMed]
Wiffen 2014
- Wiffen Philip J, Derry S, Moore RA, Kalso Eija A. Carbamazepine for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd, 2014, issue 4. [DOI: 10.1002/14651858.CD005451.pub3; CD005451] [DOI] [PMC free article] [PubMed]
Woolf 1999
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 1999;353(9168):1959‐64. [DOI] [PubMed] [Google Scholar]