

Summary
Introduction
The relationship between blood metabolites and hemoglobin degradation products (BMHDPs) formed in the cerebrospinal fluid and the development of vasospasm and delayed cerebral ischemia (DCI) after aneurysmal subarachnoid hemorrhage (aSAH) has been the focus of several previous studies, but their molecular and cellular targets remain to be elucidated.
Methods
Because BMHDP‐induced changes in Cav2.3 channel function are thought to contribute to DCI after aSAH, we studied their modulation by unconjugated bilirubin (UCB) in an organotypical neuronal network from wild‐type (WT) and Cav2.3‐deficient animals (KO). Murine retinae were isolated from WT and KO and superfused with nutrient solution. Electroretinograms were recorded before, during, and after superfusion with UCB. Transretinal signaling was analyzed as b‐wave, implicit time, and area under the curve (AUC).
Results
Superfusion of UCB significantly attenuated the b‐wave amplitude in the isolated retina from wild‐type mice by 14.9% (P < 0.05), followed by gradual partial recovery (P = 0.09). Correspondingly, AUC decreased significantly with superfusion of UCB (P < 0.05). During washout, the b‐wave amplitude returned to baseline (P = 0.2839). The effects of UCB were absent in Cav2.3‐deficient mice, lacking the expression of Cav2.3 as proofed on the biochemical level.
Conclusions
Ex vivo neuronal recording in the murine retina is able to detect transient impairment of transretinal signaling by UCB in WT, but not in KO. This new model may be useful to further clarify the role of calcium channels in neuronal signal alteration in the presence of BHMDPs.
Keywords: bilirubin, blood degradation products, ex vivo, isolated vertebrate retina, murine ERG
1. INTRODUCTION
In the central nervous system, voltage‐gated calcium channels (VGCCs) regulate a variety of processes, including hormonal communication, modulation of cell migration, triggering of muscle contraction, and many other processes.1 They contribute to synaptic transmission and calcium homeostasis in different neuronal networks including the additional processes like disruption of neuronal networks.2, 3 Moreover, there is convincing evidence that subarachnoid hemorrhage (SAH) can alter the expression profile of calcium channels in cerebral arteries. Experimental SAH has been shown to trigger expression of Cav2.3 (R‐type) calcium channels in rat and rabbit cerebral smooth muscle cells, thereby reducing their sensitivity to L‐type VGCC antagonists.4 Anti‐inflammatory agents of VGCC also reduced constriction of isolated cerebral arteries but not in control animals and improved cerebral blood flow after experimental SAH in rats.5 Interestingly, prolonged exposure of rabbit cerebral arteries to oxyhemoglobin mimicked the ability of SAH to induce Cav2.3 channel expression, suggesting that the emergence of these channels is linked to lysis of red blood cells within the subarachnoid space.6 Products formed during degradation of hemoglobin—mainly unconjugated bilirubin (UCB) and its oxidation products—have been investigated for their role in the development of cerebral vasospasm (CVS).7, 8 The participation of UCB in the processes underlying delayed cerebral ischemia (DCI) by, for example, altering Ca2+ influx into neuronal cells, would be conceivable.
The aim of the present work was to assess the potential role of Cav2.3 channels for effects propagated by UCB—as present in the context of aneurysmal SAH—in an organotypical neuronal network. To this end, we studied the action of UCB on scotopic electroretinograms (ERGs) recorded in the isolated and superfused retina from Cav2.3‐deficient and wild‐type mice. We hypothesized that b‐wave amplitude of the Cav2.3‐competent retina is transiently reduced during continuous bilirubin superfusion, while the ERGs of Cav2.3‐deficient mice are not influenced by such a superfusion.
2. METHODS
2.1. Material and reagents
Unless noted otherwise, reagents and albumin fraction V ≥ 98% were used without further purification (Sigma‐Aldrich Chemie GmbH, Schnelldorf, Germany; Carl Roth GmbH, Karlsruhe, Germany). Solutions were prepared in autoclaved glass bottles with deionized, double‐distilled water or type I ultrapure water dispensed from an ELGA LabWater system (Purelab Flex 2, ELGA LabWater, High Wycombe, UK), respectively.
2.2. Animals
Male mice (12‐15 weeks old, 25‐31 g) were housed at a constant temperature (20‐22°C) in Makrolon type II cages, with light on from 7 am to 7 pm (light intensity at the surface of the animal cages was between 5 and 10 lux) and ad libitum access to food and water. The cacna1e gene encoding Cav2.3 was disrupted in vivo by agouti‐colored Cav2.3(fl|+) and deleter mice expressing Cre recombinase constitutively.9 Thus, exon 2 was ablated by Cre‐mediated recombination. Cav2.3‐deficient mice were fertile, exhibited no obvious behavioral abnormalities, and had the same lifespan as control mice. Parallel breeding of parental inbred mouse lines of Cav2.3‐deficient and control mice ensured their identical background. The institutional committee on animal care approved the animal experimentation described in the text (4.17.007) and conducted following accepted standards of humane animal care.
2.3. Preparation of unconjugated bilirubin (UCB)
Fresh stock solutions of bilirubin (1 mmol/L UCB in 10 mmol/L NaOH) were prepared immediately prior and diluted into the test solution to yield the required concentration. All solutions containing UCB were stored in the dark and protected from illumination during the experiments. All experiments were performed in the presence of albumin. UCB was added to test solutions containing 10 μmol/L bovine serum albumin (BSA) to give U/A ratios of 0.5. Solutions for control recordings and washout always contained the same volume of NaOH without bilirubin and (for recordings with albumin) the same concentration of BSA as in the corresponding test solution. For the isolated and superfused murine retina, 5 μmol/L UCB was added to the test solution.
2.4. Retina isolation and ERG recordings
Mice were dark‐adapted overnight and killed by cervical dislocation under dim red light, and the eyes were extirpated immediately. A 27‐gauge needle (Sterican, size 20, 0.4 × 20 mm Bl/LB) was employed to puncture the cornea of the extirpated eye bulb. Enucleated eyes were protected from light and transferred into carbogen‐saturated (95% O2/5% CO2) modified Ames medium.10 The whole retina was isolated immediately under dim red light using superfine scissors and ultrafine suturing forceps (WPI, Berlin, Germany) and transferred to the recording chamber as described previously.11 The electroretinogram was recorded via two silver/silver‐chloride electrodes on either side of the isolated retina, with the recording chamber containing a retina placed in an electrically and optically isolated air thermostat. From the dark‐adapted retina, electroretinograms in response to a single white light flash were recorded at intervals of 3 minutes at 27.5°C and with a constant superfusion of modified Ames medium at 2 mL/min. The duration of light stimulation was 500 ms. The prestimulus delay was 380 ms. The flash intensity was set to 63 mlux at the retinal surface using calibrated neutral density filters.
The ERG was amplified and bandpass‐limited between 0.3 and 300 Hz (PowerLab 8/35, Animal Bio Amp FE136, ADInstruments, Oxford, UK). Light flash, heating unit, fan, and roller pump were automatically controlled (BNC‐2120, DASY‐Lab V8.0; National‐Instruments, Austin, TX, USA). For each experiment, a new retina from independent mouse was transferred to the recording chamber. The retina was superfused with 10 μmol/L albumin and stimulated repetitively until the responses had reached a stable level (after 90 minutes of superfusion), followed by UCB/albumin (U/A) molar ratio of 0.5 and again with 10 μmol/L albumin (bilirubin washout), each for 45 minutes. Switching from one solution to another was performed with a three‐way valve to prevent disturbance of the experimental conditions.
2.5. Isolation of microsomal membranes and immunoblotting
Retinal and neocortical microsomes were isolated according to standard procedures using a method, which is suitable to deal with minute amounts of tissue samples.12 In short, in the retinal tissue homogenate, microsomes containing integral membrane proteins were separated from nonintegral only adherent membrane proteins by washing stepwise first with a high‐salt buffer (containing 2 mol/L NaCl, 10 mmol/L HEPES‐NaOH, pH 7.4, 1 mmol/L EDTA) and next with a carbonate buffer (0.1 mol/L Na2CO3 and 1 mmol/L EDTA) at a pH of 11.3. Six retinas from 3 mice of each genotype were isolated under dim light and immediately placed in high‐salt buffer and homogenized, followed by ultracentrifugation at 110.000 g for 30 minutes. The supernatant was discarded and the pellet rehomogenized in the alkaline carbonate buffer followed by a second ultracentrifugation at 110.000 g for 30 minutes. The supernatant was again discarded and the pellet rehomogenized in small volumes of a sucrose‐containing buffer (0.25 mol/L sucrose, 100 mmol/L NaCl, 10 mmol/L HEPES‐NaOH, pH 7.4, and 1 mmol/L EDTA). All buffers for microsome isolation contained freshly added protease inhibitors (protease inhibitor cocktail set III, EDTA‐free with final concentration of 0.5% (v/v), Calbiochem®), and all buffers were used at low temperature of 4°C. Aliquots of microsomal membranes were stored at −20°C. After polyacrylamide gel electrophoresis (PAGE) and immunoblotting, the peptide‐directed antibody anti‐α1E‐com was used, which was directed against the peptide Nast‐195 (SGILEGFDPPHPCGVQG[C]. The peptide‐specific antibody was designed to recognize an epitope between the extracellular loop IS5 and the pore region common to all cloned splice variants of Cav2.313 (Figure 1).
Figure 1.
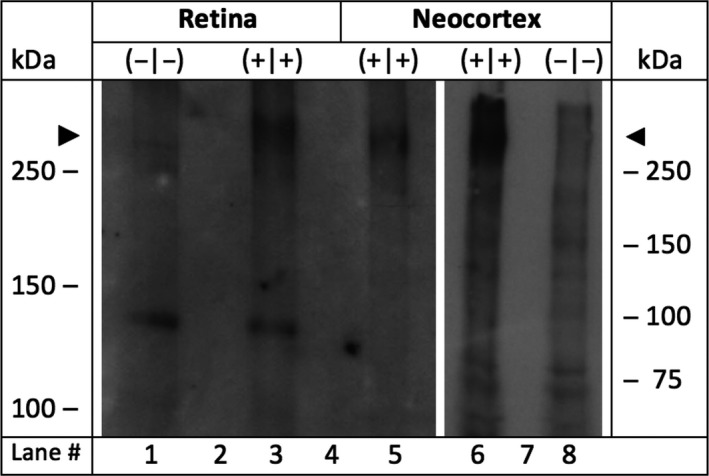
Expression analysis of Cav2.3 after SDS‐polyacrylamide gel electrophoresis and Western blot. Microsomal membranes were isolated from the retina and the neocortex of Cav2.3 null mutants and wild‐type littermates.12 Lane 1: 49 μg protein from Cav2.3‐deficient retina (−|−). Lane 3: 34 μg protein from Cav2.3‐competent retinae (+|+). Microsomal membranes from murine neocortices of Cav2.3‐deficient mice were used as negative (−|−) and from Cav2.3‐competent mice as positive controls (+|+), respectively. Lane 5: 32 μg protein from Cav2.3‐competent neocortical membranes. Lane 6 and 8, 72 μg of neocortical microsomal protein from Cav2.3‐competent or Cav2.3‐deficient mice, respectively. In lanes 2, 4, and 7, no protein was loaded. Peptide‐directed antibodies directed against a common epitope in all Cav2.3 splice variants (anti‐a1E‐com) were used as the primary antibody during Western blot analysis.9 Note that the predicted size of the full length Cav2.3 protein (262 kDa) is marked by the arrowheads
2.6. Data analysis of ERGs
Analysis of ERGs was carried out according to the parameters depicted in Figure 2. For quantification of transretinal signaling, we calculated the b‐wave amplitudes and their implicit time. The b‐wave amplitude was measured from the trough of the a‐wave to the peak of the b‐wave; t (50%), time from flashlight to the point of the signal regression of 50% of the amplitude of b‐wave; full width at half maximum (FWHM) of the b‐wave. In addition, the response of b‐wave was quantified by determination of the area under the curve until maximum and from maximum until return to baseline (AUC1 and AUC2).
Figure 2.
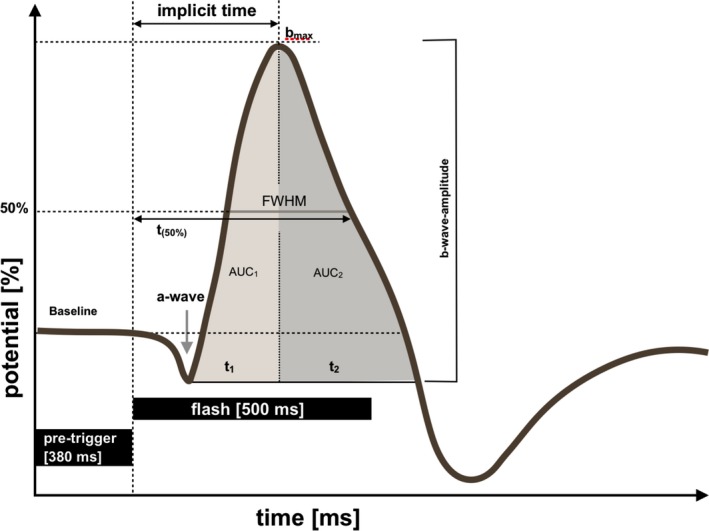
Schematic representation of an electroretinogram (ERG) recorded from the isolated and superfused murine retina and the parameters quantified for analysis. Latency, implicit times of the b‐wave; FWHM, full width at half maximum of the b‐wave; t (50%), time (ms) after the flash required for the b‐wave amplitude to decay to 50% of its maximal value; AUC1 and AUC2, area under the curve (AUC) of the b‐wave
Quantitative, normally distributed data are presented as mean ± standard deviation (SD) or median and interquartiles. In case of categorical variables, data are performed as numbers and percentage. Nonparametric tests are demonstrated as median [1.quartile‐3.quartile]. Two‐sided, paired Student's t test was used for comparison of quantitative parameters in case of normal distribution. If not applicable, Wilcoxon test was used instead.
According to the purpose of the study, we controlled in the primary analysis the main parameter “b‐wave amplitude” for multiple comparisons using Holm‐Bonferroni method. We considered 6 comparisons for the mentioned parameter: 3 for WT and 3 for KO group (Table 1). Comparisons of other ERG parameters were carried out the explorative way without correction for multiple comparisons to show tendencies of the ERG course in the groups and to evaluate additional characteristics of ERG, describing UCB exposure in wild‐type and Cav2.3‐deficient mice. Statistical significance was set at P < 0.05, and statistical results with P < 0.1 were accepted as a trend. All analyses were performed with IBM® SPSS® Statistics v 22.0 (IBM, Chicago, IL, USA) and (GraphPad Prism®, GraphPad Software, Inc, La Jolla, CA, USA).
Table 1.
Systematical comparisons of ERG traces before and after UCB exposure in wild‐type and Cav2.3‐deficient mice (explorative testing without correction for multiple comparisons)
| Parameters | Nutrient | Wild type (WT) | Knockout (KO) | ||||
|---|---|---|---|---|---|---|---|
| n | Median [1.quartile‐3.quartile] | P‐value* | n | Median [1.quartile‐3.quartile] | P‐value* | ||
| b‐wave amplitude, μV | Equilibrium | 16 | 34 [22‐38] | 0.0060 § | 14 | 27 [16‐46] | 0.7780 § |
| UCB1 | 16 | 31 [16‐36] | 14 | 28 [14‐45] | |||
| UCB1 | 16 | 31 [16‐36] | 0.0300 § | 14 | 28 [14‐45] | 0.1770 § | |
| UCB2 | 16 | 31 [21‐38] | 14 | 27 [16‐45] | |||
| Equilibrium | 15 | 34 [21‐39] | 0.3630 § | 14 | 27 [16‐46] | 0.0353 § | |
| Washout | 15 | 36 [21‐39] | 14 | 26 [14‐43] | |||
| Implicit time of b‐wave, ms | Equilibrium | 16 | 300 [273‐320] | 0.8489 | 14 | 275 [258‐320] | 0.9971§ |
| UCB1 | 16 | 300 [280‐345] | 14 | 275 [260‐318] | |||
| UCB1 | 16 | 300 [280‐345] | 0.5254 | 14 | 275 [260‐318] | 0.3916§ | |
| UCB2 | 16 | 305 [280‐330] | 14 | 280 [268‐333] | |||
| Equilibrium | 15 | 300 [270‐320] | 0.2519 | 14 | 275 [258‐320] | 0.2329§ | |
| Washout | 15 | 310 [280‐350] | 14 | 275 [268‐328] | |||
| AUC1, μV*ms | Equilibrium | 16 | 4032 [2806‐5162] | 0.0110 | 14 | 3342 [1944‐4936] | 0.7609§ |
| UCB1 | 16 | 3453 [1619‐5306] | 14 | 2814 [1700‐6223] | |||
| UCB1 | 16 | 3453 [1619‐5306] | 0.0792 | 14 | 2814 [1700‐6223] | 0.8077§ | |
| UCB2 | 16 | 3340 [2027‐5205] | 14 | 3134 [2018‐5954] | |||
| Equilibrium | 15 | 4214 [2732‐5245] | 0.6760 | 14 | 3342 [1944‐4936] | 0.2676§ | |
| Washout | 15 | 3971 [2567‐6136] | 14 | 3323 [1880‐5056] | |||
| AUC2, μV*ms | Equilibrium | 15 | 7485 [4259‐9861] | 0.0323 | 13 | 5844 [3169‐9496] | 0.3396§ |
| UCB1 | 15 | 6717 [3218‐9276] | 13 | 5052 [3151‐9210] | |||
| UCB1 | 15 | 6717 [3218‐9276] | 0.0818 | 13 | 5052 [3151‐9210] | 0.1909§ | |
| UCB2 | 15 | 7547 [4068‐9614] | 13 | 5133 [3533‐9413] | |||
| Equilibrium | 15 | 7485 [3334‐9861] | 0.4346 | 13 | 5844 [3169‐9496] | 0.3054§ | |
| Washout | 15 | 7338 [3428‐10 209] | 13 | 5576 [3044‐9468] | |||
| AUC, μV*ms | Equilibrium | 15 | 10 800 [7289‐16 526] | 0.0074 | 13 | 8992 [5406‐15 068] | 0.4973§ |
| UCB1 | 15 | 11 011 [5509‐14 717] | 13 | 7866 [4817‐15 433] | |||
| UCB1 | 15 | 11 011 [5509‐14 717] | 0.0300 | 13 | 7866 [4817‐15 433] | 0.4143§ | |
| UCB2 | 15 | 11 108 [6682‐15 825] | 13 | 8181 [5746‐15 882] | |||
| Equilibrium | 15 | 10 800 [5464‐16 526] | 0.4622 | 13 | 8992 [5406‐15 068] | 0.2734§ | |
| Washout | 15 | 11 839 [5995‐14 725] | 13 | 8473 [4898‐14 087] | |||
| t 1, ms | Equilibrium | 15 | 230 [210‐260] | 0.1437 | 14 | 210 [195‐240] | 0.5469§ |
| UCB1 | 15 | 220 [180‐260] | 14 | 215 [198‐230] | |||
| UCB1 | 15 | 220 [180‐260] | 0.0180 | 13 | 215 [198‐230] | 0.0469§ | |
| UCB2 | 15 | 240 [190‐260] | 13 | 230 [200‐260] | |||
| Equilibrium | 15 | 210 [210‐260] | 0.1263 | 13 | 210 [195‐240] | 0.8750 | |
| Washout | 15 | 220 [210‐290] | 13 | 210 [198‐248] | |||
| t 2, ms | Equilibrium | 15 | 450 [380‐530] | 0.4132 | 13 | 390 [310‐440] | 0.0207 |
| UCB1 | 15 | 470 [360‐580] | 13 | 410 [320‐490] | |||
| UCB1 | 15 | 470 [360‐580] | 0.8454 | 13 | 410 [320‐490] | 0.0301 | |
| UCB2 | 15 | 460 [360‐550] | 13 | 390 [295‐490] | |||
| Equilibrium | 15 | 460 [380‐530] | 0.7901 | 13 | 390 [310‐440] | 0.0108 | |
| Washout | 15 | 440 [310‐520] | 13 | 440 [325‐500] | |||
| t 1+2, ms | Equilibrium | 16 | 680 [560‐780] | 0.8318 | 13 | 590 [520‐685] | 0.0902 |
| UCB1 | 16 | 730 [540‐810] | 13 | 620 [520‐740] | |||
| UCB1 | 15 | 730 [540‐810] | 0.6824 | 13 | 620 [520‐740] | 0.6578 | |
| UCB2 | 15 | 700 [570‐820] | 13 | 620 [515‐720] | |||
| Equilibrium | 15 | 680 [560‐780] | 0.7000 | 13 | 590 [520‐685] | 0.0153 | |
| Washout | 15 | 680 [540‐790] | 13 | 650 [510‐735] | |||
| FWHM, ms | Equilibrium | 16 | 310 [268‐355] | 0.0591 | 14 | 270 [245‐290] | 0.2581 |
| UCB1 | 16 | 285 [263‐348] | 14 | 285 [258‐333] | |||
| UCB1 | 16 | 285 [263‐348] | 0.1370 | 14 | 285 [258‐333] | 0.7801 | |
| UCB2 | 16 | 290 [260‐358] | 14 | 280 [248‐313] | |||
| Equilibrium | 15 | 310 [260‐360] | 0.0891 | 14 | 270 [245‐290] | 0.1201 | |
| Washout | 15 | 320 [280‐380] | 14 | 285 [250‐348] | |||
| t (50%), ms | Equilibrium | 16 | 510 [445‐565] | 0.1350 | 14 | 445 [410‐470] | 0.8652§ |
| UCB1 | 16 | 490 [435‐550] | 14 | 455 [418‐510] | |||
| UCB1 | 16 | 490 [435‐550] | 0.1995 | 14 | 455 [418‐510] | 0.0225§ | |
| UCB2 | 16 | 490 [465‐558] | 14 | 425 [390‐453] | |||
| Equilibrium | 15 | 520 [440‐570] | 0.1649 | 14 | 445 [410‐470] | 0.0723§ | |
| Washout | 15 | 530 [480‐580] | 14 | 455 [435‐525] | |||
In one wild‐type experiment, air bubbles were observed after washout. Due to the signal interference, these data were not included (b‐wave amplitude, implicit time, and AUC1). Furthermore, in one wild‐type experiment and one experiment in Cav2.3‐deficient retina, b‐wave responses did not achieve the 0 μV level after light flash. The parameters AUC2, t 2, and t 1+2 could not be calculated.
FWHM, full width at half maximum; t (50%), time after the flash required for the b‐wave amplitude to decay to 50% of its maximal value. AUC, area under the curve.
§Wilcoxon test.
*Paired t test unless otherwise noted.
Bold values indicate the main parameter “b‐wave amplitude” for multiple comparisons using Holm‐Bonferroni method.
3. RESULTS
To assess the potential role of Cav2.3 channels for effects propagated by UCB, its retinal expression was confirmed by SDS‐gel electrophoresis and Western blot analysis (Figure 1). No protein was detected in the retina and neocortex of Cav2.3‐deficient mice.
ERGs recorded under scotopic conditions in response to repetitive stimulation of the isolated and superfused retina are shown in Figure 3. In sixteen independent experiments in wild‐type mice, application of 5 μmol/L UCB after equilibration with 10 μmol/L albumin significantly reduced b‐wave amplitudes by 14.9% (P < 0.05, corrected for multiple comparisons, Figures 3A and 4A,B), area under the curve (AUC1: 4032 μV*ms [2806‐5162] vs 3453 μV*ms [1619‐5306], P = 0.0110; AUC2: 7485 μV*ms [4259‐9861] vs 6717 μV*ms [3218‐9276], P < 0.05; AUC: 10 800 μV*ms [7289‐16 526] vs 11 011 μV*ms [5509‐14 717], P < 0.01), and the FWHM (310 ms [268‐355] vs 285 ms [263‐348]; P < 0.1) within approximately 18‐24 minutes.
Figure 3.
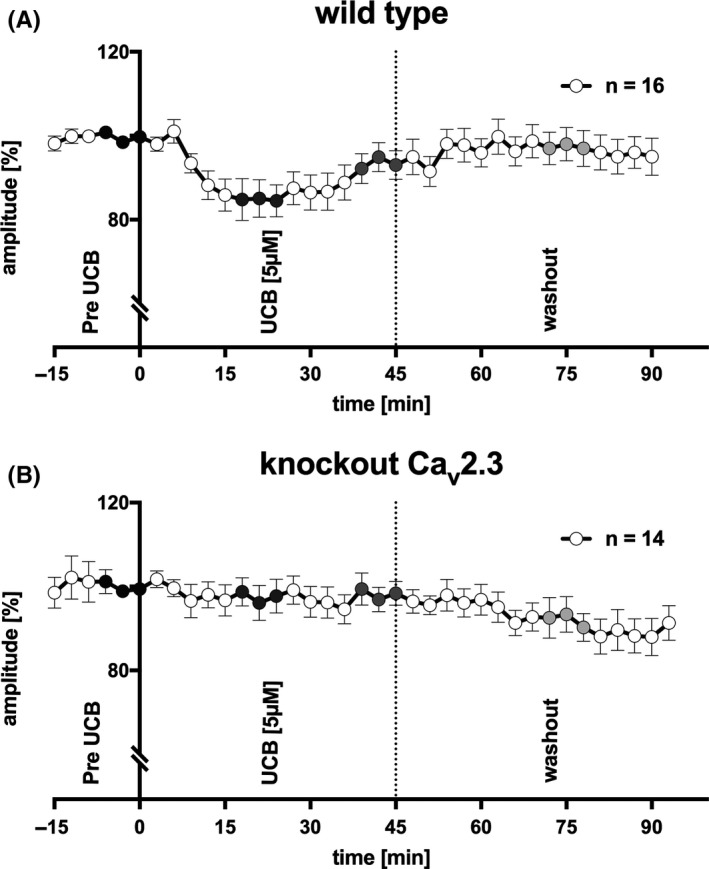
ERG recordings from superfused murine retinae. (A) B‐wave amplitude in response to repetitive light stimulation every 3 min in wild‐type mice (n = 16). UCB was only added after reaching an equilibrium of b‐wave amplitude with albumin (pre‐UCB, black circles). Superfusion with UCB 5 μmol/L lasted 45 min and was followed by a washout of the retina by albumin only. UCB significantly decreased the b‐wave amplitude after approximately 18‐24 min (P < 0.05, corrected with Holm‐Bonferroni, dark gray circles), with partial recovery toward the end of UCB superfusion (P = 0.09, corrected, gray circles). After washout, the b‐wave amplitude recovered completely (n = 16; wild type, light gray circles). (B) UCB effects were not observed in Cav2.3‐deficient mice (n = 14). After washout, the b‐wave amplitude tended to decrease (P = 0.09, corrected)
Figure 4.
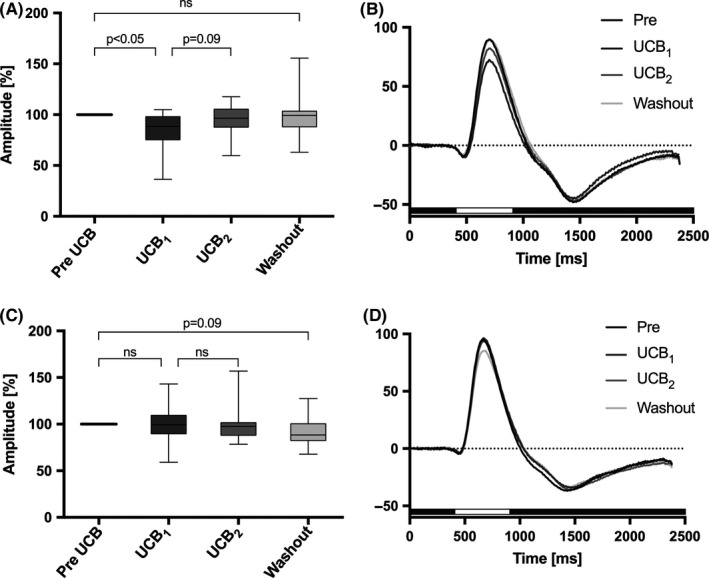
ERG responses of the isolated and superfused murine retina. Data analysis of the selected ERGs shown in Figure 3 before and after superfusion with unconjugated bilirubin (UCB) in the presence of albumin 5 μmol/L/10 μmol/L. (A) Effect of UCB on the transretinal signaling in wild‐type mice (n = 16). The b‐wave amplitude decreased significantly after 18‐24 min superfusion with UCB by 14.9% (UCB1, P < 0.05, corrected with Holm‐Bonferroni) and spontaneously increased again after 39‐45 min (UCB2, P = 0.09, corrected). After washout with albumin 10 μmol/L only, the b‐wave amplitude returned to baseline. (B) Corresponding ERG traces from the normalized data presented in panel A. Superfusion with UCB (dark gray curve) decreased the (b)‐wave after 18‐24 min, followed by partial recovery (light gray curve) after 39‐45 min. (C) Effect of UCB on the transretinal signaling in Cav2.3‐deficient mice (n = 14). The b‐wave amplitude was not affected by UCB (P = 0.8077). After washout, the b‐wave tended to decrease by 11.6% (P = 0.09, corrected), independent from UCB (Figure 2C). (D) Corresponding ERG traces for normalized data are presented in panel C. After superfusion with UCB, the b‐wave amplitude was unaffected (UCB1, dark gray curve vs UCB2, light gray curve). The duration of light stimulus was always 500 ms, and it is indicated by a white bar beyond the traces
B‐wave implicit time (ms), t 1 (ms), t 2 (ms), t 1+2 (ms), and t (50%) (ms) were comparable before and during application of UCB (P = 0.8489; P = 0.1437; P = 0.4132; P = 0.8318; P = 0.1350) (Table 1). The effect of UCB on the b‐wave amplitude was attenuated after 39‐45 minutes of continuous superfusion (UCB2, P = 0.09, after correction, Figures 3A and 4A,B; n = 14, wild type). After washout, the effect of UCB was eliminated completely (P = 0.2839).
The effect of UCB was not observed in Cav2.3‐deficient mice (UCB1, P = 0.7780; UCB2, P = 0.1770; AUC1, P = 0.7609; AUC2, P = 0.3396; AUC, P = 0.4973; FWHM, P = 0.2581), but after washout, the b‐wave amplitude tended to decrease in this genotype by 11.6% (P = 0.09, after correction, Figures 3B and 4C,D; n = 14, knockout).
Exploratory usage of albumin superfusion in knockout animals showed comparable effects to superfusion with UCB (Figure 3B), and the b‐wave amplitude spontaneously decreased by 11.7 ± 4.8% (n = 3). Control experiments in WT using albumin alone showed comparable effects on the b‐wave, 12.4 ± 3.2% (n = 3), (Figure S1).
4. DISCUSSION
The relationship between blood metabolites and hemoglobin degradation products (BMHDPs) formed in the cerebrospinal fluid and the development of vasospasm and delayed cerebral ischemia (DCI) has been the focus of several previous studies,14 but the molecular and cellular targets of BMHDPs remain to be identified.15 However, inhibition of arterial smooth muscle contraction alone did not translate into superior outcome, putting previous concepts of aneurysmal SAH treatment partially into question. DCI can occur even in the absence of angiographic vasospasm,16 supporting the hypothesis of a multifactorial cascade of adverse events after aneurysmal SAH. In the present study, we investigated the role of Cav2.3 channels as a direct target of BMHDPs.
4.1. BMHDPs after aneurysmal SAH and the relevance of VGCCs in neuronal tissue
Lysis of erythrocytes begins almost immediately after aneurysmal SAH, resulting in an increase in oxyhemoglobin (Oxy‐Hb) concentration in the CSF that peak after 24‐48 hours with gradual decrease thereafter, reflecting degradation and metabolism of hemoglobin into heme, ferrous and ferric iron, carbon monoxide, biliverdin, and bilirubin.14 Previous investigations have found bilirubin levels in CSF to be 5‐fold increase in aneurysmal SAH patients with vasospasm compared to healthy controls.17 BMHDPs can affect various pathological processes, among others, the expression of voltage‐gated calcium channels (VGCCs) such as P‐/Q‐type and R‐type calcium channels (Cav2.3 channels),6, 18 the latter previously assumed to be associated with delayed cerebral vasospasm.4 Furthermore, mediation of vasoconstriction in smaller arteries (100‐200 μm) by Cav2.3 channels is also assumed after SAH.19 VGCCs play a protective role after ischemic neuronal injury,20 and most of these channels, including R‐type Ca2+ channels,21 are also present in the neuronal network of the vertebrate retina (embryological part of the brain), where they play a major role in signal transduction and its regulation, but the final proof comparing UCB‐induced signaling changes was missing.
In the present study, we investigated the role of a single VGCC, the Cav2.3/R‐type Ca2+‐channel, for bilirubin evoked changes of signal‐transduction in a well‐defined neuronal network.22 The vertebrate retina is emerging as a suitable model system for elucidating mechanisms of neural signal processing and propagation.
Within the retinal network, VGCC can be modulated by drugs, toxins, or metabolites and can be inactivated on the gene level to deduce its function. The interneuronal network contains more than 30 different types of amacrine cells, the major inhibitory interneurons in the retina. They influence bipolar cells, ganglion cells, and other amacrine cells by releasing inhibitory neurotransmitters that activate ionotropic and metabotropic postsynaptic receptors. Roughly, half of the amacrine cells release GABA and the other half glycine.23, 24, 25, 26 These reciprocal synapses are thought to play an important role in tuning bipolar cell output to the dynamic range of ganglion cells.27, 28 Gene inactivation of Cav2.3 (R‐type) or of Cav3.2 (T‐type calcium channel) has partially elucidated its role during reciprocal inhibition in the murine retina.29 Concerning signal processing in the inner retina, the b‐wave amplitude and implicit time were taken as a measure of the response of bipolar cells. Comparing light‐evoked responses from retinas of wild‐type and Ca+2 channel‐deficient mice enables us to define the effect of drugs, toxins, and metabolites on Cav2.3‐mediated signal transduction.
4.2. UCB‐induced modulation of ex vivo retinal signaling in wild‐type and Cav2.3‐deficient mice
In ex vivo experiments, most studies favor the application of bilirubin in the presence of albumin to reduce its toxicity.30 Based on this recommendation, UCB/albumin ratio in our setup has been chosen as a molar ratio of smaller than one to achieve physiological conditions. Acute administrations of high UCB concentration without albumin cause Ca2+ overload and subsequent neuronal vulnerability primarily by targeting P/Q‐type Ca2+ channels on auditory brain regions, via Ca2+ and calmodulin‐dependent processes.18 Such high concentration of UCB was prevented in the present study.
To test the relevance of Cav2.3 channels in a complex system containing native Cav2.3 channels as well as other potential targets, we employed ERG recordings in the isolated and superfused murine retina. Using both wild‐type and Cav2.3‐deficient mice allowed us to differentiate effects mediated by Cav2.3 from effects mediated by other variables.
While the ERGs of Cav2.3‐deficient mice before and after superfusion with UCB were comparable, the b‐wave amplitudes from the Cav2.3‐competent retinas were transiently reduced during continuous bilirubin superfusion. Such a transient effect could reflect the initial slow increase during wash‐in of bilirubin concentration in the recording chamber, but may also result from activation of signaling cascades downstream of Cav2.3 channels. Thus, a high‐affinity target mediates inhibition of the b‐wave amplitude. The subsequent partial recovery of b‐wave amplitudes might conversely be attributable to astrocytes, which are capable of protecting neuronal cells against the cytotoxic effects for short‐term exposure to low concentration of UCB.31 Within 15 seconds, the initial interaction of bilirubin with a neuronal membrane seems to create other effects compared to reaching an equilibrium after 15 minutes.32 As signaling must include the R‐type channel, this inhibition must be caused by a transient activation of R‐type currents concomitant with increase in GABA and/or Glycine release on bipolar cells. In Cav2.3‐deficient mice, a decrease in the b‐wave amplitude was observed after washout of UCB, which was likely related to differences in the spontaneous decline of the b‐wave amplitude even under optimized recording conditions.10 This observation was replicated during an exploratory control experiment with albumin alone. The slow spontaneous decline of the b‐wave amplitude could reflect time‐dependent changes in retinal hemostasis due to lack of the pigment epithelium or other metabolic changes. This phenomenon was evident in both genotypes but less evident in wild‐type retinae due to residual UCB‐induced stimulation of Cav2.3 channels.
Regardless of the exact mechanisms shaping the time course of UCB effects, our findings show that—despite the presence of numerous potential targets for UCB in the retina—genetic inactivation of Cav2.3 channels is sufficient to prevent UCB effects on the b‐wave amplitude completely. The explanation of the transient effect must be related to a single target, the Cav2.3/R‐type channel complex. In addition, our results illustrate that UCB‐induced changes in Cav2.3 channel function can lead to rapid changes in normal neurotransmission. Therefore, the involvement of Cav2.3/R‐type VGCCs could represent a potential target for early interventions, aimed at ameliorating the response to blood degradation products mediated by these channels.33 The ratio of U/A in our study [5/10 μmol/L] seems to be physiological, but we cannot exclude the breaking and disturbance of the membrane architecture and cytolysis in the phospholipid bilayer.34 Maybe, the fast and transient inhibition of the b‐wave amplitude was caused by a direct high‐affinity binding of UCB to the R‐type channel protein, and phosphobilayer disturbance initiated the later relief, but specific to the R‐type calcium channel.
4.3. Bilirubin and the toxicity of its oxidation products
Catalytically, hemoglobin is converted by heme oxygenases into ferrous iron, carbon monoxide, and biliverdin, which is subsequently reduced by biliverdin reductases to bilirubin. Nonenzymatically, heme, biliverdin, and bilirubin are degraded in the presence of reactive oxygen species to a series of products, including the monopyrroles Z‐BOX A and Z‐BOX B as well as other bilirubin oxidation end products (BOX).35 BOXes have been shown to inhibit a voltage‐ and Ca2+‐dependent K+ channel (Slo1 BK channel), which was proposed to contribute to the development of delayed cerebral vasospasm after aneurysmal SAH.36 Investigation of bilirubin is hampered by its low water solubility and possible oxidation. We have controlled UCB stability by recording absorption spectra before and after applying on the retina, and we did not observe any changes in the spectrum from 200 to 800 nm.
SNX‐482‐sensitive voltage‐gated Cav2.3/R‐type Ca2+ channel in rats is upregulated after experimental SAH, as shown at the transcript level by semiquantitative RT‐PCR.4 Consequently, Cav2.3/R‐type Ca2+ channels were proposed as a novel therapeutic target for the treatment of aSAH‐induced cerebral vasospasm.19 Studies have shown that bilirubin is produced in a time course parallel to the development of vasospasm. However, contraction of smooth muscle due to bilirubin could be shown neither in vitro nor in vivo,37 possibly reflecting the lack of Cav2.3 channel expression in healthy arteries.
We have found that UCB at a physiological U/A molar ratio of 0.5 significantly alters neurotransmission in the isolated and superfused retina from wild‐type but not Cav2.3‐deficient mice, indicating an acute and early action mediated by selective modulation of Cav2.3 channels.
5. CONCLUSIONS
The ex vivo neuronal setup of the isolated and superfused murine retina demonstrates a transient effect of bilirubin on neuronal signaling only when Cav2.3/R‐type channels are present. Modulation of Cav2.3/R‐type Ca2+ channels may contribute to the pathophysiological cascades of vasospasm or DCI. The described model may facilitate further investigation of BMHDPs on calcium channels to determine their exact role in aSAH.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We would like to especially thank Mrs. Renate Clemens for her dedication and hard work in this project.
Albanna W, Neumaier F, Lüke JN, et al. Unconjugated bilirubin modulates neuronal signaling only in wild‐type mice, but not after ablation of the R‐type/Cav2.3 voltage‐gated calcium channel. CNS Neurosci Ther. 2018;24:222–230. 10.1111/cns.12791
Funding information
This study was funded by the START program of the Faculty of Medicine, RWTH Aachen.
Part of this study was presented at the 68th Annual Meeting of the German Society of Neurosurgery (DGNC) 2017, Magdeburg, Germany.
Walid Albanna, Felix Neumaier, Toni Schneider & Gerrit Alexander Schubert contributed equally to this work*.
*[Correction added on 01 February 2018, after first online publication: The corresponding author would like to acknowledge that Walid Albanna, Felix Neumaier, Toni Schneider & Gerrit Alexander Schubert contributed equally to this work.]
REFERENCES
- 1. Lazniewska J, Weiss N. Glycosylation of voltage‐gated calcium channels in health and disease. Biochim Biophys Acta. 2017;1859:662‐668. [DOI] [PubMed] [Google Scholar]
- 2. Ricoy UM, Frerking ME. Distinct roles for Cav 2.1‐2.3 in activity‐dependent synaptic dynamics. J Neurophysiol. 2014;111:2404‐2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wormuth C, Lundt A, Henseler C, et al. Review: Cav2.3 R‐type voltage‐gated Ca2 + channels ‐ functional implications in convulsive and non‐convulsive seizure activity. Open Neurol J. 2016;10:99‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ishiguro M, Wellman TL, Honda A, Russell SR, Tranmer BI, Wellman GC. Emergence of a R‐type Ca2+ channel (CaV 2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ Res. 2005;96:419‐426. [DOI] [PubMed] [Google Scholar]
- 5. Wang Z, Wang KY, Wu Y, Zhou P, Sun XO, Chen G. Potential role of CD34 in cerebral vasospasm after experimental subarachnoid hemorrhage in rats. Cytokine. 2010;52:245‐251. [DOI] [PubMed] [Google Scholar]
- 6. Link TE, Murakami K, Beem‐Miller M, Tranmer BI, Wellman GC. Oxyhemoglobin‐induced expression of R‐type Ca2+ channels in cerebral arteries. Stroke. 2008;39:2122‐2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clark JF, Sharp FR. Bilirubin oxidation products (BOXes) and their role in cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26:1223‐1233. [DOI] [PubMed] [Google Scholar]
- 8. Joerk A, Seidel RA, Walter SG, et al. Impact of heme and heme degradation products on vascular diameter in mouse visual cortex. J Am Heart Assoc. 2014;3:e001220 10.1161/JAHA.114.001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pereverzev A, Mikhna M, Vajna R, et al. Disturbances in glucose‐tolerance, insulin‐release, and stress‐induced hyperglycemia upon disruption of the Ca(v)2.3 (alpha 1E) subunit of voltage‐gated Ca(2+) channels. Mol Endocrinol. 2002;16:884‐895. [DOI] [PubMed] [Google Scholar]
- 10. Albanna W, Lueke JN, Sjapic V, et al. Electroretinographic assessment of inner retinal signaling in the isolated and superfused murine retina. Curr Eye Res. 2017;42:1‐9. [DOI] [PubMed] [Google Scholar]
- 11. Albanna W, Banat M, Albanna N, et al. Longer lasting electroretinographic recordings from the isolated and superfused murine retina. Graefes Arch Clin Exp Ophthalmol. 2009;247:1339‐1352. [DOI] [PubMed] [Google Scholar]
- 12. Wisniewski RJ. Protocol to enrich and analyze plasma membrane proteins. Methods Mol Biol. 2009;528:127‐134. [DOI] [PubMed] [Google Scholar]
- 13. Pereverzev A, Klockner U, Henry M, et al. Structural diversity of the voltage‐dependent Ca2+ channel alpha1E‐subunit. Eur J Neurosci. 1998;10:916‐925. [DOI] [PubMed] [Google Scholar]
- 14. Ishiguro M, Murakami K, Link T, et al. Acute and chronic effects of oxyhemoglobin on voltage‐dependent ion channels in cerebral arteries. Acta Neurochir Suppl. 2008;104:99‐102. [DOI] [PubMed] [Google Scholar]
- 15. Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22:971‐982. [DOI] [PubMed] [Google Scholar]
- 16. Woitzik J, Dreier JP, Hecht N, et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2012;32:203‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pyne‐Geithman GJ, Morgan CJ, Wagner K, et al. Bilirubin production and oxidation in CSF of patients with cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:1070‐1077. [DOI] [PubMed] [Google Scholar]
- 18. Liang M, Yin XL, Shi HB, et al. Bilirubin augments Ca2+ load of developing bushy neurons by targeting specific subtype of voltage‐gated calcium channels. Sci Rep. 2017;7:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishiguro M, Wellman GC. Cellular basis of vasospasm: role of small diameter arteries and voltage‐dependent Ca2+ channels. Acta Neurochir Suppl. 2008;104:95‐98. [DOI] [PubMed] [Google Scholar]
- 20. Toriyama H, Wang L, Saegusa H, et al. Role of Ca(v) 2.3 (alpha1E) Ca2+ channel in ischemic neuronal injury. NeuroReport. 2002;13:261‐265. [DOI] [PubMed] [Google Scholar]
- 21. Xu HP, Zhao JW, Yang XL. Expression of voltage‐dependent calcium channel subunits in the rat retina. Neurosci Lett. 2002;329:297‐300. [DOI] [PubMed] [Google Scholar]
- 22. Alnawaiseh M, Albanna W, Banat M, Abumuaileq R, Hescheler J, Schneider T. Electroretinographic recordings from the isolated and superfused murine retina In Belulsic G, ed. Electroretinograms. Croatia: InTech; 2011. ISBN 978‐953‐307‐383‐5. [Google Scholar]
- 23. Diamond JS. Inhibitory interneurons in the retina: types, circuitry, and function. Annu Rev Vis Sci. 2017;3:1‐24. [DOI] [PubMed] [Google Scholar]
- 24. Diamond JS, Lukasiewicz PD. Amacrine cells: seeing the forest and the trees. Vis Neurosci. 2012;29:1‐2. [DOI] [PubMed] [Google Scholar]
- 25. Chavez AE, Grimes WN, Diamond JS. Mechanisms underlying lateral GABAergic feedback onto rod bipolar cells in rat retina. J Neurosci. 2010;30:2330‐2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siapich SA, Banat M, Albanna W, Hescheler J, Luke M, Schneider T. Antagonists of ionotropic gamma‐aminobutyric acid receptors impair the NiCl2‐mediated stimulation of the electroretinogram b‐wave amplitude from the isolated superfused vertebrate retina. Acta Ophthalmol. 2009;87:854‐865. [DOI] [PubMed] [Google Scholar]
- 27. Hartveit E. Reciprocal synaptic interactions between rod bipolar cells and amacrine cells in the rat retina. J Neurophysiol. 1999;81:2923‐2936. [DOI] [PubMed] [Google Scholar]
- 28. Vigh J, von Gersdorff H. Prolonged reciprocal signaling via NMDA and GABA receptors at a retinal ribbon synapse. J Neurosci. 2005;25:11412‐11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alnawaiseh M, Albanna W, Chen CC, et al. Two separate Ni(2+) ‐sensitive voltage‐gated Ca(2+) channels modulate transretinal signalling in the isolated murine retina. Acta Ophthalmol. 2011b;89:e579‐e590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hulzebos CV, Dijk PH. Bilirubin‐albumin binding, bilirubin/albumin ratios, and free bilirubin levels: where do we stand? Semin Perinatol. 2014;38:412‐421. [DOI] [PubMed] [Google Scholar]
- 31. Chuniaud L, Dessante M, Chantoux F, Blondeau JP, Francon J, Trivin F. Cytotoxicity of bilirubin for human fibroblasts and rat astrocytes in culture. Effect of the ratio of bilirubin to serum albumin. Clin Chim Acta. 1996;256:103‐114. [DOI] [PubMed] [Google Scholar]
- 32. Vazquez J, Garcia‐Calvo M, Valdivieso F, Mayor F, Mayor F Jr. Interaction of bilirubin with the synaptosomal plasma membrane. J Biol Chem. 1988;263:1255‐1265. [PubMed] [Google Scholar]
- 33. Kim KJ, Ramiro Diaz J, Iddings JA, Filosa JA. Vasculo‐neuronal coupling: retrograde vascular communication to brain neurons. J Neurosci. 2016;36:12624‐12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Noy N, Leonard M, Zakim D. The kinetics of interactions of bilirubin with lipid bilayers and with serum albumin. Biophys Chem. 1992;42:177‐188. [DOI] [PubMed] [Google Scholar]
- 35. Ritter M, Seidel RA, Bellstedt P, et al. Isolation and Identification of Intermediates of the Oxidative Bilirubin Degradation. Org Lett. 2016;18:4432‐4435. [DOI] [PubMed] [Google Scholar]
- 36. Hou S, Xu R, Clark JF, Wurster WL, Heinemann SH, Hoshi T. Bilirubin oxidation end products directly alter K+ channels important in the regulation of vascular tone. J Cereb Blood Flow Metab. 2011;31:102‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duff TA, Feilbach JA, Yusuf Q, Scott G. Bilirubin and the induction of intracranial arterial spasm. J Neurosurg. 1988;69:593‐598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials